

Abstract
Although gallstone and alcohol use have been considered the most common causes of acute pancreatitis, hundreds of frequently prescribed medications are associated with this disease state. The true incidence is unknown since there are few population based studies available. The knowledge of drug induced acute pancreatitis is limited by the availability and the quality of the evidence as the majority of data is extrapolated from case reports. Establishing a definitive causal relationship between a drug and acute pancreatitis poses a challenge to clinicians. Several causative agent classification systems are often used to identify the suspected agents. They require regular updates since new drug induced acute pancreatitis cases are reported continuously. In addition, infrequently prescribed medications and herbal medications are often omitted. Furthermore, identification of drug induced acute pancreatitis with new medications often requires accumulation of post market case reports. The unrealistic expectation for a comprehensive list of medications and the multifactorial nature of acute pancreatitis call for a different approach. In this article, we review the potential mechanisms of drug induced acute pancreatitis and provide the perspective of deductive reasoning in order to allow clinicians to identify potential drug induced acute pancreatitis with limited data.
Keywords: Drug-induced pancreatitis, Mechanism
Core tip: The knowledge of drug-induced acute pancreatitis (DIAP) is limited by the availability and the quality of the evidence. Potential publication bias may also impact our knowledge of DIAP. Several causative agent classification systems have been proposed, but they require regular updates. In addition, Infrequent prescribed medications and herbal medications are often omitted from those summarized lists. We review the potential mechanisms of DIAP and provide the perspective of deductive reasoning in order to allow clinicians to identify potential DIAP with limited data.
INTRODUCTION
Acute pancreatitis (AP) is an acute inflammatory condition of the pancreas that may extend to local and distant extra-pancreatic tissues. The annual incidence of AP in the United States is approximately 17 cases per 100000. Acute pancreatitis results in 100000 hospitalizations per year, based on previous reports[1]. An average of 2000 patients per year die from complications related to AP. Although gallstones and alcohol are responsible for more than 90% of all cases in adults, medications have been recognized as a potential cause of AP[2]. Since the first reported case with chlorthalidone and cortisone in the 1950s, hundreds of commonly prescribed medications from different classes have been reported to induce pancreatic damage. It is expected that the list of drug induced acute pancreatitis (DIAP) will continue to expand with newly approved medications, new cases identified for older agents, and the alternative medicines which have less clinical research support in general. While medications are considered as a common cause of AP, reports of DIAP range from 0.1%-2% of overall cases[2,3].
It is not clear if the true incidence of DIAP has been established due to a lack of mandatory adverse drug report (ADR) system to clinicians, potential publication bias, and the challenge to associate AP with medications. Data from clinical trials of new drugs usually are not informative due to the idiosyncratic character of DIAP. In general, idiosyncratic adverse drug reactions occur with a frequency lower than 1:10000[4]. It is extremely difficult to identify adverse reactions in phase I to phase III clinical investigational trials. Incretin mimetics have been recently introduced in the treatment of diabetes and are widely used in many countries. Incretin mimetics, including exenatide and sitagliptin, were reported to induce AP shortly after those were approved which resulted in warranted an FDA drug safety communication regarding those agents. However, a meta-analysis of randomized clinical trials of most incretin mimetics including sitagliptin did not show any relevant effect on the incidence of pancreatitis. Overall, the incidence was only 0.1% (22 pancreatitis cases found in a pool of 20312 patients). This is an example of the limitation of clinical trials in finding adverse event with low incidence such as DIAP[5].
The knowledge of DIAP is also limited by the availability and the quality of the evidence. It can be difficult to rule out other causes of DIAP, especially in patients who have multiple comorbidities, medications, and underlying risk factors. Since all reports depend on the judgment of the clinicians to exclude other possible causes, reporting more severe ADR has also lead to publication bias. Due to its rarity, most of the evidence comes from case reports of individual drugs and few from case control studies. With a lack of standard ADR reporting format, inadequate data collection in several domains, such as the drug dose, onset of DIAP relative to the use of the medication, and exclusions of other causes, makes it difficult to establish a true causality. In addition, a causal relationship between the agents and DIAP may be difficult to establish due to ethical and practical considerations of re-challenge with the suspected agents. Therefore, the definite relationship between DIAP and medications has only been established in no more than 6% of the agents that have been shown to cause DIAP[6]. Since the identification of DIAP has relied mostly on individual case reports, specific drugs instead of the entire class are usually noted, which makes it even more challenging to identify possible cases in a timely manner.
Potential publication bias may also impact our understanding of DIAP and influence how DIAP is being managed. New drugs or medications with known severe side effects are usually more closely monitored than those that have been in existence for a long time, infrequently prescribed, or considered harmless (i.e., over the counter medications or herbal supplements). Despite the low incidence of drug-induced AP, it is associated with higher morbidity, extended hospital stays, and increased healthcare cost[7]. Approximately 25% of the cases may require intensive care treatment[8]. Developing a systemic approach of identifying potential DIAP is warranted. The aim of this review is to offer a different perspective of approaching DIAP by examining the potential mechanisms of DIAP in order to allow clinicians to identity possible cases with limited data.
ETIOLOGY OF ACUTE PANCREATITIS
Acute pancreatitis is an inflammatory process of the pancreas with varying involvement of other regional tissues or remote organ systems. Gallstone and alcohol use have been considered the most common causes of acute pancreatitis. Gallstone-associated AP is mainly identified by imaging. Previous association with tobacco use is directly linked to alcohol abuse. More evidence associates tobacco use as another toxin that can be directly linked to both acute and recurrent pancreatitis. Other potential mechanical etiologies include periampullary pathologies including intraductal tumor or parasites that are possible in developing countries. In addition to the most common causes, other etiological risk factors for acute pancreatitis are associated with mechanical factors including pancreas divisum, endoscopic retrograde cholangiopancreatography and manometry, as well as trauma or surgical procedures near the pancreas.
Metabolic or systemic process such as hyperlipidemia, infection, and chronic hypercalcemia are well known causes of pancreatitis as well[9]. Infections and toxins, including viral etiologies: mumps, coxsackievirus, hepatitis B, cytomegalovirus, varicella-zoster, herpes simplex virus, human immunodeficiency virus. Bacteria such as Mycoplasma, Legionella, Leptospira, Salmonella, Aspergillus, Toxoplasma, Cryptosporidium and Ascaris are potential causes of AP. Last, but not least, vascular diseases and pregnancy are also described as causes for pancreatitis.
AP can occur if there is damage to the acinar cells and/or injury to the pancreatic duct that leads to inappropriate accumulation and activation of proenzymes within the pancreas. The activated pancreatic enzymes digest the cell membranes of the pancreas and activate an inflammatory response, which increases the vascular permeability of the pancreas. Hemorrhage, edema, ischemia, and necrosis can result[1,9]. Data from animal studies show that reduced exocytosis and premature fusion of zymogen granules to lysosomes in pancreatic exocrine cells may activate pancreatic proenzymes and lead to cellular autodigestion.
CLASSIFICATION OF CAUSATIVE AGENTS
It is difficult to determine if the effects are intrinsic for all members of a drug class despite reports of DIAP incidence within the class. Several classification systems have been proposed. A substantial number of medications are known to cause AP, however, the underlying mechanism is still not well understood. The classification systems rely on summarized lists of medications from previously published reviews to help make the diagnosis of DIAP. Mallory and Kern in 1980s classified drugs that may cause pancreatitis into three groups: definite, probable, or possible association with pancreatitis[5,9]. In order to improve the quality of evidence, different classification systems have also been proposed that categorized DIAP in classes based on the number of reports and re-challenge results[5,10].
Badalov et al[11] in 2007 expanded the classification system to five categories: Ia, Ib, II, III, and IV (Table 1). Classifications are based on the published reports from 1955 to 2005. Class Ia includes drugs with at least one case report, evidence of a positive re-challenge, and exclusion of other causes of AP. Class Ib is similar to class Ia, except that other causes of AP could not be ruled out. Criteria for class II drugs include at least four case reports with a consistent latency period for at least 75% of the cases. Class III drugs have at least two case reports but do not have re-challenge data or a consistent latency period. Finally, class IV drugs have one case report without re-challenge data. This classification provides a quick reference of potential causative agents based on the available data at the time of the review. However, regular updates of existing classification are needed since new cases of DIAP are reported continuously. Furthermore, infrequently prescribed medications and alternative medications are often omitted from these summarized lists.
Table 1.
Classification system of drug-induced acute pancreatitis according to Badalov et al[11]
| Definition | Example | |
| Class I drug | Ia: at least one case report, evidence of a positive re-challenge, and exclusion of other causes of AP | Codeine, cytarabine, dapsone, enalapril, furosemide, isoniazid, mesalamine, metronidazole, pentamidine, pravastatin, procainamide, simvastatin, sulfamethoxazole, sulindac, tetracycline, valproic acid |
| Ib: similar to class Ia, except that other causes of AP could not be ruled out | Amiodarone, azathioprine, dexamethasone, ifosfaide, lamivudine, losartan, 6-MP, premarin, TMP-SMZ | |
| Class II drugs | Include at least four case reports with a consistent latency period for at least 75% of the cases | Acetaminophen, Clozapine, DDI, erythromycin, estrogen, l-asparaginase, propofol, tamoxifen |
| Class III drug | At least two case reports but do not have re-challenge data or a consistent latency period | Alendronate, carbamazepine, ceftriaxone, clarithromycin, cyclosporin, hydrochlorothiazide, interferone/ribavirin, metformin, minocycline, naproxen, paclitaxel, prednisone, prednisolone |
| Class IV drug | One case report without re-challenge data | Ampicillin, cisplatin, colchicine, cyclophosphamide, diclofenac, doxorubicin, interleukin-2, octreotide, propoxyphene, rifampin, risperidone, sertaline, tacrolimus, vincristine |
AP: Acute pancreatitis; 6-MP: 6-mercaptopurine; TMP-SMZ: Trimethoprim and sulfamethoxazole.
Mechanism of DIAP
The majority of the reported DIAP cases seem to have an idiosyncratic character. Idiosyncratic reactions to drugs are adverse effects that are not directly related to pharmacodynamic mechanisms of the drugs. These adverse events can occur unpredictably via abnormal interactions between the drugs and the organism, which is usually mediated by immunologic or cytotoxic effects triggered by the drug or its metabolites in a specific organ, in this case, the pancreas[11]. Although the exact mechanism of DIAP is not always known, the pathogenesis should not differ from other causes of AP. It is believed that the pathogenesis of AP differs only in the injury mechanism. It consists of three steps: (1) premature activation of trypsin in acinar cells; (2) intrapancreatic inflammation; and (3) extrapancreatic inflammation[5]. Several mechanisms have been hypothesized including immune-mediated, direct pancreatic toxicity, pancreatic-duct constriction, influence of medication on the bile flow, thrombosis, metabolic effects, and hypersensitivity[12,13]. Mechanisms of DIAP is showed in Figure 1.
Figure 1.
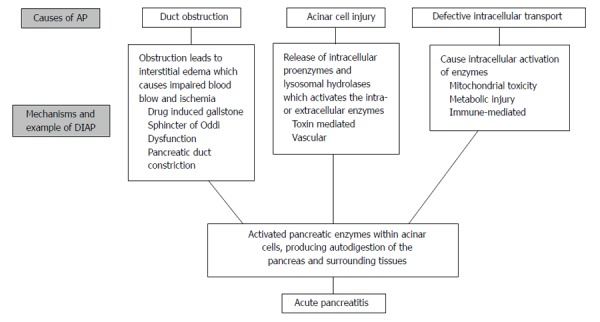
Mechanism of drug-induced pancreatitis. AP: Acute pancreatitis; DIAP: Drug-induced acute pancreatitis.
Researchers have also used latency to classify the potential mechanisms of DIAP[5]. It is hypothesized that direct immunological effects are usually observed within the first month of drug exposure, whereas toxic effects are noted after a few months of treatment. Potential mechanisms of DIAP include hypersensitivity (onset after four to eight weeks of use), accumulation of a toxic metabolite (onset after several months of use), hypertriglyceridemia (onset after several months of use), and intrinsic toxicity, which is sometimes related to overdose (onset may be almost immediate)[14]. However, there are exceptions that exist. Clinicians should not ignore long lasting medications while DIAP is a concern.
The following reviews summarize five major types of mechanisms of DIAP (Table 2), namely structural, toxins, metabolic, vascular, and other.
Table 2.
Mechanism of drug induced pancreatitis with drugs associated with acute pancreatitis
| Mechanism of DIAP | Drugs with a definite relationship or with class I/II to AP | Probable | Similar structure/class/mechanism with reported cases |
| Structural | Cholestatic liver injury | Rofecoxib | |
| Azathioprine | |||
| Cytarabine | |||
| Spasm of the sphincter of Oddi | Octreotide | Opium | |
| Opioids | Marcolides | ||
| Codeine | |||
| Erythromycin | |||
| Obstruction | ACE-inhibitors | ||
| Enalapril-angioedema | |||
| Duct constriction | NSAIDs | ||
| Sulindac | |||
| Stone | Ceftriaxone | ||
| Dipyridamole | |||
| Toxins | Acetaminophen | Metformin | Minocycline |
| Didanosine | Tigecycline | ||
| Isoniazid | Doxycycline | ||
| Metronidazole | NRTI | ||
| Valproic acid | HMG-CoA reductase inhibitors | ||
| Mesalamine | |||
| Pentamidine | |||
| Asparaginase | |||
| Sitaliptin | |||
| Exenatide | |||
| Tetracycline | |||
| Pravastatin | |||
| Metabolic | Hypertriglyceridemia | Hydrochlorothiazide | Isotretinoin |
| Estrogens | Interferon alfa | Retinoid derivaties | |
| Corticosteroids | Propofol | Protease inhibitors | |
| Furosemide | Tamoxifen | Saw palmetto | |
| β-blocker | Ethacrynic acid | ||
| Clomiphene | Anti-psychotics (aripiprazole, clozapine, olanzapine, quetiapine, risperidone) | ||
| Hypercalcemia | IV calcium | ||
| Vitamin D | |||
| Vascular | Contrast media - iopamidol Procainamide | ||
| Immune-mediated | Azathioprine/mercaptopurine sulfasalazine |
AP: Acute pancreatitis; NRTI: Nucleoside reverse transcriptase inhibitor; NSAIDs: Nonsteroidal anti-inflammatory drugs.
Structural
The structural damage such as compression, obstruction, or inflammation of the pancreatic duct may lead to AP. The most common cause for obstruction is choledochlithiasis, or gallstones. Obstruction can also be caused by duodenal inflammation in Crohn’s disease[1].
Medications with risk of gallstones: Ceftriaxone, a third-generation cephalosporin that is excreted from bile duct, has been associated with the development of sludge or stones in the gallbladders for some patients treated with this medication. Secondary pancreatitis has been considered in association with ceftriaxone-induced pseudolithiasis[15]. Unlike ceftriaxone, the kidney pathway is the major means of elimination for most of cephalosporins. It could potentially explain why DIAP has not been reported as class wide induced disease.
Based on an increased amount of cholesterol secreted in bile, causing an increased risk of gallstones which may explain the mechanism of 3-hydroxy-3-methyl-glutaryl-CoA reductase inhibitor-induced AP[10]. Long-term administration of dipyridamole and octreotide can form insoluble substances that precipitate in the gallbladder bile to promote gallstone formation as they are highly excreted from the bile[16,17].
Medications that Cause Sphincter of Oddi Dysfunction: As another example of structural disturbance, the Sphincter of Oddi (SO) is situated at the junction of the bile and pancreatic ducts where they enter the duodenum and serves to regulate the flow of bile and pancreatic juices as well as preventing reflux of duodenal contents into the pancreatobiliary system. SO dysfunction refers to two possible conditions-papillary stenosis (edema or hypertrophy) and dyskinesia (tachyoddia, induced spasm) that lead to partial or complete obstruction of the pancreatic duct resulting in pancreatitis[18]. SO dysfunction is implicated as a cause of various forms of AP including gallstone pancreatitis, pancreatitis secondary to alcohol, scorpion envenomation, and organophosphate poisoning. Medications such as octreotide, opioids, opium, and codeine reportedly induce AP in association with SO dysfunction[19,20]. Erythromycin can cause DIAP due to its prokinetic effect on the smooth muscle of the gastrointestinal track and the gallbladder subsequently increasing the pressure of the SO[21]. Since all macrolide antibiotics have prokinetic effect of different degrees, it is reasonable to consider that AP could potentially be drug related when patients are treated with these agents. As an example, clarithromycin and azithromycin have been reported to be associated with AP[6].
The mechanism of action of the class of drug is also an important factor when evaluating the relatedness of the adverse event to the drug. The probable mechanism of aspirin- or nonsteroidal anti-inflammatory drug (NSAID)- induced pancreatitis is due to inhibition of prostaglandins that otherwise may cause pancreatic duct constriction[12]. Aspirin is shown to increase pancreatic duct permeability in animal models. It increases calcium secretion from the pancreas, which is considered a marker of pancreatic damage. Experimental studies suggested that prostaglandins may have a protective effect on pancreatic cells[22]. Membrane stabilization of pancreatic cells may be the mechanism behind the cytoprotection conferred by prostaglandins. In NSAID-associated AP, sulindac seems to stand out as the individual drug from the class with the highest number of published cases[23-26].
Toxins
Cumulative dose-dependent effect of toxic metabolites is also hypothesized in drugs showing a consistent long latency (more than 30 d) at the onset of the first episode of DIAP such as valproic acid[5]. Below we discussed a few classic examples of toxin-mediated DIAP.
Nucleoside reverse transcriptase inhibitor: The leading hypothesis of nucleoside reverse transcriptase inhibitor (NRTI)-associated pancreatitis involves mitochondrial toxicity caused by the inhibition of human mitochondrial DNA polymerase-gamma[27]. This inhibition leads to impaired oxidative phosphorylation and failure to synthesize ATP, which is vital for energy-requiring reactions within the cell. Tissues with the highest energy demand appear to be most susceptible. Mitochondrial toxicity is shared among nucleoside analogues and AP attributed to these agents has been described[28]. The degree of mitochondrial impairment and the resultant tissue-specific clinical manifestations vary depending on the NRTI. Among non-nucleoside reverse transcriptase inhibitors, nevirapine is associated with pancreas-related toxicities, whereas efavirenz is not[29,30].
Metronidazole: One speculative mechanism of metronidazole-induced pancreatitis is that under aerobic conditions, it may undergo redox cycling and yield hydrogen peroxide, superoxide, and other free radicals, which can be toxic to pancreatic beta cells and induce pancreatitis[31].
Pentamidine: Pentamidine has a cytotoxic effect on pancreatic β-cells isle and can cause hypoglycemia or hyperglycemia[32]. Same effect can be expected on acinar pancreatic cells.
L-asparaginase: Animal models suggested that L-asparaginase -induced pancreatic injury can involve disruption of the plasma amino acid balance. Disruption of protein synthesis in acinar cells can cause inhibition of exocytosis following the histologic morphologic changes[33].
Tetracycline: Medications in the tetracycline class, including tetracycline, minocycline, and oxytetracycline, are also associated with AP[34-37]. Tetracycline-induced fatty metamorphosis of the liver usually accompanies evidence of pancreatitis, but pancreatitis without evidence of liver disease has also been observed after administration of tetracycline. Steinberg hypothesized that accumulation of an unidentified toxic metabolite may be the cause of tetracycline-induced pancreatitis[38]. Others suggest that high biliary concentration of tetracycline may be associated with tetracycline-induced pancreatitis[39]. Bile concentrations of minocycline after a 200 mg loading dose followed by one single 100 mg dose were observed to be more than 10 times higher than concurrent serum concentrations (mean serum concentration 0.65 mcg/mL, range 0.07-1.85 mcg/mL)[40]. Tigecycline, the first available member of the glycylcycline group, is a derivate of minocycline and can share similar side effects. Concentrations of tigecycline in bile (median 75.2 mg/L, range 15.9-1150 mg/L) were also found to be several logs greater than concurrent serum concentration (median 0.112 mg/L, range 0.042-0.25 mg/L) after a single 100 mg dose. The mean and median bile-to-serum 24-h area under the concentration-time curve (AUC0-24) ratios were 537 and 368 respectively[41]. It is reasonable to suspect DIAP as a possible complication in tigecycline treated patients. Several cases have been reported previously[42].
Metabolic
Hypertriglyceridemia: It is generally accepted that levels of triglycerides (TG) greater than 1000 mg/dL may increase the risk of precipitating an episode of pancreatitis[43]. The breakdown products of TG are probably responsible for inducing pancreatitis. When lipase in the pancreatic capillary bed acts on the high levels of TG in serum, toxic free fatty acids are generated. The endothelial lining of small pancreatic blood vessels is the first site of injury. Damages of small blood vessels lead to recruitment of inflammatory cells and thrombosis. Hyperlipidemic pancreatitis may be associated with normal serum amylase but with elevated serum lipase levels[44]. With excessive TG, local ischemia and acidemia may occur due to capillary obstruction[45]. This damage exposes TG to pancreatic lipases, which impact degradation of TG[46]. Hydrolysis of TG by pancreatic lipase, excessive formation of free fatty acids with inflammatory changes, capillary injury, and hyperviscosity are postulated to account for the development of hypertriglyceridemia-induced pancreatitis.
Drugs including estrogens, isotretinoin, propofol, retinoid derivatives, HIV protease inhibitors, β-blockers, thiazides, and furosemide are thought to induce AP owing to hypertriglyceridemia. Estrogen is the most well studied drug in this manner. Exogenous estrogens increase serum TG and fatty acids primarily by reducing levels of lipoprotein and hepatic lipases, which subsequently decrease clearance and aggravate insulin resistance[47]. Typically, estrogen-related pancreatitis occurs within the first months following estrogen initiation. Obese patients with underlying glucose intolerance or fasting hypertriglyceridemia are at greater risk[44]. However, reports have also shown that estrogen-associated DIAP can happen without elevated serum lipid concentrations[48,49]. It is thought that arteriolar thrombosis may be another potential mechanism of action[48,50]. Tamoxifen and clomiphene are synthetic estrogen analogues with mixed agonist-antagonist actions. Cases of tamoxifen- or clomiphene-associated AP have been reported with mechanisms similar to that of estrogen.
Dibenzodiazepine-derived atypical antipsychotics (i.e., clozapine, olanzapine, and quetiapine) may also be a potential cause of DIAP. Both risperidone and ziprasidone are non-dibenzodiazepine atypical antipsychotics and appear to have minimal effect on serum lipids[51]. This is another example where clinicians can apply the general knowledge of each medication when evaluating the likelihood of DIAP for the newer medications.
The previous section discussed that tetracyclines-associated DIAP due to its toxic metabolite and high biliary concentrations. Elmore and Rogge[36] also proposed a tetracycline-induced hypertriglyceridemia mechanism with subsequent pancreatitis. Tetracycline inhibits protein synthesis by binding to the 30S ribosomal subunit in the messenger ribonucleic acid (mRNA) translation complex. Blockage of protein synthesis could result in accumulation of defective proteins within hepatocytes. This inhibits the release of TG from the liver, which may lead to pancreatitis.
Hypercalcemia: Calcium is identified as the most important intracellular element in acinar cell stimulus-secretion coupling[52]. Disruption in the secretory process could be the mechanism by which hypercalcemia induces pancreatitis. Based on experimental studies, increase in extracellular calcium leads to a functional secretory block with dose-dependent characteristics[53]. Acinar cell stimulation induces spikes in cytosolic calcium concentration by repetitively releasing calcium from intracellular stores, which activates the normal secretory process of digestive enzymes from intracellular zymogen stores. Excessive extracellular calcium concentration leads to sustained increases in cytosolic calcium. It results in vacuole formation and trypsinogen activation and eventually leads to edematous or necrotizing pancreatitis[54]. Research indicates that hypercalcemia is associated with an increase in serum enzymes[44]. Intravenous calcium administration has been associated with pancreatitis in at least two published reports. Additionally, pancreatitis has been correlated to cases of vitamin D poisoning and to patients receiving total parenteral nutrition[55]. It is suspected that all drugs which can cause hypercalcemia carry risk of inducing AP.
Thiazides, a class with hypertriglyceridemia potential, could also induce hypercalcemia and hypophosphatemia. Thiazide-induced reductions in blood pressure may lead to pancreatic ischemia. They may act directly on the pancreas or indirectly by altering calcium metabolism. Therefore, there are multiple mechanisms exhibited by thiazides that could potentially lead to AP.
Vascular
Ischemia is an uncommon cause of AP. Pancreatic infarcts may occur in patients with underlying atherosclerotic vascular disease, but they are unusual because the pancreas is richly perfused from several different arterial sources. Cholesterol emboli may cause pancreatitis, cholecystitis, or bowel ulceration or infarction, and should be suspected when AP occurs after vascular interventions such as cardiac catheterization. Patients may have associated evidence of renal, gut, or peripheral cholesterol emboli. Ischemic pancreatic and hepatic injury may be associated with malignant hypertension, low flow states due to severe heart failure, or administration of potent vasoconstrictors. Vasculitis may cause pancreatitis associated with systemic autoimmune diseases. Acute pancreatitis secondary to drug-induced lupus syndrome has also been described[56].
Contrast-induced pancreatitis may be related to decreased oxygenation and impaired circulation of the pancreas. Iopamidol has a viscosity of 9.4 cP at 37 degrees centigrade versus human plasma of 1.72 cP at hematocrit of 43%. A similar pathophysiologic process has been proposed in contrast-induced kidney injury. Cholesterol crystal embolization may be another mechanism that results in occlusion of small arteries[57].
Immune-mediated reaction
Direct immunological effects are usually observed within the first month of drug exposure, whereas toxic effects are noted after a few months of treatment[45]. Researchers have also considered AP an immune-mediated reaction if relapse occurs rapidly after re-challenge as seen with sulfonamides and aminosalicylates (e.g., sulfasalazine and mesalazine)[58-60]. The latency between initiation of the drug and the onset of DIAP is usually one week to a month, but reexposure can lead to a new episode in one to three days[5]. Cases of azathioprine or the thiopurine bases mercaptopurine-induced pancreatitis are well documented. Studies have shown patients with decreased levels of thiopurine metabolizing enzyme inosine triphosphate pyrophosphatase may be at an increased risk of developing thiopurine-induced AP. However, 6-tioguanine-induced pancreatitis is less common than conventional thiopurine. Only 1% of inflammatory bowel disease (IBD) patients previously intolerant to the conventional thiopurines are reported to have 6-tioguanine-induced AP after treatment[61]. A strong correlation with immune disorders, mainly Crohn’s disease and HIV infections, implies an immune-mediated reaction as a chief causative factor of the disease.
Angiotensin-converting-enzyme inhibitors: Captopril, enalapril, lisinopril, perindopril, benazepril, and quinapril have all been associated with AP[25,52]. Pancreatic duct obstruction by local angioedema may be the mechanism by which angiotensin-converting-enzyme-inhibitors cause pancreatitis. Others propose a direct toxic effect on pancreatic cells. Since captopril is structurally dissimilar to enalapril and lisinopril, an allergic reaction seems less likely. Angiotensin receptor blockers may share a similar mechanism for pancreatitis, at this point definitive cases are not described in literature[10].
Alternative medicines including herbal medication
There are very limited data of DIAP associated with herbal or over the counter medications when compared to prescription medications. Although a mechanism for saw palmetto-induced AP has not been thoroughly established, cases of saw palmetto-induced cholestatic hepatitis associated with AP have been reported. Currently, there are only two reported cases of saw palmetto induced AP. Another theory suggests that it occurs through its estrogenic effects by stimulating estrogen receptors and then induces a hypercoagulable state that leads to pancreatic necrosis[2]. This information should prompt clinicians to consider saw palmetto a potential cause of AP.
A WORK UP FOR ACUTE PANCREATITIS
Since no specific test for establishing the diagnosis of DIAP is available, the diagnosis is usually based on excluding all other common causes. Pancreatitis is suspected when a patient presents with clinical features including acute onset of persistent and severe epigastric abdominal pain, and is then confirmed by laboratory and imaging studies that exclude other serious intra-abdominal conditions. Most patients will have elevations in serum levels of amylase or lipase within a few hours of the onset of symptoms. Lipase tends to remain elevated longer than amylase. Amylase and lipase levels above three times the upper limit of normal are mostly associated with pancreatitis. Once these levels are elevated, serial measurements are of no clinical significance for prognosis or outcomes. They should not be obtained routinely after the initial measurements are obtained.
Imaging studies are used to establish the diagnosis, but also to determine etiology and prognosis. Both abdominal ultrasound and abdominal computed tomography (CT) can be used interchangeably; however, the latter is preferred as it can provide alternative diagnoses.
As part of the investigation for potential causes of AP, a history of alcohol/tobacco use, previous biliary colic, medication history, family history, and recent trauma should be elicited. Gallstone-associated pancreatitis should be suspected if stones are seen on imaging studies or if liver chemistries are abnormal and then improve over a few days. A three-fold elevation of ALT has a high predictive value for gallstone-associated AP. Hypertriglyceridemia, especially with levels above 1000 mg/dL, and hypercalcemia can be evaluated based on laboratory data. Infections, including viral etiologies, are potential causes of AP as well.
A suspected drug etiology should be considered after the exclusion of more common causes of illness. As mentioned above, it is challenging to establish the causality between medications and associated AP. The use of classification systems may be useful as the first screening tool. Since the mean interval between initial drug administration and start of the symptoms is approximately 5 wk, with a range of 2 to 36 wk[46], clinicians can target those medications when reviewing the medication profiles. If the mediations are not listed on these summarized lists, clinicians should identify if similar structured medications have been associated with DIAP and evaluate the possibility of sharing a similar mechanism of inducing pancreatitis. Once the target agent is identified, the offending agent should be discontinued, preferably one at the time to avoid confounders. Most reactions are reversible and resolve on their own within 3-7 d after the offending agent has been discontinued. Due to the nature of the disease state and ethical consideration, re-challenge of the suspected drug is usually not possible. Often times, the medication can be just a possible/probable cause of AP. If re-challenge of the suspect drug is considered necessary, the patient’s written informed consent should be obtained. An algorithm of identifying a potential case of drug induced AP is presented in Figure 2.
Figure 2.
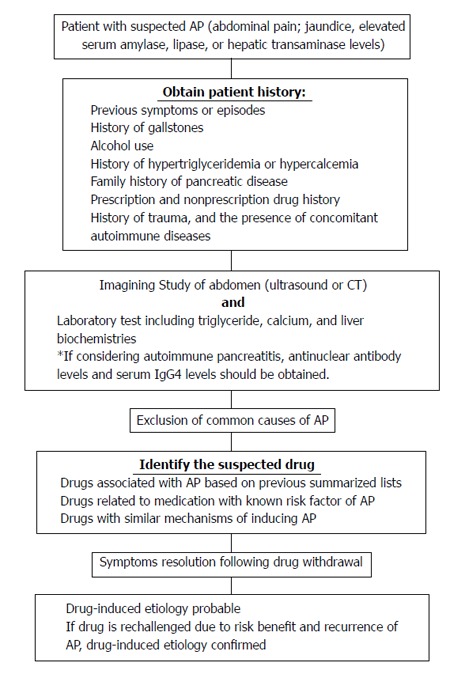
Algorithm of Identifying A potential case of drug induced acute pancreatitis. AP: Acute pancreatitis.
DISCUSSION
Hundreds of medications have been suggested to be the potential cause of AP, although the true incidence of DIAP is unknown. Evidence associating drugs with AP is largely based on individual cases. It is unrealistic to expect a comprehensive list that includes all agents associated with AP due to continuously reported new cases. Although relapse of pancreatitis after controlled re-challenge confirms a causal relationship, such proof is uncommon. Furthermore, re-challenge is only ethical when the same treatment is absolutely necessary for the patient. It leads to only few causal relationships for the reported agents.
Few causative agent classifications have been proposed. These classifications have helped clinicians understand the quality of evidence behind each potential causative agent. However, with the exception of a few agents with a definite relationship confirmed by re-challenge, it depends on each individual report to exclude all other possible causes, especially drug effects that may be difficult to separate from the underlying conditions. Clinically, certain subpopulations such as children, women, the elderly, patients with IBD and patients with HIV appear to be at a higher risk[2]. Mesalazine, azathioprine, and corticosteroids, for instance, are used in the treatment of IBD which itself increases the risk of AP. Anti-retroviral agents is another example as HIV is an independent risk factor of AP. A study that compared patients with and without HIV infection found a drug-related etiology in 41% and 5% of the patients with AP respectively[62]. The use of NRTIs such as didanosine, stavudine, and lamivudine and co-administration of other medications such as pentamidine, cotrimoxazole, antimycobacterial therapy, or cytotoxic chemotherapy for at least 6 mo were found to be a significant risk factor for at least a three-fold increase in serum pancreatic enzymes (P < 0.05). Certain medications, such as proton pump inhibitors and histamine2-receptor antagonists as well as NSAIDs, may be initiated in response to early symptoms of unrecognized pancreatitis. This may have led to erroneously attributing the pancreatitis to these medications[63,64]. Repeated cases of DIAP are more likely to be published or even diagnosed than those without prior reports. Due to underreporting incidence rates from spontaneous reports and potential publication bias with only reporting severe cases, it has further complicated the assessment of the causal relationship between drugs and AP based on current proposed classification.
Efforts have been devoted to improve drug safety surveillance strategies. Vilar et al[65] have shown promising results of detecting adverse drug events related to pancreatitis by developing molecular fingerprint-based models. The models were based on the premise that similar molecules can have comparable biological properties. For example, tigecycline is structurally related to minocycline and shares similar pharmacokinetic properties and side effects with tetracyclines. Not surprisingly, cases of tigecycline-induced AP were reported soon after its introduction to the market[42]. Nevertheless, DIAP is generally not considered as a drug class effect, so specific drugs are usually noted instead of the entire class[14]. It is suggested that clinicians take the potential mechanism of DIAP into account. For example, ceftriaxone has different pharmacokinetic properties than other cephalosporins and may lead to secondary pancreatitis caused by only ceftriaxone induced pseudolithiasis.
Limited data exist regarding the mechanisms of DIAP. The pathogenesis is not completely understood. Nevertheless, DIAP should not have unique features that distinguish it from AP due to other causes. Drugs may lead to pancreatitis by inducing known risk factors of AP such as structural (e.g., cholestatic liver injury, spasm of the SO, duct obstruction/constriction, and stones), metabolic (e.g., hypertriglyceridemia and hypercalcemia), and vascular effects. Some drugs or drug metabolites may theoretically have a direct toxic effect on the pancreas. Other than known mechanisms of toxicity such as mitochondrial toxicity and protein synthesis inhibition, the high level of gastrointestinal drug concentration may be needed to cause cytotoxic damage. Drugs with a definite causal relationship to AP including isoniazid, metronidazole, valproic acid, mesalamine, and tetracycline share similar pharmacokinetic properties by extensive hepatic metabolism. If other potential causes of DIAP have been ruled out, drugs that are highly concentrated in the gastrointestinal tract could be potential suspects of DIAP. For other drugs, an immunoallergic idiosyncratic reaction is more likely. Re-challenge with these drugs usually leads to prompt recurrence of symptoms in a dose-independent manner. In an animal study, the results suggest that DIAP is multifactorial and may explain why the incidence of DIAP is low[18].
Establishing a definitive causal relationship between a drug and AP poses a challenge to clinicians. Depending on the agents, the time from the initiation of therapy to the onset of pancreatitis symptoms varies. Pancreatitis can occur within a short time after administration of the first dose to years after therapy begins for most of the drugs. The unrealistic expectation of the comprehensive list and the multifactorial natures of the causes of AP call for a different approach. This article reviews the potential mechanisms of DIAP and provides the perspective of deductive reasoning in order to identify potential DIAP.
Footnotes
P- Reviewer: Nihalani D, Sakata N S- Editor: Song XX L- Editor: A E- Editor: Wang CH
References
- 1.Tenner S, Baillie J, DeWitt J, Vege SS. American College of Gastroenterology guideline: management of acute pancreatitis. Am J Gastroenterol. 2013;108:1400–1415; 1416. doi: 10.1038/ajg.2013.218. [DOI] [PubMed] [Google Scholar]
- 2.Balani AR, Grendell JH. Drug-induced pancreatitis: incidence, management and prevention. Drug Saf. 2008;31:823–837. doi: 10.2165/00002018-200831100-00002. [DOI] [PubMed] [Google Scholar]
- 3.Lankisch PG, Dröge M, Gottesleben F. Drug induced acute pancreatitis: incidence and severity. Gut. 1995;37:565–567. doi: 10.1136/gut.37.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Urbanek K, Vinklerova I, Krystynik O, Prochazka V. Acute pancreatitis induced by drugs. Acute pancreatitis. 2011;(2):17–34. [Google Scholar]
- 5.Monami M, Dicembrini I, Martelli D, Mannucci E. Safety of dipeptidyl peptidase-4 inhibitors: a meta-analysis of randomized clinical trials. Curr Med Res Opin. 2011;27 Suppl 3:57–64. doi: 10.1185/03007995.2011.602964. [DOI] [PubMed] [Google Scholar]
- 6.Drug-Induced Pancreatitis. Pharmacist’s Letter/Prescriber’s Letter. April 2013. Available from: http://prescribersletter.therapeuticresearch.com/home.aspx?cs=&s=PRL&AspxAutoDetectCookieSupport=1.
- 7.Neoptolemos JP, Raraty M, Finch M, Sutton R. Acute pancreatitis: the substantial human and financial costs. Gut. 1998;42:886–891. doi: 10.1136/gut.42.6.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sekimoto M, Takada T, Kawarada Y, Hirata K, Mayumi T, Yoshida M, Hirota M, Kimura Y, Takeda K, Isaji S, et al. JPN Guidelines for the management of acute pancreatitis: epidemiology, etiology, natural history, and outcome predictors in acute pancreatitis. J Hepatobiliary Pancreat Surg. 2006;13:10–24. doi: 10.1007/s00534-005-1047-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soran A, Chelluri L, Lee KK, Tisherman SA. Outcome and quality of life of patients with acute pancreatitis requiring intensive care. J Surg Res. 2000;91:89–94. doi: 10.1006/jsre.2000.5925. [DOI] [PubMed] [Google Scholar]
- 10.Fagenholz PJ, Castillo CF, Harris NS, Pelletier AJ, Camargo CA. Increasing United States hospital admissions for acute pancreatitis, 1988-2003. Ann Epidemiol. 2007;17:491–497. doi: 10.1016/j.annepidem.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 11.Badalov N, Baradarian R, Iswara K, Li J, Steinberg W, Tenner S. Drug-induced acute pancreatitis: an evidence-based review. Clin Gastroenterol Hepatol. 2007;5:648–651; quiz 644. doi: 10.1016/j.cgh.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 12.Zaccara G, Franciotta D, Perucca E. Idiosyncratic adverse reactions to antiepileptic drugs. Epilepsia. 2007;48:1223–1244. doi: 10.1111/j.1528-1167.2007.01041.x. [DOI] [PubMed] [Google Scholar]
- 13.Underwood TW, Frye CB. Drug-induced pancreatitis. Clin Pharm. 1993;12:440–448. [PubMed] [Google Scholar]
- 14.Tenner S, Steinberg WM. Acute Pancreatitis. In: Feldman M, Friedman LS, Brandt LJ, editors. 9th ed. St. Louis, MO: Saunders: Sleisenger & Fordtran’s Gastrointestinal and Liver Disease; 2010. [Google Scholar]
- 15.Zinberg J, Chernaik R, Coman E, Rosenblatt R, Brandt LJ. Reversible symptomatic biliary obstruction associated with ceftriaxone pseudolithiasis. Am J Gastroenterol. 1991;86:1251–1254. [PubMed] [Google Scholar]
- 16.Trendle MC, Moertel CG, Kvols LK. Incidence and morbidity of cholelithiasis in patients receiving chronic octreotide for metastatic carcinoid and malignant islet cell tumors. Cancer. 1997;79:830–834. doi: 10.1002/(sici)1097-0142(19970215)79:4<830::aid-cncr20>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 17.Tauber JP, Poncet MF, Harris AG, Barthel HR, Simonetta-Chateauneuf C, Buscail L, Bayard F. The impact of continuous subcutaneous infusion of octreotide on gallstone formation in acromegalic patients. J Clin Endocrinol Metab. 1995;80:3262–3266. doi: 10.1210/jcem.80.11.7593435. [DOI] [PubMed] [Google Scholar]
- 18.Chen JW, Thomas A, Woods CM, Schloithe AC, Toouli J, Saccone GT. Sphincter of Oddi dysfunction produces acute pancreatitis in the possum. Gut. 2000;47:539–545. doi: 10.1136/gut.47.4.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vidal J, Sacanella E, Muñoz E, Miro JM, Navarro S. Acute pancreatitis related to octreotide in a patient with acquired immunodeficiency syndrome. Pancreas. 1994;9:395–397. doi: 10.1097/00006676-199405000-00021. [DOI] [PubMed] [Google Scholar]
- 20.Soule S, Conway G, Hatfield A, Jacobs H. Effectiveness and tolerability of slow release lanreotide treatment in active acromegaly: six-month report on an Italian multicentre study. J Clin Endocrinol Metab. 1996;81:4502–4503. doi: 10.1210/jcem.81.12.8954072. [DOI] [PubMed] [Google Scholar]
- 21.Berger TM, Cook WJ, O’Marcaigh AS, Zimmerman D. Acute pancreatitis in a 12-year-old girl after an erythromycin overdose. Pediatrics. 1992;90:624–626. [PubMed] [Google Scholar]
- 22.Chen HM, Chen JC, Ng CJ, Chiu DF, Chen MF. Melatonin reduces pancreatic prostaglandins production and protects against caerulein-induced pancreatitis in rats. J Pineal Res. 2006;40:34–39. doi: 10.1111/j.1600-079X.2005.00271.x. [DOI] [PubMed] [Google Scholar]
- 23.Goldstein J, Laskin DA, Ginsberg GH. Sulindac associated with pancreatitis. Ann Intern Med. 1980;93:151. doi: 10.7326/0003-4819-93-1-151_1. [DOI] [PubMed] [Google Scholar]
- 24.Siefkin AD. Sulindac and pancreatitis. Ann Intern Med. 1980;93:932–933. doi: 10.7326/0003-4819-93-6-932_2. [DOI] [PubMed] [Google Scholar]
- 25.Memon AN. Pancreatitis and sulindac. Ann Intern Med. 1982;97:139. doi: 10.7326/0003-4819-97-1-139_1. [DOI] [PubMed] [Google Scholar]
- 26.Lerche A, Vyberg M, Kirkegaard E. Acute cholangitis and pancreatitis associated with sulindac (clinoril) Histopathology. 1987;11:647–653. doi: 10.1111/j.1365-2559.1987.tb02675.x. [DOI] [PubMed] [Google Scholar]
- 27.Brinkman K, ter Hofstede HJ, Burger DM, Smeitink JA, Koopmans PP. Adverse effects of reverse transcriptase inhibitors: mitochondrial toxicity as common pathway. AIDS. 1998;12:1735–1744. doi: 10.1097/00002030-199814000-00004. [DOI] [PubMed] [Google Scholar]
- 28.Dalton SD, Rahimi AR. Emerging role of riboflavin in the treatment of nucleoside analogue-induced type B lactic acidosis. AIDS Patient Care STDS. 2001;15:611–614. doi: 10.1089/108729101753354608. [DOI] [PubMed] [Google Scholar]
- 29.Wester CW, Thomas AM, Bussmann H, Moyo S, Makhema JM, Gaolathe T, Novitsky V, Essex M, deGruttola V, Marlink RG. Non-nucleoside reverse transcriptase inhibitor outcomes among combination antiretroviral therapy-treated adults in Botswana. AIDS. 2010;24 Suppl 1:S27–S36. doi: 10.1097/01.aids.0000366080.91192.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kesselring AM, Wit FW, Sabin CA, Lundgren JD, Gill MJ, Gatell JM, Rauch A, Montaner JS, de Wolf F, Reiss P, et al. Risk factors for treatment-limiting toxicities in patients starting nevirapine-containing antiretroviral therapy. AIDS. 2009;23:1689–1699. doi: 10.1097/QAD.0b013e32832d3b54. [DOI] [PubMed] [Google Scholar]
- 31.Sura ME, Heinrich KA, Suseno M. Metronidazole-associated pancreatitis. Ann Pharmacother. 2000;34:1152–1155. doi: 10.1345/aph.10021. [DOI] [PubMed] [Google Scholar]
- 32.Assan R, Perronne C, Assan D, Chotard L, Mayaud C, Matheron S, Zucman D. Pentamidine-induced derangements of glucose homeostasis. Determinant roles of renal failure and drug accumulation. A study of 128 patients. Diabetes Care. 1995;18:47–55. doi: 10.2337/diacare.18.1.47. [DOI] [PubMed] [Google Scholar]
- 33.Suzuki M, Shimizu T, Kudo T, Shoji H, Ohtsuka Y, Yamashiro Y. Octreotide prevents L-asparaginase-induced pancreatic injury in rats. Exp Hematol. 2008;36:172–180. doi: 10.1016/j.exphem.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 34.Nicolau DP, Mengedoht DE, Kline JJ. Tetracycline-induced pancreatitis. Am J Gastroenterol. 1991;86:1669–1671. [PubMed] [Google Scholar]
- 35.Elmore MF, Rogge JD. Tetracycline-induced pancreatitis. Gastroenterology. 1981;81:1134–1136. [PubMed] [Google Scholar]
- 36.Kunelis CT, Peters JL, Edmondson HA. Fatty liver of pregnancy and its relationship to tetracycline therapy. Am J Med. 1965;38:359–377. doi: 10.1016/0002-9343(65)90145-2. [DOI] [PubMed] [Google Scholar]
- 37.Steinberg WM. Acute drug and toxin induced pancreatitis. Hosp Pract (Off Ed) 1985;20:95–102. doi: 10.1080/21548331.1985.11703057. [DOI] [PubMed] [Google Scholar]
- 38.Gilson M, Moachon L, Jeanne L, Dumaine V, Eyrolle L, Morand P, Ben m’Rad M, Salmon D. Acute pancreatitis related to tigecycline: case report and review of the literature. Scand J Infect Dis. 2008;40:681–683. doi: 10.1080/00365540801938949. [DOI] [PubMed] [Google Scholar]
- 39.Macdonald H, Kelly RG, Allen ES, Noble JF, Kanegis LA. Pharmacokinetic studies on minocycline in man. Clin Pharmacol Ther. 1973;14:852–861. doi: 10.1002/cpt1973145852. [DOI] [PubMed] [Google Scholar]
- 40.Wyeth Pharmaceutics. Tygacil® (package insert) Philadelphia, PA: Wyeth Pharmaceutics; 2009. [Google Scholar]
- 41.Hung WY, Kogelman L, Volpe G, Iafrati M, Davidson L. Tigecycline-induced acute pancreatitis: case report and literature review. Int J Antimicrob Agents. 2009;34:486–489. doi: 10.1016/j.ijantimicag.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 42.Athyros VG, Giouleme OI, Nikolaidis NL, Vasiliadis TV, Bouloukos VI, Kontopoulos AG, Eugenidis NP. Long-term follow-up of patients with acute hypertriglyceridemia-induced pancreatitis. J Clin Gastroenterol. 2002;34:472–475. doi: 10.1097/00004836-200204000-00020. [DOI] [PubMed] [Google Scholar]
- 43.Frick TW, Spycher MA, Heitz PU, Largiadèr F, Goodale RL. Hypercalcaemia and pancreatic ultrastructure in cats. Eur J Surg. 1992;158:289–294. [PubMed] [Google Scholar]
- 44.Kimura W, Mössner J. Role of hypertriglyceridemia in the pathogenesis of experimental acute pancreatitis in rats. Int J Pancreatol. 1996;20:177–184. doi: 10.1007/BF02803766. [DOI] [PubMed] [Google Scholar]
- 45.Wilde PJ, Chu BS. Interfacial & amp; colloidal aspects of lipid digestion. Adv Colloid Interface Sci. 2011;165:14–22. doi: 10.1016/j.cis.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 46.Perseghin G, Scifo P, Pagliato E, Battezzati A, Benedini S, Soldini L, Testolin G, Del Maschio A, Luzi L. Gender factors affect fatty acids-induced insulin resistance in nonobese humans: effects of oral steroidal contraception. J Clin Endocrinol Metab. 2001;86:3188–3196. doi: 10.1210/jcem.86.7.7666. [DOI] [PubMed] [Google Scholar]
- 47.Foster ME, Powell DE. Pancreatitis, multiple infarcts and oral contraception. Postgrad Med J. 1975;51:667–669. doi: 10.1136/pgmj.51.599.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mungall IP, Hague RV. Pancreatitis and the pill. Postgrad Med J. 1975;51:855–857. doi: 10.1136/pgmj.51.602.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murley RS. Pancreatitis from oral contraceptives. BMJ. 1974;1:161. [Google Scholar]
- 50.Meyer JM. A retrospective comparison of weight, lipid, and glucose changes between risperidone- and olanzapine-treated inpatients: metabolic outcomes after 1 year. J Clin Psychiatry. 2002;63:425–433. doi: 10.4088/jcp.v63n0509. [DOI] [PubMed] [Google Scholar]
- 51.Petersen OH. Stimulus-excitation coupling in plasma membranes of pancreatic acinar cells. Biochim Biophys Acta. 1982;694:163–184. doi: 10.1016/0304-4157(82)90023-5. [DOI] [PubMed] [Google Scholar]
- 52.Frick TW, Mithöfer K, Fernández-del Castillo C, Rattner DW, Warshaw AL. Hypercalcemia causes acute pancreatitis by pancreatic secretory block, intracellular zymogen accumulation, and acinar cell injury. Am J Surg. 1995;169:167–172. doi: 10.1016/s0002-9610(99)80127-5. [DOI] [PubMed] [Google Scholar]
- 53.Frick TW. The role of calcium in acute pancreatitis. Surgery. 2012;152:S157–S163. doi: 10.1016/j.surg.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 54.Khan AS, Latif SU, Eloubeidi MA. Controversies in the etiologies of acute pancreatitis. JOP. 2010;11:545–552. [PubMed] [Google Scholar]
- 55.Gorges R, Ghalayini W, Zughaib M. A case of contrast-induced pancreatitis following cardiac catheterization. J Invasive Cardiol. 2013;25:E203–E204. [PubMed] [Google Scholar]
- 56.Falko JM, Thomas FB. Letter: Acute pancreatitis due to procainamide-induced lupus erythematosus. Ann Intern Med. 1975;83:832–833. doi: 10.7326/0003-4819-83-6-832. [DOI] [PubMed] [Google Scholar]
- 57.Widdison AL, Karanjia ND. Pancreatic infection complicating acute pancreatitis. Br J Surg. 1993;80:148–154. doi: 10.1002/bjs.1800800208. [DOI] [PubMed] [Google Scholar]
- 58.Schaad UB, Wedgwood-Krucko J, Tschaeppeler H. Reversible ceftriaxone-associated biliary pseudolithiasis in children. Lancet. 1988;2:1411–1413. doi: 10.1016/s0140-6736(88)90596-x. [DOI] [PubMed] [Google Scholar]
- 59.Niemelä S, Lehtola J, Karttunen T, Lähde S. Pancreatitis in patients with chronic inflammatory bowel disease. Hepatogastroenterology. 1989;36:175–177. [PubMed] [Google Scholar]
- 60.de Boer NK, Derijks LJ, Gilissen LP, Hommes DW, Engels LG, de-Boer SY, den Hartog G, Hooymans PM, Mäkelburg AB, Westerveld BD, et al. On tolerability and safety of a maintenance treatment with 6-thioguanine in azathioprine or 6-mercaptopurine intolerant IBD patients. World J Gastroenterol. 2005;11:5540–5544. doi: 10.3748/wjg.v11.i35.5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bruminhent J, Carrera P, Li Z, Amankona R, Roberts IM. Acute pancreatitis with saw palmetto use: a case report. J Med Case Rep. 2011;5:414. doi: 10.1186/1752-1947-5-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cappell MS, Marks M. Acute pancreatitis in HIV-seropositive patients: a case control study of 44 patients. Am J Med. 1995;98:243–248. doi: 10.1016/S0002-9343(99)80370-2. [DOI] [PubMed] [Google Scholar]
- 63.Eland IA, Alvarez CH, Stricker BH, Rodríguez LA. The risk of acute pancreatitis associated with acid-suppressing drugs. Br J Clin Pharmacol. 2000;49:473–478. doi: 10.1046/j.1365-2125.2000.00196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Evans JM, McMahon AD, Steinke DT, McAlpine RR, MacDonald TM. Do H2-receptor antagonists cause acute pancreatitis? Pharmacoepidemiol Drug Saf. 1998;7:383–388. doi: 10.1002/(SICI)1099-1557(199811/12)7:6<383::AID-PDS377>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 65.Vilar S, Harpaz R, Santana L, Uriarte E, Friedman C. Enhancing adverse drug event detection in electronic health records using molecular structure similarity: application to pancreatitis. PLoS One. 2012;7:e41471. doi: 10.1371/journal.pone.0041471. [DOI] [PMC free article] [PubMed] [Google Scholar]