

Abstract
Alzheimer’s disease (AD) is the most common form of dementia. At the diagnostic stage, the AD brain is characterized by the accumulation of extracellular amyloid plaques, intracellular neurofibrillary tangles and neuronal loss. Despite the large variety of therapeutic approaches, this condition remains incurable, since at the time of clinical diagnosis, the brain has already suffered irreversible and extensive damage. In recent years, it has become evident that AD starts decades prior to its clinical presentation. In this regard, transgenic animal models can shed much light on the mechanisms underlying this “pre-clinical” stage, enabling the identification and validation of new therapeutic targets. This paper summarizes the formidable efforts to create models mimicking the various aspects of AD pathology in the rat. Transgenic rat models offer distinctive advantages over mice. Rats are physiologically, genetically and morphologically closer to humans. More importantly, the rat has a well-characterized, rich behavioral display. Consequently, rat models of AD should allow a more sophisticated and accurate assessment of the impact of pathology and novel therapeutics on cognitive outcomes.
Keywords: Alzheimer’s disease, Rat, Transgenic, Behavior, Early pathology, Amyloid, Intracellular, Mature plaques, Tau, Neuronal loss
Introduction
Despite important advances in our understanding of the molecular basis of Alzheimer’s disease (AD) neuropathology and the large variety of novel therapeutic approaches attempted, this condition remains incurable. The recent failure of passive immunotherapy clinical trials (Bapineuzumab and Solanezeumab) best illustrates the challenges that lay ahead [1]. It seems that the main obstacle to an effective therapy is that, at the time of clinical diagnosis, the brain has already suffered extensive and perhaps irreparable damage.
At the diagnostic stage, the AD brain is characterized by abundant senile amyloid plaques, formed by extracellular aggregates of amyloid-beta (Aβ) peptides, and by neurofibrillary tangles (NFTs) consisting of intracellular aggregates of abnormally phosphorylated tau (p-tau) protein (reviewed in [2-4]). The neurodegenerative changes in the central nervous system (CNS) neurons and synapses ultimately lead to the progressive cognitive decline characteristic of AD [5].
There is also substantive evidence indicating that CNS pro-inflammatory mechanisms contribute to cognitive impairment [6,7]. Inflammatory processes in AD have been classically regarded as an Aβ plaque-induced event, characterized by activation of microglia and astrocytes surrounding the plaques [8,9].
In recent years, it has become evident that AD starts decades prior to its clinical presentation, based on biomarker studies in familial and sporadic forms of the disease [10-12]. Exploring the neuropathology of AD in such human “pre-clinical” stages is not an easy task. Transgenic animal models can shed much light on the many factors impacting or accelerating disease progression. They can also be of value to identify new biomarkers or potential new therapeutic targets. Transgenic rat models of the AD-like pathology offer distinctive advantages, as will be discussed below.
Transgenic models of AD
Our knowledge of the molecular mechanisms underlying AD pathogenesis has made a leap forward with the creation of the first transgenic AD mouse models in the mid 1990s [13-15]. Based on the identification of the mutations in the amyloid precursor protein (APP), presenilin 1 (PS1) and presenilin 2 (PS2) genes involved in the familial forms of AD, these models reflect various aspects of AD including amyloid accumulation, abnormal tau phosphorylation, inflammation or behavioral deficits [13-17].
Transgenic mouse models have made an important contribution to testing the so-called “Amyloid Hypothesis”. Furthermore, they have played a role in assessing a large variety of disease-modifying compounds. Given the progressive and predictable evolution of the pathology, the use of transgenic models also offers the opportunity to find reliable biomarkers, crucial in detecting the disease at the early, “silent”, asymptomatic stage, during which therapeutic interventions would have much greater possibilities of success.
Although transgenic mouse models have proven highly valuable in elucidating the mechanisms involved in AD, the recent failure of AD immunotherapy highlights the necessity of developing superior models of the AD pathology. The ideal transgenic model should mimic multiple aspects of the disease including its etiology and a time-dependent progression of the pathology, involving similar structures and cells similar to the human pathology. Most importantly, however, the model should provide reliable, longitudinal readings about the status of higher brain function by applying suitable learning and memory tests, analysis of body fluids, such as cerebrospinal fluid, and imaging. Such models should be free of confounding factors such as impairments in visual or motor systems, which can be falsely interpreted as AD deficits provoked by nonspecific transgene- or strain-effects.
Advantages of using rats: the rat as a model for AD
Historically, mice were preferred over rats for transgenesis mainly due to technical reasons. Compared to mice, rat one-cell embryos have less visible pronuclei and more flexible plasma and pronuclear membranes, making transgene injection in pronuclei more difficult [18]. The low survival of embryos following injection also contributes to making rat transgenesis more demanding and time-consuming [18]. Additionally, tools for manipulation of the rat genome are less readily available [19]. Until recently, embryonic stem (ES) cell-based targeting technology, a powerful tool allowing gene replacement (knock-in) or loss of function mutations (knock-out), was not available, as viable rat ES cells had been difficult to obtain [20].
Despite these drawbacks, rats offer numerous advantages compared to mice. The rat is physiologically, genetically and morphologically closer to humans than mice [21-23]. Its larger body and brain size facilitates intrathecal administration of drugs, microdialysis, multiple sampling of cerebrospinal fluid, in vivo electrophysiology, as well as neurosurgical and neuroimaging procedures [19]. Of relevance for AD modeling, similarly to humans, the rat contains 6 isoforms of tau [24], although the ratio of 4R/3R tau isoforms is different (9:1 in rats; 1:1 in humans). In addition, there is good homology between the rat and human apoE amino acid sequences (73.5% with human apoE3, 73.9% with apoE4) [25,26]. However, while its sequence is more similar to apoE4, rat apoE displays the biophysical behavior of apoE3 [27].
Another major advantage of this species is that it is behaviorally well characterized. Rats have finer and more accurate motor coordination than mice and exhibit a richer behavioral display. They also display a more complex social behavior. They display juvenile play fighting and courtship as well as low levels of aggression [28]. Since the rat is a terrestrial, aquatic and arboreal mammal, it is more competent and less stressed in water-based navigation tasks such as the Morris water maze (MWM) [28]. These behavioral differences may be accounted for by the fact that rats, like humans, and opposed to mice, have a post-natal brain development that would lead to a greater number of synapses and a more complex synaptic organization [28]. Consequently, rat models of AD should allow a more sophisticated characterization at the behavioral level and thus enable a more accurate assessment of the impact of the pathology on cognitive outcomes. They should also enable a better assessment of the effects of potential therapeutics on cognition in longitudinal studies.
Based on these advantages, rats are increasingly and successfully used to mimic key pathological hallmarks of neurodegenerative diseases including Alzheimer’s (as discussed in this review), Parkinson’s (PD) [29-31], Huntington’s (HD) [32], amyotrophic lateral sclerosis [33] and tauopathies [34,35].
Importantly, it has been reported that some transgenic rat models offer a more accurate representation of the human disease compared to mice bearing the same transgene. This has been exemplified in hypertension [36] and atherosclerosis [37], as well as in models of neurodegenerative diseases. Thus, mouse models of HD can only mimic juvenile HD pathological changes whereas HD transgenic rats allow study of the common adult type of the disease [32]. Also, no significant loss of dopaminergic neurons is observed in the human alpha-synuclein transgenic mouse model of PD, but severe loss of the dopaminergic integrity is reported in human alpha-synuclein transgenic rats [31].
Early rat models of AD
Rats have played a prominent role in the modeling of AD, well before the advent of transgenesis. However, most of the models summarized in this section do not represent accurate model systems for AD as they do not exhibit neuritic plaques, NFTs or neuron loss. This is the case, for example, in aged rats, which reflect only some aspects of human aging, such as learning and memory impairments and moderate deficits in cortical cholinergic and dopaminergic function [38-40].
Chemical and lesion-induced rat models have been extensively used, particularly to test the cholinergic hypothesis of AD. This hypothesis states that CNS cholinergic deficits in elderly adults and demented patients are the main factors responsible for their cognitive impairments [41-43], and has led to the well-established, symptomatic, anticholinesterase therapies (for review see [44]).
A large variety of compounds have been used to induce AD-like cortical cholinergic neuronal loss with varying degrees of specificity. These include the relatively non-specific scopolamine [45,46] and the p75NTR-specific immunotoxin for cholinergic neurons, 192-IgG-saporin [47,48]. Several rat lesion models have been used, and include models of brain trauma [49], bilateral transection of the hippocampal fimbria-fornix [50], ovariectomy [51] and hyperthermia on the post-ischemic brain [52], the last creating AD-like pathology [52].
The finding that Aβ is central to the development of plaques [53] and is neurotoxic [54] has led to studies investigating the impact of Aβ on brain function in vivo. However, although cerebral infusion of Aβ in naïve rats can recapitulate some key features of human AD including cholinergic dysfunction, Aβ deposits, ventricular enlargement, neuron loss and behavior deficits, it can not simulate the progressive neurodegeneration characteristic of AD [55-58]. In addition, there is great inconsistency between Aβ infusion models, likely due to differences in methodology such as identity, type (fresh, presumably oligomeric, versus fibrillar forms) and the concentration of peptide administered, duration of the treatment and site of infusion. Therefore, while such models might be of value to examine specific aspects of the amyloid pathology, they fail to reproduce the full spectrum of AD neuropathological hallmarks.
Virus-mediated rat models of AD
More recently, the advance of virus-mediated gene transfer technology has allowed for the expression of human APP (hAPP)695 bearing the Swedish mutation in the adult rat hippocampus. This induced Aβ42 immunoreactivity and learning deficits in the MWM up to 12 months post-injection. However, the injection did not lead to Aβ plaque deposition, gliosis or neural loss [59]. Virally-mediated gene transfer of human Aβ42 and Aβ40 peptides bearing the British mutation into the hippocampus of adult Wistar rats has also helped to establish the contribution of each Aβ species in AD [60].
Transgenic rat models of AD
Since the early 2000s, a wide array of transgenic rats has been created based on the expression of human genes relevant for early-onset familial AD such as wild-type or mutated APP and mutated PS1. These models offer a large heterogeneity in their phenotype, which arises from several factors. First, the expression of these transgenes is controlled by different neuronal promoters resulting in varying expression strengths and patterns. In addition, these models were produced and stabilized in both inbred (Lewis and Fischer-344) or outbred (Sprague Dawley and Wistar) genetic backgrounds [18]. It is now well established that genetic background has a great influence on pathogenesis. For example, it was demonstrated that rat strain SHR72 expressing human truncated tau (151–391, 4R) under the control of the Thy1 promoter (Table 1) displayed different NFT load and neuroinflammation markers depending on the background in which it was stabilized [61]. Differences in expression strength can also result from the method used to introduce the transgene in fertilized eggs (pronuclear injection of DNA or lentiviral delivery). Most importantly, differences in phenotypes may be explained by the introduction of single, double or triple transgenes.
Table 1.
Transgenic rat models of Alzheimer’s disease
| Name | Transgene | Background | Amyloid pathology | Tau pathology | Other | References |
|---|---|---|---|---|---|---|
| UKUR28 |
hAPP751 Swe, Ind PDGF promoter |
Wistar (outbred) |
iAβ in cortex and hippocampus from 6 mo No plaques |
N/A |
Increased pERK2 |
[62,63] |
| UKUR25 |
hAPP751 Swe, Ind Human PS1 (M146L) PDGF promoter |
Wistar (outbred) |
iAβ in cortex and hippocampus from 6 mo No plaques |
Increased ptau (PHF-1) at 9 mo, no tangles |
Increased pERK2, decreased p- p90RSK Altered proteome (SELDI-TOFF MS) Altered subcellular compartments |
[62-65] |
| TgAPPswe |
hAPP751 Swe, PDGF promoter |
Fisher-344 (inbred) |
Increased APP mRNA (56%) and Aβ-40 and Aβ-42 peptides, no plaques |
N/A |
Better cognitive performance in MWM and STFP |
[68] |
| Tg6590 |
hAPP695 Swe, UbiquitinC promoter |
Sprague-Dawley (outbred) |
Increased APP products Cerebrovascular deposits at 15 mo Few diffuse plaques |
Increased ptau (PHF-1) at 15 mo, no tangles. |
Impairment in MWM and open-field |
[75,76] |
| hAPP695 |
hAPP695 wild-type, UbiquitinC promoter |
Wistar (outbred) |
Increased APP/ Aβ levels (2 fold) in cortex and hippocampus no plaques |
N/A |
Smaller infarct volume impairment in MWM and BWT after MCAO |
[72] |
| APP21 APP31 |
hAPP695 Swe, Ind, UbiquitinC promoter Lentiviral delivery |
Fisher-344 (inbred) |
Increased APP products and APP mRNA in brain (2.9 fold), kidneys and lungs Increased Aβ-40 and Aβ-42 in serum No plaques |
N/A |
|
[73,74] |
| PSAPP Tg478/ Tg1116/ Tg11587 |
hAPP695 Swe, Rat synapsin I promoter hAPP695 Swe, Lon, PDGFβ promoter Human PS1 (M146V), Rat synapsin I promoter |
Sprague-Dawley (inbred) |
Mostly diffuse plaques Few compact plaques in hippocampus. No vascular Aβ deposits Aβ load confirmed with (F-18) FDDNP microPET |
Increased ptau (AT8, PHF-1), no tangles |
Impairment in LTP and in MWM performance Activation of astrocytes and few microglia particularly around plaques No neuronal loss |
[77-79] |
| McGill-R-Thy1-APP |
hAPP751 Swe, Ind, Mouse Thy1.2 promoter |
Wistar (outbred) |
Progressive accumulation of iAβ in cortex and hippocampus from 1 week post-natal Aβ plaques starting at 6-9 mo |
N/A |
Dystrophic neurites and astrogliosis around plaques Progressive learning deficits (MWM) Altered metabolites (MRS) |
[81,85] |
| TgF344-AD |
hAPP695 Swe, Human PS1ΔE9 Mouse PrP promoter |
Fisher-344 (inbred) |
Progressive accumulation of iAβ, Aβ-40 and Aβ-42 and Aβ plaques |
Increased ptau (CP-13, pTau-PADRE and others) and Gallyas-positive NFT |
Deficits in open-field, NOR, BM Presence of dystrophic neurites, activated astrocytes and microglia around plaques. Neuronal loss |
[91] |
| SHR72 and SHR318 |
Human tau truncated (151-391, 4R) Mouse Thy1 promoter |
SHR (inbred) |
N/A |
Increased ptau (AT8) Tangles in brainstem |
Deficits in MWM and BWT Impaired reflex responses no neuronal loss in brain, axonal damage in the brain stem and spinal cord, decreased lifespan |
[35,95,96] |
| SHR24 | Human tau truncated (151-391, 3R) Mouse Thy1 promoter |
SHR (inbred) | N/A | Increased ptau (DC11 and others) Tangles in cortex |
No neuronal loss in cortex and hippocampus, decreased lifespan | [97] |
BM Barnes maze, BWT beam walking test, IHC immunohistochemistry, MCAO middle cerebral artery occlusion, mo months-old, MRS magnetic resonance spectroscopy, MWM Morris water maze, N/A information not available, NFT neurofibrillary tangles, NOR novel object recognition, PET positron emission tomography, STFP social transmission of food preference.
Rat models of amyloid pathology devoid of plaques
The earliest transgenic rat models of AD showed accumulation of intracellular Aβ (iAβ) but no senile plaques. It was suggested that this was due to inadequate Aβ levels, since higher concentrations are required to initiate the deposition process. Some of these models however, did display synaptic dysfunction (LTP and behavior) supporting the view that cognitive deficits are independent of plaque formation but correlate better with Aβ oligomers and other Aβ species.
Our first attempts to generate rat models of AD failed to reproduce the classical AD pathological hallmarks (Table 1) [62-65]. This can be attributed to moderate levels of APP gene expression. However, UKUR25 and UKUR28 transgenic rat strains showed an important accumulation of intracellular Aβ (iAβ)-immunoreactive material in pyramidal neurons of the neocortex and in CA2 and CA3 regions of the hippocampus. These models significantly contributed in supporting the role of iAβ in the amyloid cascade at the early, pre-plaque phase of the amyloid pathology. Indeed, they confirmed in vivo that the accumulation of iAβ material, in the absence of plaques, induces deregulated ERK2 activation [62,63] as previously demonstrated in vitro[66,67]. Furthermore, they also demonstrate that iAβ is sufficient to trigger the initial steps of the tau-phosphorylation cascade, learning impairments in the MWM task [62,63], significant changes in the hippocampal proteome, particularly in synaptic proteins implicated in learning and memory formation [65] and morphological alterations in the Golgi apparatus, lysosomes and lipofuscin bodies [64].
Intracellular iAβ accumulation was observed in other rat models of amyloid pathology. Similarly to our model, TgAPPswe rats do not develop extracellular plaques or NFTs up to 18 months of age [68]. These rats show a mild increase in APP mRNA (56.8% at 12 months). In contrast with UKUR25, these rats perform better at 6 and 12 months old in two hippocampus-dependent tasks, the MWM and the social transmission of food preference task when compared with non-transgenic animals. This discrepancy can be explained by differences in the genetic background and the transgene expressed (Table 1), as TgAPPswe rats do not carry the APP Indiana and the PS1 Finn mutations, which could result in lower iAβ levels. Accordingly, the authors suggest a dose-dependent effect of APP, which would play a role in normal learning and memory processes at low doses but would lead to neurodegeneration and cognitive decline at higher doses [68].
Models expressing wild-type or mutated hAPP isoform 695 (hAPP695) have also been attempted. This choice is justified, as it is regarded as the isoform preferentially expressed by neurons. In addition, it was reported that there is a selective loss of APP695 transcripts in the brain of AD-affected patients [69]. However, it has been shown in vitro and in transgenic mouse models that overexpression of hAPP751 causes more AD-like pathology and cognitive impairments than hAPP695 [70,71].
Trangenesis with wild-type hAPP695 results in a two-fold increase of APP/Aβ fragments in cortex and hippocampus compared to non-transgenic animals, leading to behavioral impairments after middle cerebral artery occlusion [72]. Higher levels of APP expression (2.9 fold more APP mRNA) were first achieved in the brain of APP21 and APP31 inbred models which were created by injecting recombinant lentivirus carrying the hAPP695 with the Swedish and Indiana mutations into zygotes (Table 1) [73]. These models have substantial quantities of Aβ40 and 42 in serum, and especially so in homozygous animals. Circulating Aβ most likely does not arise from the brain, as the models also express high levels of APP mRNA in the kidneys, heart and lungs. Desspite of the high levels of human APP in neurons of the cortex and hippocampus, they do not develop extracellular deposits of Aβ [74]. However, senile plaques and cerebral Aβ angiopathy can be observed 9 months after the cerebral injection of dilute brain extracts from AD patients into APP21 animals, suggesting that Aβ deposition can be exogenously seeded if the host expresses human Aβ [74].
A third rat model expressing hAPP695 with the Swedish mutation (Table 1) has shown an accumulation of iAβ in neurons of the cortex, hippocampus and cerebellum and an increased amount of soluble Aβ material. At 9 months old, Tg6590 exhibits impaired spatial learning in the MWM and altered spontaneous activity in the open-field [75]. In addition, magnetic resonance imaging (MRI) suggests a tendency towards enlargement of the lateral ventricles at 11 months old [75]. At 15 months of age, these rats show Aβ cerebrovascular deposits, rare diffuse plaques, and tau hyperphosphorylation at PHF-1 site without the formation of mature plaques or NFTs even by the age of 22 months [75,76].
Rat models of amyloid pathology with mature plaques
The first model to develop amyloid plaques was achieved in homozygous double transgenic rats Tg478/Tg1116 expressing hAPP695 carrying the Swedish and Swedish/London mutations. These rats show increased APP, Aβ40 and Aβ42 load and developed diffuse plaques by 17–18 months of age [77]. The age of plaque onset was accelerated to 9 months by introducing a third transgene carrying a human mutated presenilin gene [77,78]. From the age of 9 months-old, PSAPP rats (also named Tg478/Tg1116/Tg11587) display abundant diffuse plaques in the cortex, hippocampus, olfactory bulb, thalamus and hypothalamus but not in the cerebellum or brain-stem (Table 1). However, only few compact plaques are detectable in the hippocampus even at 22 months-old and no vascular Aβ deposits are observed. The progressive accumulation of Aβ plaques was confirmed with the use of [F-18]FDDNP micro positron emission tomography [79]. Astrocytic and light microglial activation and tau hyperphosphorylation is present around compact plaques. These rats also show impaired LTP accompanied by progressive behavior deficits in the MWM task, detectable at 7 months of age, in the absence of plaques. Behavior deficits correlate with Aβ42 load in the hippocampus. However, these rats lack neurofibrillary pathology or neuronal loss [78]. Its use as an efficient AD model is also hampered by a tendency towards premature death related to kidney disease, hypertension and immunosuppression, which are likely a consequence of the genetic disturbance caused by the presence of the triple transgenes [80].
The McGill-R-Thy1-APP rat model is the only model able to reproduce extensive AD-like amyloid pathology with a single transgene (Figure 1) [81]. This model expresses the hAPP751, bearing the Swedish and Indiana mutations under the control of the murine Thy1.2 promoter. In the McGill-R-Thy1-APP transgenic rat, a single transgene is able to produce human APP expression specifically in AD-relevant areas of the brain without cerebellar and peripheral tissue expression. The presence of a single transgene with a low copy number makes of this rat the least genetically aggressive AD transgenic model developed so far.
Figure 1.
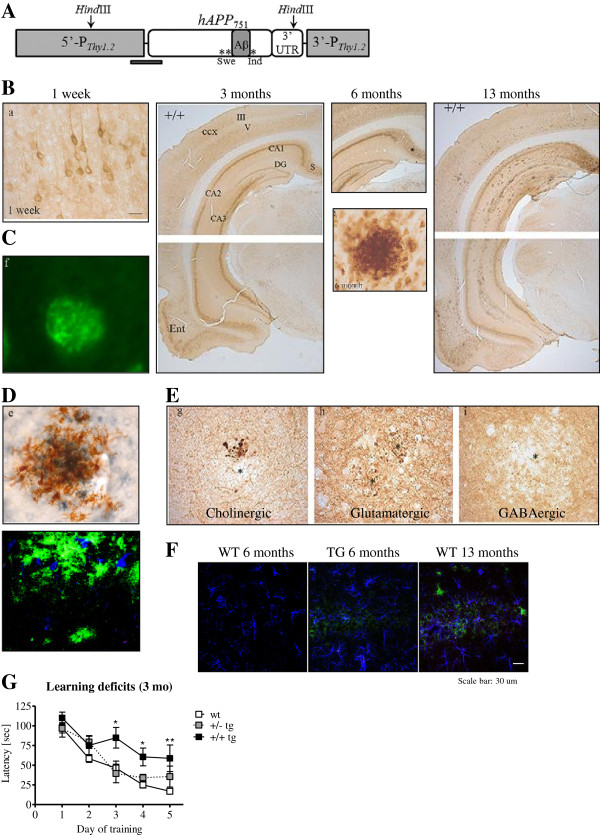
The McGill-R-Thy1-APP transgenic rat phenotype. (A) The McGill-R-Thy1-APP transgenic rat expresses the human APP751, bearing the Swedish and Indiana mutations under the control of the murine Thy1.2 promoter. Its phenotype is fairly similar to the human pathology reported in AD and MCI. (B) We observe intraneuronal Aβ accumulation starting at 1 week post-natal, as determined with our murine monoclonal antibody (McSA1) against the N-terminus of the Aβ peptide. The development of plaques follows the same anatomical sequence as in humans. Mature amyloid plaques are Thioflavin S-positive (C) and are surrounded by activated microglia as observed with MHCII- (brown) and Aβ-specific antibodies (McSA1-blue) and also with Iba-1(blue) and McSA1(blue) (D). Plaques are also accompanied by dystrophic neurites (E) and astrogliosis (GFAP-blue, McSA1-green) (F). (G) These rats already show learning deficits in the Morris water maze task at the pre-plaque stage (3 months old) and these deficits progress with amyloid accumulation. Images adapted from [81] with the publisher’s permission and from [83].
McGill-R-Thy1-APP rats display iAβ as soon as one week post natal in the cortex and hippocampus in both hemi and homozygous animals. The oligomeric nature of the iAβ material was confirmed using the NU-1 monoclonal antibody that specifically recognizes soluble aggregates of Aβ [82]. The pathology is dose-dependent as, in homozygous animals, iAβ accumulation leads to progressive neuritic plaque deposition starting from 6–9 months old. Hemizygous rats develop no or very few plaques at much later stages. The anatomical spreading of plaques coincides with that observed in human AD, starting from the subiculum and expanding to the entorhinal cortex and hippocampus. The AD-like pathological phenotype also includes the presence of transmitter-specific dystrophic neurites (Figure 1) [81]. Moreover, we have recently observed a recruitment of microglia towards iAβ-burdened hippocampal neurons and a remarkable pre-plaque astrogliosis [83]. It is of significance that the McGill-R-Thy1-APP rat presents progressive behavior impairments in the MWM starting at 3 months of age while no amyloid plaques are yet present. The deficits are transgene-dose-dependent and they correlate with the abundance of the 12kDa 6E10-immureactive band likely corresponding to a combination of Aβ trimers and the APP C-terminal fragment, C99 [81]. This observation reinforces the hypothesis on the impact of oligomeric iAβ in cognition [84].
These rats were also used to study metabolite levels by magnetic resonance spectroscopy in the dorsal hippocampus and frontal cortex. The findings demonstrate complex metabolite alterations during the progression of the amyloid pathology, different from those observed during normal aging [85]. MRI imaging on these rats also showed marked brain shrinkage, which is more evident for the hippocampal complex and resting-state connectivity impairment [86-88]. Consistent with these observations, McGill-R-Thy1-APP rats display impairments in firing rates for place discrimination of spatial context [89] and a very compelling in vivo impairment in hippocampal LTP formation at preplaque stages [90].
More recently, a bigenic TgF344-AD rat has been reported (Table 1) [91]. These rats express hAPP695, with the Swedish mutation, and PS1ΔE9 under the control of the strong murine PrP promoter. These rats demonstrate strong age-dependent accumulation of iAβ, soluble and insoluble Aβ40 and Aβ42 peptides and thioflavin-positive amyloid plaques. The amyloidosis is associated with hyperactivity in the open-field as well as age-dependent deficits in spatial learning and memory as assessed with the novel object recognition and the Barnes maze tasks. Surprisingly, as it was never seen before in other transgenic rat models of AD and even in transgenic AD mouse models expressing APP and PS1 mutations, by 16 months of age these rats present Gallyas-positive structures resembling NFTs seen in human AD. These structures contain p-tau as detected with several p-tau antibodies. The observed amyloidosis and tauopathy are accompanied by neuronal loss. These rats also present glial activation as early as 6 months old, before appreciable extracellular Aβ deposition [91].
Rat models of tau pathology
Several transgenic mouse models expressing mutated forms of human tau develop neurofibrillary degeneration [92,93]. Because the rat contains 6 tau isoforms, as do humans, rat models of tau pathology were also created (Table 1) [35,94-97]. Overexpression of human non-mutated truncated tau encompassing 4 repeat domains (151–391, 4R) in neurons leads to a hyperphosphorylation of tau and the development of neurofibrillary degeneration similar to that reported in AD [35]. Behavior analysis highlighted a progressive cognitive decline in spatial navigation in MWM, as well as disturbances in sensorimotor and reflex responses [95]. These impairments correlate with the progressive accumulation of argyrophilic NFTs and mature sarcosyl-insoluble tau complexes and extensive axonal damage in the brain stem and spinal cord. However, although hyperphosphorylated tau was observed in cortex and hippocampus, no neuronal loss or tangles were observed in the brain [95]. These impairments lead to decreased lifespan [35,96]. The first rat model developing progressive NFTs in the cortex expresses a human non-mutated truncated tau encompassing 3 repeat domains (151–391, 3R). These rats develop progressive cortical neurofibrillary degeneration as early as 9 months of age [97]. Surprisingly, this rat does not show neuronal death in the cortex, the region with the largest accumulation of tangles, or the hippocampus, the region presenting the highest expression of human tau. However, the neurofibrillar pathology leads to decreased lifespan. More models of tau transgenesis are likely to appear in coming years.
Comparison of transgenic rat and mouse models of AD
It has been more complicated to achieve AD-like amyloid deposition in the brain of transgenic rats than mice. The elevation of soluble Aβ or the extent of plaque accumulation is often less in rat (Table 1) than in mouse models expressing similar constructs, resulting in less aggressive phenotypes. Accordingly, Tg6590 [75,76] (Table 1), fail to develop mature plaques despite displaying some cognitive impairments. Conversely, its equivalent in mice, Tg2576, which expresses hAPP695 bearing the Swedish mutations under the control of the PrP promoter, presents cognitive decline accompanied by numerous Aβ plaques [98]. Similarly, TgAPPswe rats show very slight increases in Aβ peptide production with no plaque development [68] while TgAPP23 mice expressing hAPP751 with the Swedish mutations under the control of the Thy-1 promoter show typical plaques by 6 months of age accompanied by neuritic and synaptic degeneration [99]. Furthermore, TgCRND8 mice [100] expressing hAPP695 with the Swedish and Indiana mutations under the control of the PrP promoter develop early and extensive plaque deposition by 3 months of age while APP21 and APP31 rats [73,74] never accumulate extracellular amyloid. In these cases, differences in phenotype might arise from differences in the promoters used in mice and rats. In support to this, TgAPP(Sw,V717F) mice [101] and UKUR28 rats [62,63] expressing hAPP751with the Swedish and Indiana mutations driven by the PDGF promoter have similar phenotypes despite an absence of Aβ42 and plaque accumulation.
However, there are other cases where the exact same construct used in the two species resulted in different phenotypes indicating that species-specific factors likely contribute to these phenotype differences. For example, McGill-Thy1-APP mice [102] and McGill-Thy1-APP rats [81] expressing exactly the same construct containing hAPP751 with the Swedish and Indiana mutations under the control of the Thy1 promoter develop a similar phenotype. However, it is far more aggressive in mice (plaques at 4 months) than in the rat (plaques at 6–9 months), the latter needing to be homozygous to develop plaques. This might be explained by the number of copies of the transgene inserted in the host DNA. While few transgene copies are present in McGill rats [81], McGill mice [102] contain more than 20 copies of the transgene. Phenotypic differences were also observed in TgF344-AD rats [91] and Tg-APPswe/PS1dE9 mice [103,104], both expressing hAPP695 with the Swedish mutations under PrP regulatory control. These models show extensive and progressive amyloid pathology accompanied by cognitive deficits and some tau pathology. However, TgF344-AD rats have greater abundance of soluble oligomeric Aβ species. Also, while TgF344-AD rats display NFT-like structures and frank neuronal loss, these are not observed in Tg-APPswe/PS1dE9 mice.
Concluding remarks and future directions
Transgenesis in rats offers great potential to decipher subtle and early aspects of AD pathology. The rat is, in many aspects, closer to humans than mice. Because of its predictable and multi-faceted behavioral display, this species is of great value for accurate cognitive assessment. The current rat transgenic models, while replicating all aspects of the amyloid pathology including inflammation and the presence of dystrophic neurites, remain incomplete models as it does not develop “bona fide” human-like neurofibrillary tangles. Despite these weaknesses, current transgenic rat models of AD are contributing significantly to our understanding of processes and mechanisms involved in the disease progression. However, as the available AD rat models display varying degrees of competence in recapitulating the characteristics and severity of the pathology, the choice of the model is crucial for the investigative objectives. Also, although the CNS of rats is closer to humans than that of the mouse, the extrapolation of conclusions from rat to humans would require a measure of caution. These transgenic models are closer to the pathology elicited in familial AD (both genetically driven), however, models with a minimal genetic invasiveness, such as the McGill-R-Thy1-APP, offer the closest available analogy to the human sporadic AD pathology. Despite their obvious limitations, transgenic rat models will be of great assistance in the search of potential biomarkers signaling an early, preclinical, pathology and in the search and validation of novel therapies. From the experimental point of view, they have already shown to be of significant advantage for in vivo electrophysiology, imaging, proteomics, epigenetics and in the future for optogenetic studies.
Abbreviations
AD: Alzheimer’s disease; Aβ: Amyloid-β peptide; APP: Amyloid precursor protein; CNS: Central nervous system; ERK: Extracellular-signal-regulated kinases; HD: Huntington’s disease; MWM: Morris water maze; MRI: Magnetic resonance imaging; NFTs: Neurofibrillary tangles; PD: Parkinson’s disease; PHF: Paired helical filaments; PS1: Presenilin 1; PS2: Presenilin 2.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SDC and ACC contributed to the writing and revising of the manuscript. Both authors read and approved the final manuscript.
Contributor Information
Sonia Do Carmo, Email: sonia.docarmo@mail.mcgill.ca.
A Claudio Cuello, Email: claudio.cuello@mcgill.ca.
Acknowledgements
This research is supported by the Canadian Institute for Health and Research grant MOP102752 to AC Cuello. AC Cuello is the holder of the McGill University Charles E. Frosst-Merck Chair in Pharmacology. S Do Carmo is the holder of the Charles E. Frosst-Merck Post-Doctoral Fellowship. The authors also acknowledge the support from Dr Alan C. Frosst and the Frosst family.
References
- Callaway E. Alzheimer’s drugs take a new tack. Nature. 2012;489:13–14. doi: 10.1038/489013a. [DOI] [PubMed] [Google Scholar]
- Cuello AC. In: Pharmacological Mechanisms in Alzheimer's Therapeutics. Cuello AC, editor. New York: Springer Science and Business Media; 2007. Overview of the Alzheimer’s Disease Pathology and Potential Therapeutic Targets; pp. 1–27. [Google Scholar]
- Hardy JA, Mann DM, Wester P, Winblad B. An integrative hypothesis concerning the pathogenesis and progression of Alzheimer’s disease. Neurobiol Aging. 1986;7:489–502. doi: 10.1016/0197-4580(86)90086-2. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron. 1991;6:487–698. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Barrientos RM, Higgins EA, Sprunger DB, Watkins LR, Rudy JW, Maier SF. Memory for context is impaired by a post context exposure injection of interleukin-1 beta into dorsal hippocampus. Behav Brain Res. 2002;134:291–298. doi: 10.1016/S0166-4328(02)00043-8. [DOI] [PubMed] [Google Scholar]
- Yaffe K, Lindquist K, Penninx BW, Simonsick EM, Pahor M, Kritchevsky S, Launer L, Kuller L, Rubin S, Harris T. Inflammatory markers and cognition in well-functioning African-American and white elders. Neurology. 2003;61:76–80. doi: 10.1212/01.WNL.0000073620.42047.D7. [DOI] [PubMed] [Google Scholar]
- Dandrea MR, Reiser PA, Gumula NA, Hertzog BM, Andrade-Gordon P. Application of triple immunohistochemistry to characterize amyloid plaque-associated inflammation in brains with Alzheimer’s disease. Biotech Histochem. 2001;76:97–106. [PubMed] [Google Scholar]
- Mehlhorn G, Hollborn M, Schliebs R. Induction of cytokines in glial cells surrounding cortical beta-amyloid plaques in transgenic Tg2576 mice with Alzheimer pathology. Int J Dev Neurosci. 2000;18:423–431. doi: 10.1016/S0736-5748(00)00012-5. [DOI] [PubMed] [Google Scholar]
- Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC. Dominantly Inherited Alzheimer Network. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR Jr, Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, Phelps CH. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:280–892. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W, Xu S, Eckman C, Younkin S, Price D, Iadecola C, Brent Clark H, Carlson G. Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron. 1995;15:1203–1218. doi: 10.1016/0896-6273(95)90107-8. [DOI] [PubMed] [Google Scholar]
- Quon D, Wang Y, Catalano R, Scardina JM, Murakami K, Cordell B. Formation of beta-amyloid protein deposits in brains of transgenic mice. Nature. 1991;352:239–241. doi: 10.1038/352239a0. [DOI] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah F, McConologue L, Montoya-Zavala M, Mucke L, Paganini L, Penniman E, Power M, Schenk D, Seubert P, Snyder B, Soriano F, Tan H. et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O’Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Charreau B, Tesson L, Soulillou JP, Pourcel C, Anegon I. Transgenesis in rats: technical aspects and models. Transgenic Res. 1996;5:223–234. doi: 10.1007/BF01972876. [DOI] [PubMed] [Google Scholar]
- Tesson L, Cozzi J, Ménoret S, Rémy S, Usal C, Fraichard A, Anegon I. Transgenic modifications of the rat genome. Transgenic Res. 2005;14:531–546. doi: 10.1007/s11248-005-5077-z. [DOI] [PubMed] [Google Scholar]
- Tong C, Li P, Wu NL, Yan Y, Ying QL. Production of p53 gene knockout rats by homologous recombination in embryonic stem cells. Nature. 2010;467:211–213. doi: 10.1038/nature09368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, Scott G, Steffen D, Worley KC, Burch PE, Okwuonu G, Hines S, Lewis L, DeRamo C, Delgado O, Dugan-Rocha S, Miner G, Morgan M, Hawes A, Gill R, Celera Holt RA, Adams MD, Amanatides PG, Baden-Tillson H, Barnstead M, Chin S, Evans CA, Ferriera S, Fosler C. et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428:493–521. doi: 10.1038/nature02426. [DOI] [PubMed] [Google Scholar]
- Jacob HJ, Kwitek AE. Rat genetics: attaching physiology and pharmacology to the genome. Nat Rev Genet. 2002;3:33–42. doi: 10.1038/nrg702. [DOI] [PubMed] [Google Scholar]
- Lin JH. Species similarities and differences in pharmacokinetics. Drug Metab Dispos. 1995;23:1008–1021. [PubMed] [Google Scholar]
- Hanes J, Zilka N, Bartkova M, Caletkova M, Dobrota D, Novak M. Rat tau proteome consists of six tau isoforms: implication for animal models of human tauopathies. J Neurochem. 2009;108:1167–1176. doi: 10.1111/j.1471-4159.2009.05869.x. [DOI] [PubMed] [Google Scholar]
- McLean JW, Fukazawa C, Taylor JM. Rat apolipoprotein E mRNA. Cloning and sequencing of double-stranded cDNA. J Biol Chem. 1983;258:8993–9000. [PubMed] [Google Scholar]
- Rajavashisth TB, Kaptein JS, Reue KL, Lusis AJ. Evolution of apolipoprotein E: mouse sequence and evidence for an 11-nucleotide ancestral unit. Proc Natl Acad Sci U S A. 1985;82:8085–8089. doi: 10.1073/pnas.82.23.8085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran TN, Kim SH, Gallo C, Amaya M, Kyees J, Narayanaswami V. Biochemical and biophysical characterization of recombinant rat apolipoprotein E: similarities to human apolipoprotein E3. Arch Biochem Biophys. 2013;529:18–25. doi: 10.1016/j.abb.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whishaw IQ, Metz GA, Kolb B, Pellis SM. Accelerated nervous system development contributes to behavioral efficiency in the laboratory mouse: a behavioral review and theoretical proposal. Dev Psychobiol. 2001;39:151–170. doi: 10.1002/dev.1041. [DOI] [PubMed] [Google Scholar]
- Taravini IR, Chertoff M, Cafferata EG, Courty J, Murer MG, Pitossi FJ, Gershanik OS. Pleiotrophin over-expression provides trophic support to dopaminergic neurons in parkinsonian rats. Mol Neurodegener. 2011;6:40. doi: 10.1186/1750-1326-6-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura Y, Watanabe S, Taguchi M, Takagi K, Kawata T, Takahashi-Niki K, Yasui H, Maita H, Iguchi-Ariga SM, Ariga H. Neuroprotective effect of a new DJ-1-binding compound against neurodegeneration in Parkinson’s disease and stroke model rats. Mol Neurodegener. 2011;6:48. doi: 10.1186/1750-1326-6-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuber S, Harmuth F, Kohl Z, Adame A, Trejo M, Schönig K, Zimmermann F, Bauer C, Casadei N, Giel C, Calaminus C, Pichler BJ, Jensen PH, Müller CP, Amato D, Kornhuber J, Teismann P, Yamakado H, Takahashi R, Winkler J, Masliah E, Riess O. A progressive dopaminergic phenotype associated with neurotoxic conversion of α-synuclein in BAC-transgenic rats. Brain. 2013;136:412–432. doi: 10.1093/brain/aws358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Hörsten S, Schmitt I, Nguyen HP, Holzmann C, Schmidt T, Walther T, Bader M, Pabst R, Kobbe P, Krotova J, Stiller D, Kask A, Vaarmann A, Rathke-Hartlieb S, Schulz JB, Grasshoff U, Bauer I, Vieira-Saecker AM, Paul M, Jones L, Lindenberg KS, Landwehrmeyer B, Bauer A, Li XJ, Riess O. Transgenic rat model of Huntington’s disease. Hum Mol Genet. 2003;12:617–624. doi: 10.1093/hmg/ddg075. [DOI] [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutantmediated amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RL, Dayton RD, Tatom JB, Diaczynsky CG, Salvatore MF. Tau expression levels from various adeno-associated virus vector serotypes produce graded neurodegenerative disease states. Eur J Neurosci. 2008;27:1615–1625. doi: 10.1111/j.1460-9568.2008.06161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilka N, Filipcik P, Koson P, Fialova L, Skrabana R, Zilkova M, Rolkova G, Kontsekova E, Novak M. Truncated tau from sporadic Alzheimer’s disease suffices to drive neurofibrillary degeneration in vivo. FEBS Lett. 2006;580:3582–3588. doi: 10.1016/j.febslet.2006.05.029. [DOI] [PubMed] [Google Scholar]
- Mullins JJ, Peters J, Ganten D. Fulminant hypertension in transgenic rats harbouring the mouse Ren-2 gene. Nature. 1990;344:541–544. doi: 10.1038/344541a0. [DOI] [PubMed] [Google Scholar]
- Herrera VL, Makrides SC, Xie HX, Adari H, Krauss RM, Ryan US, Ruiz-Opazo N. Spontaneous combined hyperlipidemia, coronary heart disease and decreased survival in Dahl salt-sensitive hypertensive rats transgenic for human choles-teryl ester transfer protein. Nat Med. 1999;5:1383–1389. doi: 10.1038/70956. [DOI] [PubMed] [Google Scholar]
- Gilad GM, Gilad VH. Age-related reductions in brain cholinergic and dopaminergic indices in two rat strains differing in longevity. Brain Res. 1987;408:247–250. doi: 10.1016/0006-8993(87)90381-7. [DOI] [PubMed] [Google Scholar]
- Greene E, Naranjo JN. Degeneration of hippocampal fibers and spatial memory deficits in the aged rat. Neurobiol Aging. 1987;8:35–43. doi: 10.1016/0197-4580(87)90055-8. [DOI] [PubMed] [Google Scholar]
- Anderson JJ, Holtz G, Baskin PP, Wang R, Mazzarelli L, Wagner SL, Menzaghi F. Reduced cerebrospinal fluid levels of alpha-secretase-cleaved amyloid precursor protein in aged rats: correlation with spatial memory deficits. Neuroscience. 1999;93:1409–1420. doi: 10.1016/S0306-4522(99)00244-4. [DOI] [PubMed] [Google Scholar]
- Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR. Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215:1237–1239. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- Quirion R, Martel JC, Robitaille Y, Etienne P, Wood P, Nair NP, Gauthier S. Neurotransmitter and receptor deficits in senile dementia of the Alzheimer type. Can J Neurol Sci. 1986;13(Suppl 4):503–510. doi: 10.1017/s0317167100037215. [DOI] [PubMed] [Google Scholar]
- Bierer LM, Haroutunian V, Gabriel S, Knott PJ, Carlin LS, Purohit DP, Perl DP, Schmeidler J, Kanof P, Davis KL. Neurochemical correlates of dementia severity in Alzheimer’s disease: relative importance of the cholinergic deficits. J Neurochem. 1995;64:749–760. doi: 10.1046/j.1471-4159.1995.64020749.x. [DOI] [PubMed] [Google Scholar]
- Giacobini E. Cholinesterase inhibitors: new roles and therapeutic alternatives. Pharmacol Res. 2004;50:433–440. doi: 10.1016/j.phrs.2003.11.017. [DOI] [PubMed] [Google Scholar]
- Pazzagli A, Pepeu G. Amnesic properties of scopolamine and brain acetylcholine in the rat. Int J Neuropharmacol. 1965;4:291–299. doi: 10.1016/0028-3908(65)90025-0. [DOI] [PubMed] [Google Scholar]
- Nitta A, Katono Y, Itoh A, Hasegawa T, Nabeshima T. Nicotine reverses scopolamine-induced impairment of performance in passive avoidance task in rats through its action on the dopaminergic neuronal system. Pharmacol Biochem Behav. 1994;49:807–812. doi: 10.1016/0091-3057(94)90227-5. [DOI] [PubMed] [Google Scholar]
- McGaughy J, Everitt BJ, Robbins TW, Sarter M. The role of cortical cholinergic afferent projections in cognition: impact of new selective immunotoxins. Behav Brain Res. 2000;115:251–263. doi: 10.1016/S0166-4328(00)00262-X. [DOI] [PubMed] [Google Scholar]
- Rossner S, Wörtwein G, Gu Z, Yu J, Schliebs R, Bigl V, Perez-Polo JR. Cholinergic control of nerve growth factor in adult rats: evidence from cortical cholinergic deafferentation and chronic drug treatment. J Neurochem. 1997;69:947–953. doi: 10.1046/j.1471-4159.1997.69030947.x. [DOI] [PubMed] [Google Scholar]
- Iwata A, Chen XH, McIntosh TK, Browne KD, Smith DH. Long-term accumulation of amyloid-beta in axons following brain trauma without persistent upregulation of amyloid precursor protein genes. J Neuropathol Exp Neurol. 2002;61:1056–1068. doi: 10.1093/jnen/61.12.1056. [DOI] [PubMed] [Google Scholar]
- Liu J, Zhang Z, Li JT, Zhu YH, Zhou HL, Liu S, Wang TH. Effects of NT-4 gene modified fibroblasts transplanted into AD rats. Neurosci Lett. 2009;466:1–5. doi: 10.1016/j.neulet.2009.09.020. [DOI] [PubMed] [Google Scholar]
- Feng Z, Cheng Y, Zhang JT. Long-term effects of melatonin or 17 beta-estradiol on improving spatial memory performance in cognitively impaired, ovariectomized adult rats. J Pineal Res. 2004;37:198–206. doi: 10.1111/j.1600-079X.2004.00158.x. [DOI] [PubMed] [Google Scholar]
- Sinigaglia-Coimbra R, Cavalheiro EA, Coimbra CG. Postischemic hyperthermia induces Alzheimer-like pathology in the rat brain. Acta Neuropathol. 2002;103:444–452. doi: 10.1007/s00401-001-0487-3. [DOI] [PubMed] [Google Scholar]
- Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/S0006-291X(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Pike CJ, Walencewicz AJ, Glabe CG, Cotman CW. Aggregation-related toxicity of synthetic beta-amyloid protein in hippocampal cultures. Eur J Pharmacol. 1991;207:367–368. doi: 10.1016/0922-4106(91)90014-9. [DOI] [PubMed] [Google Scholar]
- Harkany T, O’Mahony S, Keijser J, Kelly JP, Kónya C, Borostyánkoi ZA, Görcs TJ, Zarándi M, Penke B, Leonard BE, Luiten PG. Beta-amyloid(1–42)-induced cholinergic lesions in rat nucleus basalis bidirectionally modulate serotonergic innervation of the basal forebrain and cerebral cortex. Neurobiol Dis. 2001;8:667–678. doi: 10.1006/nbdi.2001.0398. [DOI] [PubMed] [Google Scholar]
- Li DB, Tang J, Fan XT, Song M, Xu HW, Bai Y. Comparative study of histopathology changes between the PS1/APP double transgenic mouse model and Abeta1-40 -injected rat model of Alzheimer disease. Neurosci Bull. 2006;22:52–57. [PubMed] [Google Scholar]
- Nag S, Yee BK, Tang F. Chronic intracerebroventricular infusion of beta-amyloid (1–40) results in a selective loss of neuropeptides in addition to a reduction in choline acetyltransferase activity in the cortical mantle and hippocampus in the rat. Ann N Y Acad Sci. 1999;897:420–422. doi: 10.1111/j.1749-6632.1999.tb07911.x. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Murayama N, Noshita T, Annoura H, Ohno T. Progressive brain dysfunction following intracerebroventricular infusion of beta(1–42)-amyloid peptide. Brain Res. 2001;912:128–136. doi: 10.1016/S0006-8993(01)02704-4. [DOI] [PubMed] [Google Scholar]
- Gong Y, Meyer EM, Meyers CA, Klein RL, King MA, Hughes JA. Memory-related deficits following selective hippocampal expression of Swedish mutation amyloid precursor protein in the rat. Exp Neurol. 2006;200:371–377. doi: 10.1016/j.expneurol.2006.02.136. [DOI] [PubMed] [Google Scholar]
- Lawlor PA, Bland RJ, Das P, Price RW, Holloway V, Smithson L, Dicker BL, During MJ, Young D, Golde TE. Novel rat Alzheimer’s disease models based on AAV-mediated gene transfer to selectively increase hippocampal Abeta levels. Mol Neurodegener. 2007;2:11. doi: 10.1186/1750-1326-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stozicka Z, Zilka N, Novak P, Kovacech B, Bugos O, Novak M. Genetic background modifies neurodegeneration and neuroinflammation driven by misfolded human tau protein in rat model of tauopathy: implication for immunomodulatory approach to Alzheimer’s disease. J Neuroinflammation. 2010;7:64. doi: 10.1186/1742-2094-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echeverria V, Ducatenzeiler A, Alhonen L, Janne J, Grant SM, Wandosell F, Muro A, Baralle F, Li H, Duff K, Szyf M, Cuello AC. Rat transgenic models with a phenotype of intracellular Abeta accumulation in hippocampus and cortex. J Alzheimers Dis. 2004;6:209–219. doi: 10.3233/jad-2004-6301. [DOI] [PubMed] [Google Scholar]
- Echeverria V, Ducatenzeiler A, Dowd E, Jänne J, Grant SM, Szyf M, Wandosell F, Avila J, Grimm H, Dunnett SB, Hartmann T, Alhonen L, Cuello AC. Altered mitogen-activated protein kinase signaling, tau hyperphosphorylation and mild spatial learning dysfunction in transgenic rats expressing the beta-amyloid peptide intracellularly in hippocampal and cortical neurons. Neuroscience. 2004;29:583–592. doi: 10.1016/j.neuroscience.2004.07.036. [DOI] [PubMed] [Google Scholar]
- Lopez EM, Bell KF, Ribeiro-da-Silva A, Cuello AC. Early changes in neurons of the hippocampus and neocortex in transgenic rats expressing intracellular human a-beta. J Alzheimers Dis. 2004;6:421–431. doi: 10.3233/jad-2004-6410. [DOI] [PubMed] [Google Scholar]
- Vercauteren FG, Clerens S, Roy L, Hamel N, Arckens L, Vandesande F, Alhonen L, Janne J, Szyf M, Cuello AC. Early dysregulation of hippocampal proteins in transgenic rats with Alzheimer’s disease-linked mutations in amyloid precursor protein and presenilin 1. Brain Res Mol Brain Res. 2004;132:241–259. doi: 10.1016/j.molbrainres.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Echeverria V, Ducatenzeiler A, Chen CH, Cuello AC. Endogenous beta-amyloid peptide synthesis modulates cAMP response element-regulated gene expression in PC12 cells. Neuroscience. 2005;135:1193–1202. doi: 10.1016/j.neuroscience.2005.06.057. [DOI] [PubMed] [Google Scholar]
- Grant SM, Morinville A, Maysinger D, Szyf M, Cuello AC. Phosphorylation of mitogen-activated protein kinase is altered in neuroectodermal cells overexpressing the human amyloid precursor protein 751 isoform. Brain Res Mol Brain Res. 1999;72:115–120. doi: 10.1016/S0169-328X(99)00157-6. [DOI] [PubMed] [Google Scholar]
- Ruiz-Opazo N, Kosik KS, Lopez LV, Bagamasbad P, Ponce LR, Herrera VL. Attenuated hippocampus-dependent learning and memory decline in transgenic TgAPPswe Fischer-344 rats. Mol Med. 2004;10:36–44. doi: 10.2119/2003-00044.herrera. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SA, Rogers J, Finch CE. APP-695 transcript prevalence is selectively reduced during Alzheimer’s disease in cortex and hippocampus but not in cerebellum. Neurobiol Aging. 1989;10:755–760. doi: 10.1016/0197-4580(89)90017-1. [DOI] [PubMed] [Google Scholar]
- Higgins LS, Catalano R, Quon D, Cordell B. Transgenic mice expressing human beta-APP751, but not mice expressing beta-APP695, display early Alzheimer’s disease-like histopathology. Ann N Y Acad Sci. 1993;695:224–227. doi: 10.1111/j.1749-6632.1993.tb23056.x. [DOI] [PubMed] [Google Scholar]
- Ho L, Fukuchi K, Younkin SG. The alternatively spliced Kunitz protease inhibitor domain alters amyloid beta protein precursor processing and amyloid beta protein production in cultured cells. J Biol Chem. 1996;271:30929–30934. doi: 10.1074/jbc.271.48.30929. [DOI] [PubMed] [Google Scholar]
- Clarke J, Thornell A, Corbett D, Soininen H, Hiltunen M, Jolkkonen J. Overexpression of APP provides neuroprotection in the absence of functional benefit following middle cerebral artery occlusion in rats. Eur J Neurosci. 2007;26:1845–1852. doi: 10.1111/j.1460-9568.2007.05807.x. [DOI] [PubMed] [Google Scholar]
- Agca C, Fritz JJ, Walker LC, Levey AI, Chan AW, Lah JJ, Agca Y. Development of transgenic rats producing human beta-amyloid precursor protein as a model for Alzheimer’s disease: transgene and endogenous APP genes are regulated tissue-specifically. BMC Neurosci. 2008;9:28. doi: 10.1186/1471-2202-9-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen RF, Fritz JJ, Dooyema J, Cintron AF, Hamaguchi T, Lah JJ, LeVine H 3rd, Jucker M, Walker LC. Exogenous seeding of cerebral β-amyloid deposition in βAPP-transgenic rats. J Neurochem. 2012;120:660–666. doi: 10.1111/j.1471-4159.2011.07551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloskowska E, Pham TM, Nilsson T, Zhu S, Oberg J, Codita A, Pedersen LA, Pedersen JT, Malkiewicz K, Winblad B, Folkesson R, Benedikz E. Cognitive impairment in the Tg6590 transgenic rat model of Alzheimer’s disease. J Cell Mol Med. 2010;14:1816–1823. doi: 10.1111/j.1582-4934.2009.00809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkesson R, Malkiewicz K, Kloskowska E, Nilsson T, Popova E, Bogdanovic N, Ganten U, Ganten D, Bader M, Winblad B, Benedikz E. A transgenic rat expressing human APP with the Swedish Alzheimer’s disease mutation. Biochem Biophys Res Commun. 2007;358:777–782. doi: 10.1016/j.bbrc.2007.04.195. [DOI] [PubMed] [Google Scholar]
- Flood DG, Lin YG, Lang DM, Trusko SP, Hirsch JD, Savage MJ, Scott RW, Howland DS. A transgenic rat model of Alzheimer’s disease with extracellular Abeta deposition. Neurobiol Aging. 2009;30:1078–1090. doi: 10.1016/j.neurobiolaging.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Liu L, Orozco IJ, Planel E, Wen Y, Bretteville A, Krishnamurthy P, Wang L, Herman M, Figueroa H, Yu WH, Arancio O, Duff K. A transgenic rat that develops Alzheimer’s disease-like amyloid pathology, deficits in synaptic plasticity and cognitive impairment. Neurobiol Dis. 2008;31:46–57. doi: 10.1016/j.nbd.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng E, Kepe V, Frautschy SA, Liu J, Satyamurthy N, Yang F, Chen PP, Cole GB, Jones MR, Huang SC, Flood DG, Trusko SP, Small GW, Cole GM, Barrio JR. [F-18]FDDNP microPET imaging correlates with brain Aβ burden in a transgenic rat model of Alzheimer disease: effects of aging, in vivo blockade, and anti-Aβ antibody treatment. Neurobiol Dis. 2011;43:565–575. doi: 10.1016/j.nbd.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahorsky-Reeves J, Lawson G, Chu DK, Schimmel A, Ezell PC, Dang M, Couto M. Maintaining longevity in a triple transgenic rat model of Alzheimer’s disease. J Am Assoc Lab Anim Sci. 2007;46:124. [Google Scholar]
- Leon WC, Canneva F, Partridge V, Allard S, Ferretti MT, DeWilde A, Vercauteren F, Atifeh R, Ducatenzeiler A, Klein W, Szyf M, Alhonen L, Cuello AC. A novel transgenic rat model with a full Alzheimer’s-like amyloid pathology displays pre-plaque intracellular amyloid-beta-associated cognitive impairment. J Alzheimers Dis. 2010;20:113–126. doi: 10.3233/JAD-2010-1349. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Velasco PT, Chang L, Viola KL, Fernandez S, Lacor PN, Khuon D, Gong Y, Bigio EH, Shaw P, De Felice FG, Krafft GA, Klein WL. Monoclonal antibodies that target pathological assemblies of Abeta. J Neurochem. 2007;100:23–35. doi: 10.1111/j.1471-4159.2006.04157.x. [DOI] [PubMed] [Google Scholar]
- Hanzel CE, Pichet-Binette A, Cuello AC. Early inflammatory process in a novel transgenic rat model of Alzheimer’s disease. New Orleans, USA: Society for Neuroscience 2012; 2012. [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen LH, Melø TM, Saether O, Witter MP, Sonnewald U. Altered neurochemical profile in the McGill-R-Thy1-APP rat model of Alzheimer’s disease: a longitudinal in vivo 1 H MRS study. J Neurochem. 2012;123:532–541. doi: 10.1111/jnc.12003. [DOI] [PubMed] [Google Scholar]
- Parent M, Do Carmo S, Aliaga A, Cuello AC, Gauthier S, Rosa-Neto P. Amyloid accumulation and neurodegeneration in a rat model of Alzheimer’s disease. Miami, USA: 7th Human Amyloid Imaging meeting; 2013. [Google Scholar]
- Shin M, Gauthier S, Wang S, Parent M, Fonov V, Allard S, Kang MS, Do Carmo S, Cuello AC, Rosa-Neto P. Assessment of hippocampal volumetry in transgenic AD rat. Boston, USA: Alzheimer’s Association International Conference; 2013. [Google Scholar]
- Parent M, Shin M, Do Carmo S, Aliaga A, Gauthier S, Cuello AC, Rosa-Neto P. Amyloid-induced fMRI resting-state connectivity impairment in a rat model of Alzheimer’s disease. Boston, USA: Alzheimer’s Association International Conference; 2013. [Google Scholar]
- Blackshear AL, Goetz A, Slayyeh B, Wagner S, Leutgeb JK, Cuello AC, Leutgeb S. Changes in place fields in a transgenic rat model of Alzheimer’s disease precede mnemonic behavioral deficits in the Barnes maze. New Orleans, LA: Society for Neuroscience; 2012. [Google Scholar]
- Qi Y, Klyubin I, Steffen J, Cuello AC, Fuhrmann M, Rowan MJ. Transgenic Rats Overexpressing Human Amyloid Precursor Protein Show Early-life Impairment of Long-term Potentiation in vivo. Florence, Italy: 11th International Conference on Alzheimer’s and Parkinson’s Diseases; 2013. [Google Scholar]
- Cohen RM, Rezai-Zadeh K, Weitz TM, Rentsendorj A, Gate D, Spivak I, Bholat Y, Vasilevko V, Glabe CG, Breunig JJ, Rakic P, Davtyan H, Agadjanyan MG, Kepe V, Barrio JR, Bannykh S, Szekely CA, Pechnick RN, Town T. A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric aβ, and frank neuronal loss. J Neurosci. 2013;33:6245–6256. doi: 10.1523/JNEUROSCI.3672-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocanu MM, Nissen A, Eckermann K, Khlistunova I, Biernat J, Drexler D, Petrova O, Schönig K, Bujard H, Mandelkow E, Zhou L, Rune G, Mandelkow EM. The potential for beta-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J Neurosci. 2008;28:737–748. doi: 10.1523/JNEUROSCI.2824-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden M, Kotilinek L, Forster C, Paulson J, McGowan E, SantaCruz K, Guimaraes A, Yue M, Lewis J, Carlson G, Hutton M, Ashe KH. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L) J Neurosci. 2005;25:10637–10647. doi: 10.1523/JNEUROSCI.3279-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cente M, Filipcik P, Pevalova M, Novak M. Expression of a truncated tau protein induces oxidative stress in a rodent model of tauopathy. Eur J Neurosci. 2006;24:1085–1090. doi: 10.1111/j.1460-9568.2006.04986.x. [DOI] [PubMed] [Google Scholar]
- Hrnkova M, Zilka N, Minichova Z, Koson P, Novak M. Neurodegeneration caused by expression of human truncated tau leads to progressive neurobehavioural impairment in transgenic rats. Brain Res. 2007;1130:206–213. doi: 10.1016/j.brainres.2006.10.085. [DOI] [PubMed] [Google Scholar]
- Koson P, Zilka N, Kovac A, Kovacech B, Korenova M, Filipcik P, Novak M. Truncated tau expression levels determine life span of a rat model of tauopathy without causing neuronal loss or correlating with terminal neurofibrillary tangle load. Eur J Neurosci. 2008;28:239–246. doi: 10.1111/j.1460-9568.2008.06329.x. [DOI] [PubMed] [Google Scholar]
- Filipcik P, Zilka N, Bugos O, Kucerak J, Koson P, Novak P, Novak M. First transgenic rat model developing progressive cortical neurofibrillary tangles. Neurobiol Aging. 2012;33:1448–1456. doi: 10.1016/j.neurobiolaging.2010.10.015. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Bürki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, George-Hyslop PS, Westaway D. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276:21562–21570. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- Lee KW, Lee SH, Kim H, Song JS, Yang SD, Paik SG, Han PL. Progressive cognitive impairment and anxiety induction in the absence of plaque deposition in C57BL/6inbred mice expressing transgenic amyloid precursor protein. J Neurosci Res. 2004;76:572–580. doi: 10.1002/jnr.20127. [DOI] [PubMed] [Google Scholar]
- Ferretti MT, Partridge V, Leon WC, Canneva F, Allard S, Arvanitis DN, Vercauteren F, Houle D, Ducatenzeiler A, Klein WL, Glabe CG, Szyf M, Cuello AC. Transgenic mice as a model of pre-clinical Alzheimer’s disease. Curr Alzheimer Res. 2011;8:4–23. doi: 10.2174/156720511794604561. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/S1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- Liu Y, Yoo MJ, Savonenko A, Stirling W, Price DL, Borchelt DR, Mamounas L, Lyons WE, Blue ME, Lee MK. Amyloid pathology is associated with progressive monoaminergic neurodegeneration in a transgenic mouse model of Alzheimer’s disease. J Neurosci. 2008;28:13805–13814. doi: 10.1523/JNEUROSCI.4218-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]