

Abstract
Obesity and type 2 diabetes mellitus (T2DM) — disorders of energy homeostasis and glucose homeostasis, respectively — are tightly linked and the incidences of both conditions are increasing in parallel. The CNS integrates information regarding peripheral nutrient and hormonal changes and processes this information to regulate energy homeostasis. Recent findings indicate that some of the neural circuits and mechanisms underlying energy balance are also essential for the regulation of glucose homeostasis. We propose that disruption of these overlapping pathways links the metabolic disturbances associated with obesity and T2DM. A better understanding of these converging mechanisms may lead to therapeutic strategies that target both T2DM and obesity.
Homeostasis — the maintenance of optimal internal conditions1 — is achieved through a complex set of physiological and behavioural responses to external and internal stimuli. Body temperature, blood pressure, and nutrient and energy levels all have precise homeo-static ranges. In other words, when the internal milieu is challenged, physiological responses are initiated in order to defend the homeostatic range.
The concept of energy homeostasis refers to the combined processes that manage energy intake, storage and usage to maintain stable levels of stored fuel in the form of adiposity and to enable adequate access to stored energy during times of limited food access, enhanced energy intake and storage when food is available, and appropriate metering of fuels to tissues at all times. Glucose is a specific type of energy, and the term glucose homeostasis refers to the hormonal and neural regulatory elements that specifically control glucose production and use. Glucose homeostasis maintains plasma glucose levels within a relatively small range (70–110 mg per dL), even in the face of physiological challenges, including meal ingestion, fasting and intense exercise. Generally, these two homeostatic systems have the same goal, namely to ensure adequate nutrient flow to the tissues. The balance within each system is maintained by matching the provision of fuel to the body (through food intake and glucose appearance in the blood, respectively) with fuel consumption (through energy expenditure and glucose disappearance from the blood, respectively).
Obesity is a progressive metabolic disorder of energy homeostasis, and type 2 diabetes mellitus (T2DM) is a progressive metabolic disorder of glucose homeostasis. They are physiologically linked and are respectively associated with increased levels of adiposity and glucose that are actively maintained and defended. This link is illustrated by the facts that the incidences of both conditions are increasing in parallel and that weight loss lowers plasma glucose2. In one view, obesity and T2DM are the result of dysfunctional homeostatic systems that cannot constrain adiposity and blood glucose to the normal range. In another view, obesity and T2DM result from homeostatic systems that are functioning well but that have inappropriately high ‘set-points’ that are dictated by an environment characterized by calorically dense foods and reduced exercise. Regardless of which view one takes, it is clear that a combination of increased caloric consumption and/or decreased activity, on a background of other environmental and genetic factors, results in obesity and hyperglycaemia — conditions that are defended over time.
Historically, the CNS has been considered to be the major regulator of body adiposity. This is because the evolution of our understanding of obesity has been heavily driven by the discovery of leptin and its actions in the CNS3,4. By contrast, the regulation of plasma glucose was generally considered to be a peripheral process driven by insulin-induced changes in liver and skeletal muscle glucose fluxes; it was thought that the brain only becomes involved in glucose regulation in situations of dangerously low glucose levels. However, over the past few years, this view has changed, and accumulating data suggest that the two systems interact at the level of the CNS. Recent findings, which are reviewed in this article, indicate that neuronal populations in the hypothalamus that had already been identified as being crucial for the regulation of energy balance are also essential for the regulation of glucose homeostasis. A key outstanding question is whether the neuronal circuits that are crucial for body weight regulation, and that may be dysregulated in obesity, also contribute to the poor glucose homeostasis that eventually results in T2DM.
In this Review, we first discuss the role of the neurocircuitry within the arcuate nucleus (ARC) of the hypothalamus in the regulation of both energy homeostasis and glucose homeostasis. We then focus on neuronal circuitries outside the ARC, including in the ventromedial hypothalamus (VMH), the hindbrain and the neuronal connection between the gut and the CNS, and consider whether they regulate solely glucose homeostasis or whether they regulate both glucose and energy homeostasis. Last, we discuss new methodologies that will have an important impact on our understanding of the CNS regulation of glucose homeostasis, and the clinical implications of this understanding.
CNS control of energy balance
In order to maintain energy balance, the CNS receives hormonal signals (such as insulin and leptin) that reflect the availability of long-term energy stores in adipose tissue and nutrient signals that reflect acute nutrient availability. These signals are processed in numerous brain nuclei, the best studied of which is the melanocortin system. This system is comprised of two peptide-expressing populations of neurons in the ARC and their downstream targets. One set of ARC neurons expresses the precursor peptide pro-opiomelanocortin (POMC), which is processed into the melanocortin receptor 4 (MC4R) agonist α-melanocyte-stimulating hormone, and the other expresses neuropeptide Y (NPY) and the MC4R antagonist/inverse agonist agouti-related protein (AGRP). These two populations of neurons have opposite effects on food intake and body weight: activation of POMC neurons results in decreased food intake and weight loss, whereas activation of NPY–AGRP neurons results in increased food intake and weight gain5. The downstream effects of activation of these neurons are mediated by MC4Rs and NPY receptors located in many other hypothalamic locations including, but not limited to, the paraventricular nucleus (PVN), VMH and the lateral hypothalamic area5,6. There is continuous crosstalk between the two neuronal populations in the ARC, and this includes the ability of NPY–AGRP neurons to suppress activity in POMC neurons through GABA input7.
Many nutrients and hormones act on POMC and NPY–AGRP neurons. Insulin and leptin have been hypothesized to act as ‘adiposity signals’, as their plasma levels are in direct proportion to the amount of stored fuel in adipose tissue8. Insulin receptors (IRs)9 and leptin receptors (OBRs; also known as LEPRs)10,11 are expressed throughout the hypothalamus, including on POMC and NPY–AGRP neurons in the ARC12,13, and are necessary for the normal regulation of body weight14. Indeed, a dose-dependent injection of either hormone into the third ventricle adjacent to the ARC in rodents suppresses food intake and reduces body weight7,15–18. The catabolic actions of insulin and leptin are mediated in part by increased expression of POMC and simultaneously reduced expression of NPY and AGRP19. Some amino acids (for example, leucine)20, glucose and fatty acids21,22 have been shown to regulate the activity of these neurons as well (see below). Thus, POMC and NPY–AGRP neurons in the ARC have an important role in energy homeostasis.
Given that these hypothalamic circuits are integral to the regulation of energy homeostasis, it is tempting to hypothesize that dysregulation of these circuits may underlie the increased set-point of body weight that is observed in obesity. Consistent with this, genetic loss-of-function mutations of the melanocortin system lead to obesity in animal models and humans23. However, given the rarity of these mutations in humans, the more salient question is whether dysfunction of these circuits is responsible for the greatly increased incidence of obesity that is occurring in the contemporary obesigenic environment. Exposing genetically normal animals to calorically dense, palatable high-fat diets results in increased body fat and a reduced ability of leptin and insulin to inhibit NPY–AGRP neurons and activate POMC neurons17,24. This suggests that an altered balance of activity in these two populations of ARC neurons contributes to the maintenance of a higher body weight in obesity25.
CNS regulation of glucose homeostasis
Before the discovery of insulin, the general assumption was that the CNS has a role in the control of glucose homeostasis. However, the finding that the removal of the pancreas produced diabetes led people to deduce that factors secreted from the pancreas must regulate blood glucose levels26. After the discovery of insulin, and later glucagon27,28, the focus of the field shifted to peripheral organs (specifically, the pancreas, liver and skeletal muscle) as having an exclusive role in the control of glucose homeostasis (BOX 1). In this model, peripheral organs execute precise regulation of postprandial changes in glucose levels, whereas the CNS (specifically, the autonomic nervous system) only responds to dangerously falling glucose levels, such as during prolonged fasting, insulin-induced hypoglycaemia or exercise. In the past decade, the working model of glucose homeostasis regulation has again incorporated an important role for the CNS through the whole range of glucose levels, from postprandial hyperglycaemia to hypoglycaemia.
Box 1. Peripheral control of glucose homeostasis.
After meal ingestion, nutrients are digested by the gastrointestinal tract and absorbed into the circulation via the hepatic portal vein. Thus, the liver is anatomically positioned to monitor ingested nutrients and has a pivotal role in regulating postprandial glucose levels by matching glucose appearance into the blood from meal ingestions (indicated by dashed arrows in the figure) to tissue glucose usage (see the figure). Ingested nutrients also act directly on intestinal endocrine cells to stimulate the release of incretins, which — together with the rise in blood glucose — stimulate pancreatic β-cells to release insulin. Insulin secretion is biphasic: the first phase occurs early after meal ingestion and is thought to suppress hepatic glucose production (HGP). The second plateaus within 1–2 h after the meal and stimulates glucose uptake by insulin-sensitive tissues (that is, skeletal muscle and adipose tissue)109. Insulin also indirectly regulates HGP by inhibiting the release of glucagon from pancreatic α-cells. This change in insulin and glucagon levels potently restrains postprandial glucose excursions.
The metabolic processes that directly contribute to HGP are glycogenolysis and gluconeogenesis. The balance between these processes varies with nutrient status and needs. For example, during fasting, hepatic glycogenolysis decreases as hepatic glycogen stores deplete. Subsequently, gluconeogenesis increases, fuelled by the delivery of glucose precursors to the liver. Hormonal (insulin and glucagon) and autonomic neural signals regulate HGP and tissue glucose uptake, such that blood glucose levels remain stable across a wide range of physiological conditions.
The hepatic portal vein is richly innervated with visceral sensory axons that express hormone and nutrient sensors (for example, glucose transporters and/or glucagon-like peptide 1 receptors). Portal glucose delivery increases both glucose uptake by the liver and peripheral glucose uptake through a neural mechanism109, as demonstrated by the fact that ligation of the hepatic branch of the vagal nerve blocks the effect of portal glucose levels on glucose production and uptake110. Thus, although the hormonal regulation of glucose levels is important (see the figure), the nervous system is positioned to sense and respond to acute changes in glucose and nutrient needs through innervation of the gut, the pancreas, the portal vein, the liver and all other glucose-demanding tissues.
Glucose homeostasis in type 2 diabetes mellitus
Type 2 diabetes mellitus (T2DM) results from a combination of insulin resistance and inadequate insulin secretion (see the figure). In the early stages of the disease, the first-phase insulin response is lost and the second-phase insulin response is raised for a given glucose load compared with non-diabetic controls. Thus, glucose tolerance is maintained by increased insulin levels that compensate for peripheral insulin resistance.
Furthermore, the ability of insulin to suppress HGP is reduced (a phenomenon known as hepatic insulin resistance)111. Eventually, pancreatic β-cells lose the ability to compensate by increasing insulin release, and the resulting reduced insulin levels often occur in parallel with increased glucagon levels. This further shifts the balance between glucagon and insulin levels, leading to further to increases in HGP112, specifically through increases in gluconeogenesis. Thus, changes in both glucose production and tissue glucose uptake in T2DM lead to a chronic maintenance of raised basal and postprandial glucose levels.

Many approaches have been used to study how the CNS regulates glucose and energy homeostasis. These include direct injection of substances (such as peptides, nutrients and neurotransmitters) into the brain as well as the use of genetic models in which receptors or key intracellular signalling molecules are deleted from specific neurons in otherwise normal animals or re-introduced in animals that lack these receptors or signalling molecules. With these latter models, it has become possible to assess whether specific receptors or signalling pathways are necessary and sufficient for CNS control of glucose and energy balance. A question of interest for this Review is whether chronic manipulation of one signalling component leads to simultaneous changes in body weight and glucose homeostasis. However, an interpretative problem arises here because obesity itself causes impairments in glucose handling in peripheral organs, and this makes it difficult to isolate the potential role of such a manipulation in glucose homeostasis independent of its role in energy balance. This underscores the importance of experimental designs that can differentiate between the role of the CNS in energy homeostasis versus its role in glucose homeostasis.
Glucose regulation by the ARC
Given that the melanocortin system within the ARC has an important role in the regulation of energy homeostasis, it is perhaps not surprising that this system also turns out to be important in regulating glucose homeostasis. In this section, we discuss the evidence that particular signals — insulin, leptin, nutrients and 5-hydroxy-tryptamine (5-HT; also known as serotonin) — act within the hypothalamus, in many cases specifically in the ARC, to regulate glucose homeostasis. We also consider the role of hypothalamic melanocortin receptors in these processes.
Insulin and leptin
Activating CNS IRs or OBRs at doses that decrease food intake and increase energy expenditure via modulation of the melanocortin system also modulates glucose flux. For example, acute administration of insulin into the ARC suppresses hepatic glucose production by reducing both gluconeogenesis and glycogenolysis29. Leptin administration, by contrast, increases hepatic gluconeogenesis and decreases glycogenolysis, leaving overall hepatic glucose production similar to that of saline-treated animals30. Thus, although insulin and leptin acting within the hypothalamus have similar effects on food intake and energy expenditure7,15–18, they have different effects on hepatic glucose production (FIG. 1).
Figure 1. Leptin and insulin actions in the ARC.

The arcuate nucleus (ARC) of the hypothalamus contains two main sets of neurons that either express the neuropeptide pro-opiomelanocortin (POMC) or co-express agouti-related protein (AGRP) and neuropeptide Y (NPY). Both sets of neurons express leptin receptors (OBRs) and insulin receptors (IRs). Leptin acts on POMC neurons, and the effect of these actions on glucose production is partially mediated by melanocortin receptor 4 (MC4R) neurons in extra-arcuate sites (for example, the paraventricular nucleus). Insulin acts on NPY–AGRP neurons and at least partially on POMC neurons to regulate hepatic glucose production; this effect is independent of MC4Rs. Leptin and insulin actions on POMC neurons have additive effects on the regulation of hepatic glucose production, possibly because they act on different POMC populations within the ARC (not shown). In addition, insulin actions on neuronal populations of currently unknown phenotype result in the activation of ATP-sensitive potassium (KATP) channels, mammalian target of rapapmycin (mTOR) and peroxisome proliferator-activated receptor-γ (PPARγ) to regulate hepatic glucose production. Leptin acts on PPARγ to regulate food intake, but it is unknown whether this pathway also regulates glucose homeostasis. Thus, leptin and insulin signalling in the ARC use both divergent and overlapping circuits and mechanisms.
Administration of exogenous insulin or leptin into the ARC does not necessarily reveal how endogenous activation of IRs and OBRs in this hypothalamic nucleus might normally regulate glucose homeostasis. To address this, loss-of-function experiments have been used to assess how an absence of these receptors affects glucose regulation. An intraventricular injection of an antisense oligodeoxynucleotide that knocks down IR expression in all ARC cell populations reached by the oligodeoxynucleotide increased hepatic glucose production31. This effect was independent of food intake, as it occurred even when the injected animals were pair-fed to the control animals31. This manipulation also induced an increase in NPY and AGRP mRNA levels in the ARC, and is consistent with previous data demonstrating that insulin-induced anorexia is dependent on the melanocortin system32, which may suggest that the effect of IR knockdown on glucose production was mediated by the melanocortin system. However, administration of a melanocortin receptor antagonist into the third ventricle did not block the ability of central insulin to reduce hepatic glucose production29. This suggested that the effect of centrally administered insulin on glucose homeostasis is not mediated by melanocortin receptors and, therefore, that distinct pathways mediate the regulation of energy homeostasis and glucose homeostasis by insulin.
However, more recent experiments using genetic mouse models suggest a more complex system. Studies in mice in which IRs were inactivated in either POMC or AGRP neurons indicated that these receptors are not required for the maintenance of body weight or control of food intake33, which is in contrast to the findings discussed in the paragraph above. However, in mice in which IRs had been removed from AGRP neurons, insulin was unable to suppress hepatic glucose production during a hyperinsulinaemic–euglycaemic clamp33 (see BOX 2 for a review of techniques that are used to measure glucose homeostasis). Importantly, selective reactivation of IRs on AGRP neurons restored normal hepatic glucose production33. Together, these two findings indicate that IRs on AGRP neurons are both necessary and sufficient for the central regulation of hepatic glucose production. Interestingly, inactivation of IRs on POMC neurons in mice had no effect on hepatic insulin sensitivity33, whereas IR re-activation specifically in POMC neurons in these mice increased both energy expenditure and hepatic glucose production34. This is contrary to the assumption that insulin’s catabolic actions involve increased energy expenditure and decreased hepatic glucose production. It is possible that the overall increased metabolism in POMC IR knock-in mice may have caused an increased flux of nutrients to the liver, resulting in increases in gluconeogenesis that contributed to an overall increase in glucose production.
Box 2. Measuring glucose homeostasis.
Several physiological processes are involved in the regulation of glucose homeostasis, including glucose tolerance, insulin sensitivity and insulin secretion. Here we briefly highlight techniques that have been used to assess these processes.
Glucose tolerance
A glucose tolerance test (GTT) measures the speed with which glucose is cleared from the blood. The route of glucose administration in the GTT has important implications. A bolus of glucose injected into the intraperitoneal cavity moves quickly into the vascular space and drains into the portal vein rather than passing through the gastrointestinal tract, thus circumventing gut responses. By contrast, glucose administered orally is subject to the processes that regulate glucose levels under normal conditions. The result of a GTT does not indicate whether altered insulin secretion or altered hepatic and/or skeletal muscle insulin sensitivity underlie altered glucose tolerance. Nevertheless, GTTs provide a general assessment of glucose homeostasis and are easy to perform, even in mice.
Insulin sensitivity
Insulin sensitivity can be measured in several ways. Increased overnight fasting glucose or insulin levels can be used, by themselves or incorporated into mathematical models, to estimate insulin sensitivity. In humans, overnight fasting establishes steady-state metabolic conditions but, in rodents, it reflects a nutrient-deprived, stress condition. Insulin sensitivity can also be assessed using insulin tolerance tests. However, interpretative problems arise when animals become hypoglycaemic after the insulin injection, because hypoglycaemia initiates endocrine and neural responses that independently cause insulin resistance in an attempt to restore glucose levels. This limits the use of this test in the manipulation of CNS circuits that influence both counter-regulation and insulin sensitivity. The gold standard method for measuring insulin sensitivity is the insulin clamp. In this technique, a primed continuous intravenous infusion of insulin is given concurrently with exogenous glucose aimed at maintaining basal glucose levels, so that glucose and insulin levels are similar across experimental groups. In these conditions, the overall amount of exogenous glucose infusion reflects whole-body insulin sensitivity. For example, individuals with high insulin sensitivity will need a high rate of exogenous glucose to maintain plasma glucose levels, whereas it will be low in individuals who are relatively insulin resistant. If the test includes infusion of radio- or stable-labelled glucose isotopes, it can assess glucose turnover in the liver and skeletal muscle. A lot of information can be obtained from this test, but it is certainly a pharmacologic tool. Studies should use a combination of tools in order to understand the overall implications of CNS manipulations on the system of interest.
Insulin secretion
Insulin secretion can be assessed with an intravenous GTT, in which a small glucose load is given intravenously, allowing for glucose removal from the circulation to be dictated via the mass action of glucose rather than by insulin. Blood is sampled repeatedly to measure changes in plasma insulin levels. An alternative version of this test involves a hyperglycaemic clamp. Here, a primed, continuous intravenous infusion of glucose is given to cause similarly raised glucose levels across experimental groups, and blood is sampled repeatedly to examine the insulin response to the hyperglycaemia. The hyperglycaemic clamp gives an indication of both early and late insulin responses to a glucose load.
To summarize, the involvement of the melanocortin system in insulin-induced regulation of glucose homeostasis is more complex than the involvement of this system in insulin-induced regulation of energy balance. IR signalling, specifically in AGRP neurons, is required for the regulation of hepatic glucose production but is not necessary for the regulation of body weight or food intake, whereas the role of IR signalling in POMC neurons remains somewhat unclear because of its confounding effect on energy expenditure.
Similar experimental approaches have been used to study the role of central OBRs in the regulation of glucose homeostasis. Leptin administered in the third ventricle increases hepatic gluconeogenesis and decreases glycogenolysis35. General pharmacological antagonism of melanocortin receptors by SHU9119 blocks the increased hepatic gluconeogenesis but not the decreased glycogenolysis36, suggesting that leptin’s action on regulating hepatic glucose fluxes is partly mediated by the melanocortin system. In addition, in OBR-deficient Koletsky rats, which have reduced hepatic insulin sensitivity, virally induced re-expression of OBRs specifically in the ARC restored hepatic insulin sensitivity37. Mice in which the OBR was selectively deleted from POMC neurons had a modest increase in body weight and adiposity38 and impaired whole-body glucose tolerance39 that could not be dissociated from the effect of increased adiposity on glucose tolerance. However, OBR re-activation in POMC neurons in OBR-null mouse normalized hepatic glucose production at a time point when their body weight was similar to that of whole-body OBR-null mice40, indicating that OBRs on POMC neurons indeed regulate glucose homeostasis independently of body weight. These data illustrate that leptin acts on the melanocortin system — specifically, the POMC neurons — to regulate both glucose and energy homeostasis.
As insulin and leptin (at least partially) act on the melanocortin system to regulate energy balance and glucose homeostasis, one could speculate that their actions on these neurons would lead to additive or at least interacting effects on regulating glucose homeostasis. This has been assessed in transgenic mice in which OBRs, IRs or both were deleted from POMC neurons. Deletion of OBRs on POMC neurons resulted in obesity and, counterintuitively, this effect was partially prevented when IRs were additionally removed from these neurons41. By contrast, mice lacking both receptors on POMC neurons had a greater impairment in insulin tolerance than mice lacking only OBRs (at a time point when body weights of the two types of mice were similar). In addition, only the double-knockout mice were glucose intolerant and showed hepatic insulin resistance relative to control littermates41. These data indicate that insulin and leptin signalling have opposite effects on POMC neuronal-induced regulation of body weight but additive effects on glucose homeostasis. As IRs and OBRs are not colocalized to the same POMC neurons42, the crosstalk between insulin and leptin signalling probably occurs between, rather than within, POMC neurons. The fact that insulin and leptin act on different populations of POMC neurons could explain why some effects of activation of the respective receptors have divergent effects on energy homeostasis but additive effects on glucose homeostasis. FIGURE 1 summarizes how insulin and leptin signalling in the ARC influences hepatic glucose production by acting on NPY–AGRP and POMC neurons. Importantly, MC4Rs are involved in leptin-induced but not insulin-induced regulation of glucose homeostasis.
Fuel-sensing
Circulating nutrients (specifically, glucose, the branched chain amino acid leucine and fatty acids) convey short-term fuel availability to the CNS and are thought to act within the hypothalamus to influence food intake by altering peptide expression and/or neuronal activity14,20–22. Indeed, when studied ex vivo, specific neurons within hypothalamic nuclei (such as the ARC and VMH) and the hindbrain (the nucleus of the solitary tract (NTS)) can be excited or inhibited by increasing levels of glucose43 and lipids44. Because these nutrients are ubiquitously used by cells for fuel, it is far more complicated to determine their role as signalling molecules than to study the signalling role of, for instance, a peptide. Nonetheless, the signalling capabilities of nutrients in regulating glucose homeostasis have been investigated by injecting these nutrients into the ARC. For example, in rodents, administration of glucose, lactate or long-chain fatty acids directly into the ARC potently inhibits total hepatic glucose production by reducing both gluconeo-genesis and glycogenolysis45,46. Administering glucose or fatty acids directly into the CNS does not reliably influence food intake. It is unknown why this is the case but it may be related to the fact that nutrients do not just function as signalling molecules but are also used by neurons as a fuel to maintain cellular energy requirements43. By contrast, the amino acid leucine regulates both food intake20 (by activating the mammalian target of rapamycin (mTOR) pathway (see below)) and hepatic glucose production47 (via metabolism of the amino acid by neurons in the ARC).
Another approach used to understand the role of nutrient-sensing in the CNS is to deprive animals of a specific macronutrient and investigate its effects on energy and glucose metabolism. Such studies have shown that fuel deprivation (for example, hypoglycaemia) activates the autonomic nervous system and subsequently has a profound effect on increasing food intake and stimulating hepatic glucose production43. Thus, as fuel deprivation is a physiological stress, it is difficult to distinguish the effect of the absence of a specific type of fuel on neuronal-induced regulation of glucose homeostasis from the wide array of stress responses caused by said absence.
Nutrients use diverse and sometimes overlapping receptors and signalling pathways, including ATP signalling pathways. ATP is produced through the metabolism of fats, carbohydrates and proteins, and is the common energy currency of all cells. ATP-sensitive potassium (KATP) channels respond to cellular changes in ATP48. Populations of neurons in the ARC, VMH and NTS express KATP channels, and depolarization of these channels leads to neuronal activation49 (FIG. 2). Application of KAT P channel inhibitors into the hypothalamus attenuates the changes in hepatic glucose production that are induced by centrally administered glucose, fatty acid, insulin or leucine in rodents45–47, indicating that KAT P channels mediate these effects. Indeed, introducing a mutant, dysfunctional KATP channel specifically in POMC neurons renders these neurons unable to become activated in response to glucose (although they depolarize as expected in response to leptin)48. Mice with this mutant KAT P channel have impaired glucose tolerance compared with littermate controls but normal body weight48. Taken together, these data suggest that hypothalamic KATP channels are a common pathway through which different types of nutrients regulate glucose homeostasis and that the melanocortin system (specifically, POMC neurons) is important in mediating nutrient-induced regulation of glucose homeostasis via the CNS. Evidence is currently lacking for a role of these channels in regulating energy homeostasis.
Figure 2. Fuel sensing in CNS neurons.
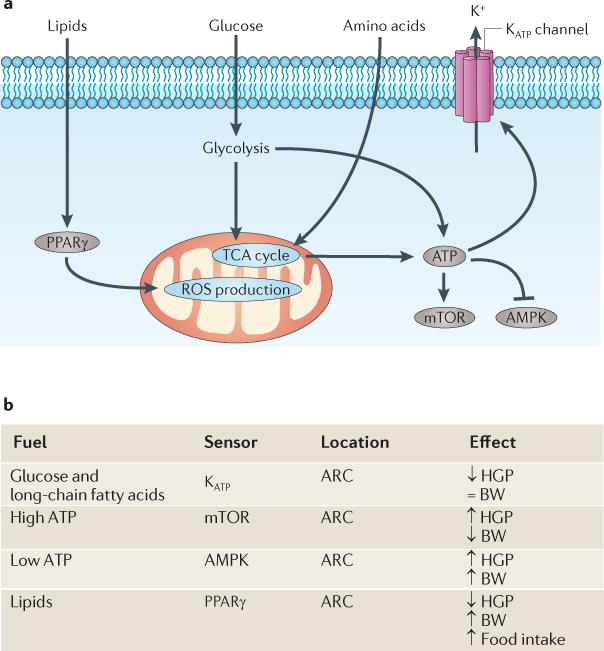
a | Various molecular machineries sense the bioavailability of glucose, amino acids and lipids, as well as ATP stores. Metabolism of glucose and amino acids (leucine) via glycolytic and tricarboxylic acid (TCA) cycle pathways results in increased ATP levels. In the hypothalamus, this in turn results in inhibition of AMP-activated protein kinase (AMPK), activation of mammalian target of rapamycin (mTOR) and activation of neuronal ATP-sensitive potassium (KATP) channels. Each of these isolated events has been associated with the regulation of hepatic glucose production (HGP). Bioavailable lipids act as a ligand for hypothalamic peroxisome proliferator-activated receptor-γ (PPARγ), the activation of which reduces HGP, possibly by modulating levels of reactive oxygen species (ROS) in hypothalamic neurons. In general, as nutrients become bioavailable, overall HGP decreases. b | Although these pathways are clearly important for regulating glucose homeostasis, only some are also involved in the regulation of energy homeostasis. For example, hypothalamic mTOR and PPARγ activation regulate both processes (albeit in opposite directions), whereas KATP channels seem to play no part in energy homeostasis. ARC, arcuate nucleus; BW, body weight.
KATP channels are located on different populations of hypothalamic neurons, and the downstream effects of their depolarization vary depending on which neurons are activated. For example, in the ARC, KATP depolarization leads to an increase in hepatic glucose production50,51, whereas depolarization of KATP channels specifically in melanin-concentrating hormone neurons of the lateral hypothalamus inhibits hepatic glucose production52. These data suggest that KATP channels are only one aspect of the neuronal nutrient-sensing that is important for regulating glucose homeostasis.
Indeed, two other ATP-dependent fuel-sensing pathways involve AMP-activated protein kinase (AMPK) and m T O R (FIG. 2). AMPK and mTOR are int racellular signalling pathways that respond to changing levels of ATP. In the hypothalamus, AMPK and mTOR signalling regulate the expression of NPY, AGRP and POMC20,53,54. In all cells, the level of activated AMPK increases in response to ATP depletion55. Thus, as one might predict, pharmacologically inducing a rise in hypothalamic AMPK levels in rodents increases food intake53, hepatic glucose production and muscle glycogen synthesis56 — which are all metabolic processes that increase energy (ATP) production. Genetic deletion of the α2 subunit of AMPK in POMC neurons leads to obesity in mice, whereas its removal from AGRP neurons results in leanness57; it is therefore not known whether the role of fuel-sensing through hypothalamic AMPK signalling in the regulation of glucose homeostasis is independent of body weight. However, electrophysiological recordings of whole-brain slices from mice lacking the α2 subunit of AMPK show that they no longer respond to glucose57.
mTOR is a highly conserved kinase that is expressed in most eukaryotic cells and has an important role in regulating cell growth58. The mTOR pathway is activated by a surplus of available energy — that is, by high ATP levels that are generated as a result of excess nutrients or the actions of anabolic hormones such as insulin58. Within the hypothalamus, mTOR expression is increased in fed and decreased in fasted rodents20. Injection of an adenovirus encoding constitutively active S6 kinase — an enzyme that is downstream of mTOR activation — directly into the hypothalamus of rats reduced obesity that had been induced by a high-fat diet. It did not affect blood glucose levels but reduced the insulin response to a glucose load59. Paradoxically, this same manipulation increased hepatic glucose production at a time point when body weight changes were not yet evident60. Thus, the mTOR pathway probably regulates both energy and glucose homeostasis in a complex manner that is dependent on diet and the timing of exposure to that diet.
KATP channels, AMPK and mTOR are ‘general’ fuel sensors — they sense changes in overall energy status via changes in ATP levels regardless of how that ATP was generated. However, molecular mechanisms exist to monitor specific types of fuel. For example, various fatty acids serve as ligands for peroxisome proliferator-activated receptors (PPARs). One specific isoform, PPARγ, has recently been established as an important CNS lipid sensor that is involved in the regulation of both energy and glucose homeostasis. This was surprising, as PPARγ was always thought to directly promote adipogenesis locally rather than indirectly via the CNS61. The activation of PPARγ in peripheral tissues directs lipid storage to adipose tissue and away from the liver and skeletal muscle, as fat accumulation in these organs would impair insulin sensitivity; this is the primary mechanism by which PPARγ agonists (thiazolidinedi-ones) are thought to improve glucose metabolism in patients with T2DM62. One downside to the use of these therapeutics is the increase in body weight they cause. In rats, central administration of PPARγ agonists or virally induced hypothalamic Pparg expression increased body weight61. Conversely, mice with neuron-specific Pparg deletion do not show PPARγ agonist-induced hyper-phagia and do not gain weight on a high-fat diet63. Interestingly, these animals also have improved glucose tolerance and hepatic, but not peripheral, insulin sensitivity compared with high-fat-fed wild-type mice63. It is possible that PPARγ agonists act within the CNS to increase insulin sensitivity and that this contributes to the improvements in hepatic insulin sensitivity that is seen in patients with T2DM treated with PPARγ agonists. CNS PPARγ may regulate food intake and glucose homeostasis by regulating the levels of reactive oxygen species64, which in turn modulate the activity of NPY–AGRP and POMC neurons64. In mice, administration of a PPARγ agonist decreases, whereas administration of PPARγ antagonists increases, levels of reactive oxygen species in POMC neurons64. Pharmacologically increasing reactive oxygen species levels in the hypothalamus increases feeding in mice, and vice versa64. Together, these data link hypothalamic fuel-sensing and cellular metabolic stress to PPARγ, and not only implicates PPARγ actions in the hypothalamus as a mechanism underlying diet-induced obesity and hepatic insulin resistance but also adds another potential mechanism for the pharmacological action of PPARγ agonists.
To summarize, the different types of fuel-sensing pathways that are used by hypothalamic neurons to regulate energy and glucose homeostasis (FIG. 2) seem to dissociate the regulation of glucose and energy homeostasis. Hypothalamic mTOR (primarily in the ARC) regulates energy homeostasis and glucose homeostasis but in opposite directions; the same is the case for hypothalamic PPARγ activation, whereas ARC KATP channels are important in regulating glucose but seem to have no role in regulating energy homeostasis. The role of AMPK in glucose homeostasis is confounded by its effect on regulating body weight. It is interesting to speculate that these fuel-sensing pathways respond to acute rather than long-term changes in fuel status, rendering them more important in regulating glucose homeostasis than in regulating energy homeostasis.
5-HT
5-HT, which is primarily synthesized by neurons in the dorsal raphe nucleus of the midbrain, also has a role in the central regulation of both food intake and glucose homeostasis65, specifically through binding to hypothalamic 5-HT2C receptors66. Fenfluramine, a drug that increases 5-HT bioavailability, suppresses food intake both in humans and rodents66, whereas global deletion of the 5-HT2C receptor in mice results in hyperphagia, obesity and obesity-associated insulin resistance with impaired glucose tolerance67. 5-HT2C receptor agonists improve glucose homeostasis at doses lower than those that suppress food intake in diet-induced obese mice68. Conversely, dogs treated with the atypical antipsychotic and 5-HT2C receptor antagonist/inverse agonist olanzapine gained body and fat mass and also had hepatic insulin resistance beyond what was seen in dietary-induced obese animals with similar body weight and adiposity69.
POMC neurons in the ARC receive dense innervation from raphe-derived 5-HT neurons70 and express 5-HT2C receptors, and 5-HT depolarizes POMC neurons in brain slices71. Furthermore, restoration of 5-HT2C receptors specifically in POMC neurons in 5-HT2C-null mice72 rescues the feeding behaviour response to fenfluramine and improves hepatic insulin sensitivity72. Leptin is also important in 5-HT-induced regulation of glucose homeostasis. For example, leptin-deficient mice that also lack 5-HT2C receptors have weight-independent impairments in fasting glucose and glucose intolerance that surpass those in leptin-deficient mice73. Leptin and 5-HT2C receptors both activate POMC neurons via transient receptor-potential C channels74,75, but they act on separate subsets of POMC neurons, and it remains unknown if the downstream projections of 5-HT and leptin activation of POMC neurons are similar74.
The effect of 5-HT on food intake is dependent on functional MC4Rs. MC4R-deficient mice do not respond to the anorexic effect of fenfluramine, but the response is restored when MC4Rs are specifically re-introduced in neurons in the PVN and amygdala — CNS regions known to regulate food intake76. Together, these data illustrate that hypothalamic serotonergic circuits regulate both energy and glucose homeostasis, and this is mediated via the melanocortin system.
Hypothalamic melanocortin receptors
POMC and NPY–AGRP neurons in the ARC project to neurons in the PVN, which express both MC3Rs and MC4Rs. MC4Rs are activated downstream of many different signalling peptides and nutrients; in addition, MC4Rs are, in and of themselves, necessary for the regulation of glucose homeostasis. For example, mice lacking MC4Rs become severely obese but are hyperinsulinaemic before the onset of obesity77, suggesting a weight-independent effect of MC4Rs on glucose homeostasis. Rats receiving chronic intracerebroventricular (ICV) administration of an MC4R agonist have better hepatic and skeletal insulin sensitivity compared with controls, whereas chronic ICV administration of a combined MC3R and MC4R antagonist impairs hepatic and skeletal muscle insulin sensitivity in a weight-independent fashion (animals were pair-fed to controls in order to abrogate any orexigenic effects of the MC3R and MC4R antagonist)78. This indicates that MC4R activation is necessary for the maintenance of normal glucose homeostasis.
To summarize, there is both convergence and divergence in the ARC mechanisms that regulate glucose homeostasis and energy homeostasis. POMC neurons and NPY–AGRP neurons in the ARC have a role in nutrient- and hormone-induced regulation of glucose homeostasis, but the processing of these signals diverges either in terms of the population of neurons they activate or in terms of the downstream intracellular events they induce. Insulin signalling and nutrient-sensing in the ARC are both dependent on KATP channels for the regulation of glucose production, but whether this is also the case for leptin is unknown. Insulin and leptin both act on POMC neurons, but these are likely to be separate populations within the ARC. Moreover, leptin actions, but not insulin actions, partially depend on downstream hypothalamic MC4Rs (FIG. 1,2). The data also demonstrate that POMC neurons and NPY–AGRP neurons in the ARC are an important site of integration for the regulation of both energy and glucose homeostasis in response to several signals, including leptin, serotonin and fuel-sensing pathways. By contrast, KATP channels on POMC neurons and IRs on AGRP neurons are necessary for the regulation of glucose homeostasis but not energy homeostasis, demonstrating divergence in the regulation of these two homeostatic systems.
Other regulatory systems
VMH
The VMH is another important CNS region that is involved in the regulation of energy homeostasis and glucose homeostasis. Early studies in rats showed that electrolytic lesions of the VMH resulted in profound obesity and hyperinsulinaemia79. The hyperinsulinaemia was at least partially due to reduced glucose-induced insulin secretion80. In addition, these animals had reduced glucagon and catecholamine levels81 during hypoglycaemia. It is important to note, however, that electrolytic lesions damage both neurons and non-synapsing fibre tracts routed through the lesioned area. To understand the role of specific VHM neurons and signalling pathways in the regulation of energy and glucose homeostasis, recent studies have genetically targeted steroidogenic factor 1 (SF1), a transcription factor that is primarily expressed in the VMH.
SF1 neurons within the VMH express IRs and OBRs82. Deletion of OBRs from SF1-containing neurons in mice results in mild obesity that is similar in magnitude to that in mice lacking OBRs on POMC neurons83,84, indicating that leptin acts within both the ARC and the VMH to regulate energy homeostasis. OBR-induced signalling in hypothalamic nuclei activates negative-feedback proteins, such as suppressor of cytokine signalling 3 (SOCS3). Increased SOCS3 signalling in the VMH could explain why the high circulating levels of leptin in obese individuals are not able to constrain food intake. Mice lacking SOCS3 signalling in SF1-containing neurons have an improved anorexic response to exogenous leptin and reduced basal food intake, but as they also have reduced energy expenditure, their body weight is similar to that of control animals85. They also have normal fasting insulin levels when exposed to a high-fat diet85, suggesting that improved leptin signalling via unconstrained STAT3 (signal transducer and activator of transcription 3) signalling — resulting from inactivation of SOCS3 in SF1-containing neurons — confers greater insulin sensitivity. Thus, although the phenotype of these mice is less extreme than that of mice lacking OBRs in POMC neurons, it suggests that OBRs on SF1 neurons play a part in regulating both energy and glucose homeostasis.
A subset of SF1 neurons that respond to insulin are not responsive to leptin82, again suggesting that IRs and OBRs are not always colocalized to the same neuronal populations. Mice lacking IRs specifically in SF1 neurons do not develop diet-induced obesity and leptin resistance when they are exposed to a high-fat diet82. Perhaps secondary to their resistance to weight gain, these mice have reduced basal insulin levels and better systemic glucose and insulin tolerance compared with control mice82. This superior glucose homeostasis is associated with increased phosphatidylinositol 3-kinase (PI3K) activity in SF1 neurons82. As PI3K is activated by IR activation, the authors of this study speculated that the hyperinsulinaemia that is associated with obesity leads to increased PI3K activity on VMH glutamatergic neurons, and these neurons in turn inhibit POMC neurons, thereby further exacerbating obesity82. However, in other studies, deletion of synaptic glutamate vesicular transporters from VMH SF1 neurons resulted in only a small increase in body weight and impaired counterregulatory responses to hypoglycaemia86, indicating that POMC neurons are not a probable target of SF1 neuronal activation in the regulation of energy and glucose homeostasis. Nevertheless, taken together, these data suggest that SF1 neurons within the VMH are part of a pathway that regulates both energy and glucose homeostasis in response to changes in insulin and leptin.
Hindbrain MC4Rs
MC4Rs are not only expressed in the hypothalamus; they are also located within the hindbrain, a CNS region that is also essential in the control of energy and glucose homeostasis87. Administration of MC4R agonists and antagonists into the hindbrain decreases and increases food intake in rats, respectively88. As mentioned above, MC4R-null mice show obesity, hyperglycaemia, hyperinsulinaemia and hepatic and skeletal muscle insulin resistance77. As these are whole-body-null mice, it is difficult to parse out the synergistic or differential roles of the hypothalamic versus hindbrain MC4Rs in these effects. However, recent studies have attempted to study the role of MC4Rs within the hindbrain. In one study, MC4Rs were re-introduced into MC4R-null mice specifically in the cholinergic preganglionic parasympathetic and sympathetic neurons of the dorsal motor nucleus of the vagus (DMV)89. In these mice, body weight gain was reduced and hyperglycaemia and hyperinsulinaemia were attenuated. In addition, hepatic but not skeletal muscle insulin sensitivity was normalized89, suggesting that MC4R-induced improvement in skeletal muscle insulin sensitivity is mediated by a separate (possibly hypothalamic) pathway. Furthermore, the fact that re-expression of MC4Rs in parasympathetic and sympathetic DMV neurons normalized hepatic glucose production but did not eliminate adiposity suggests that hindbrain MC4Rs have weight-independent effects on regulating liver glucose fluxes. By comparison, reactivation of MC4Rs in parasympathetic afferent neurons located in the nodose ganglia did not prevent the obesity, hyperglycaemia or insulin resistance but did abolish hyperinsulinaemia89, suggesting a role for the sympathetic nervous system in regulating insulin secretion but not overall glucose homeostasis. These data support the hypothesis that MC4Rs in hind-brain sympathetic neurons have a role in regulating both energy homeostasis and glucose homeostasis. It remains unknown whether hindbrain POMC neurons or ARC POMC neurons are the source of endogenous ligand for the hindbrain MC4R. Furthermore, these data differ from previous data demonstrating that the vagal nerve provides the main innervation for hepatic glucose pro-duction50,51,90,91. Given the importance of hepatic glucose production to physiological function, various signals involved in regulating nutrient status may be integrated by the autonomic nervous system and so produce a hepatic response.
Central actions of gut hormones
Nutrients are first sensed and processed in the gastrointestinal tract. The gut is richly innervated with vagal afferent neurons that directly project to sympathetic neurons in hindbrain nuclei92, and these hindbrain nuclei project to forebrain areas such as the hypothalamus. Thus, the hindbrain serves as an interface between the peripheral organs and higher brain centres. The gut informs the rest of the body, including the CNS, about current nutritional status by secreting hormones (that is, ghrelin, gastric inhibitory peptide, peptide Y Y, cholecystokinin (CCK) and glucagon-like peptide 1 (GLP1)) and neurotransmitters (for example, serotonin) that are key players in regulating glucose and energy homeostasis90,93. In particular, recent studies of CCK and GLP193,94 have expanded our understanding of the link between the gut and CNS-induced regulation of energy and glucose homeostasis.
The gut hormone CCK is secreted from the proximal intestine in response to lipid ingestion and initiates satiation through activation of its receptors on vagal afferent axons95. Recent evidence has implicated CCK in a gut–brain–liver axis; infusion of lipids96 or CCK90 into the upper intestine of rats suppressed hepatic glucose production, and this effect was mediated by NMDA receptors in the dorsal vagal complex of the hindbrain97. These studies also suggested that lipid activation of CCK receptors in the upper intestine activates protein kinase Cδ and G protein-coupled signalling pathways in duodenal enterocytes, which leads to vagal afferent neuronal activation of the CNS and, ultimately, top-down neuronal suppression of hepatic glucose production94.
The gut hormone GLP1 is secreted from the distal small intestine in response to ingested nutrients. It is also produced in a distinct population of neurons in the NTS98, where it is thought to regulate food intake. Specifically, GLP1 injected into the fourth cerebral ventricle, which lies adjacent to the NTS, suppresses food intake in rats99. GLP1 is also important in the central regulation of glucose homeostasis. Intravenous administration of GLP1 inhibits hepatic glucose production in humans100, and GLP1 infusion into the ARC101 decreases hepatic glucose production in rats (in which insulin levels were kept similar to those in saline infused rats). This suggests that activation of GLP1 receptors in the ARC causes an insulin-independent decrease in hepatic glucose production. However, ICV administration of GLP1 in rats also stimulates insulin secretion, suggesting both insulin-dependent and insulin-independent central mechanisms by which GLP1 regulates glucose homeostasis101,102. Although GLP1 has historically been considered to be a hormone, it has a short circulating half-life, as it is rapidly degraded within the endothelium by dipeptidyl peptidase 4 (REF. 103). GLP1 receptors are located on, and increase the activity of, afferent neurons in the wall of the hepatic portal vein104 and are also located on enteric neurons105, providing an additional neural site for GLP1 to regulate glucose homeostasis and food intake.
These findings suggest that the ‘gut signals’ CCK and GLP1 have a role in regulating both energy and glucose homeostasis through their actions in the CNS (FIG. 3). Notably, the intestine secretes dozens of other hormones that probably also interact with the CNS to regulate nutrient balance.
Figure 3. Overlapping CNS circuitries regulate energy balance and glucose homeostasis.

Most receptor populations and neuropeptides discussed in this Review overlap in the way they control energy balance and glucose homeostasis. The hypothalamic arcuate nucleus (ARC) melanocortin system, consisting of pro-opiomelanocortin (POMC) and neuropeptide Y (NPY)–agouti-related protein (AGRP) neurons, has a key role in regulating both energy and glucose homeostasis. On POMC neurons, activation of 5-hydroxytryptamine 2C (5-HT2C) receptors (via 5-HT released from the midbrain), leptin receptors (OBRs) or insulin receptors (IRs) regulates both energy and glucose homeostasis. In NPY–AGRP neurons, OBRs are important in the regulation of energy but not glucose homeostasis, whereas IRs only regulate glucose homeostasis. Downstream of these ARC neurons are melanocortin receptor 4 (MC4R)-expressing neurons within the paraventricular nucleus (PVN), which are also important for both energy and glucose homeostasis. Within the ARC fuel-sensing systems, both mammalian target of rapamycin (mTOR) and ATP-activated protein kinase (AMPK) seem to be important for regulating both energy and glucose homeostasis, whereas ATP-sensitive potassium (KATP) channel activation is only important in regulating glucose homeostasis. Other regulatory systems that control both energy and glucose homeostasis but that are independent of the ARC involve IRs and OBRs on steroidogenic factor 1 (SF1) neurons within the ventromedial hypothalamus (VMH), as well as MC4Rs located on the sympathetic neurons (in the nucleus of the solitary tract (NTS) in the dorsal motor nucleus of the vagus (DMV) in the hindbrain. Lastly, the gut hormones cholecystokinin (CKK) and glucagon-like peptide 1 (GLP1) act centrally by innervating NMDA receptors (NMDARs) on vagal afferents to the hindbrain. GLP1R, GLP1 receptor; SNS, sympathetic nervous system.
To summarize this section, CNS neuron populations outside the ARC have important roles in regulating glucose and energy homeostasis. These include SF1 neurons within the VMH and multiple populations of neurons within the hindbrain (MC4R-expressing neurons in the sympathetic nervous system and NMDA receptor-expressing neurons that are activated by peripheral CCK- and GLP1-expressing neurons).
Conclusions and future directions
The data reviewed here demonstrate that, with a few exceptions (namely, insulin actions on NPY–AGRP neurons, hypothalamic KATP-dependent fuel-sensing pathways and MC4R signalling on cholinergic parasympathetic neurons), most of the neurons and circuits that regulate glucose homeostasis also regulate energy homeostasis (FIG. 3). Thus, the CNS has a highly integrative role in processing peripheral nutrient, hormonal and neural signals to regulate both glucose and energy homeostasis. This supports the notion of precise coordination between signals conveying acute (for example, meals) and long-term (for example, adiposity) energy status.
New strategies to study the role of the CNS in energy and glucose homeostasis
Determining the role of the CNS in the complex physiology of glucose regulation is difficult because it is regulated by heterogenenous neuronal populations. Site-specific administration of receptor ligands or administration of adeno- or lentiviruses allows manipulation of specific regions of the CNS but may not isolate precise neuronal populations. Cell-specific genetic techniques have allowed the manipulation of gene expression in a tissue- and even neuron-specific manner, but altering gene expression may not equate to manipulations in neuronal activity.
Recently developed technologies, such as optogenetics and the DREADD (designer receptors exclusively activated by designer drugs) system, offer exciting new avenues for metabolic research. Both of these techniques offer opportunities to acutely turn on or off specific cell populations with a combination of genetic manipulation and either photostimulation (optogenetics) or pharmacotherapy (DREADD) and then examine the in vivo implications of modulating cellular activity. The melanocortin system has been manipulated using both of these techniques. Photostimulation studies found that POMC neuron activation decreases feeding, that AGRP neuron activation increases feeding and that only the former effect is dependent on melanocortin receptor signalling106. Furthermore, these experiments have shown that photostimulation106 or DREADD activation107 of AGRP neurons results in unopposed and immediate onset of hyperphagia. The latter findings106,107 contrast with the effect of complete Agrp deletion, which results in almost no phenotype108, illustrating that different conclusions are reached after complete deletion versus acute activation of a protein. These studies also highlight an important disadvantage of genetic knock-out and knock-in models: functional redundancies and rewiring of neurocircuitry resulting from genetic manipulation can potentially lead to an underestimation of the importance of the gene of interest in the regulation of energy and glucose homeostasis. The new technologies offer an exciting opportunity to more fully study the role of changes in specific neuronal activity in regulating glucose homeostasis.
Clinical implications and therapeutics
The crucial implication of the literature reviewed here is that there is considerable overlap between the neurocircuits that regulate energy homeostasis versus glucose homeostasis. In fact, there seem to be only three signalling pathways that have divergent effects on glucose versus energy homeostasis: glucose-sensing (through KATP channels on POMC neuron), insulin signalling by AGRP neurons and MC4Rs on cholinergic parasympathetic neurons are involved in the regulation of glucose but not energy homeostasis. It is also clear that the melanocortin system is important in the regulation of both energy and glucose homeostasis, although other neuronal systems are also involved. This highlights the integrative nature of physiological regulation, and this is important for understanding the role of the CNS in the link between obesity and diabetes — the regulation of fuel dynamics is an important CNS function and is disturbed in both of these metabolic disorders.
Much of what we have learned about energy homeostasis has been discovered using animal models demonstrating that dysfunction of these circuits leads to diet-induced obesity. The problem we face in linking these dysfunctional neurocircuits to diabetes is that we lack a true animal model of T2DM (BOX 3). A further complicating factor is the lack of technologies to dissect the complex aspects of CNS control of glucose homeostasis in humans. An in-depth understanding of the neurocircuitry that regulates glucose homeostasis is crucial for elucidating the relationship between obesity and its associated increased susceptibility to T2DM. In the search for new therapeutics that target these CNS circuits, we should no longer distinguish between treating obesity and treating T2DM. Rather, we should aim to finds treatments that target the CNS dysfunction underlying both conditions and thereby improve both conditions simultaneously.
Box 3. Limitations of animal models of type 2 diabetes mellitus.
The development of tools to study energy and glucose metabolism has the potential to greatly increase our understanding of the overlapping neurocircuits that link obesity and type 2 diabetes mellitus (T2DM). With respect to the neurocircuitry that regulates energy homeostasis, many of our recent advances have come from animal models that recapitulate the hyperphagia of calorie-rich foods and the resulting increased adiposity, increased circulating levels of leptin and insulin, and increased circulating lipids, triglycerides and cholesterol levels that are associated with human obesity. Food intake in a rodent is easy to manipulate and measure; weight-restricted control groups must be included to understand whether any effects of the diet on metabolism are secondary to the level of adiposity. Body weight fluctuations over time are also easy to quantify in rats, with the caveat that rats tend to gain weight steadily over time. Energy expenditure and locomotor activity can also be measured in freely moving animals, whereas this is challenging in clinical research.
It is important to realize that the ability of current animal models to recapitulate clinical T2DM may be limited. Human T2DM is diagnosed in patients having fasting hyperglycaemia (>126 mg per dL), impaired glucose tolerance, randomly sampled hyperglycaemia (>200 mg per dL) or chronic hyperglycaemia as indicated by the degree of haemoglobin glycosylation113. As discussed in BOX 1, the transition from the pre-diabetic condition to frank T2DM is indicated by an inadequate insulin response to a given glucose load, with severe liver and skeletal muscle insulin resistance. Although laboratory rodents certainly become glucose intolerant in response to high-fat diets and obesity over time, they are highly resistant to the development of frank diabetes. This has lead researchers to rely on pharmacological or genetic animal models of the disease. These models typically manipulate one but not both arms (β-cell failure and insulin resistance) of the disease. For example, administration of streptozotocin, a pharmacologic agent that destroys pancreatic β-cells, leads to a rapid onset of hyperglycaemia without causing insulin resistance. Some genetic rat and mouse models that are greatly susceptible to obesity (for example, Zucker fatty rats and leptin-deficient (ob/ob) mice) develop insulin resistance but are continually able to compensate for the greater demand for insulin with their large β-cell capacity. Other models with lesser β-cell capacity may more closely recapitulate the disease but have either other genetic modifications (for example, db/db mice, which are mice devoid of leptin receptors) or species differences (for example, obese rhesus monkeys) that make understanding the human disease challenging. Thus, although the use of these models is unlikely to lead to an understanding of the full disease process, the models enable us to study specific components of T2DM. Furthermore, the benefits of the ability to perform large-scale high-throughput screening that is not possible in clinical studies, the battery of genetic and molecular tools, as well as the depth and abundance of scientific literature and knowledge regarding mouse physiology makes rodent models necessary to move diabetes research forward.
Acknowledgments
We are grateful for the helpful comments of S.C. Woods and S.C. Benoit. The work of the laboratory is supported in part by the US National Institutes of Health (NIH) Awards DK56863, DK57900, U01CA141464, DK082480, MH069860, DK082480 and also work with Ethicon Endo-Surgery, F. Hoffman-La Roche, Pfizer and Novo Nordisk A/S. B.E.G. is also supported by NIH Award 1F32HD68103.
Glossary
- Glucose fluxes
Changes in glucose uptake, glucose production by the liver and glucose metabolism within a tissue or cell
- Melanocortin system
Neurons in the arcuate nucleus of the hypothalamus and the nucleus of the solitary tract that express pro-opiomelanocortin, agouti-related protein and neuropeptide Y with downstream actions on melanocortin receptor 3 and melanocortin receptor 4
- Antagonist/inverse agonist
A ligand that can both block a receptor’s activity and produce the opposite action of the agonist
- Gluconeogenesis
The process of making glucose from non-glucose precursors such as lactate, glycerol and alanine
- Glycogenolysis
The breakdown of glycogen to glucose
- Hepatic insulin sensitivity
The degree to which insulin suppresses glucose production by the liver
- Glucose tolerance test
(GTT). A test that measures the glucose excursion after a bolus administration of glucose it is an index of the body’s ability to tolerate and/or handle a glucose load.
- Insulin tolerance tests
Tests that measure the fall in glucose levels after an injection of a bolus of insulin
- Whole-body insulin sensitivity
The degree to which the body responds to insulin it is usually assessed by comparing the glucose infusion rate during a hyperinsulinaemic–euglcyaemic clamp in control versus experimental animals.
- Mass action of glucose
Refers to glucose uptake into tissues, which is driven by how much glucose there is in the blood
- Insulin resistance
A metabolic state in which the tissues of the body exhibit reduced responsiveness to chronically high levels of insulin in the circulation
- Negative-feedback proteins
Proteins that suppress signalling by other molecules
- Leptin resistance
A state in which the body is no longer responsive to the anorexic effect of exogenous leptin
- Nodose ganglia
Inferior ganglia of the vagus nerve
Footnotes
Competing interests statement
D.A.S and R.J.S declare competing financial interests: see Web version for details. B.E.G. declares no competing financial interests.
FURTHER INFORMATION
Darleen A. Sandoval’s homepage: http://intmed.uc.edu/contact/directory/profile.aspx?epersonID=sandovda
References
- 1.Bernard C. In: Homeostasis: Origins of the Concept, 1973. Langley LL, editor. Dowden, Hutchinson & Ross; Stroudsberg, PA: 1870. pp. 129–151. [Google Scholar]
- 2.Knowler WC, et al. 10-year follow-up of diabetes incidence and weight loss in the Diabetes Prevention Program Outcomes Study. Lancet. 2009;374:1677–1686. doi: 10.1016/S0140-6736(09)61457-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Y, et al. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz MW, Seeley RJ, Campfield LA, Burn P, Baskin DG. Identification of hypothalmic targets of leptin action. J Clin Invest. 1996;98:1101–1106. doi: 10.1172/JCI118891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hahn TM, Breininger JF, Baskin DG, Schwartz MW. Coexpression of Agrp and NPY in fasting-activated hypothalamic neurons. Nature Neurosci. 1998;1:271–272. doi: 10.1038/1082. [DOI] [PubMed] [Google Scholar]
- 6.Cone RD. Anatomy and regulation of the central melanocortin system. Nature Neurosci. 2005;8:571–578. doi: 10.1038/nn1455. An excellent review on the various players in the melanocortin system and its role in energy balance. [DOI] [PubMed] [Google Scholar]
- 7.Cowley MA, et al. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- 8.Woods SC, Seeley RJ, Porte DJ, Schwartz MW. Signals that regulate food intake and energy homeostasis. Science. 1998;280:1378–1383. doi: 10.1126/science.280.5368.1378. [DOI] [PubMed] [Google Scholar]
- 9.Brüning JC, et al. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000;289:2122–2125. doi: 10.1126/science.289.5487.2122. [DOI] [PubMed] [Google Scholar]
- 10.Halaas JL, et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 11.Pelleymounter MA, et al. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 12.Baskin DG, Breininger JF, Schwartz MW. Leptin receptor mRNA identifies a subpopulation of neuropeptide Y neurons activated by fasting in rat hypothalamus. Diabetes. 1999;48:828–833. doi: 10.2337/diabetes.48.4.828. [DOI] [PubMed] [Google Scholar]
- 13.Baskin DG, et al. Insulin and leptin: dual adipodity signals to the brain for the regulation of food intake and body weight. Brain Res. 1999;848:114–123. doi: 10.1016/s0006-8993(99)01974-5. [DOI] [PubMed] [Google Scholar]
- 14.Schwartz MW, et al. Central nervous system control of food intake. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 15.Woods SC, Lotter EC, McKay LD, Porte D. Jr Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature. 1979;282:503–505. doi: 10.1038/282503a0. [DOI] [PubMed] [Google Scholar]
- 16.Chavez M, Kaiyala K, Madden LJ, Schwartz MW, Woods SC. Intraventricular insulin and the level of maintained body weight in rats. Behav Neurosci. 1995;109:528–531. doi: 10.1037//0735-7044.109.3.528. [DOI] [PubMed] [Google Scholar]
- 17.Seeley RJ, et al. Intraventricular leptin reduces food intake and body weight of lean rats but not obese Zucker rats. Horm Metab Res. 1996;28:664–668. doi: 10.1055/s-2007-979874. [DOI] [PubMed] [Google Scholar]
- 18.Abate N, Garg A, Peshock RM, Stray-Gundersen J, Grundy SM. Relationships of generalized and regional adiposity to insulin sensitivity in men. J Clin Invest. 1995;96:88–98. doi: 10.1172/JCI118083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woods SC, Seeley RJ, Porte D, Jr, Schwartz MW. Signals that regulate food intake and energy homeostasis. Science. 1998;280:1378–1383. doi: 10.1126/science.280.5368.1378. [DOI] [PubMed] [Google Scholar]
- 20.Cota D, et al. Hypothalamic mTOR signaling regulates food intake. Science. 2006;312:927–930. doi: 10.1126/science.1124147. An early demonstration that mTOR is a fuel sensor within the hypothalamus and regulates energy balance. [DOI] [PubMed] [Google Scholar]
- 21.Clegg DJ, Wortman MD, Benoit SC, McOsker CC, Seeley RJ. Comparison of central and peripheral administration of C75 on food intake, body weight, and conditioned taste aversion. Diabetes. 2002;51:3196–3201. doi: 10.2337/diabetes.51.11.3196. [DOI] [PubMed] [Google Scholar]
- 22.Loftus TM, et al. Reduced food intake and body weight in mice treated with fatty acid synthase inhibitors. Science. 2000;288:2299–2300. doi: 10.1126/science.288.5475.2379. [DOI] [PubMed] [Google Scholar]
- 23.Vaisse C, Clement K, B GG, Froguel P. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nature Genet. 1998;20:113–114. doi: 10.1038/2407. [DOI] [PubMed] [Google Scholar]
- 24.Clegg DJ, et al. Reduced anorexic effects of insulin in obesity-prone rats fed a moderate-fat diet. Am J Physiol Regul Integr Comp Physiol. 2005;288:R981–R986. doi: 10.1152/ajpregu.00675.2004. [DOI] [PubMed] [Google Scholar]
- 25.Ryan KK, Woods SC, Seeley RJ. Central nervous system mechanisms linking the consumption of palatable high-fat diets to the defense of greater adiposity. Cell Metab. 2012;15:137–149. doi: 10.1016/j.cmet.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.von Mering J, Minkowski O. Diabetes Mellitus nach Pankreasextirpation. Zbl Klin Med. 1889;10:393–394. [Google Scholar]
- 27.Banting FG, Best CH, Collip JB, Campbell WR, Fletcher AA. Pancreatic extracts in the treatment of diabetes mellitus. Can Med Assoc J. 1922;12:141–146. [PMC free article] [PubMed] [Google Scholar]
- 28.Macleod JJ. Pancreatic extract and diabetes. Can Med Assoc J. 1922;12:423–425. [PMC free article] [PubMed] [Google Scholar]
- 29.Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nature Med. 2002;8:1376–1382. doi: 10.1038/nm1202-798. [DOI] [PubMed] [Google Scholar]
- 30.Liu L, et al. Intracerebroventricular leptin regulates hepatic but not peripheral glucose fluxes. J Biol Chem. 1998;273:31160–31167. doi: 10.1074/jbc.273.47.31160. [DOI] [PubMed] [Google Scholar]
- 31.Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nature Neurosci. 2002;5:566–572. doi: 10.1038/nn0602-861. [DOI] [PubMed] [Google Scholar]
- 32.Benoit SC, et al. The catabolic action of insulin in the brain is mediated by melanocortins. J Neurosci. 2002;22:9048–9052. doi: 10.1523/JNEUROSCI.22-20-09048.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Konner AC, et al. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 2007;5:438–449. doi: 10.1016/j.cmet.2007.05.004. An excellent study that systematically activated IRs in AGRP- and/or POMC-expressing neurons to determine the necessity of these receptors for glucose homeostasis. [DOI] [PubMed] [Google Scholar]
- 34.Lin HV, et al. Divergent regulation of energy expenditure and hepatic glucose production by insulin receptor in agouti-related protein and POMC neurons. Diabetes. 2010;59:337–346. doi: 10.2337/db09-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pocai A, et al. Central leptin acutely reverses diet-induced hepatic insulin resistance. Diabetes. 2005;54:3182–3189. doi: 10.2337/diabetes.54.11.3182. [DOI] [PubMed] [Google Scholar]
- 36.Gutierrez-Juarez R, Obici S, Rossetti L. Melanocortin-independent effects of leptin on hepatic glucose fluxes. J Biol Chem. 2004;279:49704–49715. doi: 10.1074/jbc.M408665200. [DOI] [PubMed] [Google Scholar]
- 37.German J, et al. Hypothalamic leptin signaling regulates hepatic insulin sensitivity via a neurocircuit involving the vagus nerve. Endocrinology. 2009;150:4502–4511. doi: 10.1210/en.2009-0445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Balthasar N, et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron. 2004;42:983–991. doi: 10.1016/j.neuron.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 39.Shi H, et al. Sexually different actions of leptin in proopiomelanocortin neurons to regulate glucose homeostasis. Am J Physiol Endocrinol Metab. 2008;294:E630–E639. doi: 10.1152/ajpendo.00704.2007. [DOI] [PubMed] [Google Scholar]
- 40.Berglund ED, et al. Direct leptin action on POMC neurons regulates glucose homeostasis and hepatic insulin sensitivity in mice. J Clin Invest. 2012;122:1000–1009. doi: 10.1172/JCI59816. An excellent study from the Elmquist laboratory that dissected the physiological role of leptin by manipulating OBRs in mouse models. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hill JW, et al. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. 2010;11:286–297. doi: 10.1016/j.cmet.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williams KW, et al. Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. J Neurosci. 2010;30:2472–2479. doi: 10.1523/JNEUROSCI.3118-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Levin BE. Metabolic sensing neurons and the control of energy homeostasis. Physiol Behav. 2006;89:486–489. doi: 10.1016/j.physbeh.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 44.Wang R, et al. Effects of oleic acid on distinct populations of neurons in the hypothalamic arcuate nucleus are dependent on extracellular glucose levels. J Neurophysiol. 2006;95:1491–1498. doi: 10.1152/jn.00697.2005. [DOI] [PubMed] [Google Scholar]
- 45.Lam TK, Gutierrez-Juarez R, Pocai A, Rossetti L. Regulation of blood glucose by hypothalamic pyruvate metabolism. Science. 2005;309:943–947. doi: 10.1126/science.1112085. [DOI] [PubMed] [Google Scholar]
- 46.Obici S, et al. Central administration of oleic acid inhibits glucose production and food intake. Diabetes. 2002;51:271–275. doi: 10.2337/diabetes.51.2.271. [DOI] [PubMed] [Google Scholar]
- 47.Su Y, et al. Hypothalamic leucine metabolism regulates liver glucose production. Diabetes. 2012;61:85–93. doi: 10.2337/db11-0857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parton LE, et al. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature. 2007;449:228–232. doi: 10.1038/nature06098. [DOI] [PubMed] [Google Scholar]
- 49.Levin BE, Routh VH, Kang L, Sanders NM, Dunn-Meynell AA. Neuronal glucosensing: what do we know after 50 years? Diabetes. 2004;53:2521–2528. doi: 10.2337/diabetes.53.10.2521. [DOI] [PubMed] [Google Scholar]
- 50.Pocai A, Obici S, Schwartz GJ, Rossetti L. A brain-liver circuit regulates glucose homeostasis. Cell Metab. 2005;1:53–61. doi: 10.1016/j.cmet.2004.11.001. In this paper, the Rossetti group shows that central inhibition of fat oxidation is dependent on the activation of KATP channels. [DOI] [PubMed] [Google Scholar]
- 51.Pocai A, et al. Hypothalamic KATP channels control hepatic glucose production. Nature. 2005;434:1026–1031. doi: 10.1038/nature03439. [DOI] [PubMed] [Google Scholar]
- 52.Kong D, et al. Glucose stimulation of hypothalamic MCH neurons involves KATP channels, is modulated by UCP2, and regulates peripheral glucose homeostasis. Cell Metab. 2010;12:545–552. doi: 10.1016/j.cmet.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Minokoshi Y, et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428:569–574. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- 54.Andersson U, et al. AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem. 2004;279:12005–12008. doi: 10.1074/jbc.C300557200. [DOI] [PubMed] [Google Scholar]
- 55.Hardie DG, Carling D. The AMP-activated protein kinase — fuel gauge of the mammalian cell? Eur J Biochem. 1997;246:259–273. doi: 10.1111/j.1432-1033.1997.00259.x. [DOI] [PubMed] [Google Scholar]
- 56.Perrin C, Knauf C, Burcelin R. Intracerebroventricular infusion of glucose, insulin, and the adenosine monophosphate-activated kinase activator, 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside, controls muscle glycogen synthesis. Endocrinology. 2004;145:4025–4033. doi: 10.1210/en.2004-0270. [DOI] [PubMed] [Google Scholar]
- 57.Claret M, et al. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J Clin Invest. 2007;117:2325–2336. doi: 10.1172/JCI31516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lindsley JE, Rutter J. Nutrient sensing and metabolic decisions. Comp Biochem Physiol B Biochem Mol Biol. 2004;139:543–559. doi: 10.1016/j.cbpc.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 59.Blouet C, Ono H, Schwartz GJ. Mediobasal hypothalamic p70 S6 kinase 1 modulates the control of energy homeostasis. Cell Metab. 2008;8:459–467. doi: 10.1016/j.cmet.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ono H, et al. Activation of hypothalamic S6 kinase mediates diet-induced hepatic insulin resistance in rats. J Clin Invest. 2008;118:2959–2968. doi: 10.1172/JCI34277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ryan KK, et al. A role for central nervous system PPAR-[gamma] in the regulation of energy balance. Nature Med. 2011;17:623–626. doi: 10.1038/nm.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Derosa G, Maffioli P. Effects of thiazolidinediones and sulfonylureas in patients with diabetes. Diabetes Technol Ther. 2010;12:491–501. doi: 10.1089/dia.2009.0172. [DOI] [PubMed] [Google Scholar]
- 63.Lu M, et al. Brain PPAR-γ promotes obesity and is required for the insulin-sensitizing effect of thiazolidinediones. Nature Med. 2011;17:618–622. doi: 10.1038/nm.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Diano S, et al. Peroxisome proliferation-associated control of reactive oxygen species sets melanocortin tone and feeding in diet-induced obesity. Nature Med. 2011;17:1121–1127. doi: 10.1038/nm.2421. Interesting study showing a role for hypothalamic reactive oxygen species in the modulation of the melanocortin system. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.De Fanti BA, Hamilton JS, Horwitz BA. Meal-induced changes in extracellular 5-HT in medial hypothalamus of lean (Fa/Fa) and obese (fa/fa) Zucker rats. Brain Res. 2001;902:164–170. doi: 10.1016/s0006-8993(01)02371-x. [DOI] [PubMed] [Google Scholar]
- 66.Heisler LK, et al. Activation of central melanocortin pathways by fenfluramine. Science. 2002;297:609–611. doi: 10.1126/science.1072327. A seminal paper that demonstrated that the melanocortin neurocircuitry interacts with the 5-HT system to modulate energy balance. [DOI] [PubMed] [Google Scholar]
- 67.Nonogaki K, Strack AM, Dallman MF, Tecott LH. Leptin-independent hyperphagia and type 2 diabetes in mice with a mutated serotonin 5-HT2C receptor gene. Nature Med. 1998;4:1152–1156. doi: 10.1038/2647. [DOI] [PubMed] [Google Scholar]
- 68.Zhou L, et al. Serotonin 2C receptor agonists improve type 2 diabetes via melanocortin-4 receptor signaling pathways. Cell Metab. 2007;6:398–405. doi: 10.1016/j.cmet.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ader M, et al. Metabolic dysregulation with atypical antipsychotics occurs in the absence of underlying disease: a placebo-controlled study of olanzapine and risperidone in dogs. Diabetes. 2005;54:862–871. doi: 10.2337/diabetes.54.3.862. [DOI] [PubMed] [Google Scholar]
- 70.Kiss J, Leranth C, Halasz B. Serotoninergic endings on VIP-neurons in the suprachiasmatic nucleus and on ACTH-neurons in the arcuate nucleus of the rat hypothalamus. A combination of high resolution autoradiography and electron microscopic immunocytochemistry. Neurosci Lett. 1984;44:119–124. doi: 10.1016/0304-3940(84)90068-5. [DOI] [PubMed] [Google Scholar]
- 71.Xu Y, Elmquist JK, Fukuda M. Central nervous control of energy and glucose balance: focus on the central melanocortin system. Ann NY Acad Sci. 2011;1243:1–14. doi: 10.1111/j.1749-6632.2011.06248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xu Y, et al. 5-HT2CRs expressed by pro-opiomelanocortin neurons regulate energy homeostasis. Neuron. 2008;60:582–589. doi: 10.1016/j.neuron.2008.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wade JM, et al. Synergistic impairment of glucose homeostasis in ob/ob mice lacking functional serotonin 2C receptors. Endocrinology. 2008;149:955–961. doi: 10.1210/en.2007-0927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sohn JW, et al. Serotonin 2C receptor activates a distinct population of arcuate pro-opiomelanocortin neurons via TRPC channels. Neuron. 2011;71:488–497. doi: 10.1016/j.neuron.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Qiu J, Fang Y, Ronnekleiv OK, Kelly MJ. Leptin excites proopiomelanocortin neurons via activation of TRPC channels. J Neurosci. 2010;30:1560–1565. doi: 10.1523/JNEUROSCI.4816-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xu Y, et al. A serotonin and melanocortin circuit mediates D-fenfluramine anorexia. J Neurosci. 2010;30:14630–14634. doi: 10.1523/JNEUROSCI.5412-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huszar D, et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 78.Obici S, et al. Central melanocortin receptors regulate insulin action. J Clin Invest. 2001;108:1079–1085. doi: 10.1172/JCI12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hetherington A, Ranson S. Hypothalamic lesions and adiposity in the rat. Anat Rec. 1940;78:149–172. [Google Scholar]
- 80.Rohner F, et al. Immediate effect of lesion of the ventromedial hypothalamic area upon glucose-induced insulin secretion in anaesthetized rats. Diabetologia. 1977;13:239–242. doi: 10.1007/BF01219706. [DOI] [PubMed] [Google Scholar]
- 81.Borg WP, et al. Ventromedial hypothalamic lesions in rats suppress counterregulatory responses to hypoglycemia. J Clin Invest. 1994;93:1677–1682. doi: 10.1172/JCI117150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Klockener T, et al. High-fat feeding promotes obesity via insulin receptor/PI3K-dependent inhibition of SF-1 VMH neurons. Nature Neurosci. 2011;14:911–918. doi: 10.1038/nn.2847. One of many excellent papers from the Bruning laboratory, defining insulin action in SF1 and VMH neurons. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dhillon H, et al. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron. 2006;49:191–203. doi: 10.1016/j.neuron.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 84.Bingham NC, Anderson KK, Reuter AL, Stallings NR, Parker KL. Selective loss of leptin receptors in the ventromedial hypothalamic nucleus results in increased adiposity and a metabolic syndrome. Endocrinology. 2008;149:2138–2148. doi: 10.1210/en.2007-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang R, et al. Selective inactivation of Socs3 in SF1 neurons improves glucose homeostasis without affecting body weight. Endocrinology. 2008;149:5654–5661. doi: 10.1210/en.2008-0805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tong Q, et al. Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cell Metab. 2007;5:383–393. doi: 10.1016/j.cmet.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Grill HJ. Distributed neural control of energy balance: contributions from hindbrain and hypothalamus. Obesity. 2006;14:216S–221S. doi: 10.1038/oby.2006.312. [DOI] [PubMed] [Google Scholar]
- 88.Grill HJ, Ginsberg AB, Seeley RJ, Kaplan JM. Brainstem application of melanocortin receptor ligands produces long-lasting effects on feeding and body weight. J Neurosci. 1998;18:10128–10135. doi: 10.1523/JNEUROSCI.18-23-10128.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rossi J, et al. Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metab. 2011;13:195–204. doi: 10.1016/j.cmet.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cheung GW, Kokorovic A, Lam CK, Chari M, Lam TK. Intestinal cholecystokinin controls glucose production through a neuronal network. Cell Metab. 2009;10:99–109. doi: 10.1016/j.cmet.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 91.Lam TK, et al. Hypothalamic sensing of circulating fatty acids is required for glucose homeostasis. Nature Med. 2005;11:320–327. doi: 10.1038/nm1201. [DOI] [PubMed] [Google Scholar]
- 92.DiRocco RJ, Grill HJ. The forebrain is not essential for sympathoadrenal hyperglycemic response to glucoprivation. Science. 1979;204:1112–1114. doi: 10.1126/science.451558. [DOI] [PubMed] [Google Scholar]
- 93.Hisadome K, Reimann F, Gribble FM, Trapp S. CCK stimulation of GLP-1 neurons involves α1-adrenoceptor-mediated increase in glutamatergic synaptic inputs. Diabetes. 2011;60:2701–2709. doi: 10.2337/db11-0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rasmussen BA, et al. Duodenal activation of cAMP-dependent protein kinase induces vagal afferent firing and lowers glucose production in rats. Gastroenterology. 2012;142:834–843.e3. doi: 10.1053/j.gastro.2011.12.053. [DOI] [PubMed] [Google Scholar]
- 95.Strader AD, Woods SC. Gastrointestinal hormones and food intake. Gastroenterology. 2005;128:175–191. doi: 10.1053/j.gastro.2004.10.043. [DOI] [PubMed] [Google Scholar]
- 96.Wang PY, et al. Upper intestinal lipids trigger a gut-brain-liver axis to regulate glucose production. Nature. 2008;452:1012–1016. doi: 10.1038/nature06852. [DOI] [PubMed] [Google Scholar]
- 97.Lam CK, et al. Activation of N-methyl-D-aspartate (NMDA) receptors in the dorsal vagal complex lowers glucose production. J Biol Chem. 2010;285:21913–21921. doi: 10.1074/jbc.M109.087338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shimizu I, Hirota M, Ohboshi C, Shima K. Identification and localization of glucagon-like peptide-1 and its receptor in the brain. Endocrinology. 1987;121:1076–1082. doi: 10.1210/endo-121-3-1076. [DOI] [PubMed] [Google Scholar]
- 99.Barrera JG, Sandoval DA, D’Alessio DA, Seeley RJ. GLP-1 and energy balance: an integrated model of short-term and long-term control. Nature Rev Endocrinol. 2011;7:507–516. doi: 10.1038/nrendo.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Prigeon RL, Quddusi S, Paty B, D’Alessio DA. Suppression of glucose production by GLP-1 independent of islet hormones: a novel extrapancreatic effect. Am J Physiol Endocrinol Metab. 2003;285:E701–E707. doi: 10.1152/ajpendo.00024.2003. [DOI] [PubMed] [Google Scholar]
- 101.Sandoval DA, Bagnol D, Woods SC, D’Alessio DA, Seeley RJ. Arcuate glucagon-like peptide 1 receptors regulate glucose homeostasis but not food intake. Diabetes. 2008;57:2046–2054. doi: 10.2337/db07-1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cani PD, et al. Improvement of glucose tolerance and hepatic insulin sensitivity by oligofructose requires a functional glucagon-like peptide 1 receptor. Diabetes. 2006;55:1484–1490. doi: 10.2337/db05-1360. [DOI] [PubMed] [Google Scholar]
- 103.Kieffer TJ, Habener JF. The glucagon-like peptides. Endocr Rev. 1999;20:876–913. doi: 10.1210/edrv.20.6.0385. [DOI] [PubMed] [Google Scholar]
- 104.Vahl TP, et al. Glucagon-like peptide-1 (GLP-1) receptors expressed on nerve terminals in the portal vein mediate the effects of endogenous GLP-1 on glucose tolerance in rats. Endocrinology. 2007;148:4965–4973. doi: 10.1210/en.2006-0153. [DOI] [PubMed] [Google Scholar]
- 105.Amato A, et al. Peripheral motor action of glucagon-like peptide-1 through enteric neuronal receptors. Neurogastroenterol Motil. 2010;22:664–e203. doi: 10.1111/j.1365-2982.2010.01476.x. [DOI] [PubMed] [Google Scholar]
- 106.Aponte Y, Atasoy D, Sternson SM. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nature Neurosci. 2011;14:351–355. doi: 10.1038/nn.2739. The first demonstration of the use of optogenetics in the field of energy balance. It used optogenetics to modulate AGRP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Krashes MJ, et al. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest. 2011;121:1424–1428. doi: 10.1172/JCI46229. A ground-breaking study in the field of energy balance that demonstrated the power of DREADD technology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science. 2005;310:683–685. doi: 10.1126/science.1115524. [DOI] [PubMed] [Google Scholar]
- 109.Moore MC, et al. Sources of carbon for hepatic glycogen synthesis in the conscious dog. J Clin Invest. 1991;88:578–587. doi: 10.1172/JCI115342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Xie H, Lautt WW. Insulin resistance caused by hepatic cholinergic interruption and reversed by acetylcholine administration. Am J Physiol. 1996;271:E587–E592. doi: 10.1152/ajpendo.1996.271.3.E587. [DOI] [PubMed] [Google Scholar]
- 111.Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Fructose-mediated stress signaling in the liver: implications for hepatic insulin resistance. J Nutr Biochem. 2007;18:1–9. doi: 10.1016/j.jnutbio.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 112.Bagger JI, Knop FK, Holst JJ, Vilsbøll T. Glucagon antagonism as a potential therapeutic target in type 2 diabetes. Diabetes Obes Metab. 2011;13:965–971. doi: 10.1111/j.1463-1326.2011.01427.x. [DOI] [PubMed] [Google Scholar]
- 113.American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2010;33:S62–S69. doi: 10.2337/dc10-S062. [DOI] [PMC free article] [PubMed] [Google Scholar]