

Abstract
In recent decades there has been a dramatic rise in the incidence of esophageal adenocarcinoma (EAC) in the developed world. Over approximately the same period there has also been an increase in the prevalence of obesity. Obesity, especially visceral obesity, is an important independent risk factor for the development of gastro-esophageal reflux disease, Barrett’s esophagus and EAC. Although the simplest explanation is that this mediated by the mechanical effects of abdominal obesity promoting gastro-esophageal reflux, the epidemiological data suggest that the EAC-promoting effects are independent of reflux. Several, not mutually exclusive, mechanisms have been implicated, which may have different effects at various points along the reflux-Barrett’s-cancer pathway. These mechanisms include a reduction in the prevalence of Helicobacter pylori infection enhancing gastric acidity and possibly appetite by increasing gastric ghrelin secretion, induction of both low-grade systemic inflammation by factors secreted by adipose tissue and the metabolic syndrome with insulin-resistance. Obesity is associated with enhanced secretion of leptin and decreased secretion of adiponectin from adipose tissue and both increased leptin and decreased adiponectin have been shown to be independent risk factors for progression to EAC. Leptin and adiponectin have a set of mutually antagonistic actions on Barrett’s cells which appear to influence the progression of malignant behaviour. At present no drugs are of proven benefit to prevent obesity associated EAC. Roux-en-Y reconstruction is the preferred bariatric surgical option for weight loss in patients with reflux. Statins and aspirin may have chemopreventative effects and are indicated for their circulatory benefits.
Keywords: Adipose, Body mass index, Reflux, Barrett’s esophagus
Core tip: Excess adipose tissue, particularly visceral obesity, is an important risk factor for esophageal adenocarcinoma (EAC). The mechanisms involve both the promotion of gastro-esophageal reflux and reflux-independent mechanisms. Abnormal secretion of the adipokines leptin and adiponectin from adipose tissue in obesity may promote the development of EAC. Increased leptin levels are an independent risk factor for EAC and leptin enhances proliferation and invasion and inhibits apoptosis in Barrett’s cell lines. Relative adiponectin deficiency is an independent risk factor for EAC and adiponectin blocks the cancer promoting effects of leptin in experimental models. Obesity may influence EAC development via adipokine secretion.
INTRODUCTION
Esophageal adenocarcinoma is a health problem of increasing global health significance. The overall prognosis of esophageal adenocarcinoma (EAC), the most prevalent form of esophageal cancer in the developed world, is dismal, with a 5-year survival of 15%-20% at best[1]. At the same time the incidence of this cancer has increased dramatically, by approximately 600% in the last 30 years, leading some commentators to call this an epidemic[2]. A detailed understanding of the pathogenic mechanisms leading to this malignancy is required to enable the development of strategies for both prevention and treatment. Over a similar period the prevalence of obesity has increased in the developed world and this increase in obesity has been linked with increased risks of several cancers, including oesophageal adenocarcinoma (OAC)[3,4]. Over the last 30 years rates of obesity have been increasing steadily, most obviously in the United States and Western Europe[5] but also in lower and middle income countries[6]. Estimates from The World Health Organisation suggest that 12% of the world’s population aged over 20 years old is now obese which equates to approximately 500 million adults. It is estimated that 10% of men and 14% of women are obese by standard criteria. This has doubled from the 1980s[6].
Well-designed epidemiological investigations have been instrumental in detecting and defining the association between obesity and EAC. Relevant features of the association have been explored in detail to determine features of causality and to help clarify the potential implicated mechanisms through which obesity may act. This review summarises relevant recent observational data on the association between measures of obesity and risk of EAC, gastro-esophageal reflux and Barrett’s esophagus; and experimental data of plausible mechanisms contributing to carcinogenesis and explores where and how interventions may reduce the burden of this disease.
CLINICAL MEASURES OF ADIPOSITY AND METHODOLOGICAL CONSIDERATIONS IN RELEVANT OBSERVATIONAL RESEARCH
The World Health Organization definition for overweight and obesity is abnormal or excessive fat accumulation that may impair health. Adiposity has been quantified in observational research relevant to this topic using a number of measures including anthropometric measurements and imaging. Overall adiposity is commonly measured using body mass index (BMI) [weight (kg) per height squared (m2)], and central adiposity (synonymous to visceral and abdominal adiposity) is measured using waist-to-hip ratio (WTHR), waist circumference and anterior-posterior abdominal diameter (cm); and imaging such as visceral adipose tissue area (m2) (VATA) or volume (m3) as determined by computerised tomography or magnetic resonance imaging. While BMI is a simple measure of overall adiposity that is practical for large epidemiological studies, it is crude and does not necessarily reflect the varying proportions of fat and lean (non-fat) mass or body fat distribution. Measures of central obesity have been demonstrated to vary substantially within a narrow range of BMI, whilst central obesity itself is a combination of subcutaneous obesity around the abdomen and mesenteric adipose tissue. Furthermore, as body composition changes with age, and height decreases, for example through kyphosis and loss of vertebral height, BMI may be overestimated in older participants[7]. Imaging modalities used to quantify fat distribution, can precisely estimate the size of body fat compartments. However, they are less suitable for large scale prospective studies: they are often performed at diagnosis rather than for preceding time points and could therefore underestimate associations depending on weight loss associated with the diagnosis; and their use requires convenience sampling for controls (e.g., patients undergoing investigation for other reasons), and therefore may be less representative of the population under study. These are important considerations when appraising the evidence on the risk of reflux, Barrett’s esophagus and EAC with adiposity.
OBESITY AND RISK OF ESOPHAGEAL ADENOCARCINOMA
The association between measures of obesity and risk of OAC has been extensively examined in epidemiological investigations. The most striking evidence for a potential causal association between adiposity and risk of this cancer is the wealth of consistent data to suggest the association is among the strongest than for any other malignancy with evidence of a biological gradient[4]. A systematic review of prospective studies from Europe, Australia and the Asia-Pacific region, that measured BMI at baseline and followed participants until the development of incident cancer (hence supporting a temporal relationship), included 1315 male cases of OAC and 735 female cases, demonstrated the magnitude of the association in men was stronger than for any other malignancy, from 16 sites; and in women was only second to endometrial from 19 sites. The strength of the associations (per increase in BMI by 5 kg/m2) was almost the same in both genders (RR = 1.52, 95%CI: 1.33-1.74 for men; RR = 1.51, 95%CI: 1.31-1.74 for women) with minimal heterogeneity. This implies the association between BMI and risk of EAC is consistent between well-designed prospective studies, further supporting the causality, and that sex-specific differences in the incidence of EAC are likely unrelated to adiposity as measured by BMI. Interestingly, for squamous cell carcinoma, the most common histological type of esophageal cancer worldwide, in both men and women, risk was significantly reduced with increased BMI, more strongly than for any of the included malignancies[4]. A recent meta-analysis of five observational studies, including four prospective studies[8-11] and one case-control study[12], reported a significant association between abdominal obesity (as a composite measure of VATA, WHR, AC and abdominal diameter) and risk of EAC (adjusted OR = 2.51; 95%CI: 1.56-4.04)[13]. Indeed, the effect of abdominal obesity, as measured by WHR or AD, on risk of OAC has been reported to be independent of BMI[8,10]. This implies a role of abdominal obesity in the pathogenesis EAC over and above general obesity. Furthermore, the association between general or central adiposity and risk of EAC has been demonstrated to persist despite inclusion of plausible confounders in multivariable analyses, including: symptomatic reflux, physical activity, smoking, and intakes of total energy, red meat, fruit and vegetables[10].
Although the available cohort studies have not clearly shown that visceral adiposity is associated with an increased risk of invasive neoplasia in patients with Barrett’s esophagus, two recent studies have suggested that increased visceral fat tissue[14] or total abdominal obesity[15] are associated with the progression to high-grade dysplasia. Equally a recent meta-analysis including measures of central adiposity at least 5 years before the diagnosis of EAC showed a significant increased risk of cancer with central obesity[13]. Given our understanding of the biology of Barrett’s esophagus, these data do suggest that central obesity promotes cancers in patients with Barrett’s esophagus.
OBESITY AND RISK OF GASTRO-ESOPHAGEAL REFLUX
A commonly proposed mechanical explanation for the associations between obesity and EAC is through the following sequence: increased abdominal adiposity leading to increased intra-abdominal pressure, then consequent reflux predisposing to Barrett’s esophagus and then EAC[16]. While either abdominal or central adiposity has been associated with each of these “steps”[7,17-19] it has not been possible so far to empirically demonstrate this whole sequence to be causal[17]. Measures of central obesity appear strongly associated with symptomatic reflux, independent of BMI, in a dose-dependent manner[20]. However, in patients with reflux, for each kg/m2 increase in BMI, while both intra-gastric pressure and gastro-esophageal pressure gradient (GEPG) rise[18]; increments in GEPG are not associated with acid exposure as determined by 24-h pH monitoring. Therefore, obesity does not appear to promote reflux through a purely mechanical means, which suggests alternative obesity-induced mechanisms of esophageal dysfunction are operating. While increased reflux could feasibly contribute to the increased risk of EAC observed in obese persons, other mechanisms are likely at play as obesity is strongly associated with risk of EAC, independent of symptomatic reflux. A recent well-conceived Swedish population-based case-control study, which included 189 incident cases of EAC and 816 population-based controls, demonstrated the effect sizes for overweight BMI categories (> 25 kg/m2) verses underweight BMI category (< 25 kg/m2) on risk of EAC were not significantly different in adjusted or unadjusted models for severity, frequency or duration of reflux[21]. This study demonstrated significant synergy between BMI and reflux, most strikingly for frequency (for more than 3 times per week), but also for severity and duration of reflux, on risk of EAC. Other epidemiological studies have also demonstrated the reflux-independent effects of BMI[8,22-24] and abdominal obesity[8] on the risk of EAC, however, It should be noted these studies rely on the reporting of symptomatic reflux and do not necessarily reflect the actual amount of acid reflux.
It might be the location of the fat rather than pure BMI that is important. Abdominal obesity, rather than excess weight has been suggested as the true association of the increase in GORD. The association between BMI and GORD was attenuated when adjusted for waist circumference suggesting BMI has its affect by increasing abdominal obesity[25].
Although one large cross-sectional study found no association between GERD and waist circumference or waist: hip ratio[26], a considerable body of research suggests that an enlarged waist circumference increases the risk of erosive oesophagitis[27-29]. A Korean study of 5329 subjects reported an association between abdominal visceral adipose tissue volume, but not BMI or waist circumference, and erosive esophagitis[30]. Visceral adipose tissue has been assessed by CT scan and high levels of visceral adipose tissue were significantly associated with the duration of GERD symptoms[31]. The association is most obvious in the white population, which could help explain the high levels in the developed world. It is not associated with black or Asian ethnicities[20].
ADIPOSITY AND RISK OF BARRETT’S ESOPHAGUS
In a recent pooled analysis from the BEACON consortium, including 1102 cases of long segment BE (> 3 cm) and 1400 population-based controls from four case-control studies, increasing waist circumference was significantly associated with risk of BE, independent of BMI (OR = 1.87, 95%CI: 1.22-1.32) for the highest vs lowest quartile), with evidence of a significant biological gradient (OR = 1.16, 95%CI: 1.02-1.32, per 5 cm increase in waist circumference)[19]. The effect sizes for the association between waist circumference and risk of BE were similar in both men and women and were almost unchanged after adjustment for symptomatic reflux.
A meta-analysis reported that BMI per se was not associated with BE[32] but that increased waist circumference that confers a two-fold risk for BE[33]. Further studies have reported similar findings as to the effect of visceral obesity on the risk of BE but have also shown an inverse relationship with glutofemoral obesity[34,35]. This could be due to the less metabolically active nature of glutofemoral adipose tissue which further supports the theory it is distribution of adipose tissue, not just overall increase in weight or the excess fat tissue that is a risk for BE. Recent studies have shown that even abdominal obesity is too crude a tool: it appears to be the visceral (mesenteric) component rather than the subcutaneous component that is the most important risk factor for BE[31,36].
Furthermore, the preponderance of BE in men is not explained by differential risk in men and women according to BMI alone. Although waist circumference increased the risk of BE in male and females, the association in females but not males is attenuated when adjusted for GORD symptoms[37]. It is possible that non-reflux related mechanisms contribute more to development of BE in males and these extra mechanisms could explain the higher male prevalence of BE. Abdominal subcutaneous fat was not associated with the development of BE, whereas visceral adiposity was[14].
The pathogeneses of GERD, BE and cancer are complex and multifactorial[38]. It is important to note that symptoms of GERD are fairly uniformly distributed globally (albeit generally less prevalent in Eastern countries compared to Western or Middle Eastern[38]) but the burden of erosive esophagitis, Barrett’s esophagus and adenocarcinoma becomes increasingly concentrated in white males in the Western world[38]. Whilst this has an important correlation with exactly the group with the greatest increase in visceral obesity, it does limit the global generalizability of the data; whilst the links between obesity and GERD are generally consistent worldwide, the majority of the epidemiological data related to obesity, Barrett’s and cancer, are from this most prevalent group and it is possible, although unproven that other factors may be more important in other racial groups or geographical areas.
WEIGHT LOSS AND RISK OF EAC
Unsurprisingly, to date there are is no significant body of literature on the effect of interventions to promote weight loss as a means to reduce the risk of BE or EAC. At a general population level the age-standardized incidence of EAC is relatively low (approximately 12 per 100000 per year in the United Kingdom)[2] and therefore a randomised controlled trial would require an unfeasibly large sample size to empirically demonstrate this. A clinical trial to determine whether or not a weight loss programme could reduce the risk of EAC in a group at higher risk of progression, such as those with known BE, may be more feasible. However, such a clinical trial would be problematic in interpretation as causality could not be attributed to obesity (or lack of) per se, but ascribed to the intervention designed to promote weight loss, which may plausibly act through a number of pathways (e.g., exercise and diet). There is evidence that weight loss secondary to lifestyle, dietary changes or surgery is associated with a reduction in symptomatic reflux[39]. Ascribing causality to obesity on the risk of EAC can therefore only be determined through comprehension of the available epidemiological data on the key features of the association, which are consistent with a causal relationship, and an appraisal of laboratory data.
MOLECULAR MECHANISMS
Whilst the pathogenesis of EAC is not fully defined, increasingly the molecular changes are being understood[40]. Detailed discussion of the cellular and molecular changes leading to the development and persistence of the clone(s) of cells which give rise to initially Barrett’s Esophagus and the later progression in some cases to adenocarcinoma are outside the scope of this review[41,42] but it seems clear that reflux of gastro-duodenal contents is involved in the initiation, perpetuation and progression of the esophageal changes. However there must be other factors also driving these changes. As reviewed above, there are considerable epidemiological data linking various markers of obesity with the development of EAC and several, not mutually exclusive, biologically plausible mechanisms will be explored (Figure 1). However exactly how these mechanisms associated with obesity interact to promote EAC remains unclear, but exploration of these mechanisms is likely to be fruitful in order to explore new treatment and preventative therapies.
Figure 1.
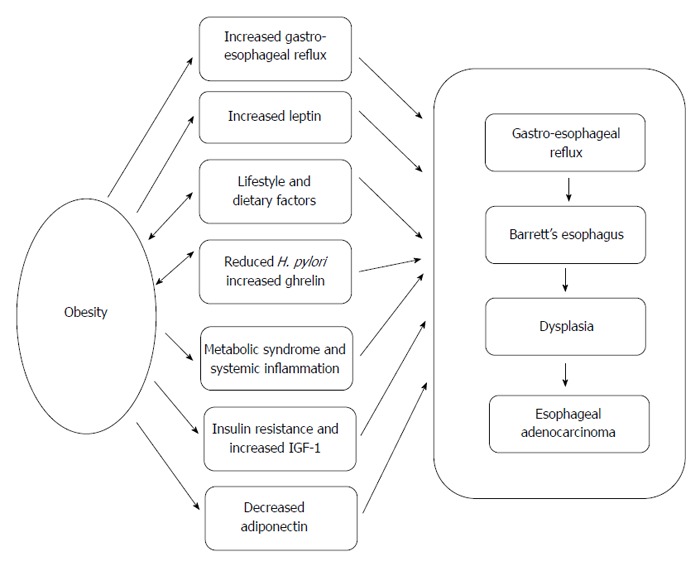
Possible mechanisms linking obesity with the development of esophageal adenocarcinoma. There are several potential and not mutually exclusive mechanisms that could link obesity and esophageal adenocarcinoma. Adipose tissue can exert both mechanical and endocrine effects that could enhance gastro-esophageal reflux and progression to adenocarcinoma. Decreased H. pylori could promote both gastro-esophageal reflux by increasing gastric acidity and increase body mass by enhancing production of the gastric appetite-stimulating peptide ghrelin.
POSSIBLE MECHANISMS LINKING OBESITY WITH ESOPHAGEAL ADENOCARCINOMA
The association is by chance
It is first necessary to consider that the association is merely chance and that obesity does not directly contribute to the pathogenesis of EAC. There have generally been parallel increases in obesity and EAC in the last few decades, and as discussed previously, obesity (especially abdominal visceral obesity) is clearly a risk factor for Barrett’s esophagus and EAC[43]. Some inconsistencies in the data deserve further comment: there have been dramatic rises in the incidence of EAC incidence Australia and Denmark but with much more modest changes in obesity. The epidemic of EAC in the United Kingdom appeared to start about 10 years before that in the United States, yet the United Kingdom was about 10 years behind the United States in the increase in obesity rates[2]. Despite these uncertainties, the vast the majority of the epidemiology showing obesity as a risk factor of EAC is compelling and obesity is also associated with the risk of many other cancers. There is biological plausibility and the relative risk of EAC with obesity is higher than other cancers, all suggesting that the association is real even if obesity is not the sole driver of EAC[3].
Lifestyle or dietary factors associated with obesity increase the risk of EAC
It is possible that specific dietary or lifestyle factors associated with obesity promote EAC development. There are many potential individual variables in but no data specifically implicating any one factor. Smoking, for instance, is a risk factor for both Barrett’s esophagus and progression to EAC[44,45] and lifestyle choices associated with significant obesity may be associated with greater proclivity to smoking but smoking is associated with a lower body mass, including patients with BE[46]. There is a complex and not completely understood inter-relationship between smoking, obesity, Barrett’s esophagus and cancer. Whilst smoking does seem to be a consistent risk factor for progression to EAC[47], the effects on the development of BE are rather more variable; positive[46] and negative[48,49] associations have been reported and a meta-analysis concluded that being an “ever-smoker” was associated with an increased risk of BE when compared to population-based (OR = 1.42, 95%CI: 1.15-1.76) or non-GERD-controls (OR = 1.44, 95%CI: 1.20-1.74) but not GERD-controls (OR = (1.18, 95%CI: 0.75-1.86)[50]. In one study the positive association between EAC and smoking was removed after adjusting co-variables[51]. There are very limited data examining the combination of measures of obesity and smoking of the risks of BE and EAC. Hardikar et al[15] reported that the increased risk of progression to EAC associated with a high WTHR was only seen in male “never smokers” and not in male regular smokers. In a case-control study of endoscopy patients smoking was a risk factor for the development of BE: there was a suggestion that the risk associated with smoking was higher in the more obese (in those with BMI > 30, OR = 5.6, 95%CI: 1.7-18.3) than those of lower body weight (BMI < 30, OR = 3.0, 95%CI: 1.5-6.1)[46], but these were not statistically significant differences. Other studies have failed to show any interaction[23,51] or have not specifically explored any possible interaction[48,49,52]. The decline in the prevalence of smoking has occurred over the same period as EAC has increased and smoking would not explain the racial differences in EAC incidence. Thus although cigarette smoking itself seems to be a risk factor for BE and progression to EAC, there are insufficient data to implicate smoking as direct line between markers of obesity and development of EAC.
Although moderate-severe exercise acutely can precipitate gastro-esophageal reflux, regular exercise is associated with a lower rate of erosive oesophagitis and also protects against obesity[53]. It is possible certain dietary substances may promote both obesity and relaxation of the lower esophageal sphincter (LOS) so promoting reflux disease and EAC. Although there are few convincing data implicating any specific dietary constituents, several possibilities exist: it seems a high calorie content of meals independently of fat content is most likely to provoke reflux[54] and chocolate promotes LOS-relaxation[53]. EAC is also associated with increased meat intake and reduced fruit and vegetable intake[55] and there are many other putative dietary components that could directly or indirectly promote EAC development in obese patients.
Increased gastro-esophageal reflux as the link between obesity and EAC
The link between EAC and fat tissue is much stronger for visceral obesity than overall obesity[43]. Perhaps the most obvious pathogenic link is that the visceral fat tissue exerts mechanical effects on the upper GI tract to promote gastro-esophageal reflux directly and hence the Barrett’s-cancer sequence indirectly. There are considerable experimental data showing that acid and/or bile exert effects on the esophageal epithelium that would be expected to promote cancer (including stimulation of proliferation, inhibition of apoptosis, generation of free radicals[56-58], hence factors that provoke reflux would be expected to enhance the development and progression of Barrett’s esophagus. There are some data to support this hypothesis: obesity is indeed associated with an increased prevalence and severity of reflux[59-61] and also with size of hiatus hernia[62], greater esophageal acid exposure[25], and increased transient lower esophageal relaxations[63]. However it seems likely that visceral fat tissue exerts both direct and indirect effects on the promotion of esophageal carcinogenesis, the majority of the data show that obesity is associated with Barrett’s oesophagus and/or EAC independently of measures of reflux[21,31,64,65].
A separate factor or factors have increased EAC and obesity
One alternative hypothesis is that a separate factor or mechanism has promoted both obesity and EAC independently of each other. There are some data implicating Helicobacter pylori (H. pylori) infection in this situation. Infection of the stomach with H. pylori, particularly the CagA positive strains that provoke more intense gastric mucosal inflammation is inversely associated with both erosive esophagitis and BE[66]. The most plausible explanation for this is that infection and the resulting inflammation of the gastric body leads to a reduction in gastric acid secretion due to either local cytokine production[67] or more irreversible process due to the subsequent development of gastric atrophy[68]. Thus H. pylori infection would be associated with less reflux severe reflux disease due to relatively decreased gastric acid secretion. The prevalence of H. pylori infection has fallen, whilst the incidence of EAC has increased[69] over the last century. Weight gain is common after H. pylori eradication and hence a reduced prevalence of H. pylori could directly provoke more severe reflux disease and an overall increase in body mass. In a recent meta-analysis infection with CagA positive H. pylori was associated with a significantly lower risk of esophageal adenocarcinoma 0.74 (95%CI: 0.57-0.97), although no significant relationship was seen with CagA negative strains or between H. pylori and esophageal squamous cancer[70].
Changes in dyspeptic symptoms could underlie the weight gain but a more direct link between these two has been postulated via the role of gastric ghrelin. Ghrelin is a peptide hormone produced in, and secreted from, the P/D1 cells in the gastric body. Ghrelin stimulates appetite. H. pylori infection is associated with lower levels of gastric mucosa ghrelin and these mucosal levels increase after H. pylori eradication[71,72]. Hence it possible that lower levels of H. pylori infection are directly linked to obesity by increasing appetite. Whilst this is an attractive hypothesis and serum ghrelin levels have been shown increase after successful H. pylori eradication[73], this link between H. pylori status and circulating ghrelin has not be found consistently[74,75] to reliably increase after H. pylori eradication and higher plasma ghrelin levels have themselves been associated with both a lower incidence of erosive oesophagitis (possibly by enhancing gastric emptying[76] and also protection against esophageal adenocarcinoma[77]. However, more in keeping with this hypothesis high plasma ghrelin levels have been shown to be positively associated with the development of Barrett’s esophagus[76]. The time course of H. pylori prevalence is not completely consistent with the changing epidemiology of EAC. The prevalence of H. pylori had been falling steadily throughout the 20th century well before the upsurge in EAC[69] and the increase in EAC incidence in Sweden seemed to begin in the early 1990s, well after the discovery and active treatment of H. pylori. The beginning of the upsurge in EAC in the United Kingdom began in the 1960s, well before the discovery of H. pylori. Hypothesizing that decreased gastric H. pylori infection as a direct cause of both obesity and EAC is also unable to explain the clear gender and racial differences in EAC[2] Therefore H. pylori infection and gastric ghrelin seem not be major contributors to the link between obesity and EAC but these potential mechanisms do outline the potential importance of factors influencing both appetite and mucosal biology.
Meta-inflammation and the metabolic syndrome
Adipose tissue is now recognised as a complex metabolically-active tissue, which secretes a variety of mediators that can have effects throughout the body. These mediators can conveniently, if rather simplistically be grouped into two: those relatively specific for adipose tissue, generally called adipokines or adipocytokines which include several important mediators including leptin, adiponectin, resistin and omentin, these are generally primarily involved in energy balance homeostasis and a second group of systemic cytokines that can be produced by a variety of tissues not limited to fat cells[3,78,79]. Most commentators now accept that obesity is a state of chronic low-grade, systemic inflammation, also termed “meta-inflammation”. This is predominantly caused by the secretion of a variety of pro-inflammatory mediators by the fat tissue. These include tumour necrosis factor-α (TNF-α), IL-1, IL-6, IL-8, interferon-β, MCP-1, VEGF and it is believed these mediators contribute not only to the development of the metabolic syndrome, with resulting insulin resistance and the related complications but also the increased risk of many cancers associated with obesity[79]. Systemic inflammation is recognised as a classical precursor to cancer, it is not completely understood how this systematic inflammatory state promotes cancer although, simplistically, many of these mediators promote cell proliferation, inhibit apoptosis and stimulate angiogenesis, all of which would be expected to promote cancer.
Faecal calprotectin, which is a marker of luminal inflammation, is increased in obesity[80]. There is an increased incidence of most gastrointestinal cancers associated with this obesity-induced inflammatory state[3], but the relative risk of EAC is higher than other cancers. Exactly how this meta-inflammation promotes EAC in the face of what would seem to be more severe and prolonged esophageal inflammation driven separately by acid and bile reflux is uncertain, although it again underlines the potential effects of circulating fat-derived mediators.
The meta-inflammation associated with obesity is associated with insulin resistance and increased circulating concentrations of both insulin and insulin growth factor-1 (IGF-1). This increase in insulin-related factors is at least partly driven by the secretions from metabolically-active visceral adipose tissue. As discussed above, a feature of obesity, and more specifically visceral obesity, is increased levels of inflammatory cytokine and mediators, including free fatty acids, TNF-α, leptin and resistin[81,82] and reduced secretion of adiponectin[83]. Insulin stimulates the production of IGF-1 and decreases production of the major serum proteins which bind insulin and IGF-1, insulin growth factor binding proteins 1 (IGFBP-1) and 3 (IGFBP-3)[84]. The overall effect is to increase the bioavailable levels of IGF-1. Both insulin and IGF-1 can bind to the insulin growth factor receptor complex, stimulating pathways that promote cellular proliferation. In a Barrett’s cohort this insulin resistance has been associated with progression to adenocarcinoma[43]. Insulin and IGF-1 are mitogenic for many tissues, including Barrett’s esophageal cells, which express IGF-1 receptors[85]. IGF-1 receptor expression is increased in EAC specimens resected from viscerally obese patients[86]. However the available data are conflicting on the role of IGF-1 and insulin as risk factors for malignant progression in BE. An increase in risk of cancer or BE have been reported[84,86], but other studies have failed to demonstrate any association between the metabolic syndrome and the risk of EAC [43] or between serum IGF-1 or IGFBP3 (the predominant serum binding protein) levels and progression of Barrett’s[87].
Adipokines as effectors of the esophageal mucosal changes
These general inflammatory changes may be important in the development of EAC, but the specific role of adipokines is attracting considerable attention. Leptin and adiponectin have been examined in some detail and it is possible they are a direct mechanistic link between obesity and progression to EAC (Figure 2). There are other adipokines such as resistin and omentin[88]: there no cell line or in vitro mechanistic studies examining the effects of these on esophageal tissues and there is a single epidemiological study showing that circulating resistin levels were higher in those with gastro-esophageal reflux disease than either Barrett’s esophagus or controls[83] but further studies are required.
Figure 2.
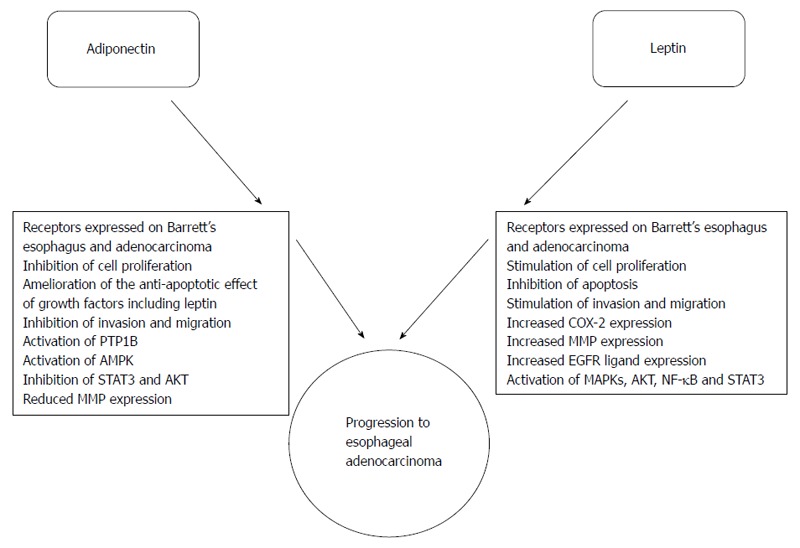
Effects of the adipokines leptin and adiponectin on Barrett’s esophagus and esophageal adenocarcinoma. Obesity, more specifically visceral obesity, is associated with increased serum leptin and decreased serum adiponectin levels. Leptin and adiponectin have a set of antagonistic pathophysiological actions on Barrett’s esophageal and adenocarcinoma cells.
LEPTIN
Leptin is the archetypal adipokine. It is secreted as a 16 kDa protein from fat cells and serum levels are proportionate to body fat mass, as might be expected from it playing an important role as a regulator of appetite, energy metabolism and body weight. In the vast majority of obese subject, serum levels are significantly elevated and leptin deficiency is very rare cause of human obesity, in contrast to the gross obesity of the naturally occurring ob/ob leptin-deficient mouse[89]. It is thought that a degree of hypothalamic hyposensitivity to leptin is a more important cause of clinical obesity[90]. Many studies have reported that increased leptin levels are an independent risk factor for many cancers including breast, colorectal, prostate, ovarian, lung and endometrial[91]. Leptin levels have be an shown to be an independent risk factor for the development of Barrett’s oesophagus[65,76,83], one study showed this effect was seen in male and not females[92] but another study confirmed this association in females[93]. Increased leptin levels have been shown to be an independent risk factor for progression to cancer in a cohort of patients with Barrett’s esophagus[43]. Consistent with these data suggesting that leptin could directly affect the esophagus, leptin receptor expression has been detected in non-dysplastic Barrett’s cell lines, esophageal adenocarcinoma cell lines, Barrett’s esophagus and EAC[94-97]. One study has reported an association between increased leptin-receptor expression and more advanced stage in EAC[96].
Leptin promotes malignant behaviour in experimental esophageal cell line models. Leptin signals via the leptin receptor and increases proliferation, inhibits apoptosis, and stimulates migration and invasion. This is accompanied by the production of the matrix metalloproteinases (MMPs) MMP-2 and MMP-9 which are involved in invasion[94,98]. In a separate study, conditioned media from visceral adipocytes stimulated production of MMP-9 from esophageal adenocarcinoma cell lines and there is a clear association between in vivo MMP-9 production by EAC tissues and visceral obesity[99], suggesting that fat-derived mediators can influence esophageal epithelial behaviours, although the latter study did not confirm a specific role of leptin.
The cell signalling pathways involved in these leptin-induced effects have been well described[94,98]. Binding of leptin to the full-length receptor stimulates phosphorylation of the receptor-associated JAK2 tyrosine kinase which subsequently leads to activation of the protein kinase B/Akt and extra-cellular signal related kinase (ERK) cascades. The p38 MAP kinase pathway is also activated in a JAK2-independent manner downstream of the leptin receptor. The NF-κB pathway is activated, predominantly via upstream Akt activation. Inhibitor studies have shown that the ERK, Akt, NF-κB and p38 pathways are all essential to the proliferative and anti-apoptotic effects of leptin. Co-ordinated activation of these pathways leads to enhanced expression of the cyclo-oxygenase-2 (COX-2) gene. This in turn enhances prostaglandin E2 (PGE2) production. PGE2 leads to by transactivation of the epidermal growth factor receptor (EGFR) and subsequent EGFR-dependant activation of the mitogen activated protein kinase cascades and late activation of c-Jun N-terminal kinase. As well as stimulating the initial steps in the pathway, leptin increases mRNA expression of the EGFR ligands and heparin binding EGF (HB-EGF) in EAC cells and immunoneutralisation of these growth factors blocks the proliferative effects of leptin, confirming their role in the pathway[95].
A separate JAK2-dependant pathway leading activation of signal transducer and activator of transcription 3 (STAT3) is also stimulated by leptin in EAC cells. Activated STAT3 also essential to the proliferation, anti-apoptotic and pro-invasive effects of leptin[98].
The experimental data show that leptin is able to stimulate malignant behaviour in Barrett’s cells bearing the leptin receptor and thus may be a direct link between obesity and progression to EAC. As discussed previously, obesity seems to promote the development of Barrett’s oesophagus and EAC through both reflux-dependent and independent mechanisms. Epidemiologically this combination of obesity and reflux is associated with a cancer risk significantly greater than either alone or when summated[21,31,43,64,65]. There are interesting parallels in the experimental cell models. The combination of exposure to leptin (as a model for obesity) and transient acid exposure (as a model of transient acid reflux) produced significantly greater (and synergistic) cell proliferation and reduction in apoptosis in EAC cell lines[56]. This acid-leptin combination resulted in synergistic activation of the Akt and ERK signalling cascades, without any further increases in leptin-receptor expression, COX-2 expression, PGE2 production and phosphorylation of either p38 MAP kinase or the EGFR[56]. In vivo, esophageal acid exposure enhances MAP kinase activation and mucosal proliferation[100] and increased AKT activation is associated with decreased apoptosis and progression to high grade dysplasia and cancer[101] Therefore it is possible that the continued exposure to the high levels of serum leptin seen in obese subjects enhances the response of Barrett’s mucosa to even physiological acid reflux and promotes malignant change.
In addition to adipocytes, leptin is also synthesised and secreted by chief cells in the gastric body and can be detected in gastric juice. The function of this luminally-secreted leptin is unclear but it is possible it is a physiological regulator of mucosal integrity or nutrient absorption. Therefore esophageal mucosa is potentially exposed to both circulating leptin and that in gastric refluxate, again suggesting some point of convergence between reflux-dependant and -independent mechanisms. Further studies are required into the possible role of gastric leptin however the presence of Barrett’s oesophagus has been associated with increased levels of gastric fundic leptin[102].
ADIPONECTIN
Adiponectin, a 30 kDa protein, is the predominant protein secreted by adipocytes. Unlike leptin, adiponectin secretion falls as obesity increases and so obesity is characterised by relative adiponectin deficiency. The exact mechanisms causing this inverse relationship between fat mass and adiponectin secretion are unclear[103]. As might be expected, in general the effects of adiponectin are to oppose those of leptin and relative adiponectin deficiency has been implicated in the pathogenesis of the metabolic syndrome and its complications, including systemic inflammation. In general low systemic adiponectin levels have been associated with an increased risk of many cancers (including breast, colorectal, prostate, endometrial and gastric)[104]. Comparison between studies is complicated by the various circulating forms of adiponectin, which may have different biological actions and are detected in different assays[105]. Adiponectin is secreted as a full-length monomer (f-adiponectin) than then aggregates into both low- molecular and high-molecular weight oligomers. A truncated form (globular adiponectin (g-adiponectin)) is also found, this is at least partly formed by breakdown of full-length adiponectin by enzymes released in inflammation and circulating levels may not accurately reflect tissue levels of g-adiponectin[106]. There at two specific cell surface adiponectin receptors: AdipoR1 which appears to be relatively globular adiponectin specific and AdipoR2 which has equal affinity for globular and full length adiponectin[107]. Adiponectin may also be able to exert cellular effects by binding to, and inhibiting the action of, HB-EGF[108].
Data are available to support relative adiponectin deficiency in the promotion of EAC (Figure 2). AdipoR1 and AdipoR2 are expressed on both non-dysplastic and neoplastic Barrett’s epithelium[97,109,110]. Circulating adiponectin levels have been shown to be inversely associated with the risk of both Barrett’s oesophagus[83,93,111], and erosive oesophagitis[112]. Increased levels of low-molecular weight adiponectin and a high low-molecular weight/total adiponectin ratio have been shown to be independently associated with a reduced risk of developing Barrett’s esophagus in patients with gastro-esophageal reflux disease[113].This relationship has not been seen in all studies[92]. Perhaps more convincingly low serum levels of adiponectin have been reported to be an independent risk factor for neoplastic progression in a cohort of Barrett’s patients[43].
In a variety of experimental studies adiponectin has been shown to exert anti-cancer effects in Barrett’s cancer cell lines. Adiponectin inhibits leptin-induced proliferation, inhibits leptin-induced invasion and migration and ameliorates the anti-apoptotic effect of leptin. Inhibition of AdipoR1 with RNA interference prevented these effects. Downstream of AdipoR1, these effects are mediated by 5’-AMP activated kinase (AMPK), which ultimately leads to blunting of leptin signalling via inhibition of the Akt pathway[110]. Further detailed studies have shown these inhibitory effects are mediated by the activation of the relatively non-specific protein tyrosine phosphatase PTP1B. Adiponectin leads to increases in both PTP1B mRNA and protein expression and also a separate activation of PTP1B enzyme activity. Activation of this tyrosine phosphatase inhibits signalling via the leptin receptor. These experimental models provide a basis to explain how leptin, adiponectin and acid may interact at the cellular level to promote either the promotion or persistence of Barrett’s epithelium or malignant behaviour in cancer cells and how obesity can remotely influence the risk so EAC[98]. Although speculative at this stage, this potential mechanism of adiponectin via PTP1B could have wider importance. PTP1B is a relatively non-specific phosphatase and would also be expected to inhibit signalling via other pathways that are believed to be important in driving malignant behaviour in Barrett’s epithelium, such as EGFR ligands, IL-6 and bile acids or even those pathways leading to cdx2 expression, which are believed to be central to the development of the Barrett’s phenotype[41,114,115]. Hence relative adiponectin deficiency in obesity could contribute to the development and progression of Barrett’s esophagus at many steps.
The different types of adipose tissue have different hormonal effects. As discussed previously EAC and Barrett’s are most clearly associated with abdominal rather than general obesity[31]. Even within this abdominal obesity there are variable contributions from the separate visceral and subcutaneous fat tissues. More specifically, excess visceral fat being specifically associated with Barrett’s esophagus. Gluteofemoral fat (“hips”) (which is subcutaneous fat) does not seem to be a specific risk factor of BE and may even be protective[34] It is thought gluteofemoral and subcutaneous fat is even less metabolically active and has less effect on progression of Barrett’s oesophagus. In light of this, it is believed that visceral, rather than subcutaneous, fat is usually the predominant source of circulating adiponectin[116-118] and this might explain how reduction in adiponectin secretion from visceral fat probably specifically contributes to the Barrett’s-carcinoma sequence.
IMPLICATIONS FOR THERAPY
The fact that obesity is a risk factor for both BE and EAC is established. This is already being translated into the clinical arena: for example the British Society of Gastroenterology guidelines now suggest that screening and case finding for Barrett’s esophagus be considered in males with reflux symptoms and at least two other risk factors (Caucasian, obesity, smoker), this has the advantage of detecting premalignant cases of Barrett’s esophagus that may be amenable to surveillance and endoscopic therapies if required[119]. A broader question is how may our understanding of the pathophysiological links between obesity and EAC be translated into useful therapeutic gains for prevention or treatment?
The mechanisms linking obesity and esophageal are undoubtedly complex and likely multifactorial and are likely to differ depending on the histological stage of the esophageal mucosa. Experimental and epidemiological studies support a role of the adipokines leptin and adiponectin in the progression to EAC but further mechanistic and clinical studies are still required. At the present time, these pathophysiological insights have suggested several new areas of therapy.
Although it is accepted that gastro-esophageal reflux plays a central role in the pathogenesis of BE and EAC and appears to accentuate the risks associated with obesity, profound acid suppression with either proton pump inhibitors or anti-reflux surgery have not conclusively been shown to have chemopreventative effects. The large United Kingdom AspECT trial comparing placebo, aspirin and standard- and very high dose-esomeprazole in a randomized trial may provide clarity on this issue when data become available[120].
At both a population and individual level weight loss with dietary and behavioural modifications remains the first line approach for obese patients. Gastric bands tend to accentuate reflux and for those patients with reflux symptoms and significant obesity[121], a Roux-en-Y gastric bypass appears to be the preferable procedure, although it cannot be advocated purely to prevent esophageal cancer. Interestingly, as well as a reduction in body mass and visceral fat, and reducing symptoms from gastro-esophageal reflux, this procedure is associated with potentially beneficial metabolic effects including higher serum adiponectin[39,122].
There may yet be some developments in therapies aimed to improve the metabolic/endocrine profile of adipose tissue that may translate into useful clinical interventions. Antagonists of CB1 receptors, such as rimonabant reduce visceral fat[123] and the PPAR-α agonists such as rosiglitazone enhance adiponectin release from visceral fat[118]. Unfortunately at the present time the adverse effects; psychiatric problems with rimonabant and bladder cancer and the increased cardiovascular mortality with PPAR-α agonists preclude their wider use. A variety of other agents have been shown to usefully increase serum adiponectin levels: these include PPAR-α agonists, inhibitors of the renin-angiotensin system, calcium channel modulators and some beta-receptor antagonists[124] and various phyochemical such as catechin[125]. All these deserve further study, although at this time, data are limited and these drugs and their effect on adipokine profiles have not been investigated in the context of esophageal disease[124]. Preclinical development of adiponectin-analogues[126] and leptin-receptor antagonists[127] is continuing but these are some way off clinical use.
Metformin seems to have some potential as a chemopreventative agent in the context of obesity-associated EAC. Metformin is a direct activator of AMPK kinase and exerts potentially useful anti-cancer effects[128]. In Barrett’s cell line studies, the inhibitory effects of adiponectin are also mediated via activation of AMPK[110]. In case-control studies, metformin use is associated with a reduced incidence of many cancers including esophageal cancer[129]. Metformin is inexpensive, has a low incidence of side effects and could be promising chemopreventative agent, although more studies specifically in EAC are needed.
There appear to be more data to recommend aspirin and statins (HMG-CoA reductase inhibitors) as the most appropriate potential chemopreventative agents to reduce the incidence of EAC associated with, or indeed without obesity. Several lines of experimental data show that cyclooxygenase inhibitors, such as aspirin, reduce malignant behaviours such as proliferation and apoptosis-inhibition in EAC and non-neoplastic Barrett’s cells lines. Non-specific and COX-2 selective inhibitors block the effects of leptin in cell line models[130-132]. Definitive conclusions on the preventative effects of aspirin may have to wait until the UK AspECT trial has reported[120]. Observational studies and meta-analyses show that aspirin use is associated with a reduced incidence of both Barrett’s esophagus and EAC[133,134]. Statins exert potent anti-cancer effects in EAC cells line models. By inhibiting the post-translational modification (prenylation) of small signalling G protein of the Ras/Rho family and so ameliorating pro-carcinogenic signalling from growth factor receptors, statins inhibit cell growth and induce apoptosis[135]. Experimentally the effect of inhibition of the COX-2/PGE2 pathway, by using a variety of small molecule COX-inhibitors, inhibition of microsomal PGES-1 or RNA interference, and the effect statins were additive[131,135]. A similar magnitude of reduced risk has been reported in two separate meta-analyses of Barrett’s cohorts, where the combination of COX-inhibitor and statin was associated with a 85% reduction in EAC incidence[133,136]. Statins may also have beneficial effects by increasing increase serum adiponectin levels[124]. It is probably premature to advocate aspirin and statin therapy as primary preventative therapy for all. It is essential to consider that cardiovascular disease and not EAC is predominant cause of death in Barrett’s cohorts and hence that statins and aspirin should be utilised for the beneficial effects on circulatory diseases pending further clarification of the chemopreventative actions[131].
CONCLUSION
Overall a large amount of epidemiological data shows that obesity is likely to be causally associated with esophageal adenocarcinoma. This cancer is strongly associated with an increase in BMI, in fact more so than for other cancers. There are also strong associations between measures of adiposity and gastro-esophageal reflux including the more serious sequelae, reflux esophagitis and Barrett’s esophagus. Abdominal, and in particular visceral, obesity is likely to play a key role in its pathogenesis though both reflux-dependent and -independent mechanisms. Leptin and adiponectin are adipokines secreted by visceral fat cells and both an increased serum leptin decreased serum adiponectin have been reported to be risk factors for progression to EAC. Experimentally, leptin enhances, and adiponectin inhibits malignant behaviour in Barrett’s cell lines, consistent with these mediators having a direct role in the pathogenesis of EAC. No specific chemopreventative strategies are of proven benefit, but appropriate weight loss in overweight subjects seems appropriate. Aspirin and statins seem to have the most potential as chemopreventative actions and should be utilized in patients with Barrett’s esophagus according to the cardiovascular risk profile.
Footnotes
P- Reviewer: Dalamaga M, Efstathiou S S- Editor: Ji FF L- Editor: A E- Editor: Wang CH
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Edgren G, Adami HO, Weiderpass E, Nyrén O. A global assessment of the oesophageal adenocarcinoma epidemic. Gut. 2013;62:1406–1414. doi: 10.1136/gutjnl-2012-302412. [DOI] [PubMed] [Google Scholar]
- 3.Alemán JO, Eusebi LH, Ricciardiello L, Patidar K, Sanyal AJ, Holt PR. Mechanisms of obesity-induced gastrointestinal neoplasia. Gastroenterology. 2014;146:357–373. doi: 10.1053/j.gastro.2013.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008;371:569–578. doi: 10.1016/S0140-6736(08)60269-X. [DOI] [PubMed] [Google Scholar]
- 5.Nguyen DM, El-Serag HB. The epidemiology of obesity. Gastroenterol Clin North Am. 2010;39:1–7. doi: 10.1016/j.gtc.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Popkin BM, Adair LS, Ng SW. Global nutrition transition and the pandemic of obesity in developing countries. Nutr Rev. 2012;70:3–21. doi: 10.1111/j.1753-4887.2011.00456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Snijder MB, van Dam RM, Visser M, Seidell JC. What aspects of body fat are particularly hazardous and how do we measure them? Int J Epidemiol. 2006;35:83–92. doi: 10.1093/ije/dyi253. [DOI] [PubMed] [Google Scholar]
- 8.Corley DA, Kubo A, Zhao W. Abdominal obesity and the risk of esophageal and gastric cardia carcinomas. Cancer Epidemiol Biomarkers Prev. 2008;17:352–358. doi: 10.1158/1055-9965.EPI-07-0748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacInnis RJ, English DR, Hopper JL, Giles GG. Body size and composition and the risk of gastric and oesophageal adenocarcinoma. Int J Cancer. 2006;118:2628–2631. doi: 10.1002/ijc.21638. [DOI] [PubMed] [Google Scholar]
- 10.O’Doherty MG, Freedman ND, Hollenbeck AR, Schatzkin A, Abnet CC. A prospective cohort study of obesity and risk of oesophageal and gastric adenocarcinoma in the NIH-AARP Diet and Health Study. Gut. 2012;61:1261–1268. doi: 10.1136/gutjnl-2011-300551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steffen A, Schulze MB, Pischon T, Dietrich T, Molina E, Chirlaque MD, Barricarte A, Amiano P, Quirós JR, Tumino R, et al. Anthropometry and esophageal cancer risk in the European prospective investigation into cancer and nutrition. Cancer Epidemiol Biomarkers Prev. 2009;18:2079–2089. doi: 10.1158/1055-9965.EPI-09-0265. [DOI] [PubMed] [Google Scholar]
- 12.Beddy P, Howard J, McMahon C, Knox M, de Blacam C, Ravi N, Reynolds JV, Keogan MT. Association of visceral adiposity with oesophageal and junctional adenocarcinomas. Br J Surg. 2010;97:1028–1034. doi: 10.1002/bjs.7100. [DOI] [PubMed] [Google Scholar]
- 13.Singh S, Sharma AN, Murad MH, Buttar NS, El-Serag HB, Katzka DA, Iyer PG. Central adiposity is associated with increased risk of esophageal inflammation, metaplasia, and adenocarcinoma: a systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2013;11:1399–1412.e7. doi: 10.1016/j.cgh.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nelsen EM, Kirihara Y, Takahashi N, Shi Q, Lewis JT, Namasivayam V, Buttar NS, Dunagan KT, Prasad GA. Distribution of body fat and its influence on esophageal inflammation and dysplasia in patients with Barrett’s esophagus. Clin Gastroenterol Hepatol. 2012;10:728–734; quiz e61-62. doi: 10.1016/j.cgh.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hardikar S, Onstad L, Blount PL, Odze RD, Reid BJ, Vaughan TL. The role of tobacco, alcohol, and obesity in neoplastic progression to esophageal adenocarcinoma: a prospective study of Barrett’s esophagus. PLoS One. 2013;8:e52192. doi: 10.1371/journal.pone.0052192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lagergren J. Influence of obesity on the risk of esophageal disorders. Nat Rev Gastroenterol Hepatol. 2011;8:340–347. doi: 10.1038/nrgastro.2011.73. [DOI] [PubMed] [Google Scholar]
- 17.Anggiansah R, Sweis R, Anggiansah A, Wong T, Cooper D, Fox M. The effects of obesity on oesophageal function, acid exposure and the symptoms of gastro-oesophageal reflux disease. Aliment Pharmacol Ther. 2013;37:555–563. doi: 10.1111/apt.12208. [DOI] [PubMed] [Google Scholar]
- 18.de Vries DR, van Herwaarden MA, Smout AJ, Samsom M. Gastroesophageal pressure gradients in gastroesophageal reflux disease: relations with hiatal hernia, body mass index, and esophageal acid exposure. Am J Gastroenterol. 2008;103:1349–1354. doi: 10.1111/j.1572-0241.2008.01909.x. [DOI] [PubMed] [Google Scholar]
- 19.Kubo A, Cook MB, Shaheen NJ, Vaughan TL, Whiteman DC, Murray L, Corley DA. Sex-specific associations between body mass index, waist circumference and the risk of Barrett’s oesophagus: a pooled analysis from the international BEACON consortium. Gut. 2013;62:1684–1691. doi: 10.1136/gutjnl-2012-303753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corley DA, Kubo A, Zhao W. Abdominal obesity, ethnicity and gastro-oesophageal reflux symptoms. Gut. 2007;56:756–762. doi: 10.1136/gut.2006.109413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lagergren J, Mattsson F, Nyrén O. Gastroesophageal reflux does not alter effects of body mass index on risk of esophageal adenocarcinoma. Clin Gastroenterol Hepatol. 2014;12:45–51. doi: 10.1016/j.cgh.2013.07.027. [DOI] [PubMed] [Google Scholar]
- 22.Chow WH, Blot WJ, Vaughan TL, Risch HA, Gammon MD, Stanford JL, Dubrow R, Schoenberg JB, Mayne ST, Farrow DC, et al. Body mass index and risk of adenocarcinomas of the esophagus and gastric cardia. J Natl Cancer Inst. 1998;90:150–155. doi: 10.1093/jnci/90.2.150. [DOI] [PubMed] [Google Scholar]
- 23.Whiteman DC, Sadeghi S, Pandeya N, Smithers BM, Gotley DC, Bain CJ, Webb PM, Green AC. Combined effects of obesity, acid reflux and smoking on the risk of adenocarcinomas of the oesophagus. Gut. 2008;57:173–180. doi: 10.1136/gut.2007.131375. [DOI] [PubMed] [Google Scholar]
- 24.Lindblad M, Rodríguez LA, Lagergren J. Body mass, tobacco and alcohol and risk of esophageal, gastric cardia, and gastric non-cardia adenocarcinoma among men and women in a nested case-control study. Cancer Causes Control. 2005;16:285–294. doi: 10.1007/s10552-004-3485-7. [DOI] [PubMed] [Google Scholar]
- 25.El-Serag HB, Ergun GA, Pandolfino J, Fitzgerald S, Tran T, Kramer JR. Obesity increases oesophageal acid exposure. Gut. 2007;56:749–755. doi: 10.1136/gut.2006.100263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen T, Lu M, Wang X, Yang Y, Zhang J, Jin L, Ye W. Prevalence and risk factors of gastroesophageal reflux symptoms in a Chinese retiree cohort. BMC Gastroenterol. 2012;12:161. doi: 10.1186/1471-230X-12-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tai CM, Lee YC, Tu HP, Huang CK, Wu MT, Chang CY, Lee CT, Wu MS, Lin JT, Wang WM. The relationship between visceral adiposity and the risk of erosive esophagitis in severely obese Chinese patients. Obesity (Silver Spring) 2010;18:2165–2169. doi: 10.1038/oby.2010.143. [DOI] [PubMed] [Google Scholar]
- 28.Ha NR, Lee HL, Lee OY, Yoon BC, Choi HS, Hahm JS, Ahn YH, Koh DH. Differences in clinical characteristics between patients with non-erosive reflux disease and erosive esophagitis in Korea. J Korean Med Sci. 2010;25:1318–1322. doi: 10.3346/jkms.2010.25.9.1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang MS, Park DI, Oh SY, Yoo TW, Ryu SH, Park JH, Kim HJ, Cho YK, Sohn CI, Jeon WK, et al. Abdominal obesity is an independent risk factor for erosive esophagitis in a Korean population. J Gastroenterol Hepatol. 2007;22:1656–1661. doi: 10.1111/j.1440-1746.2006.04518.x. [DOI] [PubMed] [Google Scholar]
- 30.Nam SY, Choi IJ, Ryu KH, Park BJ, Kim HB, Nam BH. Abdominal visceral adipose tissue volume is associated with increased risk of erosive esophagitis in men and women. Gastroenterology. 2010;139:1902–1911.e2. doi: 10.1053/j.gastro.2010.08.019. [DOI] [PubMed] [Google Scholar]
- 31.El-Serag HB, Hashmi A, Garcia J, Richardson P, Alsarraj A, Fitzgerald S, Vela M, Shaib Y, Abraham NS, Velez M, et al. Visceral abdominal obesity measured by CT scan is associated with an increased risk of Barrett’s oesophagus: a case-control study. Gut. 2014;63:220–229. doi: 10.1136/gutjnl-2012-304189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cook MB, Greenwood DC, Hardie LJ, Wild CP, Forman D. A systematic review and meta-analysis of the risk of increasing adiposity on Barrett’s esophagus. Am J Gastroenterol. 2008;103:292–300. doi: 10.1111/j.1572-0241.2007.01621.x. [DOI] [PubMed] [Google Scholar]
- 33.Kramer JR, Fischbach LA, Richardson P, Alsarraj A, Fitzgerald S, Shaib Y, Abraham NS, Velez M, Cole R, Anand B, et al. Waist-to-hip ratio, but not body mass index, is associated with an increased risk of Barrett’s esophagus in white men. Clin Gastroenterol Hepatol. 2013;11:373–381.e1. doi: 10.1016/j.cgh.2012.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rubenstein JH, Morgenstern H, Chey WD, Murray J, Scheiman JM, Schoenfeld P, Appelman HD, McMahon L, Metko V, Kellenberg J, et al. Protective role of gluteofemoral obesity in erosive oesophagitis and Barrett’s oesophagus. Gut. 2014;63:230–235. doi: 10.1136/gutjnl-2012-304103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edelstein ZR, Farrow DC, Bronner MP, Rosen SN, Vaughan TL. Central adiposity and risk of Barrett’s esophagus. Gastroenterology. 2007;133:403–411. doi: 10.1053/j.gastro.2007.05.026. [DOI] [PubMed] [Google Scholar]
- 36.Akiyama T, Yoneda M, Inamori M, Iida H, Endo H, Hosono K, Yoneda K, Fujita K, Koide T, Tokoro C, et al. Visceral obesity and the risk of Barrett’s esophagus in Japanese patients with non-alcoholic fatty liver disease. BMC Gastroenterol. 2009;9:56. doi: 10.1186/1471-230X-9-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kendall BJ, Macdonald GA, Hayward NK, Prins JB, O’Brien S, Whiteman DC. The risk of Barrett’s esophagus associated with abdominal obesity in males and females. Int J Cancer. 2013;132:2192–2199. doi: 10.1002/ijc.27887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boeckxstaens G, El-Serag HB, Smout AJ, Kahrilas PJ. Symptomatic reflux disease: the present, the past and the future. Gut. 2014;63:1185–1193. doi: 10.1136/gutjnl-2013-306393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Groot NL, Burgerhart JS, Van De Meeberg PC, de Vries DR, Smout AJ, Siersema PD. Systematic review: the effects of conservative and surgical treatment for obesity on gastro-oesophageal reflux disease. Aliment Pharmacol Ther. 2009;30:1091–1102. doi: 10.1111/j.1365-2036.2009.04146.x. [DOI] [PubMed] [Google Scholar]
- 40.Gibson MK, Dhaliwal AS, Clemons NJ, Phillips WA, Dvorak K, Tong D, Law S, Pirchi ED, Räsänen J, Krasna MJ, et al. Barrett’s esophagus: cancer and molecular biology. Ann N Y Acad Sci. 2013;1300:296–314. doi: 10.1111/nyas.12252. [DOI] [PubMed] [Google Scholar]
- 41.Dvorak K, Goldman A, Kong J, Lynch JP, Hutchinson L, Houghton JM, Chen H, Chen X, Krishnadath KK, Westra WM. Molecular mechanisms of Barrett’s esophagus and adenocarcinoma. Ann N Y Acad Sci. 2011;1232:381–391. doi: 10.1111/j.1749-6632.2011.06062.x. [DOI] [PubMed] [Google Scholar]
- 42.Fang Y, Chen X, Bajpai M, Verma A, Das KM, Souza RF, Garman KS, Donohoe CL, O’Farrell NJ, Reynolds JV, et al. Cellular origins and molecular mechanisms of Barrett’s esophagus and esophageal adenocarcinoma. Ann N Y Acad Sci. 2013;1300:187–199. doi: 10.1111/nyas.12249. [DOI] [PubMed] [Google Scholar]
- 43.Duggan C, Onstad L, Hardikar S, Blount PL, Reid BJ, Vaughan TL. Association between markers of obesity and progression from Barrett’s esophagus to esophageal adenocarcinoma. Clin Gastroenterol Hepatol. 2013;11:934–943. doi: 10.1016/j.cgh.2013.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cook MB, Shaheen NJ, Anderson LA, Giffen C, Chow WH, Vaughan TL, Whiteman DC, Corley DA. Cigarette smoking increases risk of Barrett’s esophagus: an analysis of the Barrett’s and Esophageal Adenocarcinoma Consortium. Gastroenterology. 2012;142:744–753. doi: 10.1053/j.gastro.2011.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pohl H, Wrobel K, Bojarski C, Voderholzer W, Sonnenberg A, Rösch T, Baumgart DC. Risk factors in the development of esophageal adenocarcinoma. Am J Gastroenterol. 2013;108:200–207. doi: 10.1038/ajg.2012.387. [DOI] [PubMed] [Google Scholar]
- 46.Balasubramanian G, Gupta N, Giacchino M, Singh M, Kanakadandi V, Gaddam S, Wani SB, Higbee AD, Rastogi A, Bansal A, et al. Cigarette smoking is a modifiable risk factor for Barrett’s oesophagus. United European Gastroenterol J. 2013;1:430–437. doi: 10.1177/2050640613504917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coleman HG, Bhat S, Johnston BT, McManus D, Gavin AT, Murray LJ. Tobacco smoking increases the risk of high-grade dysplasia and cancer among patients with Barrett’s esophagus. Gastroenterology. 2012;142:233–240. doi: 10.1053/j.gastro.2011.10.034. [DOI] [PubMed] [Google Scholar]
- 48.Yates M, Cheong E, Luben R, Igali L, Fitzgerald R, Khaw KT, Hart A. Body mass index, smoking, and alcohol and risks of Barrett’s esophagus and esophageal adenocarcinoma: a UK prospective cohort study. Dig Dis Sci. 2014;59:1552–1559. doi: 10.1007/s10620-013-3024-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thrift AP, Kramer JR, Richardson PA, El-Serag HB. No significant effects of smoking or alcohol consumption on risk of Barrett’s esophagus. Dig Dis Sci. 2014;59:108–116. doi: 10.1007/s10620-013-2892-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andrici J, Cox MR, Eslick GD. Cigarette smoking and the risk of Barrett’s esophagus: a systematic review and meta-analysis. J Gastroenterol Hepatol. 2013;28:1258–1273. doi: 10.1111/jgh.12230. [DOI] [PubMed] [Google Scholar]
- 51.Cooper S, Menon S, Nightingale P, Trudgill Nj. Risk factors for the development of oesophageal adenocarcinoma in Barrett’s oesophagus: a UK primary care retrospective nested case-control study. United European Gastroenterol J. 2014;2:91–98. doi: 10.1177/2050640614523596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steevens J, Schouten LJ, Driessen AL, Huysentruyt CJ, Keulemans YC, Goldbohm RA, van den Brandt PA. A prospective cohort study on overweight, smoking, alcohol consumption, and risk of Barrett’s esophagus. Cancer Epidemiol Biomarkers Prev. 2011;20:345–358. doi: 10.1158/1055-9965.EPI-10-0636. [DOI] [PubMed] [Google Scholar]
- 53.Falk GW, Jacobson BC, Riddell RH, Rubenstein JH, El-Zimaity H, Drewes AM, Roark KS, Sontag SJ, Schnell TG, Leya J, et al. Barrett’s esophagus: prevalence-incidence and etiology-origins. Ann N Y Acad Sci. 2011;1232:1–17. doi: 10.1111/j.1749-6632.2011.06042.x. [DOI] [PubMed] [Google Scholar]
- 54.Fox M, Barr C, Nolan S, Lomer M, Anggiansah A, Wong T. The effects of dietary fat and calorie density on esophageal acid exposure and reflux symptoms. Clin Gastroenterol Hepatol. 2007;5:439–444. doi: 10.1016/j.cgh.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 55.Navarro Silvera SA, Mayne ST, Risch HA, Gammon MD, Vaughan T, Chow WH, Dubin JA, Dubrow R, Schoenberg J, Stanford JL, et al. Principal component analysis of dietary and lifestyle patterns in relation to risk of subtypes of esophageal and gastric cancer. Ann Epidemiol. 2011;21:543–550. doi: 10.1016/j.annepidem.2010.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beales IL, Ogunwobi OO. Leptin synergistically enhances the anti-apoptotic and growth-promoting effects of acid in OE33 oesophageal adenocarcinoma cells in culture. Mol Cell Endocrinol. 2007;274:60–68. doi: 10.1016/j.mce.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 57.Dvorak K, Payne CM, Chavarria M, Ramsey L, Dvorakova B, Bernstein H, Holubec H, Sampliner RE, Guy N, Condon A, et al. Bile acids in combination with low pH induce oxidative stress and oxidative DNA damage: relevance to the pathogenesis of Barrett’s oesophagus. Gut. 2007;56:763–771. doi: 10.1136/gut.2006.103697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huo X, Juergens S, Zhang X, Rezaei D, Yu C, Strauch ED, Wang JY, Cheng E, Meyer F, Wang DH, et al. Deoxycholic acid causes DNA damage while inducing apoptotic resistance through NF-κB activation in benign Barrett’s epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2011;301:G278–G286. doi: 10.1152/ajpgi.00092.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jacobson BC, Somers SC, Fuchs CS, Kelly CP, Camargo CA. Body-mass index and symptoms of gastroesophageal reflux in women. N Engl J Med. 2006;354:2340–2348. doi: 10.1056/NEJMoa054391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Peura DA, Pilmer B, Hunt B, Mody R, Perez MC. The effects of increasing body mass index on heartburn severity, frequency and response to treatment with dexlansoprazole or lansoprazole. Aliment Pharmacol Ther. 2013;37:810–818. doi: 10.1111/apt.12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nocon M, Labenz J, Jaspersen D, Meyer-Sabellek W, Stolte M, Lind T, Malfertheiner P, Willich SN. Association of body mass index with heartburn, regurgitation and esophagitis: results of the Progression of Gastroesophageal Reflux Disease study. J Gastroenterol Hepatol. 2007;22:1728–1731. doi: 10.1111/j.1440-1746.2006.04549.x. [DOI] [PubMed] [Google Scholar]
- 62.Pandolfino JE, El-Serag HB, Zhang Q, Shah N, Ghosh SK, Kahrilas PJ. Obesity: a challenge to esophagogastric junction integrity. Gastroenterology. 2006;130:639–649. doi: 10.1053/j.gastro.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 63.Wu JC, Mui LM, Cheung CM, Chan Y, Sung JJ. Obesity is associated with increased transient lower esophageal sphincter relaxation. Gastroenterology. 2007;132:883–889. doi: 10.1053/j.gastro.2006.12.032. [DOI] [PubMed] [Google Scholar]
- 64.Lagergren J, Bergström R, Nyrén O. Association between body mass and adenocarcinoma of the esophagus and gastric cardia. Ann Intern Med. 1999;130:883–890. doi: 10.7326/0003-4819-130-11-199906010-00003. [DOI] [PubMed] [Google Scholar]
- 65.Garcia JM, Splenser AE, Kramer J, Alsarraj A, Fitzgerald S, Ramsey D, El-Serag HB. Circulating inflammatory cytokines and adipokines are associated with increased risk of Barrett’s esophagus: a case-control study. Clin Gastroenterol Hepatol. 2014;12:229–238.e3. doi: 10.1016/j.cgh.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rubenstein JH, Inadomi JM, Scheiman J, Schoenfeld P, Appelman H, Zhang M, Metko V, Kao JY. Association between Helicobacter pylori and Barrett’s esophagus, erosive esophagitis, and gastroesophageal reflux symptoms. Clin Gastroenterol Hepatol. 2014;12:239–245. doi: 10.1016/j.cgh.2013.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Beales IL, Calam J. Interleukin 1 beta and tumour necrosis factor alpha inhibit acid secretion in cultured rabbit parietal cells by multiple pathways. Gut. 1998;42:227–234. doi: 10.1136/gut.42.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.El-Omar EM, Oien K, El-Nujumi A, Gillen D, Wirz A, Dahill S, Williams C, Ardill JE, McColl KE. Helicobacter pylori infection and chronic gastric acid hyposecretion. Gastroenterology. 1997;113:15–24. doi: 10.1016/s0016-5085(97)70075-1. [DOI] [PubMed] [Google Scholar]
- 69.Banatvala N, Mayo K, Megraud F, Jennings R, Deeks JJ, Feldman RA. The cohort effect and Helicobacter pylori. J Infect Dis. 1993;168:219–221. doi: 10.1093/infdis/168.1.219. [DOI] [PubMed] [Google Scholar]
- 70.Nie S, Chen T, Yang X, Huai P, Lu M. Association of Helicobacter pylori infection with esophageal adenocarcinoma and squamous cell carcinoma: a meta-analysis. Dis Esophagus. 2014;27:645–653. doi: 10.1111/dote.12194. [DOI] [PubMed] [Google Scholar]
- 71.Osawa H. Ghrelin and Helicobacter pylori infection. World J Gastroenterol. 2008;14:6327–6333. doi: 10.3748/wjg.14.6327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Boltin D, Niv Y. Ghrelin, Helicobacter pylori and body mass: is there an association? Isr Med Assoc J. 2012;14:130–132. [PubMed] [Google Scholar]
- 73.Nwokolo CU, Freshwater DA, O’Hare P, Randeva HS. Plasma ghrelin following cure of Helicobacter pylori. Gut. 2003;52:637–640. doi: 10.1136/gut.52.5.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gokcel A, Gumurdulu Y, Kayaselcuk F, Serin E, Ozer B, Ozsahin AK, Guvener N. Helicobacter pylori has no effect on plasma ghrelin levels. Eur J Endocrinol. 2003;148:423–426. doi: 10.1530/eje.0.1480423. [DOI] [PubMed] [Google Scholar]
- 75.Isomoto H, Ueno H, Nishi Y, Wen CY, Nakazato M, Kohno S. Impact of Helicobacter pylori infection on ghrelin and various neuroendocrine hormones in plasma. World J Gastroenterol. 2005;11:1644–1648. doi: 10.3748/wjg.v11.i11.1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rubenstein JH, Morgenstern H, McConell D, Scheiman JM, Schoenfeld P, Appelman H, McMahon LF, Kao JY, Metko V, Zhang M, et al. Associations of diabetes mellitus, insulin, leptin, and ghrelin with gastroesophageal reflux and Barrett’s esophagus. Gastroenterology. 2013;145:1237–1244.e1-5. doi: 10.1053/j.gastro.2013.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.de Martel C, Haggerty TD, Corley DA, Vogelman JH, Orentreich N, Parsonnet J. Serum ghrelin levels and risk of subsequent adenocarcinoma of the esophagus. Am J Gastroenterol. 2007;102:1166–1172. doi: 10.1111/j.1572-0241.2007.01116.x. [DOI] [PubMed] [Google Scholar]
- 78.Donohoe CL, O’Farrell NJ, Doyle SL, Reynolds JV. The role of obesity in gastrointestinal cancer: evidence and opinion. Therap Adv Gastroenterol. 2014;7:38–50. doi: 10.1177/1756283X13501786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yeung CY, Tso AW, Xu A, Wang Y, Woo YC, Lam TH, Lo SV, Fong CH, Wat NM, Woo J, et al. Pro-inflammatory adipokines as predictors of incident cancers in a Chinese cohort of low obesity prevalence in Hong Kong. PLoS One. 2013;8:e78594. doi: 10.1371/journal.pone.0078594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kant P, Fazakerley R, Hull MA. Faecal calprotectin levels before and after weight loss in obese and overweight subjects. Int J Obes (Lond) 2013;37:317–319. doi: 10.1038/ijo.2012.38. [DOI] [PubMed] [Google Scholar]
- 81.Eksteen JA, Scott PA, Perry I, Jankowski JA. Inflammation promotes Barrett’s metaplasia and cancer: a unique role for TNFalpha. Eur J Cancer Prev. 2001;10:163–166. doi: 10.1097/00008469-200104000-00008. [DOI] [PubMed] [Google Scholar]
- 82.Ryan AM, Healy LA, Power DG, Byrne M, Murphy S, Byrne PJ, Kelleher D, Reynolds JV. Barrett esophagus: prevalence of central adiposity, metabolic syndrome, and a proinflammatory state. Ann Surg. 2008;247:909–915. doi: 10.1097/SLA.0b013e3181612cac. [DOI] [PubMed] [Google Scholar]
- 83.Mokrowiecka A, Daniel P, Jasinska A, Pietruczuk M, Pawlowski M, Szczesniak P, Orszulak-Michalak D, Malecka-Panas E. Serum adiponectin, resistin, leptin concentration and central adiposity parameters in Barrett’s esophagus patients with and without intestinal metaplasia in comparison to healthy controls and patients with GERD. Hepatogastroenterology. 2012;59:2395–2399. doi: 10.5754/hge12587. [DOI] [PubMed] [Google Scholar]
- 84.Greer KB, Thompson CL, Brenner L, Bednarchik B, Dawson D, Willis J, Grady WM, Falk GW, Cooper GS, Li L, et al. Association of insulin and insulin-like growth factors with Barrett’s oesophagus. Gut. 2012;61:665–672. doi: 10.1136/gutjnl-2011-300641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Doyle SL, Donohoe CL, Finn SP, Howard JM, Lithander FE, Reynolds JV, Pidgeon GP, Lysaght J. IGF-1 and its receptor in esophageal cancer: association with adenocarcinoma and visceral obesity. Am J Gastroenterol. 2012;107:196–204. doi: 10.1038/ajg.2011.417. [DOI] [PubMed] [Google Scholar]
- 86.Donohoe CL, Doyle SL, McGarrigle S, Cathcart MC, Daly E, O’Grady A, Lysaght J, Pidgeon GP, Reynolds JV. Role of the insulin-like growth factor 1 axis and visceral adiposity in oesophageal adenocarcinoma. Br J Surg. 2012;99:387–396. doi: 10.1002/bjs.8658. [DOI] [PubMed] [Google Scholar]
- 87.Siahpush SH, Vaughan TL, Lampe JN, Freeman R, Lewis S, Odze RD, Blount PL, Ayub K, Rabinovitch PS, Reid BJ, et al. Longitudinal study of insulin-like growth factor, insulin-like growth factor binding protein-3, and their polymorphisms: risk of neoplastic progression in Barrett’s esophagus. Cancer Epidemiol Biomarkers Prev. 2007;16:2387–2395. doi: 10.1158/1055-9965.EPI-06-0986. [DOI] [PubMed] [Google Scholar]
- 88.Ohashi K, Shibata R, Murohara T, Ouchi N. Role of anti-inflammatory adipokines in obesity-related diseases. Trends Endocrinol Metab. 2014;25:348–355. doi: 10.1016/j.tem.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 89.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 90.Vansaun MN. Molecular pathways: adiponectin and leptin signaling in cancer. Clin Cancer Res. 2013;19:1926–1932. doi: 10.1158/1078-0432.CCR-12-0930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Garofalo C, Surmacz E. Leptin and cancer. J Cell Physiol. 2006;207:12–22. doi: 10.1002/jcp.20472. [DOI] [PubMed] [Google Scholar]
- 92.Kendall BJ, Macdonald GA, Hayward NK, Prins JB, Brown I, Walker N, Pandeya N, Green AC, Webb PM, Whiteman DC. Leptin and the risk of Barrett’s oesophagus. Gut. 2008;57:448–454. doi: 10.1136/gut.2007.131243. [DOI] [PubMed] [Google Scholar]
- 93.Thompson OM, Beresford SA, Kirk EA, Bronner MP, Vaughan TL. Serum leptin and adiponectin levels and risk of Barrett’s esophagus and intestinal metaplasia of the gastroesophageal junction. Obesity (Silver Spring) 2010;18:2204–2211. doi: 10.1038/oby.2009.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ogunwobi O, Mutungi G, Beales IL. Leptin stimulates proliferation and inhibits apoptosis in Barrett’s esophageal adenocarcinoma cells by cyclooxygenase-2-dependent, prostaglandin-E2-mediated transactivation of the epidermal growth factor receptor and c-Jun NH2-terminal kinase activation. Endocrinology. 2006;147:4505–4516. doi: 10.1210/en.2006-0224. [DOI] [PubMed] [Google Scholar]
- 95.Ogunwobi OO, Beales IL. Leptin stimulates the proliferation of human oesophageal adenocarcinoma cells via HB-EGF and Tgfalpha mediated transactivation of the epidermal growth factor receptor. Br J Biomed Sci. 2008;65:121–127. doi: 10.1080/09674845.2008.11732814. [DOI] [PubMed] [Google Scholar]
- 96.Howard JM, Beddy P, Ennis D, Keogan M, Pidgeon GP, Reynolds JV. Associations between leptin and adiponectin receptor upregulation, visceral obesity and tumour stage in oesophageal and junctional adenocarcinoma. Br J Surg. 2010;97:1020–1027. doi: 10.1002/bjs.7072. [DOI] [PubMed] [Google Scholar]
- 97.Mokrowiecka A, Sokolowska M, Luczak E, Dudojc M, Wieczfinska J, Kacprzak D, Wierzchniewska-Lawska A, Pawliczak R, Malecka-Panas E. Adiponectin and leptin receptors expression in Barrett’s esophagus and normal squamous epithelium in relation to central obesity status. J Physiol Pharmacol. 2013;64:193–199. [PubMed] [Google Scholar]
- 98.Beales IL, Garcia-Morales C, Ogunwobi OO, Mutungi G. Adiponectin inhibits leptin-induced oncogenic signalling in oesophageal cancer cells by activation of PTP1B. Mol Cell Endocrinol. 2014;382:150–158. doi: 10.1016/j.mce.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 99.Allott EH, Lysaght J, Cathcart MC, Donohoe CL, Cummins R, McGarrigle SA, Kay E, Reynolds JV, Pidgeon GP. MMP9 expression in oesophageal adenocarcinoma is upregulated with visceral obesity and is associated with poor tumour differentiation. Mol Carcinog. 2013;52:144–154. doi: 10.1002/mc.21840. [DOI] [PubMed] [Google Scholar]
- 100.Souza RF, Shewmake K, Terada LS, Spechler SJ. Acid exposure activates the mitogen-activated protein kinase pathways in Barrett’s esophagus. Gastroenterology. 2002;122:299–307. doi: 10.1053/gast.2002.30993. [DOI] [PubMed] [Google Scholar]
- 101.Beales IL, Ogunwobi O, Cameron E, El-Amin K, Mutungi G, Wilkinson M. Activation of Akt is increased in the dysplasia-carcinoma sequence in Barrett’s oesophagus and contributes to increased proliferation and inhibition of apoptosis: a histopathological and functional study. BMC Cancer. 2007;7:97. doi: 10.1186/1471-2407-7-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Francois F, Roper J, Goodman AJ, Pei Z, Ghumman M, Mourad M, de Perez AZ, Perez-Perez GI, Tseng CH, Blaser MJ. The association of gastric leptin with oesophageal inflammation and metaplasia. Gut. 2008;57:16–24. doi: 10.1136/gut.2007.131672. [DOI] [PubMed] [Google Scholar]
- 103.Li FY, Lam KS, Xu A. Therapeutic perspectives for adiponectin: an update. Curr Med Chem. 2012;19:5513–5523. doi: 10.2174/092986712803833173. [DOI] [PubMed] [Google Scholar]
- 104.Kelesidis I, Kelesidis T, Mantzoros CS. Adiponectin and cancer: a systematic review. Br J Cancer. 2006;94:1221–1225. doi: 10.1038/sj.bjc.6603051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dalamaga M, Diakopoulos KN, Mantzoros CS. The role of adiponectin in cancer: a review of current evidence. Endocr Rev. 2012;33:547–594. doi: 10.1210/er.2011-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Waki H, Yamauchi T, Kamon J, Kita S, Ito Y, Hada Y, Uchida S, Tsuchida A, Takekawa S, Kadowaki T. Generation of globular fragment of adiponectin by leukocyte elastase secreted by monocytic cell line THP-1. Endocrinology. 2005;146:790–796. doi: 10.1210/en.2004-1096. [DOI] [PubMed] [Google Scholar]
- 107.Yamauchi T, Iwabu M, Okada-Iwabu M, Kadowaki T. Adiponectin receptors: a review of their structure, function and how they work. Best Pract Res Clin Endocrinol Metab. 2014;28:15–23. doi: 10.1016/j.beem.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 108.Wang Y, Lam KS, Xu JY, Lu G, Xu LY, Cooper GJ, Xu A. Adiponectin inhibits cell proliferation by interacting with several growth factors in an oligomerization-dependent manner. J Biol Chem. 2005;280:18341–18347. doi: 10.1074/jbc.M501149200. [DOI] [PubMed] [Google Scholar]
- 109.Konturek PC, Burnat G, Rau T, Hahn EG, Konturek S. Effect of adiponectin and ghrelin on apoptosis of Barrett adenocarcinoma cell line. Dig Dis Sci. 2008;53:597–605. doi: 10.1007/s10620-007-9922-1. [DOI] [PubMed] [Google Scholar]
- 110.Ogunwobi OO, Beales IL. Globular adiponectin, acting via adiponectin receptor-1, inhibits leptin-stimulated oesophageal adenocarcinoma cell proliferation. Mol Cell Endocrinol. 2008;285:43–50. doi: 10.1016/j.mce.2008.01.023. [DOI] [PubMed] [Google Scholar]
- 111.Rubenstein JH, Dahlkemper A, Kao JY, Zhang M, Morgenstern H, McMahon L, Inadomi JM. A pilot study of the association of low plasma adiponectin and Barrett’s esophagus. Am J Gastroenterol. 2008;103:1358–1364. doi: 10.1111/j.1572-0241.2008.01823.x. [DOI] [PubMed] [Google Scholar]
- 112.Kato M, Watabe K, Hamasaki T, Umeda M, Furubayashi A, Kinoshita K, Kishida O, Fujimoto T, Yamada A, Tsukamoto Y, et al. Association of low serum adiponectin levels with erosive esophagitis in men: an analysis of 2405 subjects undergoing physical check-ups. J Gastroenterol. 2011;46:1361–1367. doi: 10.1007/s00535-011-0453-3. [DOI] [PubMed] [Google Scholar]
- 113.Rubenstein JH, Kao JY, Madanick RD, Zhang M, Wang M, Spacek MB, Donovan JL, Bright SD, Shaheen NJ. Association of adiponectin multimers with Barrett’s oesophagus. Gut. 2009;58:1583–1589. doi: 10.1136/gut.2008.171553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang HY, Zhang Q, Zhang X, Yu C, Huo X, Cheng E, Wang DH, Spechler SJ, Souza RF. Cancer-related inflammation and Barrett’s carcinogenesis: interleukin-6 and STAT3 mediate apoptotic resistance in transformed Barrett’s cells. Am J Physiol Gastrointest Liver Physiol. 2011;300:G454–G460. doi: 10.1152/ajpgi.00458.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Beales IL, Ogunwobi OO. Glycine-extended gastrin inhibits apoptosis in Barrett’s oesophageal and oesophageal adenocarcinoma cells through JAK2/STAT3 activation. J Mol Endocrinol. 2009;42:305–318. doi: 10.1677/JME-08-0096. [DOI] [PubMed] [Google Scholar]
- 116.Snijder MB, Dekker JM, Visser M, Bouter LM, Stehouwer CD, Yudkin JS, Heine RJ, Nijpels G, Seidell JC. Trunk fat and leg fat have independent and opposite associations with fasting and postload glucose levels: the Hoorn study. Diabetes Care. 2004;27:372–377. doi: 10.2337/diacare.27.2.372. [DOI] [PubMed] [Google Scholar]
- 117.Lysaght J, van der Stok EP, Allott EH, Casey R, Donohoe CL, Howard JM, McGarrigle SA, Ravi N, Reynolds JV, Pidgeon GP. Pro-inflammatory and tumour proliferative properties of excess visceral adipose tissue. Cancer Lett. 2011;312:62–72. doi: 10.1016/j.canlet.2011.07.034. [DOI] [PubMed] [Google Scholar]
- 118.Motoshima H, Wu X, Sinha MK, Hardy VE, Rosato EL, Barbot DJ, Rosato FE, Goldstein BJ. Differential regulation of adiponectin secretion from cultured human omental and subcutaneous adipocytes: effects of insulin and rosiglitazone. J Clin Endocrinol Metab. 2002;87:5662–5667. doi: 10.1210/jc.2002-020635. [DOI] [PubMed] [Google Scholar]
- 119.Fitzgerald RC, di Pietro M, Ragunath K, Ang Y, Kang JY, Watson P, Trudgill N, Patel P, Kaye PV, Sanders S, et al. British Society of Gastroenterology guidelines on the diagnosis and management of Barrett’s oesophagus. Gut. 2014;63:7–42. doi: 10.1136/gutjnl-2013-305372. [DOI] [PubMed] [Google Scholar]
- 120.Das D, Chilton AP, Jankowski JA. Chemoprevention of oesophageal cancer and the AspECT trial. Recent Results Cancer Res. 2009;181:161–169. doi: 10.1007/978-3-540-69297-3_15. [DOI] [PubMed] [Google Scholar]
- 121.Cunneen SA, Brathwaite CE, Joyce C, Gersin K, Kim K, Schram JL, Wilson EB, Schwiers M, Gutierrez M. Clinical outcomes of the Realize Adjustable Gastric Band-C at 2 years in a United States population. Surg Obes Relat Dis. 2013;9:885–893. doi: 10.1016/j.soard.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 122.Umeda LM, Pereira AZ, Carneiro G, Arasaki CH, Zanella MT. Postprandial adiponectin levels are associated with improvements in postprandial triglycerides after Roux-en-Y gastric bypass in type 2 diabetic patients. Metab Syndr Relat Disord. 2013;11:343–348. doi: 10.1089/met.2012.0042. [DOI] [PubMed] [Google Scholar]
- 123.Després JP, Ross R, Boka G, Alméras N, Lemieux I. Effect of rimonabant on the high-triglyceride/ low-HDL-cholesterol dyslipidemia, intraabdominal adiposity, and liver fat: the ADAGIO-Lipids trial. Arterioscler Thromb Vasc Biol. 2009;29:416–423. doi: 10.1161/ATVBAHA.108.176362. [DOI] [PubMed] [Google Scholar]
- 124.Lim S, Quon MJ, Koh KK. Modulation of adiponectin as a potential therapeutic strategy. Atherosclerosis. 2014;233:721–728. doi: 10.1016/j.atherosclerosis.2014.01.051. [DOI] [PubMed] [Google Scholar]
- 125.Cho SY, Park PJ, Shin HJ, Kim YK, Shin DW, Shin ES, Lee HH, Lee BG, Baik JH, Lee TR. (-)-Catechin suppresses expression of Kruppel-like factor 7 and increases expression and secretion of adiponectin protein in 3T3-L1 cells. Am J Physiol Endocrinol Metab. 2007;292:E1166–E1172. doi: 10.1152/ajpendo.00436.2006. [DOI] [PubMed] [Google Scholar]
- 126.Otvos L, Haspinger E, La Russa F, Maspero F, Graziano P, Kovalszky I, Lovas S, Nama K, Hoffmann R, Knappe D, et al. Design and development of a peptide-based adiponectin receptor agonist for cancer treatment. BMC Biotechnol. 2011;11:90. doi: 10.1186/1472-6750-11-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rene Gonzalez R, Watters A, Xu Y, Singh UP, Mann DR, Rueda BR, Penichet ML. Leptin-signaling inhibition results in efficient anti-tumor activity in estrogen receptor positive or negative breast cancer. Breast Cancer Res. 2009;11:R36. doi: 10.1186/bcr2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Martin-Castillo B, Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Metformin and cancer: doses, mechanisms and the dandelion and hormetic phenomena. Cell Cycle. 2010;9:1057–1064. doi: 10.4161/cc.9.6.10994. [DOI] [PubMed] [Google Scholar]
- 129.Franciosi M, Lucisano G, Lapice E, Strippoli GF, Pellegrini F, Nicolucci A. Metformin therapy and risk of cancer in patients with type 2 diabetes: systematic review. PLoS One. 2013;8:e71583. doi: 10.1371/journal.pone.0071583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Beales IL, Ogunwobi OO. Microsomal prostaglandin E synthase-1 inhibition blocks proliferation and enhances apoptosis in oesophageal adenocarcinoma cells without affecting endothelial prostacyclin production. Int J Cancer. 2010;126:2247–2255. doi: 10.1002/ijc.24875. [DOI] [PubMed] [Google Scholar]
- 131.Fang D, Das KM, Cao W, Malhotra U, Triadafilopoulos G, Najarian RM, Hardie LJ, Lightdale CJ, Beales IL, Felix VN, et al. Barrett’s esophagus: progression to adenocarcinoma and markers. Ann N Y Acad Sci. 2011;1232:210–229. doi: 10.1111/j.1749-6632.2011.06053.x. [DOI] [PubMed] [Google Scholar]
- 132.Ogunwobi OO, Beales IL. Glycine-extended gastrin stimulates proliferation via JAK2- and Akt-dependent NF-kappaB activation in Barrett’s oesophageal adenocarcinoma cells. Mol Cell Endocrinol. 2008;296:94–102. doi: 10.1016/j.mce.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 133.Beales IL, Hensley A, Loke Y. Reduced esophageal cancer incidence in statin users, particularly with cyclo-oxygenase inhibition. World J Gastrointest Pharmacol Ther. 2013;4:69–79. doi: 10.4292/wjgpt.v4.i3.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Omer ZB, Ananthakrishnan AN, Nattinger KJ, Cole EB, Lin JJ, Kong CY, Hur C. Aspirin protects against Barrett’s esophagus in a multivariate logistic regression analysis. Clin Gastroenterol Hepatol. 2012;10:722–727. doi: 10.1016/j.cgh.2012.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ogunwobi OO, Beales IL. Statins inhibit proliferation and induce apoptosis in Barrett’s esophageal adenocarcinoma cells. Am J Gastroenterol. 2008;103:825–837. doi: 10.1111/j.1572-0241.2007.01773.x. [DOI] [PubMed] [Google Scholar]
- 136.Singh S, Singh AG, Singh PP, Murad MH, Iyer PG. Statins are associated with reduced risk of esophageal cancer, particularly in patients with Barrett’s esophagus: a systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2013;11:620–629. doi: 10.1016/j.cgh.2012.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]