

Abstract
All living organisms are continually exposed to agents that damage their DNA, which threatens the integrity of their genome. As a consequence, cells are equipped with a plethora of DNA repair enzymes to remove the damaged DNA. Unfortunately, situations nevertheless arise where lesions persist, and these lesions block the progression of the cell’s replicase. Under these situations, cells are forced to choose between recombination-mediated “damage avoidance” pathways, or use a specialized DNA polymerase (pol) to traverse the blocking lesion. The latter process is referred to as Translesion DNA Synthesis (TLS). As inferred by its name, TLS not only results in bases being (mis)incorporated opposite DNA lesions, but also downstream of the replicase-blocking lesion, so as to ensure continued genome duplication and cell survival. Escherichia coli and Salmonella typhimurium possess five DNA polymerases, and while all have been shown to facilitate TLS under certain experimental conditions, it is clear that the LexA-regulated and damage-inducible pols II, IV and V perform the vast majority of TLS under physiological conditions. Pol V can traverse a wide range of DNA lesions and performs the bulk of mutagenic TLS, whereas pol II and pol IV appear to be more specialized TLS polymerases.
Keywords: DNA polymerase II; DNA polymerase IV; DNA polymerase V; RecA, LexA, β-clamp; Y-family DNA polymerase; Mutagenesis; Replication Restart; Recombination; SOS response
HISTORICAL OVERVIEW
We now know that TLS is largely facilitated by specialized DNA polymerases that can accommodate bulky adducts in their active sites. Such properties are usually associated with a dramatic decrease in replication fidelity and as a consequence, TLS is often error-prone, leading to an increase in cellular mutagenesis. Significant progress in understanding the molecular mechanisms of TLS have been achieved in the past decade, following the successful purification and biochemical characterization of a number of TLS polymerases. However, prior to the characterization of the highly purified proteins, the pioneering studies on TLS were largely focused on its genetic endpoints, namely its mutagenic consequences. Indeed, the first hint that damage-induced mutagenesis is not a passive process came in the early ‘50s when Jean Weigle reported that mutagenesis of irradiated bacteriophage λ was significantly enhanced, if the host bacterium had also been irradiated (247). However, it was not until the pioneering studies of Evelyn Witkin working with bacteria that could be killed by UV irradiation, but not mutated, that the concept of damage inducible error-prone translesion DNA synthesis was born (249–251, 253). These ideas were further expanded in 1970 by Miroslav Radman in a privately circulated letter in which he suggested that “SOS replication” was the result of the induction of an error-prone DNA polymerase under the control of the recA and lexA genes (reproduced in (30)). (This idea subsequently evolved into the “SOS repair hypothesis”(194, 252) (See Eco-Sal III: Module 5.4.3. The SOS Regulatory Network, and Module 7.2.8. The SOS Response).
Interestingly, damage-induced mutagenesis was not only dependent upon chromosomally encoded bacterial genes, but could be dramatically increased if the host bacterium harbored certain self-transmissible R-plasmids, such as ColIb (96), or R-Utrecht (R205) (143). Indeed, Donald MacPhee even presented evidence in the early ‘70s that R-Utrecht encoded for an error-prone DNA polymerase (142). The ability of R-plasmids to increase cellular mutagenesis prompted Ames and colleagues to introduce pKM101 into Salmonella strains they had developed to detect carcinogens, so as to increase the sensitivity of their assays (147). Further support for the notion of so-called “mutagenesis proteins” was provided shortly thereafter, when Kato and Shinoura (105) and independently Steinborn (219), isolated umu/uvm strains of E. coli that were specifically defective for damage-induced mutagenesis. Interestingly, DNA sequence analysis of the mutagenesis-promoting genes from pKM101 (called mucAB) revealed that they were closely related to the E. coli umuDC genes (57, 109, 185, 186, 212). During the late ‘70s and early ‘80s, many other R-plasmids from various incompatibility groups were also shown to possess mutagenesis-promoting potential (7, 159, 190, 237, 239). Many of these plasmids have now been sequenced in their entirety and similar to pKM101, shown to harbor orthologs of the E. coli umuDC genes (see more detailed information below).
The precise mechanism of Umu-dependent mutagenesis remained elusive for many years, largely because the UmuC protein is insoluble when overexpressed in E. coli (260). Based upon genetic experiments, it was hypothesized that the UmuDC proteins acted to coerce the cells main replicase, pol III, to perform TLS in a two-step process that involved a RecA-dependent (mis)insertion event opposite the lesion, followed by a second UmuDC-dependent extension step (Fig. 1A) (31, 32, 257). However, with the advent of novel expression and purification protocols, the Umu proteins were finally purified in sufficient quantities for detailed biochemical analysis (36). These studies revealed that rather than being accessory factors of pol III, the Umu proteins assemble into a complex that possesses intrinsic polymerase activity and is capable of carrying out unassisted lesion bypass (Fig. 1B). As the fifth E. coli polymerase reported in the literature, the Umu complex subsequently became known as pol V (200, 232).
Figure 1. Two-step model For UV-mutagenesis; then and now.
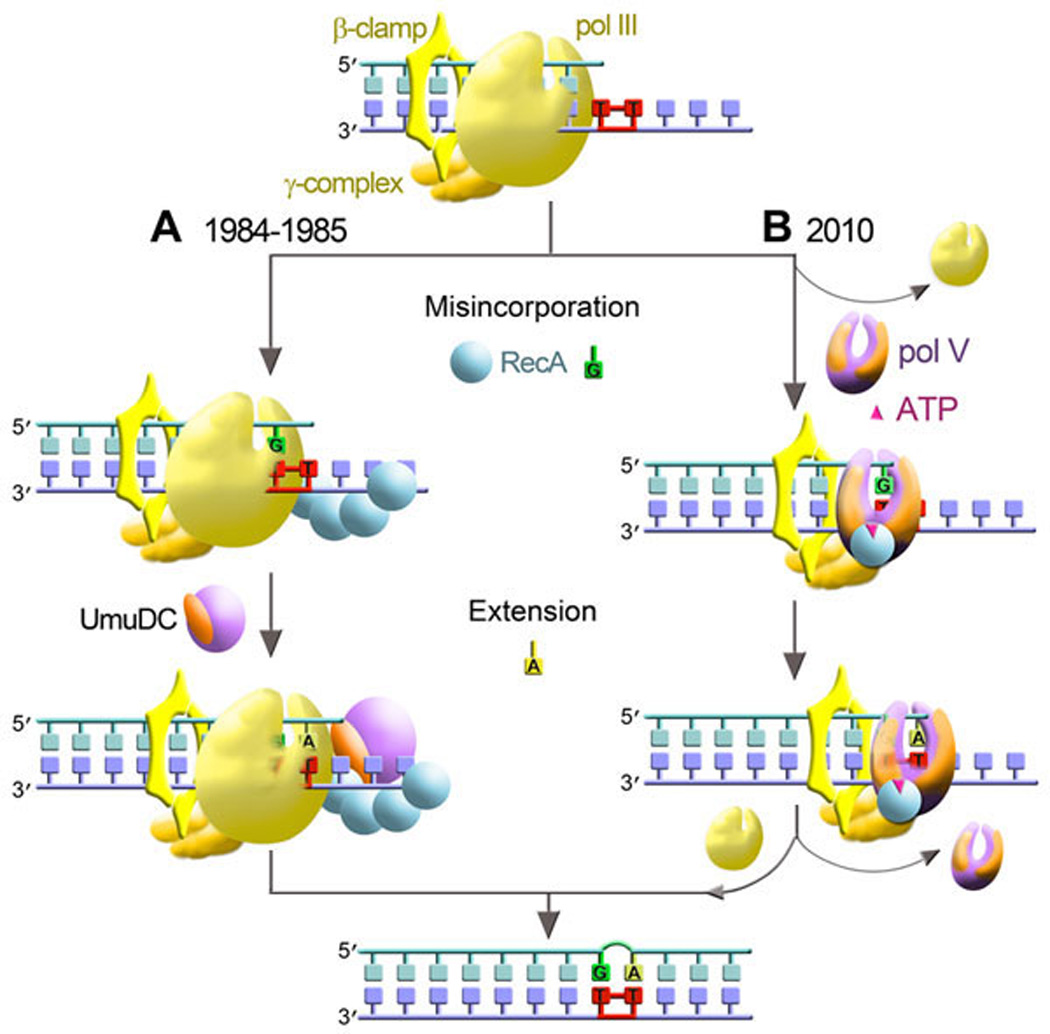
A. The original “two-step” model for UV-mutagenesis reported in 1984/1985 assumed that TLS in vivo is performed by the replicative pol III holoenzyme [consisting of catalytic core (α, θ, ε, shown here and in figures 5–7 as a yellow shape resembling a “right hand”), β-clamp (shown in figures 1, 3, 5 and 9 as yellow hexagonal shape) and γ-clamp loading complex (shown as light orange five-subunit shape)]. The first step, nucleotide misincorporation opposite a 3' T of a CPD, was hypothesized to be mediated by the RecA protein (represented as blue sphere in figures 1, 2, and 9). The misincorporated base was subsequently fixed as a mutation in a second elongation/bypass step that depended upon the UmuC (shown in purple) and UmuD (orange shape) proteins (31–33). B. Subsequent studies revealed that rather than being accessory factors of pol III, the products of umuDC genes encode a bona fide DNA polymerase, pol V (shown here and in figures 3, 7, 8, and 9 as a purple UmuC and two orange UmuD subunits assembled in a shape of a “right hand”), that executes TLS in vivo (200, 232). Thus, at a replication blocking lesion, such as a CPD, pol III is replaced by pol VMut (UmuD'2C-RecA-ATP complex) (104), which can carry out both the (mis)insertion and extension steps of TLS past the CPD. After traversing the damaged DNA, pol VMut is replaced by pol III holenzyme, which resumes high-fidelity chromosomal duplication (178).
We now know that pol V belongs to the much larger Y-family of DNA polymerases that are found in all domains of life (171). Interestingly, many organisms possess multiple Y-family polymerases. E. coli and S.typhimurium are no exception, since in addition to multiple pol V homologs (see below), they also contain the dinB-encoded pol IV (243) (Fig. 2), another Y-family DNA polymerase. The primary cellular role of Y-family polymerases appears to be TLS. While pol V is predominantly error-prone when promoting TLS, other Y-family TLS polymerases, such as E. coli pol IV, are remarkably accurate when bypassing certain DNA lesions, such as N2-dG adducts (85, 103, 158, 265), which perhaps explains why its TLS properties went largely undetected for close to 20 years after the dinB gene was first identified (106). In a similar vein, a role for E. coli’s sole B-family polymerase, pol II (Fig. 2), in TLS also went unnoticed for nearly three decades after its initial discovery in the early 1970s (84, 115). However, by using strains specifically lacking pol II and plasmids harboring site-directed DNA lesions, it has been shown that, like pol IV and pol V, pol II can promote both error-free and error-prone TLS by itself, or in combination with other TLS polymerases (6, 17, 76, 79, 116, 117). While pols II, IV and V perform the vast majority of TLS under physiological conditions, pol III, the cell’s main replicase, nevertheless also has the capacity to facilitate TLS, especially when its 3'–5' exonucleolytic proofreading function is inactivated (178). Indeed, E. coli pol III holoenzyme can bypass a synthetic abasic site in vitro with high efficiency (234) and in the absence of pol I, pol II, pol IV and pol V, an exonuclease-deficient pol III can actually facilitate efficient TLS of a cyclobutane thymine dimer in vivo, which is quite remarkable, given the constraints of its small active site (26).
Figure 2. Domain organization of E. coli polymerases.
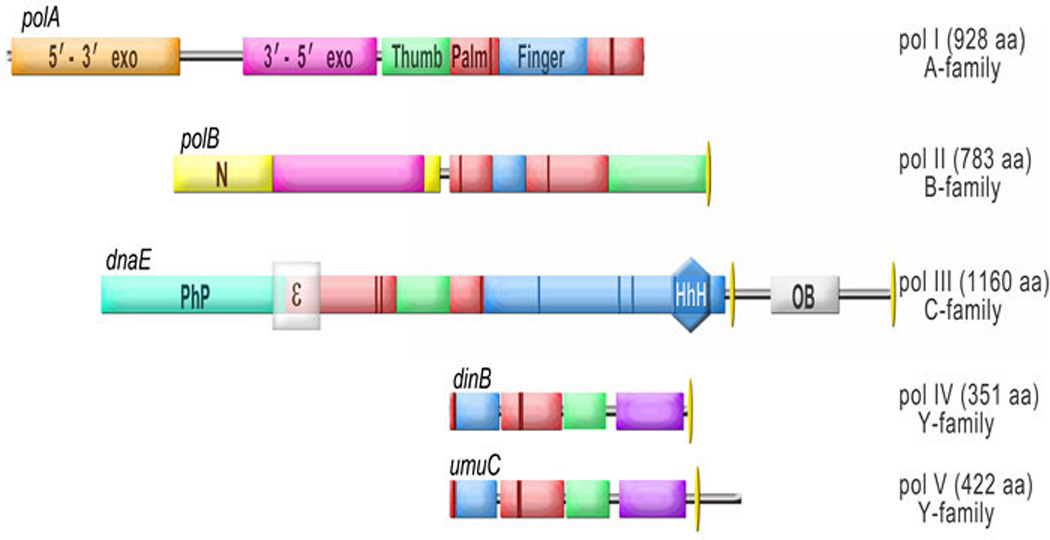
The name and phylogenetic family relationship of each of E. coli’s five DNA polymerases along with number of amino acid residues in each polymerase is indicated on the right, while the name of the gene encoding the polymerase is noted on the left above the domain structure of each respective polymerase. The structural domains present in all polymerases are color coded as shown for pol I (palm - red, thumb - green, finger - blue). The palm domain consists of two segments separated by the insertion of the finger domain for all polymerases except pol III (α-catalytic subunit), in which thumb domain separates these two segments. The five DNA polymerases have been aligned to the smaller segment of the palm domain. Other domains are: 5'-3' exonuclease of pol I– orange; 3'-5' exonuclease of pol I, and pol II– violet; N-terminal domain of pol II – yellow, little finger of pol IV and pol V – purple; PhP (Polymerases and Histidinol Phosphatase) domain- light blue. The region of the DnaE that binds to the ε-(3'-5' proofreading) subunit of pol III is indicated by a semitransparent grey box labeled “ε“. Other regions of DnaE that mediate protein-protein and protein-DNA interactions are the “helix-hairpin-helix” (HhH, shown as a dark blue hexagon) and Oligonucleotide/oligosaccharide Binding (OB, shown as a grey box) domains. The HhH domain provides binding to dsDNA, while the OB domain is involved in preferential interaction with ssDNA. The finger domain of DnaE has four sub-domains (marked by dark blue vertical bars). The acidic catalytic residues of each DNA polymerase are indicated in the diagram as dark red vertical sticks. The narrow yellow ovals mark the region of each polymerase that mediates their respective binding to the β-clamp.
REGULATION OF TLS POLYMERASES
LexA Regulation
All three E. coli TLS polymerases are regulated at the transcriptional level by the LexA repressor (8, 49, 106) (and see Eco-Sal III: Module 5.4.3. The SOS Regulatory Network, and Module 7.2.8. The SOS Response (Fig.3). LexA binds to specific sequences in the operators of genes it regulates, thereby diminishing transcription of the downstream gene. The consensus LexA-binding site is 5'-TACTG(TA)5CAGTA-3' and genes with a close match to the consensus sequence are said to have a low Heterology Index (H.I.) (131), while those with a more divergent LexA-binding site have a high H.I. Upon DNA damage, LexA undergoes a RecA-mediated cleavage reaction leading to its inactivation as a transcriptional repressor (137, 214). As the intracellular levels of intact LexA drops, LexA-regulated genes with binding-sites with a high H.I. are the first to be derepressed, while those with a lower H.I. are the last to become fully derepressed. Interestingly, the LexA-binding sites in the promoter region of both, polB, encoding pol II, and dinB, encoding pol IV, have relatively high H.I. values of 12.09 and 12.84, respectively (65), and as a consequence, both enzymes are expressed at relatively high basal levels. It is estimated that there are ~50 molecules of pol II per undamaged cell (193) and these levels increase approximately 7-fold upon DNA damage (24) (Table 1). Similarly, pol IV is expressed at high basal levels, with an estimated intracellular concentration of ~250 molecules per undamaged cell and these levels increase a further 10-fold in a lexA(Def) strain (108) (Table 1). In contrast, pol V is much more tightly regulated by LexA. Indeed, the LexA binding site in the umuDC operator has a close match to the consensus binding-site with an H.I. value of just 2.77, meaning that it is one of the last LexA-regulated genes/operons to be derepressed after DNA damage (49, 65). Despite tight regulation by LexA, it is estimated that there are nevertheless ~200 molecules of dimeric UmuD in an undamaged cell. However, levels of UmuC are much lower and are essentially undetectable (258). Undamaged cells harboring a lexA(Def) mutation encoding a defective LexA repressor, express ~2400 molecules of UmuD2 and about 200 molecules of UmuC, with the latter being roughly equivalent to basal levels of pol IV (108, 258) (Table 1).
Figure 3. Transcriptional and posttranslational regulation of TLS polymerases.
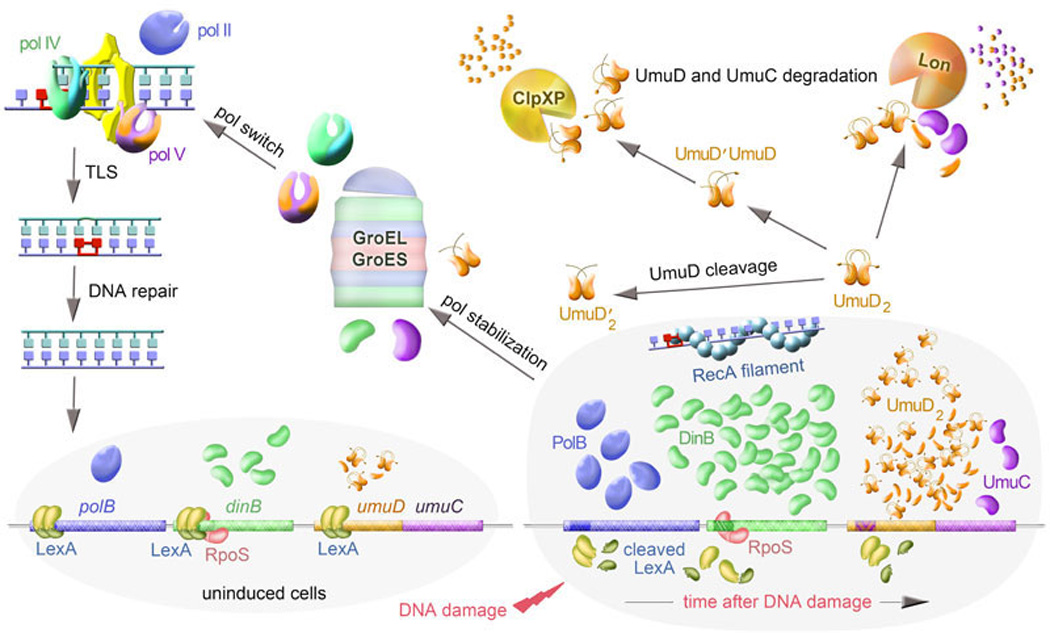
In uninduced cells, LexA (shown as yellow homodimer in which each monomer consists of an N-terminal DNA-binding domain and a C-terminal dimerization domain) represses expression of TLS polymerases (pol II represented in figures 3, 7, and 8 as a dark blue "right hand" shape and pol IV shown here and in figures 5–7 as a green "right hand" shape with a blue ““little finger” domain, as well as both, UmuC and UmuD subunits of pol V) to a different extend depending on Heterology Index (H.I.) of the LexA-binding site in the respective operator/promoter region of each polymerase. The H.I. also determines how soon after LexA cleavage expression of each gene is derepressed. DNA damage-induced formation of a RecA nucleoprotein filament on ssDNA mediates autocleavage of LexA and the derepression of genes in the LexA-regulon. It also mediates autocleavage of UmuD to UmuD'. Uncleaved and cleaved UmuD together with UmuC form different complexes of which UmuD'2C is the most stable. UmuD' is targeted for degradation by ClpXP through preferential formation of UmuD/UmuD' heterodimers. Both UmuC and UmuD are unstable and rapidly degraded by the Lon serine protease. However, UmuC is stabilized by the GroEL and GroES molecular chaperones. The groE gene products also stabilize pol IV. In addition to SOS induction, pol IV expression is also stimulated by RpoS (represented by a pink arched shape) in stressed cells. Upon repair of DNA damage, the disappearance of the RecA filament allows the newly synthesized LexA molecules to block transcription of all LexA-regulated genes and as a consequence, the intracellular concentrations of TLS polymerases return to their basal uninduced levels.
Table 1.
Intracellular concentration of DNA polymerases in E. coli
| −SOS | +SOS | |
|---|---|---|
| pol I (PolA) | 4001 | 400 |
| pol II (PolB) | 50 | 3502 |
| pol III (holoenzyme) | ~15 | 453 |
| pol IV (DinB) | 250 | 25004,8 |
| pol V (UmuD'C complex) | ND5 | 606 |
| UmuD | 200 | 24004 |
| UmuD' | ND | 25007,8 |
| UmuC | ND | 2004,8/ 7007,8 |
From reference 114.
Based upon the catalytic activity of pol II purified from SOS-induced cells (24).
Based upon the catalytic activity of pol III purified from SOS-induced cells (24)
Estimate based upon steady-state levels in strains harboring a lexA(Def) allele that leads to constitutive and full derepression of all LexA-regulated genes (108, 258).
Not detectable
Peak concentration observed in wild-type E. coli strains 45 mins after exposure to ultraviolet light (217).
Estimate based upon steady-state levels in strains harboring a lexA(Def) allele that leads to constitutive and full derepression of all LexA-regulated genes and a recA730 allele that leads to efficient and constitutive conversion of UmuD to UmuD' (258).
These values are almost certainly overestimates because the levels of expression of SOS genes in a lexA(Def) cell are far higher than that occurring during an SOS response. Indeed, Friedman et al., (74) demonstrated that the “graded” increased expression of SOS genes with greater DNA damage is caused by more transcriptional bursts of the same small magnitude over time, and that SOS genes are never fully derepressed as in lexA(Def) cells.
RecA-mediated cleavage of UmuD
Although the Umu proteins are constitutively expressed in lexA(Def) strains, the cells do not exhibit high levels of pol V-dependent mutagenesis (58), suggesting that TLS may be subject to additional levels of control, over and above simple transcriptional regulation by LexA. Clues to the mechanism of regulation came with the DNA sequence analysis of the E. coli umuDC and pKM101 mucAB operons (109, 185). Quite remarkably, both UmuD and MucA exhibited sequence similarity to the C-terminus of the LexA repressor, including conserved active site and cleavage site residues, suggesting that like LexA, UmuD and MucA might also be subject to RecA-mediated cleavage (185). Indeed, RecA-mediated cleavage of UmuD was subsequently shown to occur in vivo (211) and in vitro (39). However, unlike LexA cleavage, which inactivates repressor functions, UmuD cleavage generates a product, UmuD'2, that is activated for mutagenic TLS (168) (Fig.3). The molecular basis for this activation appears to reside in the respective abilities of UmuD2 and UmuD'2 to interact with UmuC. While it is thought that intact UmuD is capable of an interaction with UmuC, such interactions have yet to be shown definitively (209). In contrast, UmuD'2 interacts with UmuC (260) (to form UmuD'2C = pol V) that can be isolated and purified as a complex (36, 80, 230).
RecA does not directly act as a protease to cleave LexA and UmuD, rather it appears to act like a co-protease that helps modify the respective structure of the two proteins, such that they undergo a self-cleavage reaction that is mechanistically similar to signal peptidases (39, 136, 175). While cleavage of LexA is thought to occur via an intramolecular reaction (215), cleavage of dimeric UmuD occurs via an intermolecular reaction (150). That is, cleavage of one UmuD protomer in a UmuD dimer, occurs in the active site of the other protomer of the dimer (Fig. 4). While RecA-mediated cleavage of E. coli UmuD to UmuD' is quite slow, cleavage of S. typhimurium UmuD to UmuD' is very efficient (259) and the rapid cleavage can be attributed to a single amino acid change (Q23P) juxtaposed to the active site of the S.typhimurium UmuD protein (151). Interestingly, despite the efficient cleavage of UmuD to UmuD' that is required for mutagenesis, S.typhimurium is poorly mutable when compared to E. coli (204, 205, 216). Such differences may be attributed to amino acid substitutions in S. typhimurium UmuC that lead to an attenuation of UmuC activity (112).
Figure 4. Structural basis for intermolecular UmuD cleavage.
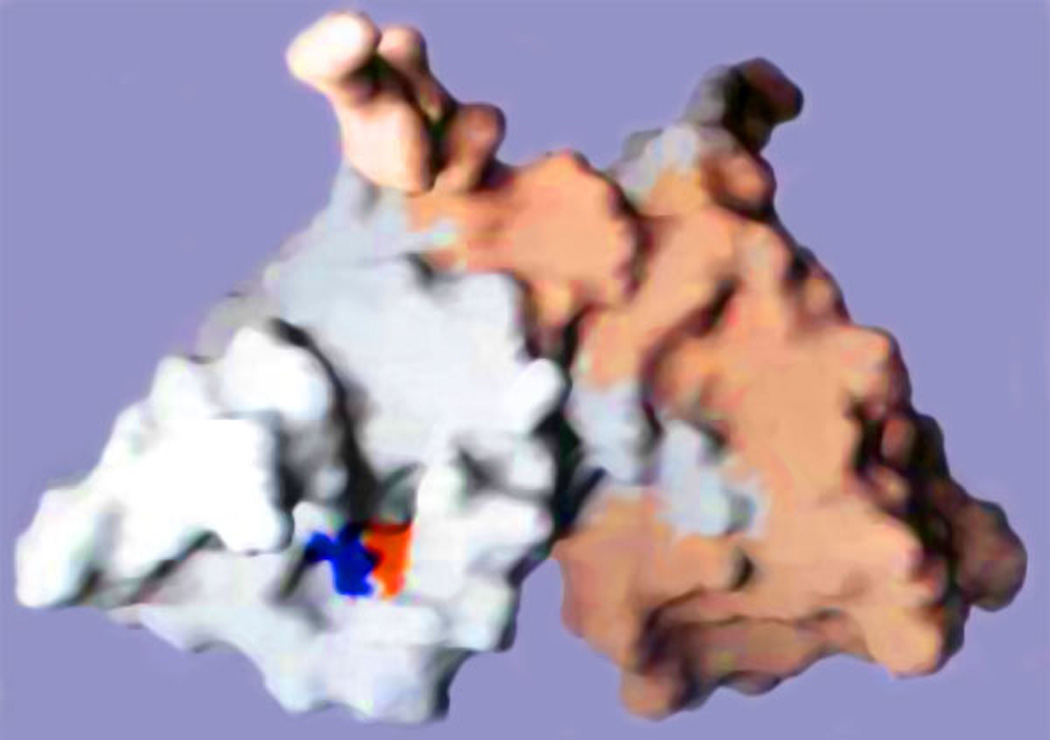
Surface representation of a UmuD' dimer (PDB: 1UMU). One protomer is colored pale gray and another one is colored pale orange. In this orientation, the active site (Ser60 is shown in red and Lys97 is in blue) of the gray protomer is clearly visible at the end of a cleft in the UmuD' protein. By forming such a dimer, the N-terminal tail of the pale orange UmuD' protomer is brought into relatively close proximity with the active site of its gray dimer mate (64, 183). It is thought that a similar configuration is assumed during RecA-mediated cleavage of UmuD and that such protein-protein interactions promote intermolecular UmuD cleavage (21, 152). The figure was generated using the program GRASP (166) and is reproduced with permission from McDonald et al (152).
Regulation of pol V-mediated mutagenesis via proteolysis
While both UmuD and UmuD' proteins can form homodimers in solution (39), when mixed in vitro, they preferentially form UmuD/UmuD' heterodimers (12). Interestingly, when non-cleavable variants of UmuD were expressed in vivo in excess of cleavable UmuD, there was a dramatic reduction in the amount of damage-inducible mutagenesis (12), suggesting that the UmuD/D' heterodimer may, in some way, attenuate mutagenesis. Possible clues to the mechanisms underlying the attenuation came from studies investigating how the Umu proteins might be regulated via proteolysis (72). Both UmuD and UmuC are rapidly degraded in vivo (72) and in vitro (86) by the Lon serine protease (Fig. 3). However, heterodimeric UmuD/D' is largely resistant to Lon, but is, instead, degraded by another serine protease, ClpXP (72, 87, 165). Interestingly, only the UmuD' protomer of the UmuD/D' heterodimer is degraded (165). Furthermore, UmuD' is only degraded by ClpXP when associated with UmuD, and is largely resistant to ClpXP-mediated proteolysis in its homodimeric form. Thus, by forming heterodimers, UmuD' is specifically targeted for degradation, thereby dramatically reducing the intracellular concentration of homodimeric UmuD' available in the cell to interact with UmuC and form mutagenic pol V. Such regulation therefore serves two purposes. First, it helps postpone mutagenic pol V-dependent TLS until the damage-inducing signal is sufficiently elevated to promote more rapid RecA-mediated conversion of UmuD to UmuD', and second, it provides a mechanism whereby intracellular levels of pol V are returned to basal levels once the cellular damage has been repaired and the inducing signal mitigated. (72).
Multiple roles for RecA in TLS
It was known for many years that RecA protein plays multiple roles in pol V-dependent TLS. The first two roles have been described above and are related to RecA's ability to mediate cleavage of LexA, so as to derepress the umu operon, and to mediate cleavage of UmuD, so as to generate the mutagenically active UmuD' protein. However, ΔrecA strains of E. coli expressing recombinant UmuD'C are rendered non-mutable (168), suggesting that RecA is required for a third, possibly direct role, in TLS. Indeed, a direct role for RecA in TLS was confirmed when Devoret and colleagues isolated the recA1730 (F117S) missense allele that was functional for all of RecA's known activities, yet was specifically unable to promote pol V-dependent TLS (55). Furthermore, studies by Sweasy and Witkin demonstrated that even in the presence of overproduced UmuD'C, the level of spontaneous mutagenesis observed in lexA(Def) cells was dependent upon the particular allele of recA in the cell (227). In particular, mutant recA alleles encoding proteins that spontaneously form nucleoprotein filaments (often referred to as RecA*) in the absence of DNA damage, showed the highest levels of spontaneous mutagenesis. Clearly, RecA* actively participates in TLS, yet its “third role” in TLS remained enigmatic for close to two decades. Recent biochemical studies have, however, revealed that the third role of RecA* is to physically interact with UmuD'2C and stimulate the catalytic activity of the polymerase (see below) (104).
Additional levels of regulation imposed on pol IV (DinB)
Like pol V, pol IV is also subject to multiple levels of regulation. In addition to transcriptional control by LexA, the intracellular level of pol IV is modulated by the stress-induced sigma factor, RpoS (Fig. 3), since the expression of pol IV (DinB) is reduced 2-fold in rpoS defective cells (125). Furthermore, Pol IV expression also appears to be induced in a LexA- and RpoS-independent manner by the inhibition of cell wall synthesis (184). In addition, it is likely that pol IV, similar to UmuC (54), is stabilized through an interaction with the groE gene products (126) (Fig. 3). Although there is no direct evidence of posttranslational modification of DinB, there is an indication that the activity and/or fidelity of pol IV might be subject to control beyond transcriptional regulation. For example, it has been proposed that the properties of pol IV are modulated by the ppk gene, encoding polyphosphate kinase (222), which catalyzes the polymerization of inorganic phosphate into long chains of polyphosphate. Indeed, both pol IV dependent stationary-phase adaptive mutagenesis and growth-dependent pol IV mutagenesis decrease in ppk-deficient cells (222). Very recently, DinB has been reported to interact with NusA, which modulates RNA polymerase and it has been hypothesized that such interactions help couple TLS to transcription at sites of DNA damage by targeting pol IV to stalled transcription complexes (46). It has also been shown that uncleaved UmuD protein can physically interact with DinB and in doing so, increases the fidelity of the normally error-prone pol IV (71, 85), possibly by suppressing pol IV-dependent primer-template slippage. Indeed, it may be no mere coincidence that the two proteins (UmuD and DinB) are expressed at similar intracellular levels in both undamaged and damaged cells (Table 1).
Interactions of the TLS polymerases with the β-clamp
All five E. coli DNA polymerases bind the cells homodimeric replicative sliding-clamp β and this interaction is required for both processive synthesis in vitro and the respective enzyme's cellular functions (16, 25, 38, 51, 130, 140, 188, 241). An interaction of the TLS polymerases with the β-clamp is absolutely required for their biological functions in vivo, since mutations in the β-binding motif, or deletion of the motif in either umuC or dinB leads to a complete loss of TLS (16). Several genetic studies have indicated the existence of a usage hierarchy among E. coli's five DNA polymerases that appears to depend on the relative affinity of the polymerases for the β-clamp in the following order: pol III > pol I > pol II > pol IV > pol V (53, 94, 223, 224). Furthermore, the TLS polymerases and replicative polymerases compete with each other for a common docking site in a hydrophobic cleft on the surface near the C-terminal tail of the β-clamp. Within this region, the main interactions occur at overlapping, although distinct sites of each β-protomer (139). The primary β-clamp interacting site of different DNA polymerases is described by the eubacterial clamp-binding motif with a consensus sequence of QL(S/D)LF (51). In pol II, the motif spans residues 778–783 (QLGLF) at the very carboxyl terminus of the polymerase. Similarly, the β-binding motif in pol IV is also located at the C-terminus of DinB and spans residues 346–350 (QLVLG). In contrast, in UmuC, the motif is located approximately 60 amino acids from the C-terminus and spans residues 357–361 (QLNLF) (51). Non-canonical regions of interaction with the β-clamp have also been described for E. coli pol IV. This secondary protein-protein interaction interface involves the Glu93 and Leu98 residues on the rim of the clamp and the C-terminal part of the polymerase that is substantially larger than the main clamp-binding motif (240).
Since the β-clamp is a homodimeric structure, it can likely bind two polymerases at the same time in a manner analogous to a “toolbelt” (176) (Fig. 5). In agreement with this hypothesis, concurrent binding of catalytic subunit of pol III and pol IV to the β-clamp has been demonstrated (98). Furthermore, since at least one TLS polymerase, pol IV, has been shown to possess two binding interfaces with the β-clamp (38, 240), it is likely that two different polymerases can concurrently interact with the same β-clamp chain (93). Thus, during processive DNA replication pol III is bound to the β-clamp at a hydrophobic cleft, while pol IV is kept in close proximity by interacting with the rim of the same molecule (Fig. 5). Different modes of interaction with the β-clamp have also been identified in UmuC (20), suggesting that the proposed “toolbelt” model for the recruitment of TLS polymerases to the progressing replication fork could be universal. Based upon all the supporting data, the current paradigm assumes that β-clamp plays an essential role in coordinating the switch between replicative and TLS polymerases. In addition, by increasing the processivity and catalytic activity of TLS polymerases (19, 80, 231, 241), the β-clamp facilitates synthesis of “TLS patches” which are long enough to prevent premature switching to the replicative pol III and subsequent proofreading/re-synthesis that results in futile cycling (78).
Figure 5. Tool-belt model for polymerase switching.
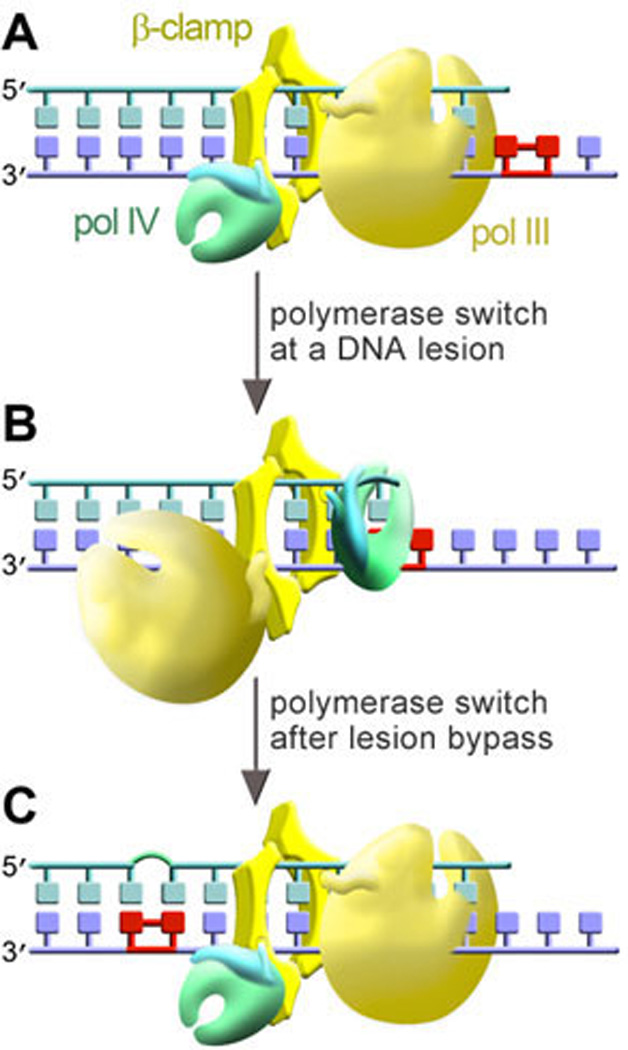
The cartoon depicts pol III (yellow) and pol IV (green with a blue little finger) simultaneously bound to the dimeric β-clamp. In the top panel, the main replicase, pol III, is engaged at the primer terminus. At a DNA lesion (middle panel), pol III disengages from the primer terminus and pol IV rotates into position so as to promote TLS. After TLS has occurred (bottom panel), pol IV is replaced by pol III, which resumes genome duplication. Cartoon modified from Pages and Fuchs (176).
PHENOTYPES OF E. COLI STRAINS WITH DELETIONS OF POL II, IV OR V, OR OVERPRODUCTION OF POL II, IV OR V
Improved evolutionary fitness
Deletion alleles of all three TLS polymerases have been generated in both E. coli and S.typhimurium, indicating that they are not absolutely required for cell viability. Indeed, long-term growth and death rates of E. coli strains with individual deletions of either polB, dinB or umuDC are virtually identical to that of the TLS-proficient wild-type strains, when each strain is incubated separately in rich medium (263). Interestingly, however, when co-cultured under the same conditions in competition with a wild-type strain, the TLS-defective strains exhibit a reduction in fitness and are rapidly replaced by the wild-type strain. However, none of the strains lacking individual SOS-inducible polymerase displayed any clear advantage or disadvantage versus other mutant strains (263).
Similar to the wild-type cells, E. coli strains with individual deletions of polB, dinB or umuDC express the GASP (Growth Advantage in Stationary Phase) phenotype when they undergo long-term stationary-phase starvation. Indeed in all three mutant strains, aged cells out-competed the co-cultured non-aged parents. However, the GASP phenotype in these strains is disrupted at least to some extent when they compete with the wild type non-aged cells (263). Pol V mutants show the greatest reduction in fitness, suggesting that it plays a more significant role in the acquisition of the GASP phenotype. Thus, while not essential, the TLS polymerases are nevertheless critical for long-term cell survival during stationary phase and overall evolutionary fitness of the bacterium.
Replication Restart
The cellular role of pol II remained enigmatic for many years after its initial discovery in the early ‘70s. However, by pulse-labeling DNA with 3H thymidine and comparing the rates of post-UV DNA synthesis in various strains, Rangarajan et al., concluded that pol II plays a critical role in restarting replication after cells have incurred DNA damage (195, 196). In wild-type cells, there is usually a transient inhibition of DNA replication for ~5–10 mins after DNA damage. In contrast, in E. coli strains lacking pol II, the period of inhibition is significantly prolonged to ~50 mins and the resumption of DNA synthesis coincides with the appearance of high levels of pol V. Based upon these observations, Rangarajan et al., hypothesized that pol II plays a major role in the early stages of “Replication Restart”, while pol V appears to act in a back-up process, presumably by promoting TLS of the replication-blocking lesions and continued cell survival. Using a similar approach, but slightly different assay conditions, Courcelle et al., observed no effect of pol II or pol IV on the kinetics of replication restart, but instead, found that replication restart was dependent upon nucleotide excision repair (uvrA) and pol V-dependent TLS (48). In addition, in an independent study Rudolph et al., found that post-UV replication does not change in the absence of pol II, thereby questioning pol II's role, if any, in replication restart (201).
Effects of overproducing TLS polymerases on potential damage-induced replication checkpoints and recombination
As noted earlier, the intracellular concentrations of the TLS polymerases are quite low and at a maximum, increase 7- to 10-fold upon DNA damage (Table 1). In addition, the TLS polymerase activity is regulated by a plethora of protein-protein interactions (e.g. pol II with the β– clamp; pol IV with β– clamp, UmuD and chaperones; and pol V with UmuD2, UmuD/D', UmuD'2, β–clamp, RecA and various proteases). It is not surprising, therefore, that there are often pleotropic effects when the TLS polymerases are significantly overproduced compared to their normally low intracellular levels. Given the fact the levels of the TLS polymerases are clearly strictly regulated, even a small increase in intracellular concentration may lead to phenotypic differences. Indeed, overproduction of pol II and pol IV have been shown to slow pol III-dependent fork progression (81, 97, 236) and overproduction of pol V leads to a cell-cycle checkpoint (161, 174) (See Eco-Sal III: Module 5.4.3. “The SOS Regulatory Network”), as well as inhibiting RecA-dependent recombination (27, 217). Such phenotypes can be attributed to the overproduced polymerases sequestering essential components normally needed for replication and recombination (such as the β–clamp and RecA). However, the physiological relevance of these phenotypes remains in question. In the case of pol V-dependent inhibition of recombination, inhibition was only observed when RecA levels were kept to a minimum, while pol V was artificially elevated. At high cellular concentrations of RecA, no inhibition of recombination was observed (27). In a normal SOS response, just a few hundred molecules of pol V accumulate some 45 mins after DNA damage, when there is an estimated 70,000 molecules of RecA per cell (218). As a consequence, pol V is unlikely to have any significant effect on RecA-dependent recombination under normal SOS-inducing conditions. (See Eco-Sal III: Module 7.2.8. “The SOS Response”, for more discussion on interpreting the phenotypes observed when TLS polymerases are overproduced).
Contributions of TLS polymerases to mutagenesis in vivo
Spontaneous mutagenesis in actively dividing cells
Neither pol IV, nor pol V, possess intrinsic 3'–5' proofreading activity and are therefore considered to be low-fidelity DNA polymerases when they duplicate undamaged DNA. Indeed, in SOS-induced cells overexpressing DinB, there is a dramatic increase in untargeted mutations in bacteriophage λ (34), as well as –1 frameshift mutations in exponentially growing cells (1, 107, 119, 244). As noted above, pol IV is expressed at moderately high basal levels even in an undamaged cell, and could, potentially, contribute to significant number of spontaneous mutations on the E. coli chromosome. However, it appears that basal levels of pol IV are unable to compete with the cell’s replicase, pol III so as to gain access to a primer terminus, since the rates and spectra of spontaneous mutations arising in actively dividing cells does not change in dinB+ or ΔdinB cells (119, 153, 154, 229, 254). Nevertheless, these data do not exclude the possibility that pol IV is involved in processing of rare spontaneous DNA lesions. Indeed, while the mutation frequency remained unchanged in ΔdinB strains defective for repair of abasic sites, bulky DNA adducts, and oxidative DNA damage, inactivation of the glycosylases responsible for excision of 3-methyladenine (3-meA) and 3-methylguanine (3-meG) in cells lacking dinB resulted in a significant increase in spontaneous mutagenesis. Thus, is appears that not only does pol IV replicate DNA with spontaneous alkylation base damage, but actually does it in error-free manner (22). The role of pol IV in the bypass of spontaneously arising alkylation damage could not be detected in cells with functional alkylation repair because the number of alkyl lesions in the genome is probably too low to influence the overall rate of spontaneous mutagenesis. However, the study by Bjedov et al., clearly implies that even at basal steady-state levels, pol IV can successfully compete with pol III for access to the replication fork (22).
The role of pol V in promoting spontaneous mutagenesis is more obvious. While pol V is tightly regulated so as to keep its activity on undamaged DNA to a minimum, the enzyme can nevertheless compete with pol III and gain access to genomic DNA, as the number of spontaneously arising his+ revertants drops 5 to10-fold in a ΔumuDC strain compared to the isogenic umuDC+ parent (256). Pol V-dependent spontaneous mutagenesis increases dramatically in strains with missense mutations in recA that lead to constitutive co-protease functions, such a recA730 (66, 227) and is further enhanced in strains with an impaired replicase (66, 238). For many years, it was thought that the so-called “SOS mutator” activity was entirely dependent upon the umuC gene product (43). However, it now appears that roughly 40–50% of the spontaneously arising mutations are also dependent upon pol IV (50, 120). The simplest interpretation of this observation, is that the mutations arise through a two-step process (33, 257), (Fig. 1B) in which pol V makes the first error-prone misinsertion and this is subsequently extended either by pol V itself, or by pol IV.
Of the three bacterial TLS polymerases, only pol II has intrinsic 3'–5' proofreading (Fig. 2) and therefore exhibits much higher fidelity than pol IV or pol V. Indeed, rather than promoting spontaneous mutagenesis, it is believed that pol II helps maintain E. coli's genomic integrity by removing replication errors generated by pols III, IV and V (9, 50, 83).
Mutagenesis in stationary phase cells
During conditions of starvation, E. coli cells employ the survival strategy of “stress-induced” adaptive mutagenesis, which requires expression of SOS-induced DNA polymerases, even in the absence of exogenous DNA damage (68, 82). The mutagenesis is clearly dependent upon error-prone pol IV, since it is abolished in cells with a deletion of dinB (153, 235) and is also reduced in rpoS cells where the cellular level of pol IV is diminished (125, 138, 191, 221). Historically, stress induced mutagenesis has been followed through the reversion of a +1 frameshift allele lacZ. However, stress induced mutagenesis that is independent of lac reversion can also be observed in ampD leading to increased antibiotic resistance (187), as well as reversion of a tet gene (37, 47, 67, 191). Stress induced mutagenesis appears to be initiated when double strand breaks are repaired (70, 90, 91). Although pol IV plays a major role in stress-induced mutagenesis, other polymerase can also participate in the process. Indeed, it has recently been shown that pol II is responsible for roughly 15% of frameshift reversions that are pol IV-independent (75). In addition, pols I, III and V may reduce pol IV-dependent stress-induced mutagenesis by simply competing with pol IV at a nascent primer terminus generated during double strand break repair (69, 92, 191).
Damage-induced mutagenesis
Since the TLS polymerases are not essential for viability, it has been possible to generate strains of E. coli and S.typhimurium, which carry deletions of each polymerase, either individually, or in combination. Conversely, cloning of the genes encoding the TLS polymerases into low-copy vectors allows for over-expression of respective polymerases. By using such strains, one can analyze the ability of each respective polymerase to promote error-free, or error-prone TLS events after exposing cells to different kinds of environmental stress (117), or to different DNA damaging agents. Using such an approach, Takehiko Nohmi and colleagues reported the effect of chemically induced frameshift mutagenesis in S. typhimurium strains either lacking (113), or overexpressing (146) one or more TLS polymerase. These studies revealed that for many lesions a combination of polymerases is required in order to successfully complete TLS and many lesions could be bypassed by more than one TLS polymerase. For example, it was shown that pol IV and pol V have distinct, but partly overlapping substrate specificities, yet they can cooperate with pol II in the two-polymerase mechanism of TLS, and when overexpressed, both polymerases are able to assume TLS functions normally performed by pol III holoenzyme. Interestingly, overexpression of E. coli pol I reduced Y-family polymerase-dependent chemically induced mutagenesis to the levels observed in the strains lacking all TLS polymerases possibly by suppressing the access of these polymerases to the replication fork (146). Given the fact that each chemical compound is likely to generate a range of DNA lesions in vivo, it is hardly surprising that more than one polymerase is involved in a cellular response to a single DNA damaging agent.
While examination of TLS in randomly damaged cells provides valuable knowledge about polymerase selectivity, more precise information regarding the exact role for each polymerase in TLS has been gained through the use of vectors containing a single defined lesion. The first such studies of this type were undertaken by Chris Lawrence and colleagues, who assayed TLS of a single cis-syn cyclobutane pyrimidine dimer (CPD) in vivo (11). These were quickly followed by studies on other DNA lesions in the single-stranded substrate, including an abasic site (apurinic/apyrimidinic site generated spontaneously, or as a byproduct of chemical or radiation DNA damage) (123) and a UV-induced trans-syn CPD (10) and T-T and T-C 6-4 photoproducts (6-4PP) (95, 127). In each instance, the defined lesion gave rise to a unique mutagenic signature. In all cases, mutagenesis increased in SOS-induced cells and was largely dependent upon pol V since bypass was greatly reduced in a ΔumuDC strain (124, 228). In agreement with the in vivo data, in vitro reconstitution of TLS has shown that in the presence of accessory proteins, pol V is the most efficient among SOS-inducible polymerases in the TLS of CPDs, 6-4PP, and abasic site (24, 144, 232). Furthermore, the nucleotide incorporation specificity of pol V opposite these lesions is in excellent agreement with mutations observed in vivo. For example, pol III and pol IV favor the non-mutagenic incorporation of dA opposite a 3'T of the 6-4PP in vitro, while pol V catalyzes the preferential incorporation of dG, which would result in the T to C transitions often observed in vivo (127, 188, 189). Compared with the 6-4PP, the CPD is a much less mutagenic lesion when replicated by pol V. Interestingly, when E. coli pol V was substituted by R-plasmid encoded orthologs (see below) MucAB (pol RI), or RumAB (pol V(R391)), the efficiency of T-T CPDs bypass and mutagenic spectrum changed considerably, even though the overall mutagenic frequency remained low (124, 170).
In subsequent years, a variety of DNA lesions have been introduced into single-stranded vectors. This includes (but is not limited to) N2-acetylaminofluorene (AAF) and N2-aminofluorene (AF) modified guanines (80, 162, 177, 226), benzo[a]pyrene diol epoxide (BaP) guanine and adenine adducts (129, 162, 208), 8-oxoguanine (8-oxoG) and other guanine oxidation products (141, 163, 164), oxidized abasic lesions (118), N2-dG and other acrolein-induced and related adducts (102, 103, 157, 265), butadiene-induced guanine intrastrand and N2-N2-guanine interstrand crosslinks (41, 121), and alkylated bases (52). By using the single-lesion containing vectors in in vitro reconstituted reactions and by introducing them into strains lacking one or more TLS polymerase, it is straightforward to determine the contribution of each polymerase to the efficiency and accuracy of in vivo lesion bypass. Although pol V is responsible for the bulk of mutagenic TLS, it is not surprising that some lesions require the presence of other SOS-inducible TLS polymerases. For example, only pol IV is capable of the efficient nucleotide incorporation opposite N2-guanine linked DNA–peptide crosslinks and this incorporation proceeds with high fidelity (Fig. 6) (157). Consistent with in vitro data, pol IV-deficient cells lack the ability to replicate DNA vectors with N2-dG–peptide crosslinks. Similar results were obtained with modified DNA containing other bulky N2-dG adducts and N2-N2-guanine interstrand crosslinks (103, 121, 265). Interestingly, pol IV is particularly efficient at extension after dC incorporation opposite the damaged guanine catalyzing the first nucleotide addition after the lesion with up to 25-fold greater catalytic efficiency than after undamaged guanine (102). The exceptional ability of pol IV to accurately copy DNA containing N2-dG damaged bases suggests that this type of lesions is the cognate substrate for pol IV. Pol IV is also involved in the TLS of BaP-induced N2-dG adducts, however in this case pol V and, to a lesser extent, pol II are also likely to play additional roles in TLS (129, 162, 208) (Fig. 7). Moreover, even the cell’s replicase, pol III, is able to carry out limited synthesis past the dG-BaP in a particular specific sequence context (208).
Figure 6. Pol IV can promote unassisted error-free TLS of certain N2 dG lesions.
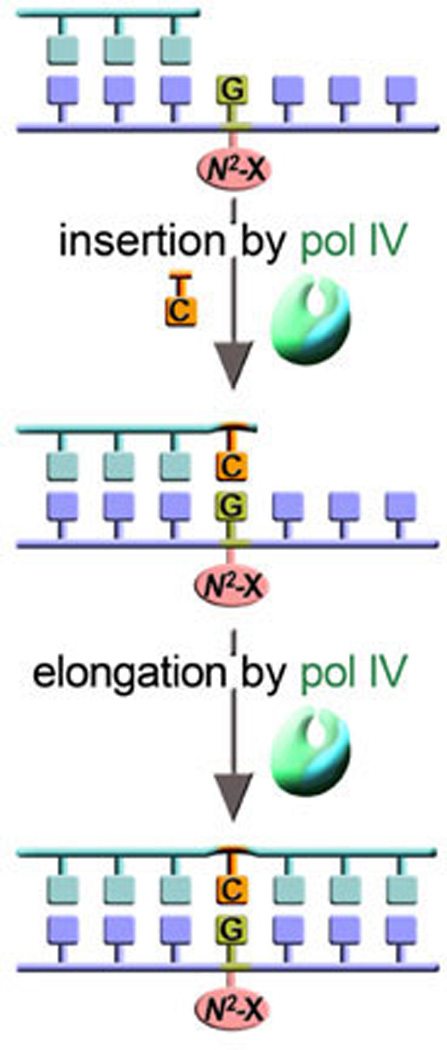
Recent data suggests that pol IV can efficiently and accurately insert nucleotides opposite 1, N2-dG adducts, such as those generated by exposure to nitrofurazone, as well as to extend from the resulting primer termini. As a consequence, cells devoid of dinB are much more sensitive to the killing effects of nitrofurazone-induced adducts (102).
Figure 7. Mutliple TLS polymerases work together to promote BaP lesion bypass.
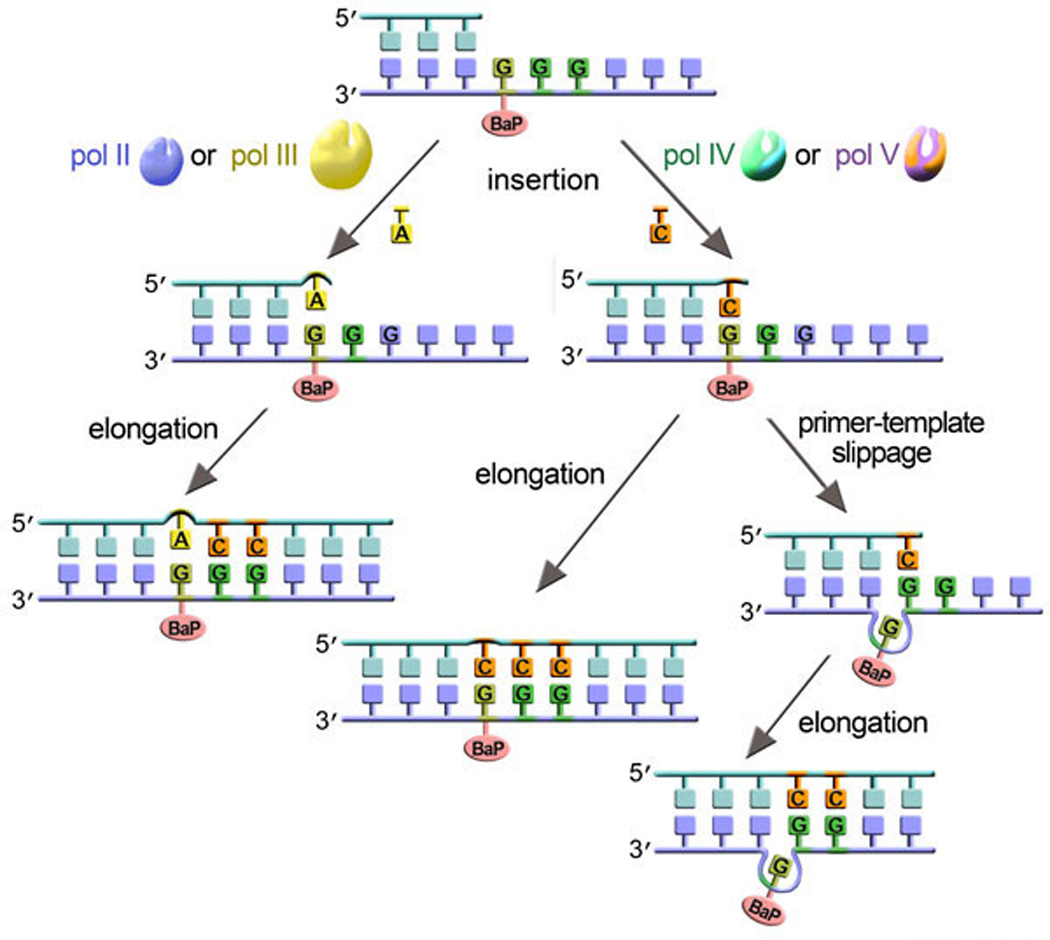
Different DNA polymerases may be involved in TLS of benzo[a]pyrene (BaP) adducts depending on the sequence context surrounding the damaged site, conformation of the adduct and identity of the damaged template base (A vs G) (129, 162, 207, 208, 264). For example, in SOS-induced cells both error-free and −1 frameshift TLS of BaP located within a short run of Gs has been shown to depend largely on pol V and pol IV (108, 134). In this case, bypass is initiated from the correct incorporation of C opposite the dG-BaP. This intermediate product could either be elongated in an error-free manner, or adopt a slipped conformation, elongation of which would result in −1 frameshift mutation. The SOS independent G to T base substitution pathway consists of misincorporation of a dA opposite the dG-BaP and elongation of an A-lesion terminus with both steps likely to be carried out by pol III and/or pol II (162, 208).
Interestingly, despite pol IV’s preference for N2-dG adducts, in vivo replication past N2-dG-AAF does not require pol IV. Instead, pol II, and pol V appear to compete for the bypass of the lesion (Fig. 8) (15, 162). When pol V catalyzes bypass of the AAF adduct, it proceeds in an error-free manner. In contrast, pol II-dependent TLS is error-prone and leads to −2bp frameshift events in vivo. It appears that the pathway is initiated by the insertion of the correct nucleotide opposite the AAF-dG lesion by either pol III or pol V. This is often followed by primer-template slippage that generates a −2 frameshift intermediate that is subsequently extended and fixed as a mutation by pol II (Fig. 8). In contrast, when pol II facilitates TLS of 5-guanidino-4-nitroimidazole (NI), lesion bypass is error-free, while pol V-catalyzed misinsertion at the lesion site causes G to T transversions (164). However, both pol V-dependent error-prone and pol II-dependent error-free TLS pathways require pol V for extension of the nascent strand after nucleotide incorporation opposite the NI lesion. Evidence for the concerted action of SOS-inducible polymerases is also found when assessing TLS of an oxidized abasic site. When the lesion is replicated in E. coli, pol V incorporates dA opposite this lesion producing full-length replication products, while pol II and pol IV utilize a dNTP-stabilized misalignment mechanism to create single-nucleotide deletion products (118).
Figure 8. Pol V and pol II can promote both error-free and error-prone TLS of AAF-dG.
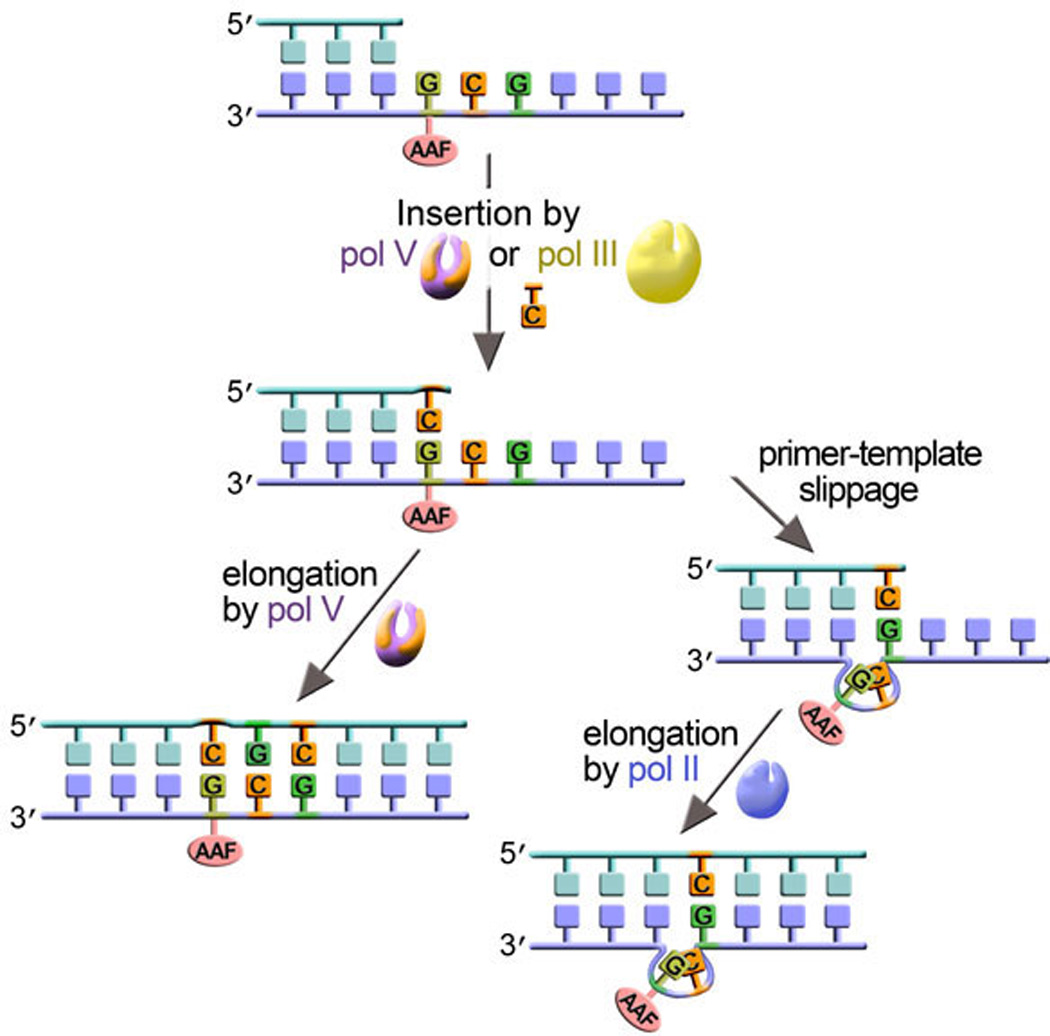
Either pol V or pol III can insert a base opposite an N-Acetyl-2-Aminofluorene (AAF)-dG lesion and both do so in a relatively error-free manner (shown on the left hand side of the figure). Pol V can extend the primer-terminus leading to error free TLS of the AAF-dG adduct. Alternatively, when the AAF-dG lesion is positioned in the 3'-GCG sequence context, and pol II performs the extension step (right-hand side), −2 base-pair frameshifts occur (76, 162).
Taken together, studies of TLS in vivo and in vitro indicate that participation of each polymerase in lesion bypass not only depends on the chemical nature of the lesion, but its local sequence context. In each particular case, cells can recruit a single, or a combination of TLS polymerases to try and accomplish TLS with maximal accuracy and efficiency. The driving goal is to ensure survival of the cell in the face of otherwise certain death and with a minimal amount of mutagenesis. Unfortunately, it’s not a perfect world and a certain amount of mutagenesis is an unavoidable consequence of TLS. Most of these mutations are likely to be deleterious, with just a small fraction ultimately improving the cellular fitness of the bacterium (6).
BIOCHEMICAL PROPERTIES OF E. COLI TLS POLYMERASES
In vitro fidelity of TLS polymerases
Pol II consists of a single 89.9-kDa polypeptide that possesses both polymerase and 3'→5' exonuclease activities (Fig. 2) (23, 40). As a consequence, pol II-dependent replication on undamaged DNA is reasonably accurate with misincorporations occurring in vitro in the range of ~1 × 10−6 (40). On damaged templates, pol II is also quite accurate, with the notable exception of AAF adducts where it promotes −2 frameshifts. Pol IV is also encoded by a single ~40 kDa polypeptide, but the protein lacks intrinsic 3'→ 5' exonuclease activity (Fig. 2) and is a low-fidelity enzyme with a misincorporation frequency in the range of 10−3 to 10−5 depending on the nature of the mispair formed (231). When replicating its cognate substrates, such as the N2-dG adducts, pol IV replicates the lesion containing DNA with the same, or perhaps even higher accuracy than the undamaged template (103). Pol V is an ~72 kDa complex comprised of dimeric UmuD' (~24 kDa) and UmuC (~48 kDa) proteins (Fig. 2). Like pol IV, the pol V complex lacks proofreading activity and is error-prone with misincorporations occurring in the range of 10−3-10−4 (231). However, when replicating various damaged substrates, pol V is generally less accurate, which leads to a clear mutagenic signature in vivo.
In addition to low-fidelity incorporation of deoxynucleoside triphosphates during TLS, both pol IV and pol V have been shown to readily incorporate oxidized deoxynucleoside triphosphates opposite undamaged template bases (262). Such properties may contribute to a significant amount of cellular mutagenesis when bacteria are exposed to environmental oxidative stress (192)
Last, but not least, both pol IV and pol V exhibit apurinic/apyrimidinic lyase activity (210), although relevance for this function in vivo has yet to be established.
Cofactor requirements that modify the fidelity and processivity of TLS polymerases in vitro
All three TLS polymerases physically interact with the cell’s replicative sliding β-clamp (51) and this helps stimulate the processivity and catalytic activity of each enzyme in vitro (19, 80, 231, 241). In the case of pol II, processivity increases from just five nucleotides in the absence of the β-clamp to greater than 1600 nucleotides per template binding event in its presence (25). The processivity of purified pol IV in vitro is very low, as it can only incorporate just a few nucleotides before dissociating (243). However, with the assistance of the β-clamp, the processivity of pol IV increases by 2- to 4-orders of magnitude enabling it to synthesize over 1 kb DNA (241). Recently, it has been suggested that UmuD physically interacts with Pol IV (85) and in doing so, helps cap the polymerase’s active site, so as to increase pol IVs fidelity (71, 85).
Pol V (UmuD'2C) is an extremely weak polymerase (200, 232). However, the catalytic activity of pol V can be greatly stimulated upon the addition of co-factors. Interestingly, pol V’s cofactor requirements in vitro are quite varied and appear to depend upon the template being replicated (80). On long single-stranded templates, the greatest stimulation of pol V’s catalytic efficiency and processivity is observed in the presence of the β-clamp, single stranded binding (SSB) protein and RecA (80). On shorter, hairpin templates, SSB is dispensable, but a requirement for RecA remains. Even though the β-clamp is not necessary for replication of short DNA templates by RecA-activated pol V, the processivity of the polymerase increases significantly when it is present in the reaction mixture (202).
Velocity of DNA synthesis
Compared with the cell’s replicase, pol III, which catalyzes elongation of DNA chains at a rate of about 1 kb per second, the velocity of nucleotide incorporation by the TLS polymerases is incredibly slow. In the presence of accessory proteins the rate of pol II-dependent DNA synthesis has been estimated to be ~20–30 nucleotides per second (25). By comparison, the velocity of pol IV-dependent synthesis is between 2–8 nucleotides per second (98, 130, 231, 242). Pol V is by far the slowest TLS polymerase, with a rate of DNA synthesis estimated to be less than 1 nucleotide per second (77) (Vaisman and Woodgate, unpublished data).
Nature of RecA filaments
Stimulation of Pol V’s activity in vitro by RecA fits nicely with the genetic requirement for RecA in pol V-dependent TLS in vivo (55, 168, 227). For many years, it was tacitly assumed that RecA stimulated pol V in the form of a nucleoprotein filament that formed in cis (Fig. 9A). That is, the RecA filament forms on the single-stranded DNA immediately 3' of the blocked replication complex. However, by using a short hairpin substrate with a 3' nucleotide overhang that is incapable of forming a RecA filament, Goodman and colleagues demonstrated that RecA activation of pol V can also occur in trans; i.e., on another DNA template with no relation to the blocked lesion-containing template (Fig.9B) (202). The fact that RecA can activate pol V in trans does not mutually exclude the possibility that RecA filaments formed in cis do activate pol V in vitro, but as yet, it has proven impossible to test this hypothesis directly, since in any in vitro reaction involving an oligonucleotide primer and a separate template, there will always be some trans activating DNA in the reaction (either from unannealed primer, if the primer is in excess, or template, if it’s in excess). Furthermore, cis-activation is also likely to be problematic for TLS, since the cis-activating RecA nucleoprotein filament would need to be displaced for DNA synthesis to occur. It is even possible that a cis-acting RecA filament could even inhibit TLS, as it was shown to interfere with pol III holoenzyme-catalyzed DNA replication (213).
Figure 9. Evolution of RecA’s “direct role” in TLS.
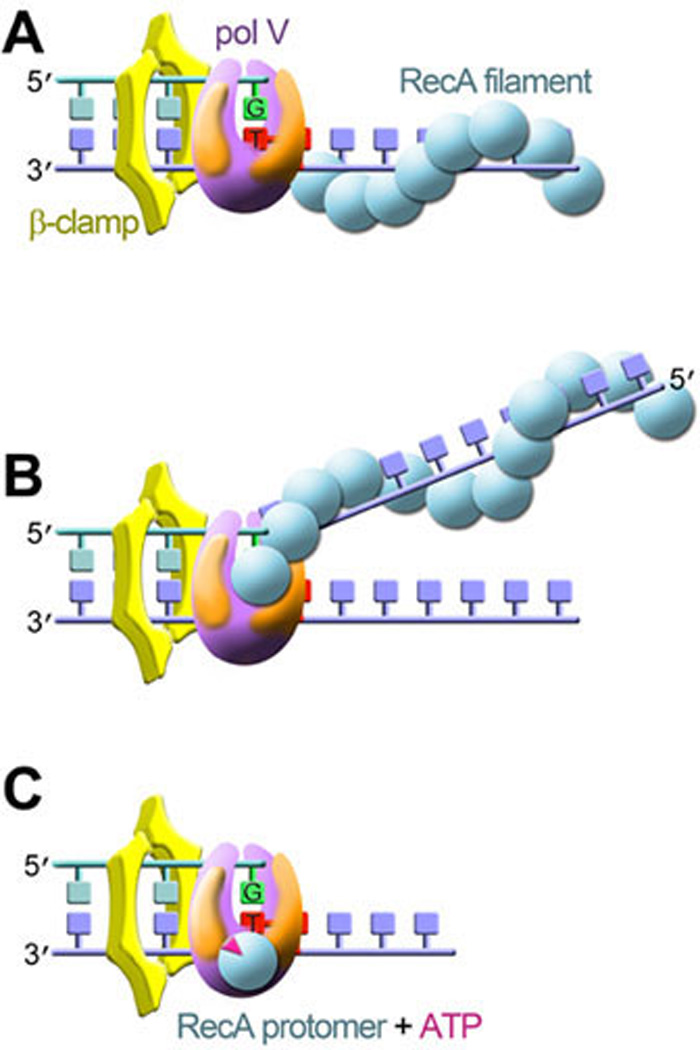
A: In 1999, TLS was reconstituted in vitro in the presence of UmuD'2C (pol V), β-sliding clamp and RecA protein (200, 232). Based upon genetic experiments in vivo (217), it was tacitly assumed that RecA stimulated pol V by forming a nucleoprotein filament on the single-stranded DNA immediately 3' of the stalled replication complex. B: However, in 2006 it was demonstrated that pol V can also be activated in trans by interacting with the 3'- tip of RecA filament formed on a separate ss DNA molecule (202). C: In 2009, it was subsequently determined that the highly active complex responsible for in vivo TLS is pol VMut, which consists of UmuD'2C•RecA•ATP and is formed by the transfer a single RecA protomer along with ATP from the 3' tip of a RecA nucleoprotein filament to pol V (104).
Cis- or trans-activation aside; what is the molecular basis of RecA’s ability to stimulate pol V in vitro? Recent studies by Goodman and colleagues have shown that the sole purpose of the RecA nucleoprotein filament is to transfer a single RecA protomer and ATP molecule from the 3' tip of the filament to pol V to generate a stable and highly active complex consisting of UmuD'2C•RecA•ATP that they called pol V Mut (Fig. 9C) (104). The term “Mut” is short for “Mutasome”, a multi-protein complex that the late Hatch Echols hypothesized some 20 years ago would facilitate TLS (56).
STRUCTURAL INSIGHTS INTO TLS
Crystal structure of pol II reveals its capacity to promote frameshift TLS events
The apo structure of pol II was deposited in the Protein Data Base (PDB) some 16 years ago (PDB Accession number: 1Q8I) (5). However, the structure of pol II in a ternary complex with DNA and incoming dNTP has only recently been solved (245). Indeed, Wang and Yang solved eight different crystal structures of pol II, including apo, binary, and ternary complexes with normal and abasic-lesion containing DNAs (Fig. 10A) (PDB accession numbers: 3K57, 3K58, 3K59, 3K5A, 3K5L, 3K5M, 3K5N, and 3K5O). Given that pol II is a B-family polymerase, it was not too surprising that the ternary structure of pol II with undamaged DNA and incoming nucleotide was virtually superimposable with structures of the related B-family φ29 replicative polymerase and RB69 polymerase, gp43 (18, 73). Unfortunately, these structures did not provide any clear insights into how pol II might facilitate TLS. Like all known polymerases, pol II has palm, finger and thumb domains (220), as well as an additional N-domain and Exo domain (Fig. 2). However, the crystal structure of pol II in a ternary complex with abasic lesion-containing DNA revealed that there were two major differences between TLS pol II and replicative gp43 polymerase. First, significant changes were observed in the DNA duplex upstream of the abasic site and in the thumb domain of pol II. These resulted in small cavities 1 and 2 bp upstream from the pol II active site that accommodated the looping out of not only the abasic site, but also the two adjacent downstream template bases. The second major difference between pol II and gp43, is that pol II has a 20 amino acid insertion in its N-domain that alters the position of the Exo-domain relative to the polymerase active site in the palm domain, so as to reduce exonucleolytic proofreading activity relative to polymerization. Thus, it appears that a combination of the small pockets in pol II that accommodate multiple template strand “acrobatics” and a weakened proofreading capacity allows pol II to facilitate TLS (245).
Figure 10. Crystal structure of TLS polymerases in complex with DNA and an incoming dNTP.
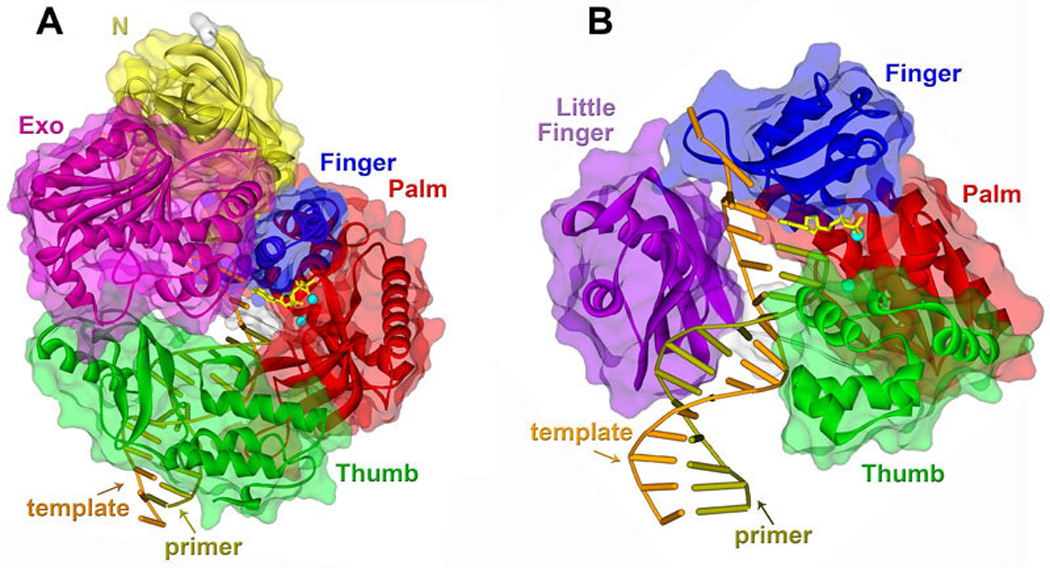
A. Structure of pol II with DNA containing abasic site and dATP (PDB: 3K5L) (245). B. Crystal structure of the Dpo4 ternary complex (PDB: 1JX4) (133). The domains of both polymerases are color-coded such as: palm - red, thumb - green, finger - blue, exonuclease of pol II– violet, N-terminal domain of pol II – yellow, little finger of Dpo4 - purple. The template strand is shown in rust color, while the primer is olive green. The incoming dATP is shown in yellow. The small blue spheres represent the two metal ions. The protein backbone is represented by ribbon surrounded by semi-transparent solvent accessible surface. The structures were created using Discovery Studio Visualizer. Comparison of these structures reveals that the active site of Dpo4 is significantly more spacious than that of pol II allowing for the accommodation of bulky DNA lesions. The active site of pol II is very similar to that of high fidelity replicative polymerases, but relaxed interactions with the upstream DNA template and an altered partitioning between the polymerase and exonuclease active sites ensures the participation of pol II in TLS.
Archaeal Dpo4 as a model for E. coli pol IV
Although pol IV can be purified in high yield and great purity, the structure of the full-length polymerase has yet to be determined. However, archaeal orthologs Dpo4 from Sulfolobus solfataricus (Fig. 10B) (PDB accession number: 1JXL)(133) and Dbh from Sulfolobus acidocladarius (PDB accession numbers: 3BQ1) (248) have been solved in a ternary complex with undamaged DNA and incoming dNTPs and have served as a model for E. coli pol IV. Despite virtually no obvious amino acid homology to DNA polymerases from other families, the crystal structures of Dpo4 and Dbh reveal a topology resembling a right hand similar to that of high fidelity polymerases with palm, thumb and finger domains (133). In addition, these proteins have another domain called the “little finger” (LF) that wraps around DNA and helps secure the polymerase to the primer-template (28, 29, 133). When compared to high fidelity polymerases, the domains are rather “stubby” and instead of having a constricted active site, the active site of Dpo4 is spacious and solvent exposed, helping to explain how the enzyme can accommodate a variety of DNA lesions in its active site (Fig. 10B). In the 10 years since the first ternary structure of Dpo4 was reported some 56 separate lesion-containing structures of Dpo4 have now been solved and deposited in the PDB (Table 2).
Table 2.
Crystal structures of various DNA lesions in the active site of Dpo4
| DNA lesion co-crystallized with Dpo4 | Number of structures |
PDB accession numbers | References |
|---|---|---|---|
| 1,N2-etheno-guanine | 5 | 2BQ3, 2BQR, 2BQU, 2BR0, 2XC9 | (100, 266) |
| 1,N2-propanodeoxyguanosine | 3 | 2R8G, 2R8H, 2R8I | (246) |
| 2,4-difluorotoluene | 3 | 2V9W, 2VA2, 2VA3 | (99) |
| 2-aminofluorene-guanine | 4 | 3KHG, 3KHH, 3KHL, 3KHR | (197) |
| 7,8-dihydro-8-oxoguanine | 16 | 2ASD, 2ASJ, 2ASL, 2C22, 2C2R, 3GII, 3GIJ, 3GIK, 3GIL, 3GIM, 2UVR, 2UVU, 2UVV, 2UVW, 2XCA, 2XCP | (60, 100, 198, 199, 267) |
| Abasic site | 5 | 1N48, 1N56, 1S0N, 1S0O, 1S10 | (134) |
| Benzo[a]pyrene diol epoxide | 1 | 1S0M | (135) |
| Cisplatin-1,2 guanine-guanine intrastrand cross-link | 3 | 3M9M, 3M9N, 3M9O | (255) |
| cis-syn thymine dimer | 2 | 1RYR, 1RYS | (132) |
| Malondialdehyde-deoxyguanosine | 2 | 2V4Q, 2V4R | (62) |
| N2N2-dimethyl-deoxyguanosine | 3 | 2W9A, 2W9B, 2W9C | (269) |
| N2-naphthyl-guanine | 2 | 2W8K, 2W8L | (268) |
| O6-benzylguanine | 4 | 2JEF, 2JEG, 2JEI, 2JEJ | (59) |
| O6-methylguanine | 3 | 2J6S, 2J6T, 2J6U | (61) |
Dpo4 (pol IV) interaction with the replicative sliding clamp
As noted above, a prerequisite for TLS is the ability of the respective TLS polymerase to interact with the β-clamp. The first structural insights into how this interaction might promote TLS came when Bunting and colleague solved the crystal structure of the E. coli pol IV LF-domain bound to the β-clamp (PDB: 1UNN) (38). Interestingly, by superimposing the full-length structure of Dpo4 onto the E. coli DinB LF- β-clamp structure, the model revealed that when the interaction occurs through the rim of the clamp, the active site of Dpo4 would not be engaged at the primer-terminus of the nascent DNA chain, but is rather “hanging off to the side” in what would be a catalytically inactive configuration (Fig. 11A). In contrast, when pol IV is flexibly attached to the clamp via the C-terminal peptide, it can simultaneously contact the DNA template, thus assuming an “active” orientation (Fig. 11B). The structure therefore provided the first evidence of a potential “toolbelt” model for TLS polymerases in which multiple enzymes could be bound to the replicative clamp simultaneously and engaged at the primer terminus only when, and if, needed (98, 176). Very recently, the crystallographic studies of Dpo4 in a complex with heterodimeric PCNA1–PCNA2 (PDB: 3FDS)(261) has shown that a flexible structure allows the polymerase to adopt one of the several different conformational states in order to optimize its contact with PCNA and/or DNA (Figs. 11C and D). In addition to the expected specific binding of Dpo4 to PCNA1 via its PCNA-interacting peptide (PIP)-box, the structure revealed additional protein-protein interactions with the clamp via the finger, thumb and little finger domains (261). Both structural studies of E. coli DinB LF with β-clamp and Dpo4 with PCNA1–PCNA2 corroborate the “toolbelt” model for polymerase switching (Fig. 5).
Figure 11. Structural/modeling studies of TLS polymerases bound to processivity clamps.
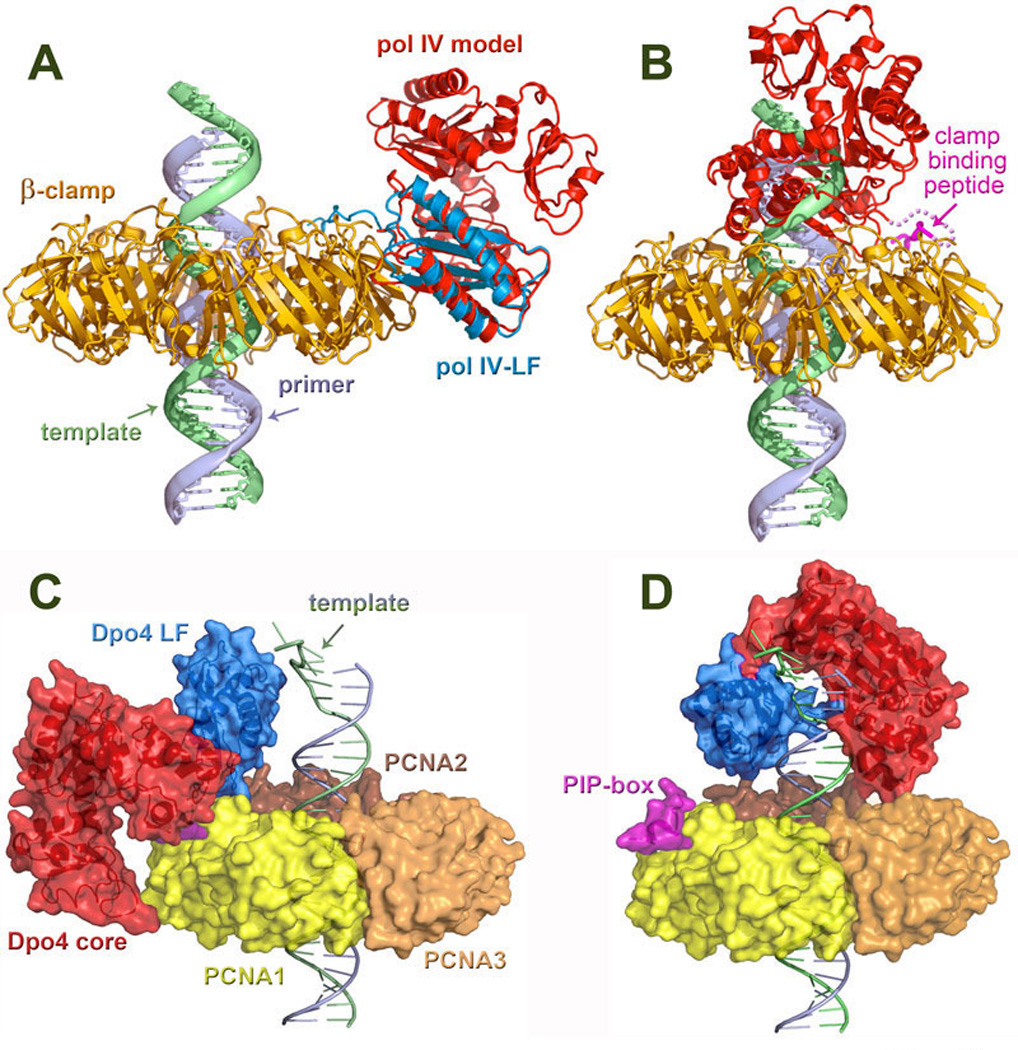
A. Model of a pol IV (shown in red) bound to the β-clamp (gold) in an inactive position. The position of the polymerase was modeled by superimposing the little finger domain from the ternary complex of Dpo4 with DNA and an incoming nucleotide (PDB code: 1JXL) (133) onto the LF of E. coli pol IV (blue) in a complex with the β-clamp (PDB: 1UNN) (38). In this position, pol IV is unable to access the primer/template terminus. B. Model of pol IV bound to the β-clamp and DNA at the primer/template junction following a polymerase switch. The position of the polymerase was modeled by superimposing the DNA from the ternary complex of Dpo4 onto the end of a DNA molecule running perpendicularly through the β-clamp. Contact with the clamp is maintained by the C-terminal clamp-binding peptide (pink), which tethers the polymerase to the replication complex. C. Dpo4 in an inactive extended form bound to PCNA (PDB: 2NTI) (261) with dsDNA passing through its central aperture. In this conformation, the core (red) and little finger (blue) of Dpo4 contact PCNA (the trimer is shown in yellow, orange, and brown) to facilitate the PIP (pink)–PCNA1 (yellow) binding. D. Model of Dpo4 in an active form bound to a DNA template and a PCNA ring. Similar to the structure shown in the panel C, in this conformation the PIP-box anchors Dpo4 to PCNA1. The DNA-bound Dpo4 is modeled onto PCNA from the type I structure (PDB: 1JX4). Panels A and B reproduced with permission from Bunting et al., (38). Panels C and D were provided through the courtesy of Dr. Hong Ling, University of Western Ontario, London, Canada. All four structures provide structural support for the proposed “tool-belt” model for polymerase switching (176).
Structure of E. coli UmuD' and modeling of E. coli UmuD
The structure of pol V (UmuD'2C) or pol V Mut (UmuD'2C•RecA•ATP) has yet to be solved, at least in part because it has been historically difficult to obtain large quantities of the enzyme for crystallographic studies. However, recombinant UmuD' is readily overexpressed in E. coli and both the crystal structure (182), and the NMR-solution structure (64) of UmuD'2 have been solved. Two dimer interfaces were observed in the crystal structure. One termed the “Molecular” dimer in which the N-terminal tail of each protomer is in opposite directions and the Ser-Lys dyad of the peptidase active site is occluded (PDB: 1AY9) (Fig. 12A). The second dimer observed in the crystal was termed the “Filament”, and in this interface, the N-terminal tails of the two protomers cross and are likely to be in close proximity to the active site Ser60-Lys97 residues of the adjacent protomer (PDB: 1UMU) (Fig. 12B). The latter structure is consistent with the observation that UmuD cleavage occurs via an intermolecular reaction (150) (Fig. 4).
Figure 12. Comparison of the crystal and NMR structure of UmuD'2.
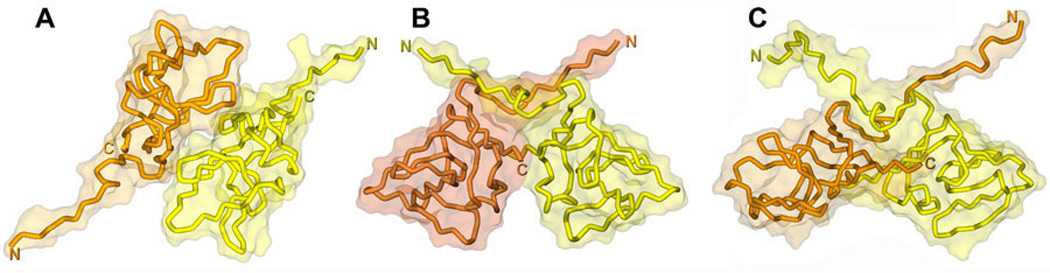
A. Crystal structure of a UmuD' “Molecular” dimer (PDB: 1AY9) in which the N-termini of each protomer are extended in diametrically opposite directions (182). B. Crystal structure of UmuD' “Filament” dimer (PDB: 1UMU) in which the N-terminal tail of each protomer cross over the globular body of its partner in a configuration that is likely to mimic the one in which the RecA-mediated cleavage of UmuD to UmuD' occurs (183). C. NMR solution structure of UmuD'2 (PDB: 1I4V) revealing a structural fold similar to the crystallographic “Filament” dimer, but in which the globular domain is somewhat “flattened” and the N-terminal tails are free in solution (64). The protein backbone is depicted as a tube that is surrounded by the semi-transparent solvent accessible surface of the dimer. One UmuD' protomer is colored yellow, while the other one is in orange. The structures were created using Discovery Studio Visualizer.
While two dimeric interfaces were observed in the crystal structure, only one, corresponding to the so-called “Filament” dimer, was observed in the NMR solution structure (PDB: accession code 1I4V) (64), and it is clearly the filament dimer that is active for mutagenesis in E. coli (155, 172). In the crystal structure, the catalytically active Ser-Lys residues are close to each other and are poised for catalysis, whereas in the NMR-solution structure they are 7Å apart (Fig. 12C). Presumably, the functional role of RecA in mediating UmuD cleavage is to induce a conformational change in UmuD such that the Ser-Lys active site residues are juxtaposed, so as to induce its self-cleavage.
Although UmuD only differs from UmuD' by the presence of a 24 amino acid N-terminal tail, the structure of UmuD has yet to be determined. Molecular models of UmuD have, however, been generated. Early models were based upon the ease with which mono-cysteine mutants of UmuD could be chemically cross-linked (89, 128) and subsequently mapped on to the NMR structure of UmuD' (64). The model was subsequently refined based upon additional data including solvent accessibility and electron paramagnetic spin resonance (EPR) studies (225). Based upon these studies, Sutton et al. proposed that the N-terminal tails of UmuD (residues 1–39) form an extended interface in the UmuD2 homodimer by folding down over the globular domains of their respective intra-dimer partners (225). While this clearly has to be the case to ensure the appropriate orientation of the cleavage site of one protomer in the active site of the adjacent protomer, recent UmuD models that are based upon the crystal structures of UmuD' and LexA (21) and hydrogen–deuterium exchange mass spectrometry (HXMS), reveal that the N-terminal tail of UmuD is actually very dynamic and highly flexible with the N-terminus solvent exposed in solution (63). One can easily imagine that it is the interaction with RecA that helps to trap these flexible tails in a “bear-hug” manner, so as to facilitate autodigestion of UmuD to UmuD'.
SIMILARITIES AND DIFFERENCES BETWEEN E. COLI AND S.TYPHIMURIUM TLS POLYMERASES
Genomic locations of TLS polymerases in E. coli and S.typhimurium
Overall, the E. coli and S.typhimurium genomes are nearly 90% identical at the nucleotide level. In particular, the pol II and DinB proteins from E. coli and S.typhimurium are 89% and 91% identical, respectively. Pol II is located at 1.4 mins of the E. coli genome and is flanked by the hepA and araD genes (xBase). S.typhimurium polB is located at a roughly similar position of the genome and is flanked by hepA and several intervening putative genes and then followed by the araD gene (Fig. 13).
Figure 13. Comparison of the genetic map locations and genomic context of translesion DNA polymerases in E. coli and S. typhimurium.
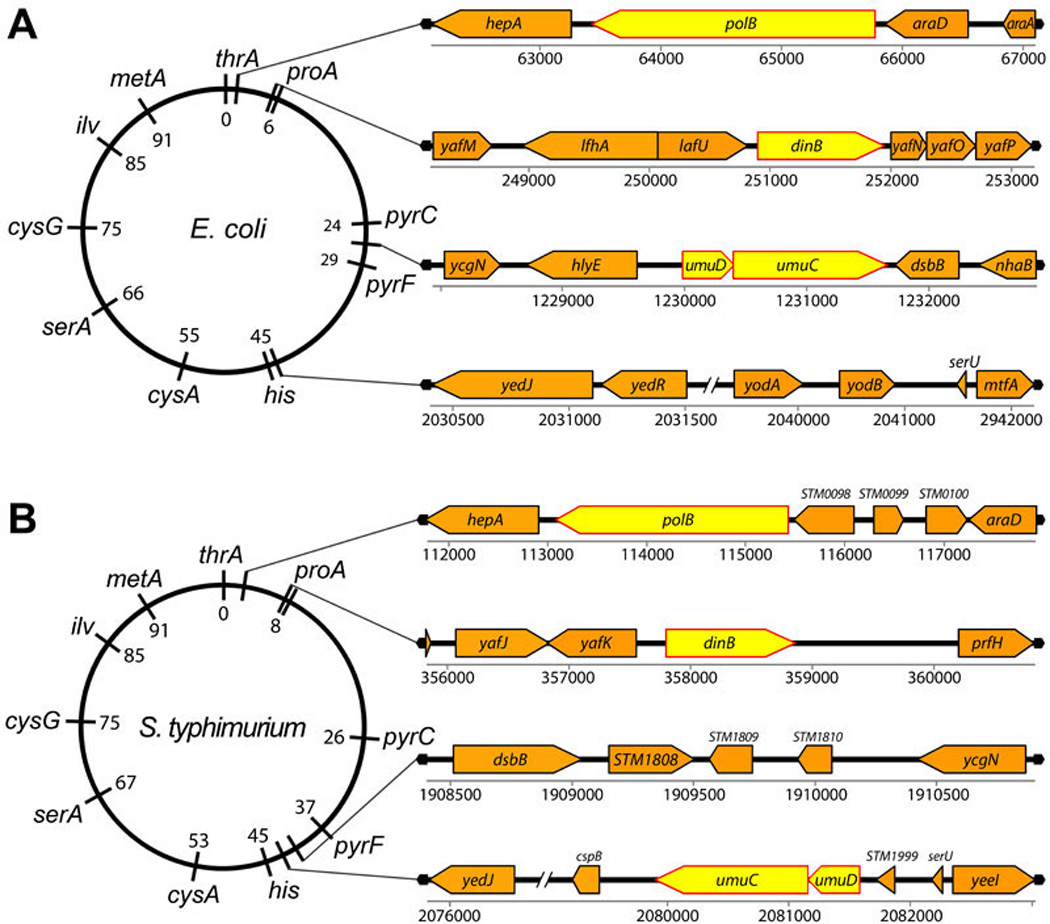
A. The genetic map locations and genomic architecture of the E. coli polB dinB and umuDC genes are depicted. B. The genetic map locations and genomic architecture of the S. typhimurium polB dinB and umuDC genes are depicted. The location and context of the E. coli and S. typhimurium polB and dinB genes are quite conserved. In contrast, the E. coli and S. typhimurium umuDC operons are located in different positions on the genetic maps, bordered by different genes and are in opposite orientations.
E. coli dinB gene is located at 5.4 mins on the chromosome. yafM gene and two pseudogenes are located upstream of dinB, while yafN, yafO and yafP are located immediately downstream of dinB with all four genes expressed as a single operon (154). S.typhimurium dinB is located at 7.2 mins and lacks the downstream yafN, yafO and yafP genes but is, instead, flanked by the yafK and prfH genes. The lack of a downstream yafP gene is intriguing as the operon arrangement is apparently conserved in a number of naturally occurring E.coli and YafP seems to work with DinB to protect cells from cytotoxic effects of exposure to nitroaromatic compounds (88). Despite the lack of an obvious dinB-yafN-yafO-yafP operon in S. typhimurium, its dinB gene is nevertheless located in a similar genomic region to E.coli dinB, since four genes upstream of the E. coli yafM gene is the yafK gene and 1.4 kb downstream of the yafN gene is a prfH-like pseudogene (Fig. 13). Thus, both the polB genes and the dinB genes from both E. coli and S.typhimurium are positioned within analogous genomic locations and architectures.
Interestingly, the UmuD and UmuC proteins from these two organisms are considerably more diverged than the rest of the genome with only 73% and 83% identity, respectively. Furthermore, their chromosomal locations are substantially different. Whereas the E. coli umu operon is located at 26.5 mins on the chromosome and flanked by hlyE and dsbB, the S.typhimurium umuDC operon is located at 42.9 mins on the map and is flanked by STM1999 and cspB and is within the large 588 kbp inversion (148), that is a major distinguishing factor between the E. coli and S.typhimurium genomes (Fig. 13). Closer analysis of the two loci reveals that the region of the S.typhimurium chromosome, which is analogous to the E. coli 26.5 minute interval is located at 41.3 minutes of the S.typhimurium genome and is identical to the E. coli interval, except that it lacks the umuDC operon (Fig. 13). Similarly, the region of the 42.9 minute interval of the S. typhimurium chromosome encompassing the umuDC operon corresponds to E. coli 44 minutes and has the same gene arrangement, except that it lacks the umuDC operon (Fig. 13). These observations, together with fact that the umuDC operon is more diverged than the rest of the E. coli and S.typhimurium genomes, suggest that the umuDC operon was possibly acquired by independent horizontal gene transfer from a plasmid-encoded transposable element instead of evolving from a single common ancestor.
An additional UmuD' ortholog in the S.typhimurium LT2 genome
BLAST analysis (2) of the S.typhimurium genome reveals another protein with similarity to E. coli UmuD. The protein annotated as STM2230 in GENBANK NC_003197.1 is 156 amino acids long and its C-terminal 128 residues are 40 and 45% identical to E. coli and S.typhimurium UmuD', respectively. Further analysis of the nucleotide sequence of SMT2230 suggests that the protein may, however, be incorrectly annotated and we suggest that the gene may actually encode a 129 amino acid protein since a consensus ribosome binding site can be identified upstream of the Methionine located at position 28 of SMT2230. This shorter protein shares highest homology with the HumD protein found on bacteriophages P1, P7, N15 and φKO2 (Fig. 14) and SMT2230 therefore appears to originate from one of the many cryptic prophages that inhabit S.typhimurium (149). Bacteriophage P1 HumD protein has previously been shown to function with E. coli UmuC (expressed from a low-copy number plasmid), so as to restore damage-induced mutagenesis to ΔumuDC strains of E. coli (156) indicating that the P1 HumD protein is a functional ortholog of UmuD'. Why S.typhimurium would retain another UmuD' ortholog in its genome remains a mystery, especially since S.typhimurium UmuD is cleaved to UmuD' very efficiently (259).
Figure 14. Homology comparison of the S. typhimurium STM2230 protein with various HumD-like proteins.
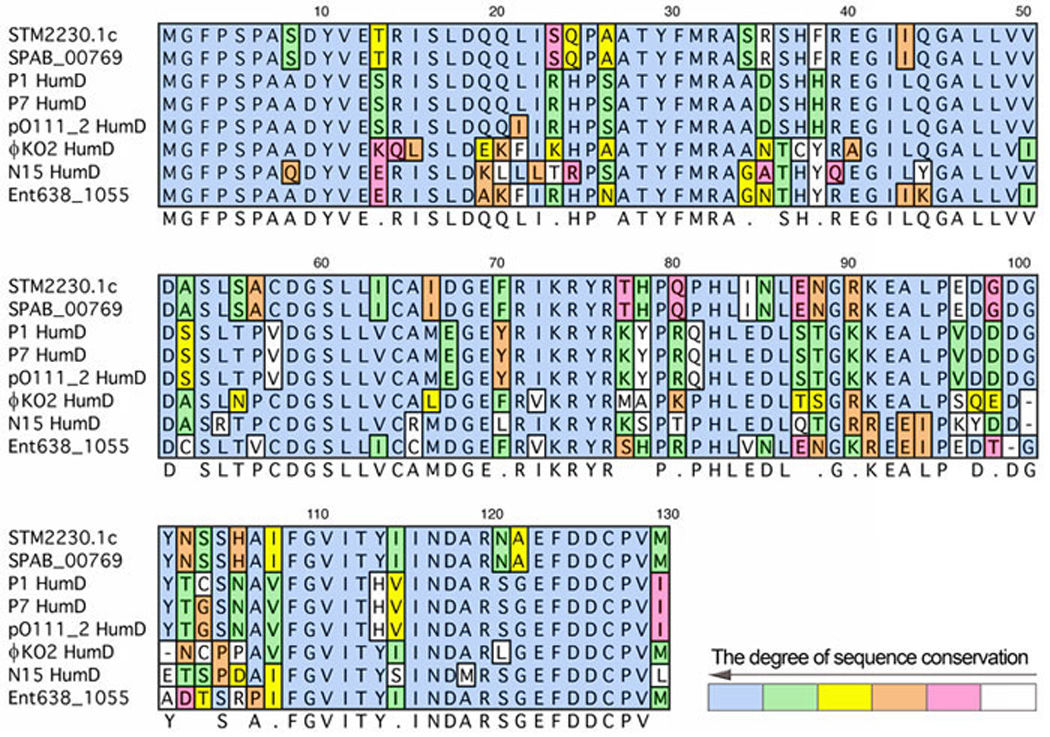
The alignment of various HumD-like proteins was performed using MacVector 11.0.4. The HumD-like proteins include: STM2230.1c (S. typhimurium; NP_461173), SPAB_00769 (S. paratyphi; YP_001587027), P1 HumD (YP_006569), P7 HumD (ZP_07505015), pO111_2 HumD (YP_003238012), fKO2 HumD (YP_006603), N15 HumD (NP_046921) and Ent638_1055 (Enterobacter sp. 638; YP_001175788). Coloration indicates the degree of similarity to the consensus sequence. Blue indicates identity to the consensus, while green indicates an amino acid residue that is highly similar to the consensus. The degree of similarity to the consensus further decreases in this order of coloration: yellow, orange, pink and white.
Many strains of S.typhimurium carry R-plasmids expressing pol V orthologs
Interestingly, in addition to possessing a genomic copy of HumD, many natural isolates of S.typhimurium harbor R-plasmids carrying orthologs of pol V. Indeed, S.typhimurium LT2 harbors the 60 MDa virulence plasmid (pSLT) (35) encoding the pol V ortholog SamAB (169). Similarly, S.typhimurium LT7 harbors the Col11a plasmid encoding pol V ortholog ImpAB (111). Quite remarkably, despite carrying multiple orthologs of low-fidelity pol V, S. typhimurium is poorly mutable compared to E. coli (205, 216, 233). It was partly for this reason that Ames and colleagues introduced yet another R-plasmid (pKM101), encoding pol V orthologs mucAB into S. typhimurium tester strains that they developed to identify potential mutagens and carcinogens (3, 4, 147).
It is now known that pol V orthologs are found on many broad-host range plasmids, including those from the “pre-antibiotic” era (206) that are capable of existing in either E. coli or S.typhimurium (Table 3). Interestingly, the genes encoding pol V orthologs are often located very close to the origin of transfer (oriT) of the R-plasmid (181) or ICE (42), meaning that they are among the first genes transferred to the host bacterium during conjugation. In this process, “rolling-circle” replication introduces a single-strand DNA into the recipient cell. The single-stranded nature of the transferred DNA prevents nucleotide excision repair of any lesion in the plasmid DNA, while the absence of an intact homolog blocks any subsequent recombinational repair of the daughter-strand gaps left after synthesis by the host bacterial replicase. While the host TLS polymerases may be able to provide assistance, it may be insufficient for the plasmid to survive. However, the damaged plasmid has a much higher chance of survival, if it quickly expresses its own pol V in the host bacterium. Plasmid survival is clearly the evolutionary driving force for the retention of pol V orthologs on the mosaic R-plasmids. This is most evident from Chris Lawrence’s TLS studies with single-stranded M13 phage (228). In the absence of pol V, survival of an M13 genome with a single cis-syn cyclobutane dimer is ~0.5%. When the same damaged DNA is introduced into a host expressing recombinant pol V, survival dramatically increased to ~70%, with only 2–5% of the survivors acquiring mutations (228). To phrase this differently, 95–98% of the survivors (that would have died in the absence of pol V) did so without any mutational consequences. Of course, once the plasmid is established in its new host, the low-fidelity plasmid encoded pol V ortholog may nevertheless contribute to antibiotic resistance and pathogenicity, particularly when the host cells are stressed (14, 44).
Table 3.
Pol V orthologs found on R-plasmids/ICEs
| Incompatibilty Group | Plasmid | Original Host | Pol V Family |
|---|---|---|---|
| IncB1 | pO111_1 | E. coli | Muc |
| IncFI | pSE11-2 | E. coli | Imp1 |
| IncFII2 | pSLT | S. typhimurium | Sam |
| IncFII | pSCV50 | S. choleraesuis | Sam |
| IncH | R27 | S. typhimurium | Muc |
| IncH | pAPEC-O1-R | E. coli | Muc |
| IncH | pAPEC-O2-R | E. coli | Imp |
| IncIa | ColIb-P9 | E. coli | Imp |
| IncIa | R64 | S. typhimurium | Imp |
| IncIa | TP110 | S. typhimurium | Imp |
| IncJ/ICE3 | R3914 | P. rettgeri | Rum |
| IncL | R471a | S. marcescens | Muc |
| IncM | R446b | P. morganii | Muc |
| IncN | R46 | S. typhimurium | Muc |
| IncN | pKM1015 | S. typhimurium | Muc |
| IncT | Rts1 | P. vulgaris | Muc/Umu6 |
| IncT | R394 | P. rettgeri | Muc |
| ICE | SXT7 | V. cholerae | Rum8 |
| ICE | ICEPmiJpn17 | P. mirabilis | Rum |
| ICE | ICEPdaSpa17 | P. damselae | Rum9 |
| ICE | ICEVflInd17 | V. fluvialis | Rum10 |
| ICE | ICEVchmex14 | V. cholerae | Rum |
| ICE | ICEVchind54 | V. cholerae | Rum11 |
| ICE | ICEVchind47 | V. cholerae | Rum11 |
| ICE | ICEVchban54 | V. cholerae | Rum11 |
| ICE | ICEPalban14 | P. alcalifaciens | Rum12 |
pSE11-2 (NC_011413); the imp-like genes are not arranged within an operon.
pSLT is believed to belong to the IncFII incompatability group (149).
Originally identified as an IncJ plasmid (45), but now considered an Integrating Conjugative Element (ICE) (13).
Belongs to the R391 ICE exclusion group (145)
pKM101 is a naturally occurring deletion derivative of R46 (NC_003292) generated during P22 transduction of R46 into S.typhimurium (160)
Rts1 (NC_003905); this plasmid harbors two polV-like operons. The mucB-like gene has a C-terminal truncation (110).
Belongs to the SXT ICE exclusion group (145)
The rumB gene of SXT is disrupted by a large 17kb insertion containing multiple antibiotic resistance genes (13).
Photobacterium damselae ICEPdaSpa1 (AJ870986); the rum operon is disrupted by a transposon.
Vibrio fluvialis ICEVflInd1 (GQ463144); the rum-like genes are not arranged within an operon.
Vibrio cholerae ICEVchind5 (GQ463142), ICEVchind4 (GQ463141) and ICEVchban5 (GQ463140); the rum-like genes are not arranged within an operon.
P. alcalifaciens ICEPalban1 (GQ463139); the rum-like genes are not arranged within an operon.
ACKNOWLEDGMENTS
This work was supported by funds from the NICHD/NIH Intramural Research Program.
Footnotes
REFERENCES
- 1.Al Mamun AA, Humayun MZ. Spontaneous mutagenesis is elevated in protease-defective cells. Mol.Microbiol. 2009;71:629–639. doi: 10.1111/j.1365-2958.2008.06551.x. [DOI] [PubMed] [Google Scholar]
- 2.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic Local Alignment Search Tool. J.Mol.Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 3.Ames BN, Lee FD, Durston WE. An improved bacterial test system for the detection and classification of mutagens and carcinogens. Proc.Natl.Acad.Sci.USA. 1973;70:782–786. doi: 10.1073/pnas.70.3.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ames BN, McCann J, Yamasaki E. Methods for detecting carcinogens and mutagens with the Salmonella/mammalian-microsome mutagencity test. Mutat.Res. 1975;31:347–364. doi: 10.1016/0165-1161(75)90046-1. [DOI] [PubMed] [Google Scholar]
- 5.Anderson WF, Prince DB, Yu H, McEntee K, Goodman MF. Crystallization of DNA polymerase II from Escherichia coli. J.Mol.Biol. 1994;238:120–122. doi: 10.1006/jmbi.1994.1765. [DOI] [PubMed] [Google Scholar]
- 6.Andersson DI, Koskiniemi S, Hughes D. Biological roles of translesion synthesis DNA polymerases in eubacteria. Mol Microbiol. 2010;77:540–548. doi: 10.1111/j.1365-2958.2010.07260.x. [DOI] [PubMed] [Google Scholar]
- 7.Babudri N, Monti-Bragadin C. Restoration of mutability in non-mutable Escherichia coli carrying different plasmids. Mol.Gen.Genet. 1977;155:287–290. doi: 10.1007/BF00272807. [DOI] [PubMed] [Google Scholar]
- 8.Bagg A, Kenyon CJ, Walker GC. Inducibility of a gene product required for UV and chemical mutagenesis in Escherichia coli. Proc.Natl.Acad.Sci.USA. 1981;78:5749–5753. doi: 10.1073/pnas.78.9.5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banach-Orlowska M, Fijalkowska IJ, Schaaper RM, Jonczyk P. DNA polymerase II as a fidelity factor in chromosomal DNA synthesis in Escherichia coli. Mol.Microbiol. 2005;58:61–70. doi: 10.1111/j.1365-2958.2005.04805.x. [DOI] [PubMed] [Google Scholar]
- 10.Banerjee SK, Borden A, Christensen RB, LeClerc JE, Lawrence CW. SOS-dependent replication past a single trans-syn T-T cyclobutane dimer gives a different mutation spectrum and increased error rate compared with replication past this lesion in uninduced cells. J.Bacteriol. 1990;172:2105–2112. doi: 10.1128/jb.172.4.2105-2112.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Banerjee SK, Christensen RB, Lawrence CW, LeClerc JE. Frequency and spectrum of mutations produced by a single cis-syn thymine-thymine dimer in a single-stranded vector. Proc.Natl.Acad.Sci.USA. 1988;85:8141–8145. doi: 10.1073/pnas.85.21.8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Battista JR, Ohta T, Nohmi T, Sun W, Walker GC. Dominant negative umuD mutations decreasing RecA-mediated cleavage suggest roles for intact UmuD in modulation of SOS mutagenesis. Proc.Natl.Acad.Sci.USA. 1990;87:7190–7194. doi: 10.1073/pnas.87.18.7190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beaber JW, Hochhut B, Waldor MK. Genomic and functional analyses of SXT, an integrating antibiotic resistance gene transfer element derived from Vibrio cholerae. J.Bacteriol. 2002;184:4259–4269. doi: 10.1128/JB.184.15.4259-4269.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beaber JW, Hochhut B, Waldor MK. SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature. 2004;427:72–74. doi: 10.1038/nature02241. [DOI] [PubMed] [Google Scholar]
- 15.Becherel OJ, Fuchs RP. Mechanism of DNA polymerase II-mediated frameshift mutagenesis. Proc.Natl.Acad.Sci.USA. 2001;98:8566–8571. doi: 10.1073/pnas.141113398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Becherel OJ, Fuchs RP, Wagner J. Pivotal role of the ®-clamp in translesion DNA synthesis and mutagenesis in E. coli cells. DNA Repair. 2002;1:703–708. doi: 10.1016/s1568-7864(02)00106-4. [DOI] [PubMed] [Google Scholar]
- 17.Berardini M, Foster PL, Loechler EL. DNA polymerase II (polB) is involved in a new DNA repair pathway for DNA interstrand cross-links in Escherichia coli. J Bacteriol. 1999;181:2878–2882. doi: 10.1093/gao/9781884446054.article.t031385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berman AJ, Kamtekar S, Goodman JL, Lazaro JM, de Vega M, Blanco L, Salas M, Steitz TA. Structures of phi29 DNA polymerase complexed with substrate: the mechanism of translocation in B-family polymerases. EMBO J. 2007;26:3494–505. doi: 10.1038/sj.emboj.7601780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bertram JG, Bloom LB, O’Donnell M, Goodman MF. Increased dNTP binding affinity reveals a nonprocessive role for Escherichia coli β clamp with DNA polymerase IV. J Biol Chem. 2004;279:33047–33050. doi: 10.1074/jbc.C400265200. [DOI] [PubMed] [Google Scholar]
- 20.Beuning PJ, Sawicka D, Barsky D, Walker GC. Two processivity clamp interactions differentially alter the dual activities of UmuC. Mol Microbiol. 2006;59:460–474. doi: 10.1111/j.1365-2958.2005.04959.x. [DOI] [PubMed] [Google Scholar]
- 21.Beuning PJ, Simon SM, Zemla A, Barsky D, Walker GC. A non-cleavable UmuD variant that acts as a UmuD' mimic. J Biol Chem. 2006;281:9633–9640. doi: 10.1074/jbc.M511101200. [DOI] [PubMed] [Google Scholar]
- 22.Bjedov I, Nag Dasgupta C, Slade D, Le Blastier S, Selva M, Matic I. Involvement of Escherichia coli DNA polymerase IV in tolerance of cytotoxic alkylating DNA lesions in vivo. Genetics. 2007;176:1431–1440. doi: 10.1534/genetics.107.072405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonner CA, Hays S, McEntee K, Goodman MF. DNA polymerase II is encoded by the DNA damage-inducible dinA gene of Escherichia coli. Proc.Natl.Acad.Sci.USA. 1990;87:7663–7667. doi: 10.1073/pnas.87.19.7663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonner CA, Randall SK, Rayssiguier C, Radman M, Eritja R, Kaplan BE, McEntee K, Goodman MF. Purification and characterization of an inducible Escherichia coli DNA polymerase capable of insertion and bypass at abasic lesions in DNA. J.Biol.Chem. 1988;263:18946–18952. [PubMed] [Google Scholar]
- 25.Bonner CA, Stukenberg PT, Rajagopalan M, Eritja R, O’Donnell M, McEntee K, Echols H, Goodman MF. Processive DNA synthesis by DNA polymerase II mediated by DNA polymerase III accessory proteins. J.Biol.Chem. 1992;267:11431–11438. [PubMed] [Google Scholar]
- 26.Borden A, O’Grady PI, Vandewiele D, Fernandez de Henestrosa AR, Lawrence CW, Woodgate R. Escherichia coli DNA polymerase III can replicate efficiently past a T-T cis-syn dimer if DNA polymerase V and the 3' to 5' exonuclease proofreading function encoded by dnaQ are inactivated. J.Bacteriol. 2002;184:2674–2681. doi: 10.1128/JB.184.10.2674-2681.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boudsocq F, Campbell M, Devoret R, Bailone A. Quantitation of the inhibition of Hfr x F recombination by the mutagenesis complex UmuD'C. J.Mol.Biol. 1997;270:201–211. doi: 10.1006/jmbi.1997.1098. [DOI] [PubMed] [Google Scholar]
- 28.Boudsocq F, Iwai S, Hanaoka F, Woodgate R. Sulfolobus solfataricus P2 DNA polymerase IV (Dpo4): an archaeal DNA polymerase with lesion-bypass properties akin to eukaryotic pol|. Nucleic Acids Res. 2001;29:4607–4616. doi: 10.1093/nar/29.22.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boudsocq F, Kokoska RJ, Plosky BS, Vaisman A, Ling H, Kunkel TA, Yang W, Woodgate R. Investigating the role of the little finger domain of Y-family DNA polymerases in low-fidelity synthesis and translesion replication. J.Biol.Chem. 2004;279:32932–32940. doi: 10.1074/jbc.M405249200. [DOI] [PubMed] [Google Scholar]
- 30.Bridges BA. Error-prone DNA repair and translesion DNA synthesis. II: The inducible SOS hypothesis. DNA Repair. 2005;4:725–739. doi: 10.1016/j.dnarep.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 31.Bridges BA, Woodgate R. Mutagenic repair in Escherichia coli, X. The umuC gene product may be required for replication past pyrimidine dimers but not for the coding error in UV mutagenesis. Mol.Gen.Genet. 1984;196:364–366. doi: 10.1007/BF00328073. [DOI] [PubMed] [Google Scholar]
- 32.Bridges BA, Woodgate R. Mutagenic repair in Escherichia coli: products of the recA gene and of the umuD and umuC genes act at different steps in UV-induced mutagenesis. Proc.Natl.Acad.Sci.USA. 1985;82:4193–4197. doi: 10.1073/pnas.82.12.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bridges BA, Woodgate R. The two-step model of bacterial UV mutagenesis. Mutat.Res. 1985;150:133–139. doi: 10.1016/0027-5107(85)90110-1. [DOI] [PubMed] [Google Scholar]
- 34.Brotcorne-Lannoye A, Maenhaut-Michel G. Role of RecA protein in untargeted UV mutagenesis of bacteriophage λ: evidence for the requirement for the dinB gene. Proc.Natl.Acad.Sci.USA. 1986;83:3904–3908. doi: 10.1073/pnas.83.11.3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown DJ, Munro DS, Platt DJ. Recognition of the cryptic plasmid, pSLT, by restriction fingerprinting and a study of its incidence in Scottish Salmonella isolates. J Hyg (Lond) 1986;97:193–197. doi: 10.1017/s0022172400065268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bruck I, Woodgate R, McEntee K, Goodman MF. Purification of a soluble UmuD'C complex from Escherichia coli: Cooperative binding of UmuD'C to single-stranded DNA. J.Biol.Chem. 1996;271:10767–10774. doi: 10.1074/jbc.271.18.10767. [DOI] [PubMed] [Google Scholar]
- 37.Bull HJ, Lombardo MJ, Rosenberg SM. Stationary-phase mutation in the bacterial chromosome: recombination protein and DNA polymerase IV dependence. Proc Natl Acad Sci U S A. 2001;98:8334–41. doi: 10.1073/pnas.151009798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bunting KA, Roe SM, Pearl LH. Structural basis for recruitment of translesion DNA polymerase Pol IV/DinB to the β-clamp. EMBO J. 2003;22:5883–5892. doi: 10.1093/emboj/cdg568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burckhardt SE, Woodgate R, Scheuermann RH, Echols H. UmuD mutagenesis protein of Escherichia coli: overproduction, purification and cleavage by RecA. Proc.Natl.Acad.Sci.USA. 1988;85:1811–1815. doi: 10.1073/pnas.85.6.1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cai H, Yu H, McEntee K, Kunkel TA, Goodman MF. Purification and properties of wild-type and exonuclease-deficient DNA polymerase II from Escherichia coli. J.Biol.Chem. 1995;270:15327–15335. doi: 10.1074/jbc.270.25.15327. [DOI] [PubMed] [Google Scholar]
- 41.Carmical JR, Kowalczyk A, Zou Y, Van Houten B, Nechev LV, Harris CM, Harris TM, Lloyd RS. Butadiene-induced intrastrand DNA cross-links: a possible role in deletion mutagenesis. J Biol Chem. 2000;275:19482–19489. doi: 10.1074/jbc.M002037200. [DOI] [PubMed] [Google Scholar]
- 42.Ceccarelli D, Daccord A, Rene M, Burrus V. Identification of the origin of transfer (oriT) and a new gene required for mobilization of the SXT/R391 family of integrating conjugative elements. J Bacteriol. 2008;190:5328–5338. doi: 10.1128/JB.00150-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ciesla Z. Plasmid pKM101-mediated mutagenesis in Escherichia coli is inducible. Mol.Gen.Genet. 1982;186:298–300. doi: 10.1007/BF00331866. [DOI] [PubMed] [Google Scholar]
- 44.Cirz RT, Chin JK, Andes DR, de Crecy-Lagard V, Craig WA, Romesberg FE. Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol. 2005;3:e176. doi: 10.1371/journal.pbio.0030176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coetzee JN, Datta N, Hedges RW. R factors from Proteus rettgeri. J.Gen.Microbiol. 1972;72:543–552. doi: 10.1099/00221287-72-3-543. [DOI] [PubMed] [Google Scholar]
- 46.Cohen SE, Godoy VG, Walker GC. Transcriptional modulator NusA interacts with translesion DNA polymerases in Escherichia coli. J Bacteriol. 2009;191:665–672. doi: 10.1128/JB.00941-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cohen SE, Walker GC. The transcription elongation factor NusA is required for stress-induced mutagenesis in Escherichia coli. Curr Biol. 2010;20:80–85. doi: 10.1016/j.cub.2009.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Courcelle CT, Belle JJ, Courcelle J. Nucleotide excision repair or polymerase V-mediated lesion bypass can act to restore UV-arrested replication forks in Escherichia coli. J Bacteriol. 2005;187:6953–6961. doi: 10.1128/JB.187.20.6953-6961.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Courcelle J, Khodursky A, Peter B, Brown PO, Hanawalt PC. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics. 2001;158:41–64. doi: 10.1093/genetics/158.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Curti E, McDonald JP, Mead S, Woodgate R. DNA polymerase switching: effects on spontaneous mutagenesis in Escherichia coli. Mol.Microbiol. 2009;71:315–331. doi: 10.1111/j.1365-2958.2008.06526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dalrymple BP, Kongsuwan K, Wijffels G, Dixon NE, Jennings PA. A universal protein-protein interaction motif in the eubacterial DNA replication and repair systems. Proc.Natl.Acad.Sci.USA. 2001;98:11627–11632. doi: 10.1073/pnas.191384398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Delaney JC, Essigmann JM. Mutagenesis, genotoxicity, and repair of 1-methyladenine, 3-alkylcytosines, 1-methylguanine, and 3-methylthymine in alkB Escherichia coli. Proc Natl Acad Sci U S A. 2004;101:14051–14056. doi: 10.1073/pnas.0403489101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Delmas S, Matic I. Interplay between replication and recombination in Escherichia coli: impact of the alternative DNA polymerases. Proc.Natl.Acad.Sci.USA. 2006;103:4564–4569. doi: 10.1073/pnas.0509012103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Donnelly CE, Walker GC. groE mutants of Escherichia coli are defective in umuDC-dependent UV mutagenesis. J.Bacteriol. 1989;171:6117–6125. doi: 10.1128/jb.171.11.6117-6125.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dutreix M, Moreau PL, Bailone A, Galibert F, Battista JR, Walker GC, Devoret R. New recA mutations that dissociate the various RecA protein activities in Escherichia coli provide evidence for an additional role for RecA protein in UV mutagenesis. J.Bacteriol. 1989;171:2415–2423. doi: 10.1128/jb.171.5.2415-2423.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Echols H, Goodman MF. Mutation induced by DNA damage: a many protein affair. Mutat.Res. 1990;236:301–311. doi: 10.1016/0921-8777(90)90013-u. [DOI] [PubMed] [Google Scholar]
- 57.Elledge SJ, Walker GC. Proteins required for ultraviolet light and chemical mutagenesis. Identification of the products of the umuC locus of Escherichia coli. J.Mol.Biol. 1983;164:175–192. doi: 10.1016/0022-2836(83)90074-8. [DOI] [PubMed] [Google Scholar]
- 58.Ennis DG, Fisher B, Edmiston S, Mount DW. Dual role for Escherichia coli RecA protein in SOS mutagenesis. Proc.Natl.Acad.Sci.USA. 1985;82:3325–3329. doi: 10.1073/pnas.82.10.3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eoff RL, Angel KC, Egli M, Guengerich FP. Molecular basis of selectivity of nucleoside triphosphate incorporation opposite O6-benzylguanine by sulfolobus solfataricus DNA polymerase Dpo4: steady-state and pre-steady-state kinetics and x-ray crystallography of correct and incorrect pairing. J Biol Chem. 2007;282:13573–13584. doi: 10.1074/jbc.M700656200. [DOI] [PubMed] [Google Scholar]
- 60.Eoff RL, Irimia A, Angel KC, Egli M, Guengerich FP. Hydrogen bonding of 7,8-dihydro-8-oxodeoxyguanosine with a charged residue in the little finger domain determines miscoding events in Sulfolobus solfataricus DNA polymerase Dpo4. J Biol Chem. 2007;282:19831–19843. doi: 10.1074/jbc.M702290200. [DOI] [PubMed] [Google Scholar]
- 61.Eoff RL, Irimia A, Egli M, Guengerich FP. Sulfolobus solfataricus DNA polymerase Dpo4 is partially inhibited by “wobble” pairing between O6-methylguanine and cytosine, but accurate bypass is preferred. J Biol Chem. 2007;282:1456–1467. doi: 10.1074/jbc.M609661200. [DOI] [PubMed] [Google Scholar]
- 62.Eoff RL, Stafford JB, Szekely J, Rizzo CJ, Egli M, Guengerich FP, Marnett LJ. Structural and functional analysis of Sulfolobus solfataricus Y-family DNA polymerase Dpo4-catalyzed bypass of the malondialdehyde-deoxyguanosine adduct. Biochemistry. 2009;48:7079–7088. doi: 10.1021/bi9003588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fang J, Rand KD, Silva MC, Wales TE, Engen JR, Beuning PJ. Conformational dynamics of the Escherichia coli DNA polymerase manager proteins UmuD and UmuD'. J Mol Biol. 2010;398:40–53. doi: 10.1016/j.jmb.2010.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ferentz AE, Opperman T, Walker GC, Wagner G. Dimerization of the UmuD' protein in solution and its implications for regulation of SOS mutagenesis. Nat.Struct.Biol. 1997;4:979–983. doi: 10.1038/nsb1297-979. [DOI] [PubMed] [Google Scholar]
- 65.Fernandez de Henestrosa AR, Ogi T, Aoyagi S, Chafin D, Hayes JJ, Ohmori H, Woodgate R. Identification of additional genes belonging to the LexA-regulon in Escherichia coli. Mol.Microbiol. 2000;35:1560–1572. doi: 10.1046/j.1365-2958.2000.01826.x. [DOI] [PubMed] [Google Scholar]
- 66.Fijalkowska IJ, Dunn RL, Schaaper RM. Genetic requirements and mutational specificity of the Escherichia coli SOS mutator activity. J.Bacteriol. 1997;179:7435–7445. doi: 10.1128/jb.179.23.7435-7445.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Foster PL. Nonadaptive mutations occur on the F’ episome during adaptive mutation conditions in Escherichia coli. J Bacteriol. 1997;179:1550–1554. doi: 10.1128/jb.179.5.1550-1554.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Foster PL. Stress-induced mutagenesis in bacteria. Crit.Rev.Biochem.Mol.Biol. 2007;42:373–397. doi: 10.1080/10409230701648494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Foster PL, Gudmundsson G, Trimarchi JM, Cai H, Goodman MF. Proofreading-defective DNA polymerase II increases adaptive mutation in Escherichia coli. Proc Natl Acad Sci U S A. 1995;92:7951–7955. doi: 10.1073/pnas.92.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Foster PL, Trimarchi JM, Maurer RA. Two enzymes, both of which process recombination intermediates, have opposite effects on adaptive mutation in Escherichia coli. Genetics. 1996;142:25–37. doi: 10.1093/genetics/142.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Foti JJ, Delucia AM, Joyce CM, Walker GC. UmuD2 inhibits a non-covalent step during DinB-mediated template slippage on homopolymeric nucleotide runs. J.Biol.Chem. 2010;285:23086–23095. doi: 10.1074/jbc.M110.115774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Frank EG, Ennis DG, Gonzalez M, Levine AS, Woodgate R. Regulation of SOS mutagenesis by proteolysis. Proc.Natl.Acad.Sci.USA. 1996;93:10291–10296. doi: 10.1073/pnas.93.19.10291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Franklin MC, Wang J, Steitz TA. Structure of the replicating complex of a pol α family DNA polymerase. Cell. 2001;105:657–667. doi: 10.1016/s0092-8674(01)00367-1. [DOI] [PubMed] [Google Scholar]
- 74.Friedman N, Vardi S, Ronen M, Alon U, Stavans J. Precise temporal modulation in the response of the SOS DNA repair network in individual bacteria. PLoS Biol. 2005;3:e238. doi: 10.1371/journal.pbio.0030238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frisch RL, Su Y, Thornton PC, Gibson JL, Rosenberg SM, Hastings PJ. Separate DNA Pol II- and Pol IV-dependent pathways of stress-induced mutation during double-strand-break repair in Escherichia coli are controlled by RpoS. J Bacteriol. 2010;192:4694–700. doi: 10.1128/JB.00570-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fuchs RP, Fujii S. Translesion synthesis in Escherichia coli: lessons from the NarI mutation hot spot. DNA Repair. 2007;6:1032–1041. doi: 10.1016/j.dnarep.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 77.Fujii S, Fuchs RP. Biochemical basis for the essential genetic requirements of RecA and the β-clamp in Pol V activation. Proc.Natl.Acad.Sci.USA. 2009;106:14825–14830. doi: 10.1073/pnas.0905855106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fujii S, Fuchs RP. Defining the position of the switches between replicative and bypass DNA polymerases. EMBO J. 2004;23:4342–4352. doi: 10.1038/sj.emboj.7600438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fujii S, Fuchs RP. Interplay among replicative and specialized DNA polymerases determines failure or success of translesion synthesis pathways. J.Mol.Biol. 2007;372:883–893. doi: 10.1016/j.jmb.2007.07.036. [DOI] [PubMed] [Google Scholar]
- 80.Fujii S, Gasser V, Fuchs RP. The biochemical requirements of DNA polymerase V-mediated translesion synthesis revisited. J Mol Biol. 2004;341:405–417. doi: 10.1016/j.jmb.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 81.Furukohri A, Goodman MF, Maki H. A dynamic polymerase exchange with Escherichia coli DNA polymerase IV replacing DNA polymerase III on the sliding clamp. J Biol Chem. 2008;283:11260–11269. doi: 10.1074/jbc.M709689200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Galhardo RS, Hastings PJ, Rosenberg SM. Mutation as a stress response and the regulation of evolvability. Crit.Rev.Biochem.Mol.Biol. 2007;42:399–435. doi: 10.1080/10409230701648502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gawel D, Pham PT, Fijalkowska IJ, Jonczyk P, Schaaper RM. Role of accessory DNA polymerases in DNA replication in Escherichia coli: analysis of the dnaX36 mutator mutant. J Bacteriol. 2008;190:1730–1742. doi: 10.1128/JB.01463-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gefter ML, Hirota Y, Kornberg T, Wechsler JA, Barnoux C. Analysis of DNA polymerases II and III in mutants of Escherichia coli thermosensitive for DNA synthesis. Proc.Natl.Acad.Sci.USA. 1971;68:3150–3153. doi: 10.1073/pnas.68.12.3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Godoy VG, Jarosz DF, Simon SM, Abyzov A, Ilyin V, Walker GC. UmuD and RecA directly modulate the mutagenic potential of the Y family DNA polymerase DinB. Mol.Cell. 2007;28:1058–1070. doi: 10.1016/j.molcel.2007.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gonzalez M, Frank EG, Levine AS, Woodgate R. Lon-mediated proteolysis of the Escherichia coli UmuD mutagenesis protein: in vitro degradation and identification of residues required for proteolysis. Genes & Dev. 1998;12:3889–3899. doi: 10.1101/gad.12.24.3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gonzalez M, Rasulova F, Maurizi MR, Woodgate R. Subunit-specific degradation of the UmuD/D’ heterodimer by the ClpXP protease: The role of trans recognition in UmuD' stability. EMBO J. 2000;19:5251–5258. doi: 10.1093/emboj/19.19.5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gutierrez A, Elez M, Clermont O, Denamur E, Matic I. Escherichia coli YafP protein modulates DNA damaging property of the nitroaromatic compounds. Nucleic Acids Res. 2011;39:4192–4201. doi: 10.1093/nar/gkr050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Guzzo A, Lee MH, Oda K, Walker GC. Analysis of the region between amino acids 30 and 42 of intact UmuD by a monocysteine approach. J.Bacteriol. 1996;178:7295–7303. doi: 10.1128/jb.178.24.7295-7303.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Harris RS, Longerich S, Rosenberg SM. Recombination in adaptive mutation. Science. 1994;264:258–260. doi: 10.1126/science.8146657. [DOI] [PubMed] [Google Scholar]
- 91.Harris RS, Ross KJ, Rosenberg SM. Opposing roles of the holliday junction processing systems of Escherichia coli in recombination-dependent adaptive mutation. Genetics. 1996;142:681–691. doi: 10.1093/genetics/142.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hastings PJ, Hersh MN, Thornton PC, Fonville NC, Slack A, Frisch RL, Ray MP, Harris RS, Leal SM, Rosenberg SM. Competition of Escherichia coli DNA polymerases I, II and III with DNA Pol IV in stressed cells. PloS one. 2010;5:e10862. doi: 10.1371/journal.pone.0010862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Heltzel JM, Maul RW, Scouten Ponticelli SK, Sutton MD. A model for DNA polymerase switching involving a single cleft and the rim of the sliding clamp. Proc Natl Acad Sci U S A. 2009;106:12664–12669. doi: 10.1073/pnas.0903460106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Heltzel JM, Scouten Ponticelli SK, Sanders LH, Duzen JM, Cody V, Pace J, Snell EH, Sutton MD. Sliding clamp-DNA interactions are required for viability and contribute to DNA polymerase management in Escherichia coli. J Mol Biol. 2009;387:74–91. doi: 10.1016/j.jmb.2009.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Horsfall MJ, Lawrence CW. Accuracy of replication past the T-C (6–4) adduct. J.Mol.Biol. 1994;235:465–471. doi: 10.1006/jmbi.1994.1006. [DOI] [PubMed] [Google Scholar]
- 96.Howarth S. Increase in frequency of ultraviolet induced mutation brought about by the colicin factor ColI in Salmonella typhimurium. Mutat.Res. 1966;3:129–134. doi: 10.1016/0027-5107(66)90026-1. [DOI] [PubMed] [Google Scholar]
- 97.Indiani C, Langston LD, Yurieva O, Goodman MF, O’Donnell M. Translesion DNA polymerases remodel the replisome and alter the speed of the replicative helicase. Proc Natl Acad Sci U S A. 2009;106:6031–6038. doi: 10.1073/pnas.0901403106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Indiani C, McInerney P, Georgescu R, Goodman MF, O’Donnell M. A sliding-clamp toolbelt binds high- and low-fidelity DNA polymerases simultaneously. Mol.Cell. 2005;19:805–815. doi: 10.1016/j.molcel.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 99.Irimia A, Eoff RL, Pallan PS, Guengerich FP, Egli M. Structure and activity of Y-class DNA polymerase DPO4 from Sulfolobus solfataricus with templates containing the hydrophobic thymine analog 2,4-difluorotoluene. J Biol Chem. 2007;282:36421–36433. doi: 10.1074/jbc.M707267200. [DOI] [PubMed] [Google Scholar]
- 100.Irimia A, Loukachevitch LV, Eoff RL, Guengerich FP, Egli M. Metal-ion dependence of the active-site conformation of the translesion DNA polymerase Dpo4 from Sulfolobus solfataricus. Acta crystallographica. 2010;66:1013–1018. doi: 10.1107/S1744309110029374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jarosz DF, Beuning PJ, Cohen SE, Walker GC. Y-family DNA polymerases in Escherichia coli. Trends Microbiol. 2007;15:70–77. doi: 10.1016/j.tim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 102.Jarosz DF, Cohen SE, Delaney JC, Essigmann JM, Walker GC. A DinB variant reveals diverse physiological consequences of incomplete TLS extension by a Y-family DNA polymerase. Proc Natl Acad Sci U S A. 2009;106:21137–21142. doi: 10.1073/pnas.0907257106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jarosz DF, Godoy VG, Delaney JC, Essigmann JM, Walker GC. A single amino acid governs enhanced activity of DinB DNA polymerases on damaged templates. Nature. 2006;439:225–228. doi: 10.1038/nature04318. [DOI] [PubMed] [Google Scholar]
- 104.Jiang Q, Karata K, Woodgate R, Cox MM, Goodman MF. The active form of DNA polymerase V is UmuD'2C-RecA-ATP. Nature. 2009;460:359–363. doi: 10.1038/nature08178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kato T, Shinoura Y. Isolation and characterization of mutants of Escherichia coli deficient in induction of mutations by ultraviolet light. Mol.Gen.Genet. 1977;156:121–131. doi: 10.1007/BF00283484. [DOI] [PubMed] [Google Scholar]
- 106.Kenyon CJ, Walker GC. DNA-damaging agents stimulate gene expression at specific loci in Escherichia coli. Proc.Natl.Acad.Sci.USA. 1980;77:2819–2823. doi: 10.1073/pnas.77.5.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kim SR, Maenhaut-Michel G, Yamada M, Yamamoto Y, Matsui K, Sofuni T, Nohmi T, Ohmori H. Multiple pathways for SOS-induced mutagenesis in Escherichia coli: an overexpression of dinB/dinP results in strongly enhancing mutagenesis in the absence of any exogenous treatment to damage DNA. Proc.Natl.Acad.Sci.USA. 1997;94:13792–13797. doi: 10.1073/pnas.94.25.13792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kim SR, Matsui K, Yamada M, Gruz P, Nohmi T. Roles of chromosomal and episomal dinB genes encoding DNA pol IV in targeted and untargeted mutagenesis in Escherichia coli. Mol.Genet.Genomics. 2001;266:207–215. doi: 10.1007/s004380100541. [DOI] [PubMed] [Google Scholar]
- 109.Kitagawa Y, Akaboshi E, Shinagawa H, Horii T, Ogawa H, Kato T. Structural analysis of the umu operon required for inducible mutagenesis in Escherichia coli. Proc.Natl.Acad.Sci.USA. 1985;82:4336–4340. doi: 10.1073/pnas.82.13.4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Koch WH, Fernandez de Henestrosa AR, Woodgate R. Identification of mucAB-like homologs on two IncT plasmids, R394 and Rts-1. Mutat.Res. 2000;457:1–13. doi: 10.1016/s0027-5107(00)00134-2. [DOI] [PubMed] [Google Scholar]
- 111.Koch WH, Henrikson E, Eisenstadt E, Cebula TA. Salmonella typhimurium LT7 and LT2 strains carrying the imp operon on ColIa. J.Bacteriol. 1995;177:1903–1905. doi: 10.1128/jb.177.7.1903-1905.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Koch WH, Kopsidas G, Meffle B, Levine AS, Woodgate R. Analysis of chimeric UmuC proteins: identification of regions in Salmonella typhimurium UmuC important for mutagenic activity. Mol.Gen.Genet. 1996;251:121–129. doi: 10.1007/BF02172909. [DOI] [PubMed] [Google Scholar]
- 113.Kokubo K, Yamada M, Kanke Y, Nohmi T. Roles of replicative and specialized DNA polymerases in frameshift mutagenesis: mutability of Salmonella typhimurium strains lacking one or all of SOS-inducible DNA polymerases to 26 chemicals. DNA Repair. 2005;4:1160–1171. doi: 10.1016/j.dnarep.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 114.Kornberg A, Baker TA. DNA replication. 2 ed. New York: W.H. Freeman & Co; 1992. [Google Scholar]
- 115.Kornberg T, Gefter ML. Purification and DNA synthesis in cell-free extracts: properties of DNA polymerase II. Proc.Natl.Acad.Sci.USA. 1971;68:761–764. doi: 10.1073/pnas.68.4.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Koskiniemi S, Andersson DI. Translesion DNA polymerases are required for spontaneous deletion formation in Salmonella typhimurium. Proc Natl Acad Sci U S A. 2009;106:10248–10253. doi: 10.1073/pnas.0904389106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Koskiniemi S, Hughes D, Andersson DI. Effect of translesion DNA polymerases, endonucleases and RpoS on mutation rates in Salmonella typhimurium. Genetics. 2010 doi: 10.1534/genetics.110.116376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kroeger KM, Kim J, Goodman MF, Greenberg MM. Replication of an oxidized abasic site in Escherichia coli by a dNTP-stabilized misalignment mechanism that reads upstream and downstream nucleotides. Biochemistry. 2006;45:5048–5056. doi: 10.1021/bi052276v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kuban W, Banach-Orlowska M, Bialoskorska M, Lipowska A, Schaaper RM, Jonczyk P, Fijalkowska IJ. Mutator phenotype resulting from DNA polymerase IV overproduction in Escherichia coli: preferential mutagenesis on the lagging strand. J.Bacteriol. 2005;187:6862–6866. doi: 10.1128/JB.187.19.6862-6866.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kuban W, Banach-Orlowska M, Schaaper RM, Jonczyk P, Fijalkowska IJ. Role of DNA polymerase IV in Escherichia coli SOS mutator activity. J.Bacteriol. 2006;188:7977–7980. doi: 10.1128/JB.01088-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kumari A, Minko IG, Harbut MB, Finkel SE, Goodman MF, Lloyd RS. Replication bypass of interstrand cross-link intermediates by Escherichia coli DNA polymerase IV. J Biol Chem. 2008;283:27433–27437. doi: 10.1074/jbc.M801237200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kurth I, O’Donnell M. EcoSal:Escherichia coli and Salmonella, Cellular and Molecular Biology. Vol. 3. Washington, DC: American Society for Microbiology; Replisome dynamics during chromosome duplication, Ch.4.4.2 . [Google Scholar]
- 123.Lawrence CW, Borden A, Banerjee SK, LeClerc JE. Mutation frequency and spectrum resulting from a single abasic site in a single-stranded vector. Nucleic Acids Res. 1990;18:2153–2157. doi: 10.1093/nar/18.8.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lawrence CW, Borden A, Woodgate R. Analysis of the mutagenic properties of the UmuDC, MucAB and RumAB proteins, using a site specific abasic lesion. Mol.Gen.Genet. 1996;251:493–498. doi: 10.1007/BF02172378. [DOI] [PubMed] [Google Scholar]
- 125.Layton JC, Foster PL. Error-prone DNA polymerase IV is controlled by the stress-response sigma factor, RpoS, in Escherichia coli. Mol Microbiol. 2003;50:549–561. doi: 10.1046/j.1365-2958.2003.03704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Layton JC, Foster PL. Error-prone DNA polymerase IV is regulated by the heat shock chaperone GroE in Escherichia coli. J Bacteriol. 2005;187:449–57. doi: 10.1128/JB.187.2.449-457.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.LeClerc JE, Borden A, Lawrence CW. The thymine-thymine pyrimidine-pyrimidone (6-4) ultraviolet light photoproduct is highly mutagenic and specifically induces 3' thymine to cytosine transitions in Escherichia coli. Proc.Natl.Acad.Sci.USA. 1991;88:9685–9689. doi: 10.1073/pnas.88.21.9685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lee MH, Ohta T, Walker GC. A monocysteine approach for probing the structure and interactions of the UmuD protein. J.Bacteriol. 1994;176:4825–4837. doi: 10.1128/jb.176.16.4825-4837.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lenne-Samuel N, Janel-Bintz R, Kolbanovskiy A, Geacintov NE, Fuchs RP. The processing of a Benzo[a]pyrene adduct into a frameshift or a base substitution mutation requires a different set of genes in Escherichia coli. Mol Microbiol. 2000;38:299–307. doi: 10.1046/j.1365-2958.2000.02116.x. [DOI] [PubMed] [Google Scholar]
- 130.Lenne-Samuel N, Wagner J, Etienne H, Fuchs RP. The processivity factor β controls DNA polymerase IV traffic during spontaneous mutagenesis and translesion synthesis in vivo. EMBO Rep. 2002;3:45–49. doi: 10.1093/embo-reports/kvf007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lewis LK, Harlow GR, Gregg-Jolly LA, Mount DW. Identification of high affinity binding sites for LexA which define new DNA damage-inducible genes in Escherichia coli. J.Mol.Biol. 1994;241:507–523. doi: 10.1006/jmbi.1994.1528. [DOI] [PubMed] [Google Scholar]
- 132.Ling H, Boudsocq F, Plosky BS, Woodgate R, Yang W. Replication of a cis-syn thymine dimer at atomic resolution. Nature. 2003;424:1083–1087. doi: 10.1038/nature01919. [DOI] [PubMed] [Google Scholar]
- 133.Ling H, Boudsocq F, Woodgate R, Yang W. Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell. 2001;107:91–102. doi: 10.1016/s0092-8674(01)00515-3. [DOI] [PubMed] [Google Scholar]
- 134.Ling H, Boudsocq F, Woodgate R, Yang W. Snapshots of replication through an abasic lesion; structural basis for base substitutions and frameshifts. Mol.Cell. 2004;13:751–762. doi: 10.1016/s1097-2765(04)00101-7. [DOI] [PubMed] [Google Scholar]
- 135.Ling H, Sayer JM, Plosky BS, Yagi H, Boudsocq F, Woodgate R, Jerina DM, Yang W. Crystal structure of a Benzo[a]pyrene Diol Epoxide adduct in a ternary complex with a DNA polymerase. Proc.Natl.Acad.Sci.U.S.A. 2004;101:2265–2269. doi: 10.1073/pnas.0308332100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Little JW. Autodigestion of LexA and phage repressors. Proc.Natl.Acad.Sci.USA. 1984;81:1375–1379. doi: 10.1073/pnas.81.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Little JW, Edmiston SH, Pacelli LZ, Mount DW. Cleavage of the Escherichia coli lexA protein by the recA protease. Proc.Natl.Acad.Sci.USA. 1980;77:3225–3229. doi: 10.1073/pnas.77.6.3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lombardo MJ, Aponyi I, Rosenberg SM. General stress response regulator RpoS in adaptive mutation and amplification in Escherichia coli. Genetics. 2004;166:669–680. doi: 10.1534/genetics.166.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lopez de Saro FJ, Georgescu RE, Goodman MF, O’Donnell M. Competitive processivity-clamp usage by DNA polymerases during DNA replication and repair. EMBO J. 2003;22:6408–6418. doi: 10.1093/emboj/cdg603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lopez de Saro FJ, O’Donnell M. Interaction of the β sliding clamp with MutS, ligase, and DNA polymerase I. Proc.Natl.Acad.Sci.USA. 2001;98:8376–8380. doi: 10.1073/pnas.121009498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lowe LG, Guengerich FP. Steady-state and pre-steady-state kinetic analysis of dNTP insertion opposite 8-oxo-7,8-dihydroguanine by Escherichia coli polymerases I exo- and II exo. Biochemistry. 1996;35:9840–9849. doi: 10.1021/bi960485x. [DOI] [PubMed] [Google Scholar]
- 142.MacPhee DG. DNA polymerase activity determined by the ultraviolet-protecting plasmid, R-Utrecht. Nature. 1974;251:432–434. doi: 10.1038/251432a0. [DOI] [PubMed] [Google Scholar]
- 143.MacPhee DG. Salmonella typhimurium hisG46 (R-Utrecht): possible use in screening mutagens and carcinogens. Appl Microbiol. 1973;26:1004–1005. doi: 10.1128/am.26.6.1004-1005.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Maor-Shoshani A, Hayashi K, Ohmori H, Livneh Z. Analysis of translesion replication across an abasic site by DNA polymerase IV of Escherichia coli. DNA Repair. 2003;2:1227–1238. doi: 10.1016/s1568-7864(03)00142-3. [DOI] [PubMed] [Google Scholar]
- 145.Marrero J, Waldor MK. The SXT/R391 family of integrative conjugative elements is composed of two exclusion groups. J Bacteriol. 2007;189:3302–3305. doi: 10.1128/JB.01902-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Matsui K, Yamada M, Imai M, Yamamoto K, Nohmi T. Specificity of replicative and SOS-inducible DNA polymerases in frameshift mutagenesis: mutability of Salmonella typhimurium strains overexpressing SOS-inducible DNA polymerases to 30 chemical mutagens. DNA Repair. 2006;5:465–478. doi: 10.1016/j.dnarep.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 147.McCann JC, Springarn NE, Kobari J, Ames BN. Detection of carcinogens as mutagens: bacterial tester strains with R factor plasmids. Proc.Natl.Acad.Sci.USA. 1975;72:979–983. doi: 10.1073/pnas.72.3.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.McClelland M, Florea L, Sanderson K, Clifton SW, Parkhill J, Churcher C, Dougan G, Wilson RK, Miller W. Comparison of the Escherichia coli K-12 genome with sampled genomes of a Klebsiella pneumoniae and three salmonella enterica serovars, Typhimurium, Typhi and Paratyphi. Nucleic Acids Res. 2000;28:4974–4986. doi: 10.1093/nar/28.24.4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, Courtney L, Porwollik S, Ali J, Dante M, Du F, Hou S, Layman D, Leonard S, Nguyen C, Scott K, Holmes A, Grewal N, Mulvaney E, Ryan E, Sun H, Florea L, Miller W, Stoneking T, Nhan M, Waterston R, Wilson RK. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature. 2001;413:852–856. doi: 10.1038/35101614. [DOI] [PubMed] [Google Scholar]
- 150.McDonald JP, Frank EG, Levine AS, Woodgate R. Intermolecular cleavage of the UmuD-like mutagenesis proteins. Proc.Natl.Acad.Sci.USA. 1998;95:1478–1483. doi: 10.1073/pnas.95.4.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.McDonald JP, Maury EE, Levine AS, Woodgate R. Regulation of UmuD cleavage: role of the amino-terminal tail. J.Mol.Biol. 1998;282:721–730. doi: 10.1006/jmbi.1998.2044. [DOI] [PubMed] [Google Scholar]
- 152.McDonald JP, Peat TS, Levine AS, Woodgate R. Intermolecular cleavage by UmuD-like enzymes: identification of residues required for cleavage and substrate specificity. J.Mol.Biol. 1999;285:2199–2209. doi: 10.1006/jmbi.1998.2433. [DOI] [PubMed] [Google Scholar]
- 153.McKenzie GJ, Lee PL, Lombardo MJ, Hastings PJ, Rosenberg SM. SOS mutator DNA polymerase IV functions in adaptive mutation and not adaptive amplification. Mol Cell. 2001;7:571–579. doi: 10.1016/s1097-2765(01)00204-0. [DOI] [PubMed] [Google Scholar]
- 154.McKenzie GJ, Magner DB, Lee PL, Rosenberg SM. The dinB operon and spontaneous mutation in Escherichia coli. J Bacteriol. 2003;185:3972–3977. doi: 10.1128/JB.185.13.3972-3977.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.McLenigan M, Peat TS, Frank EG, McDonald JP, Gonzalez M, Levine AS, Hendrickson WA, Woodgate R. Novel Escherichia coli UmuD' mutants: structure function insights into SOS mutagenesis. J.Bacteriol. 1998;180:4658–4666. doi: 10.1128/jb.180.17.4658-4666.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.McLenigan MP, Kulaeva OI, Ennis DG, Levine AS, Woodgate R. The bacteriophage P1 HumD protein is a functional homolog of the prokaryotic UmuD'-like proteins and facilitates SOS mutagenesis in Escherichia coli. J.Bacteriol. 1999;181:7005–7013. doi: 10.1128/jb.181.22.7005-7013.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Minko IG, Kozekov ID, Harris TM, Rizzo CJ, Lloyd RS, Stone MP. Chemistry and biology of DNA containing 1,N2-deoxyguanosine adducts of the α, β-unsaturated aldehydes acrolein, crotonaldehyde, and 4-hydroxynonenal. Chem Res Toxicol. 2009;22:759–778. doi: 10.1021/tx9000489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Minko IG, Yamanaka K, Kozekov ID, Kozekova A, Indiani C, O’Donnell ME, Jiang Q, Goodman MF, Rizzo CJ, Lloyd RS. Replication bypass of the acrolein-mediated deoxyguanine DNA-peptide cross-links by DNA polymerases of the DinB family. Chem Res Toxicol. 2008;21:1983–1990. doi: 10.1021/tx800174a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Molina AM, Babudri N, Tamaro M, Venturini S, Monti-Bragadin C. Enterobacteriaceae plasmids enhancing chemical mutagenesis and their distribution among incompatibility groups. FEMS Lett. 1979;5:33–37. [Google Scholar]
- 160.Mortelmans KE, Stocker BAD. Segregation of the mutator property of plasmid R46 from its ultraviolet-protecting property. Mol.Gen.Genet. 1979;167:317–327. doi: 10.1007/BF00267425. [DOI] [PubMed] [Google Scholar]
- 161.Murli S, Opperman T, Smith BT, Walker GC. A role for the umuDC gene products of Escherichia coli in increasing resistance to DNA damage in stationary phase by inhibiting the transition to exponential growth. J.Bacteriol. 2000;182:1127–1135. doi: 10.1128/jb.182.4.1127-1135.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Napolitano R, Janel-Bintz R, Wagner J, Fuchs RP. All three SOS-inducible DNA polymerases (Pol II, pol IV and pol V) are involved in induced mutagenesis. EMBO J. 2000;19:6259–6265. doi: 10.1093/emboj/19.22.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Neeley WL, Delaney JC, Henderson PT, Essigmann JM. In vivo bypass efficiencies and mutational signatures of the guanine oxidation products 2-aminoimidazolone and 5-guanidino-4-nitroimidazole. J Biol Chem. 2004;279:43568–43573. doi: 10.1074/jbc.M407117200. [DOI] [PubMed] [Google Scholar]
- 164.Neeley WL, Delaney S, Alekseyev YO, Jarosz DF, Delaney JC, Walker GC, Essigmann JM. DNA polymerase V allows bypass of toxic guanine oxidation products in vivo. J Biol Chem. 2007;282:12741–12748. doi: 10.1074/jbc.M700575200. [DOI] [PubMed] [Google Scholar]
- 165.Neher SB, Sauer RT, Baker TA. Distinct peptide signals in the UmuD and UmuD' subunits of UmuD/D’ mediate tethering and substrate processing by the ClpXP protease. Proc Natl Acad Sci U S A. 2003;100:13219–13224. doi: 10.1073/pnas.2235804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Nicholls A, Sharp KA, Honig B. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 167.Nohmi T. Environmental stress and lesion-bypass DNA polymerases. Annu Rev Microbiol. 2006;60:231–253. doi: 10.1146/annurev.micro.60.080805.142238. [DOI] [PubMed] [Google Scholar]
- 168.Nohmi T, Battista JR, Dodson LA, Walker GC. RecA-mediated cleavage activates UmuD for mutagenesis: mechanistic relationship between transcriptional derepression and posttranslational activation. Proc.Natl.Acad.Sci.USA. 1988;85:1816–1820. doi: 10.1073/pnas.85.6.1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Nohmi T, Hakura A, Nakai Y, Watanabe M, Murayama SY, Sofuni T. Salmonella typhimurium has two homologous but different umuDC operons: cloning of a new umuDC-like operon (samAB) present in a 60-megadalton cryptic plasmid of S. typhimurium. J.Bacteriol. 1991;173:1051–1063. doi: 10.1128/jb.173.3.1051-1063.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.O’Grady PI, Borden A, Ozgenc A, Vandewiele D, Woodgate R, Lawrence CW. Intrinsic polymerase activities of UmuD'2C and MucA’2B are responsible for their different mutagenic properties during bypass of a T-T cis-syn cyclobutane dimer. J.Bacteriol. 2000;182:2285–2291. doi: 10.1128/jb.182.8.2285-2291.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ohmori H, Friedberg EC, Fuchs RPP, Goodman MF, Hanaoka F, Hinkle D, Kunkel TA, Lawrence CW, Livneh Z, Nohmi T, Prakash L, Prakash S, Todo T, Walker GC, Wang Z, Woodgate R. The Y-family of DNA polymerases. Mol.Cell. 2001;8:7–8. doi: 10.1016/s1097-2765(01)00278-7. [DOI] [PubMed] [Google Scholar]
- 172.Ohta T, Sutton MD, Guzzo A, Cole S, Ferentz AE, Walker GC. Mutations affecting the ability of the Escherichia coli UmuD' protein to participate in SOS mutagenesis. J.Bacteriol. 1999;181:177–185. doi: 10.1128/jb.181.1.177-185.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ollivierre JN, Fang J, Beuning PJ. The Roles of UmuD in Regulating Mutagenesis. J Nucleic Acids. 2010 doi: 10.4061/2010/947680. 10.4061/2010/947680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Opperman T, Murli S, Smith BT, Walker GC. A model for a umuDC-dependent prokaryotic DNA damage checkpoint. Proc.Natl.Acad.Sci.USA. 1999;96:9218–9223. doi: 10.1073/pnas.96.16.9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Paetzel M, Woodgate R. UmuD and UmuD' proteins1976-1981. In: Barrett AJ, Rawlings ND, Woessner JF, editors. Handbook of Proteolytic Enzymes. Vol. 2. London: Acedemic Press, Elsevier Science; 2004. [Google Scholar]
- 176.Pages V, Fuchs RP. How DNA lesions are turned into mutations within cells? Oncogene. 2002;21:8957–8966. doi: 10.1038/sj.onc.1206006. [DOI] [PubMed] [Google Scholar]
- 177.Pages V, Fuchs RP. Uncoupling of leading- and lagging-strand DNA replication during lesion bypass in vivo. Science. 2003;300:1300–1303. doi: 10.1126/science.1083964. [DOI] [PubMed] [Google Scholar]
- 178.Pages V, Janel-Bintz R, Fuchs RP. Pol III proofreading activity prevents lesion bypass as evidenced by its molecular signature within E.coli cells. J Mol Biol. 2005;352:501–509. doi: 10.1016/j.jmb.2005.07.063. [DOI] [PubMed] [Google Scholar]
- 179.Pata JD. Structural diversity of the Y-family DNA polymerases. Biochimica et biophysica acta. 2010;1804:1124–1135. doi: 10.1016/j.bbapap.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Patel M, Jiang Q, Woodgate R, Cox MM, Goodman MF. A new model for SOS-induced mutagenesis: how RecA protein activates DNA polymerase V. Crit Rev Biochem Mol Biol. 2010;45:171–184. doi: 10.3109/10409238.2010.480968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Paterson S, More MI, Pillay G, Cellini C, Woodgate R, Walker GC, Iyer VN, Winans SC. Genetic analysis of the mobilization and leading regions of the incN plasmids pKM101 and pCU1. J.Bacteriol. 1999;181:2572–2583. doi: 10.1128/jb.181.8.2572-2583.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Peat T, Frank EG, McDonald JP, Levine AS, Woodgate R, Hendrickson WA. Structure of the UmuD' protein and its regulation in response to DNA damage. Nature. 1996;380:727–730. doi: 10.1038/380727a0. [DOI] [PubMed] [Google Scholar]
- 183.Peat TS, Frank EG, McDonald JP, Levine AS, Woodgate R, Hendrickson WA. The UmuD' protein filament and its potential role in damage induced mutagenesis. Structure. 1996;4:1401–1412. doi: 10.1016/s0969-2126(96)00148-7. [DOI] [PubMed] [Google Scholar]
- 184.Perez-Capilla T, Baquero MR, Gomez-Gomez JM, Ionel A, Martin S, Blazquez J. SOS-independent induction of dinB transcription by β-lactam-mediated inhibition of cell wall synthesis in Crit.Rev.Biochem.Mol.Biol. J Bacteriol. 2005;187:1515–1518. doi: 10.1128/JB.187.4.1515-1518.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Perry KL, Elledge SJ, Mitchell B, Marsh L, Walker GC. umuDC and mucAB operons whose products are required for UV light and chemical-induced mutagenesis: UmuD, MucA, and LexA products share homology. Proc.Natl.Acad.Sci.USA. 1985;82:4331–4335. doi: 10.1073/pnas.82.13.4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Perry KL, Walker GC. Identification of plasmid (pKM101) coded proteins involved in mutagenesis and UV resistance. Nature. 1982;300:278–281. doi: 10.1038/300278a0. [DOI] [PubMed] [Google Scholar]
- 187.Petrosino JF, Galhardo RS, Morales LD, Rosenberg SM. Stress-induced β-lactam antibiotic resistance mutation and sequences of stationary-phase mutations in the Escherichia coli chromosome. J.Bacteriol. 2009;191:5881–5889. doi: 10.1128/JB.00732-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Pham P, Bertram JG, O’Donnell M, Woodgate R, Goodman MF. A model for SOS-lesion targeted mutations in E. coli involving pol V, RecA, SSB and β sliding clamp. Nature. 2001;409:366–370. doi: 10.1038/35053116. [DOI] [PubMed] [Google Scholar]
- 189.Pham P, Rangarajan S, Woodgate R, Goodman MF. Roles of DNA polymerases V and II in SOS-induced error-prone and error-free repair in Escherichia coli. Proc.Natl.Acad.Sci.USA. 2001;98:8350–8354. doi: 10.1073/pnas.111007198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Pinney RJ. Distribution among incompatibility groups of plasmids that confer UV mutability and UV resistance. Mutat.Res. 1980;72:155–159. doi: 10.1016/0027-5107(80)90232-8. [DOI] [PubMed] [Google Scholar]
- 191.Ponder RG, Fonville NC, Rosenberg SM. A switch from high-fidelity to error-prone DNA double-strand break repair underlies stress-induced mutation. Mol Cell. 2005;19:791–804. doi: 10.1016/j.molcel.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 192.Prieto AI, Ramos-Morales F, Casadesus J. Repair of DNA damage induced by bile salts in Salmonella enterica. Genetics. 2006;174:575–584. doi: 10.1534/genetics.106.060889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Qiu Z, Goodman MF. The Escherichia coli polB locus is identical to dinA, the structural gene for DNA polymerase II. Characterization of Pol II purified from a polB mutant. J Biol Chem. 1997;272:8611–8617. doi: 10.1074/jbc.272.13.8611. [DOI] [PubMed] [Google Scholar]
- 194.Radman M. Phenomenology of an inducible mutagenic DNA repair pathway in Escherichia coli: SOS repair hypothesis128-142. In: Prakash L, Sherman F, Miller MW, Lawrence CW, Tabor HW, editors. Molecular and environmental aspects of mutagenesis. 0 ed. Springfield, Ill: Charles C. Thomas; 1974. [Google Scholar]
- 195.Rangarajan S, Woodgate R, Goodman MF. A phenotype for enigmatic DNA polymerase II: a pivotal role for pol II in replication restart in UV-irradiated Escherichia coli. Proc.Natl.Acad.Sci.USA. 1999;96:9224–9229. doi: 10.1073/pnas.96.16.9224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Rangarajan S, Woodgate R, Goodman MF. Replication restart in UV-irradiated Escherichia coli involving pols II, III, V, PriA, RecA and RecFOR proteins. Mol.Microbiol. 2002;43:617–628. doi: 10.1046/j.1365-2958.2002.02747.x. [DOI] [PubMed] [Google Scholar]
- 197.Rechkoblit O, Kolbanovskiy A, Malinina L, Geacintov NE, Broyde S, Patel DJ. Mechanism of error-free and semitargeted mutagenic bypass of an aromatic amine lesion by Y-family polymerase Dpo4. Nat Struct Mol Biol. 2010;17:379–388. doi: 10.1038/nsmb.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Rechkoblit O, Malinina L, Cheng Y, Geacintov NE, Broyde S, Patel DJ. Impact of conformational heterogeneity of OxoG lesions and their pairing partners on bypass fidelity by Y family polymerases. Structure. 2009;17:725–736. doi: 10.1016/j.str.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Rechkoblit O, Malinina L, Cheng Y, Kuryavyi V, Broyde S, Geacintov NE, Patel DJ. Stepwise translocation of Dpo4 polymerase during error-free bypass of an oxoG lesion. PLoS Biol. 2006;4:e11. doi: 10.1371/journal.pbio.0040011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Reuven NB, Arad G, Maor-Shoshani A, Livneh Z. The mutagenesis protein UmuC is a DNA polymerase activated by UmuD', RecA, and SSB and Is specialized for translesion replication. J.Biol.Chem. 1999;274:31763–31766. doi: 10.1074/jbc.274.45.31763. [DOI] [PubMed] [Google Scholar]
- 201.Rudolph CJ, Upton AL, Lloyd RG. Maintaining replication fork integrity in UV-irradiated Escherichia coli cells. DNA Repair. 2008;7:1589–1602. doi: 10.1016/j.dnarep.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 202.Schlacher K, Cox MM, Woodgate R, Goodman MF. RecA acts in trans to allow replication of damaged DNA by DNA polymerase V. Nature. 2006;442:883–887. doi: 10.1038/nature05042. [DOI] [PubMed] [Google Scholar]
- 203.Schlacher K, Goodman MF. Lessons from 50 years of SOS DNA-damage-induced mutagenesis. Nat Rev Mol Cell Biol. 2007;8:587–594. doi: 10.1038/nrm2198. [DOI] [PubMed] [Google Scholar]
- 204.Sedgwick SG, Goodwin P. Differences in mutagenic and recombinational repair in enterobacteria. Proc.Natl.Acad.Sci.USA. 1985;82:4172–4176. doi: 10.1073/pnas.82.12.4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Sedgwick SG, Ho C, Woodgate R. Mutagenic DNA repair in Enterobacteria. J.Bacteriol. 1991;173:5604–5611. doi: 10.1128/jb.173.18.5604-5611.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Sedgwick SG, Thomas SM, Hughes V, Lodwick D, Strike P. Mutagenic DNA repair genes on plasmids from the “pre-antibiotic era”. Mol.Gen.Genet. 1989;218:323–329. doi: 10.1007/BF00331285. [DOI] [PubMed] [Google Scholar]
- 207.Seo KY, Nagalingam A, Miri S, Yin J, Chandani S, Kolbanovskiy A, Shastry A, Loechler EL. Mirror image stereoisomers of the major benzo[a]pyrene N2-dG adduct are bypassed by different lesion-bypass DNA polymerases in E. coli. DNA Repair (Amst) 2006;5:515–522. doi: 10.1016/j.dnarep.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 208.Shen X, Sayer JM, Kroth H, Ponten I, O’Donnell M, Woodgate R, Jerina DM, Goodman MF. Efficiency and accuracy of SOS-induced DNA polymerases replicating benzo[a]pyrene-7,8-diol 9,10-epoxide A and G adducts. J.Biol.Chem. 2002;277:5265–5274. doi: 10.1074/jbc.M109575200. [DOI] [PubMed] [Google Scholar]
- 209.Shen X, Woodgate R, Goodman MF. Escherichia coli DNA polymerase V subunit exchange: a post-SOS mechanism to curtail error-prone DNA synthesis. J.Biol.Chem. 2003;278:52546–52550. doi: 10.1074/jbc.M310127200. [DOI] [PubMed] [Google Scholar]
- 210.Shen X, Woodgate R, Goodman MF. Lyase activities intrinsic to Escherichia coli polymerases IV and V. DNA Repair. 2005;4:1368–1373. doi: 10.1016/j.dnarep.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 211.Shinagawa H, Iwasaki H, Kato T, Nakata A. RecA protein-dependent cleavage of UmuD protein and SOS mutagenesis. Proc.Natl.Acad.Sci.USA. 1988;85:1806–1810. doi: 10.1073/pnas.85.6.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Shinagawa H, Kato T, Ise T, Makino K, Nakata A. Cloning and characterization of the umu operon responsible for inducible mutagenesis in Escherichia coli. Gene. 1983;23:167–174. doi: 10.1016/0378-1119(83)90048-3. [DOI] [PubMed] [Google Scholar]
- 213.Shwartz H, Livneh Z. RecA protein inhibits in vitro replication of single-stranded DNA with DNA polymerase III holoenzyme of Escherichia coli. Mutat Res. 1989;213:165–173. doi: 10.1016/0027-5107(89)90148-6. [DOI] [PubMed] [Google Scholar]
- 214.Simmons LA, Foti JJ, Cohen SE, Walker GC. EcoSal:Escherichia coli and Salmonella, Cellular and Molecular Biology. Vol. 3. Washington, DC: American Society for Microbiology; The SOS Regulatory Network, Ch.5.4.3 . [Google Scholar]
- 215.Slilaty SN, Rupley JA, Little JW. Intramolecular cleavage of LexA and phage λ repressors. Dependence of kinetics on repressor concentration, pH, temperature and solvent. Biochemistry. 1986;25:6866–6875. doi: 10.1021/bi00370a020. [DOI] [PubMed] [Google Scholar]
- 216.Smith CM, Koch WH, Franklin SB, Foster PL, Cebula TA, Eisenstadt E. Sequence analysis of the Salmonella typhimurium LT2 umuDC operon. J.Bacteriol. 1990;172:4964–4978. doi: 10.1128/jb.172.9.4964-4978.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Sommer S, Bailone A, Devoret R. The appearance of the UmuD'C protein complex in Escherichia coli switches repair from homologous recombination to SOS mutagenesis. Mol.Microbiol. 1993;10:963–971. doi: 10.1111/j.1365-2958.1993.tb00968.x. [DOI] [PubMed] [Google Scholar]
- 218.Sommer S, Boudsocq F, Devoret R, Bailone A. Specific RecA amino acid changes affect RecA-UmuD'C interaction. Mol.Microbiol. 1998;28:281–291. doi: 10.1046/j.1365-2958.1998.00803.x. [DOI] [PubMed] [Google Scholar]
- 219.Steinborn G. Uvm mutants of Escherichia coli K12 deficient in UV mutagenesis. I. Isolation of uvm mutants and their phenotypical characterization in DNA repair and mutagenesis. Mol.Gen.Genet. 1978;165:87–93. doi: 10.1007/BF00270380. [DOI] [PubMed] [Google Scholar]
- 220.Steitz TA. DNA polymerases: structural diversity and common mechanisms. J.Biol.Chem. 1999;274:17395–17398. doi: 10.1074/jbc.274.25.17395. [DOI] [PubMed] [Google Scholar]
- 221.Storvik KA, Foster PL. RpoS, the stress response sigma factor, plays a dual role in the regulation of Escherichia coli’s error-prone DNA polymerase IV. J Bacteriol. 2010;192:3639–3644. doi: 10.1128/JB.00358-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Stumpf JD, Foster PL. Polyphosphate kinase regulates error-prone replication by DNA polymerase IV in Escherichia coli. Mol.Microbiol. 2005;57:751–761. doi: 10.1111/j.1365-2958.2005.04724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Sutton MD, Duzen JM. Specific amino acid residues in the β sliding clamp establish a DNA polymerase usage hierarchy in Escherichia coli. DNA Repair. 2006;5:312–323. doi: 10.1016/j.dnarep.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 224.Sutton MD, Duzen JM, Maul RW. Mutant forms of the Escherichia coli β sliding clamp that distinguish between its roles in replication and DNA polymerase V-dependent translesion DNA synthesis. Mol Microbiol. 2005;55:1751–1766. doi: 10.1111/j.1365-2958.2005.04500.x. [DOI] [PubMed] [Google Scholar]
- 225.Sutton MD, Guzzo A, Narumi I, Costanzo M, Altenbach C, Ferentz AE, Hubbell WL, Walker GC. A model for the structure of the Escherichia coli SOS-regulated UmuD2 protein. DNA Repair. 2002;1:77–93. doi: 10.1016/s1568-7864(01)00006-4. [DOI] [PubMed] [Google Scholar]
- 226.Suzuki N, Ohashi E, Hayashi K, Ohmori H, Grollman AP, Shibutani S. Translesional synthesis past acetylaminofluorene-derived DNA adducts catalyzed by human DNA polymerase κ and Escherichia coli DNA polymerase IV. Biochemistry. 2002;40:15176–15183. doi: 10.1021/bi010702g. [DOI] [PubMed] [Google Scholar]
- 227.Sweasy JB, Witkin EM, Sinha N, Roegner-Maniscalco V. RecA protein of Escherichia coli has a third essential role in SOS mutator activity. J.Bacteriol. 1990;172:3030–3036. doi: 10.1128/jb.172.6.3030-3036.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Szekeres ESJ, Woodgate R, Lawrence CW. Substitution of mucAB or rumAB for umuDC alters the relative frequencies of the two classes of mutations induced by a site-specific T-T cyclobutane dimer and the efficiency of translesion DNA synthesis. J.Bacteriol. 1996;178:2559–2563. doi: 10.1128/jb.178.9.2559-2563.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Tago Y, Imai M, Ihara M, Atofuji H, Nagata Y, Yamamoto K. Escherichia coli mutator ΔpolA is defective in base mismatch correction: the nature of in vivo DNA replication errors. J Mol Biol. 2005;351:299–308. doi: 10.1016/j.jmb.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 230.Tang M, Bruck I, Eritja R, Turner J, Frank EG, Woodgate R, O’Donnell M, Goodman MF. Biochemical basis of SOS-induced mutagenesis in Escherichia coli: reconstitution of in vitro lesion bypass dependent on the UmuD'2C mutagenic complex and RecA. Proc.Natl.Acad.Sci.USA. 1998;95:9755–9760. doi: 10.1073/pnas.95.17.9755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Tang M, Pham P, Shen X, Taylor J-S, O’Donnell M, Woodgate R, Goodman M. Roles of E. coli DNA polymerases IV and V in lesion-targeted and untargeted SOS mutagenesis. Nature. 2000;404:1014–1018. doi: 10.1038/35010020. [DOI] [PubMed] [Google Scholar]
- 232.Tang M, Shen X, Frank EG, O’Donnell M, Woodgate R, Goodman MF. UmuD'2C is an error-prone DNA polymerase, Escherichia coli, DNA pol V. Proc.Natl.Acad.Sci.USA. 1999;96:8919–8924. doi: 10.1073/pnas.96.16.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Thomas SM, Crowne HM, Pidsley SC, Sedgwick SG. Structural characterization of the Salmonella typhimurium LT2 umu operon. J.Bacteriol. 1990;172:4979–4987. doi: 10.1128/jb.172.9.4979-4987.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Tomer G, Livneh Z. Analysis of unassisted translesion replication by the DNA polymerase III holoenzyme. Biochemistry. 1999;38:5948–5958. doi: 10.1021/bi982599+. [DOI] [PubMed] [Google Scholar]
- 235.Tompkins JD, Nelson JL, Hazel JC, Leugers SL, Stumpf JD, Foster PL. Error-prone polymerase, DNA polymerase IV, is responsible for transient hypermutation during adaptive mutation in Escherichia coli. J Bacteriol. 2003;185:3469–3472. doi: 10.1128/JB.185.11.3469-3472.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Uchida K, Furukohri A, Shinozaki Y, Mori T, Ogawara D, Kanaya S, Nohmi T, Maki H, Akiyama M. Overproduction of Escherichia coli DNA polymerase DinB (Pol IV) inhibits replication fork progression and is lethal. Mol Microbiol. 2008;70:608–622. doi: 10.1111/j.1365-2958.2008.06423.x. [DOI] [PubMed] [Google Scholar]
- 237.Upton C, Pinney RJ. Expression of eight unrelated Muc+ plasmids in eleven DNA repair-deficient E. coli strains. Mutat.Res. 1983;112:261–273. doi: 10.1016/0167-8817(83)90002-0. [DOI] [PubMed] [Google Scholar]
- 238.Vandewiele D, Fernandez de Henestrosa AR, Timms AR, Bridges BA, Woodgate R. Sequence analysis and phenotypes of five temperature sensitive mutator alleles of dnaE, encoding modified α-catalytic subunits of Escherichia coli DNA polymerase III holoenzyme. Mutat.Res. 2002;499:85–95. doi: 10.1016/s0027-5107(01)00268-8. [DOI] [PubMed] [Google Scholar]
- 239.Venturini S, Monti-Bragadin C. R plasmid-mediated enhancement of mutagenesis in strains of Escherichia coli deficient in known repair functions. Mutat.Res. 1978;50:1–8. doi: 10.1016/0027-5107(78)90054-4. [DOI] [PubMed] [Google Scholar]
- 240.Wagner J, Etienne H, Fuchs RP, Cordonnier A, Burnouf D. Distinct β-clamp interactions govern the activities of the Y family PolIV DNA polymerase. Mol Microbiol. 2009;74:1143–1151. doi: 10.1111/j.1365-2958.2009.06920.x. [DOI] [PubMed] [Google Scholar]
- 241.Wagner J, Fujii S, Gruz P, Nohmi T, Fuchs RP. The β clamp targets DNA polymerase IV to DNA and strongly increases its processivity. EMBO Rep. 2001;1:484–488. doi: 10.1093/embo-reports/kvd109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Wagner J, Fujii S, Gruz P, Nohmi T, Fuchs RP. The β-clamp targets DNA polymerase IV to DNA and strongly increases its processivity. EMBO Rep. 2000;1:484–488. doi: 10.1093/embo-reports/kvd109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Wagner J, Gruz P, Kim SR, Yamada M, Matsui K, Fuchs RPP, Nohmi T. The dinB gene encodes an novel Escherichia coli DNA polymerase (DNA pol IV) involved in mutagenesis. Mol Cell. 1999;4:281–286. doi: 10.1016/s1097-2765(00)80376-7. [DOI] [PubMed] [Google Scholar]
- 244.Wagner J, Nohmi T. Escherichia coli DNA polymerase IV mutator activity: genetic requirements and mutational specificity. J.Bacteriol. 2000;182:4587–4595. doi: 10.1128/jb.182.16.4587-4595.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Wang F, Yang W. Structural insight into translesion synthesis by DNA Pol II. Cell. 2009;139:1279–1289. doi: 10.1016/j.cell.2009.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Wang Y, Musser SK, Saleh S, Marnett LJ, Egli M, Stone MP. Insertion of dNTPs opposite the 1,N2-propanodeoxyguanosine adduct by Sulfolobus solfataricus P2 DNA polymerase IV. Biochemistry. 2008;47:7322–7334. doi: 10.1021/bi800152j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Weigle JJ. Induction of mutation in a bacterial virus. Proc.Natl.Acad.Sci.USA. 1953;39:628–636. doi: 10.1073/pnas.39.7.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Wilson RC, Pata JD. Structural insights into the generation of single-base deletions by the Y family DNA polymerase dbh. Mol.Cell. 2008;29:767–779. doi: 10.1016/j.molcel.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 249.Witkin EM. Mutation-proof and mutation-prone modes of survival in derivatives of Escherichia coli B differing in sensitivity to ultraviolet light. Brookhaven Symp. Biol. 1967;20:17–55. [Google Scholar]
- 250.Witkin EM. The mutability toward ultraviolet light of recombination-deficient strains of Escherichia coli. Mutat Res. 1969;8:9–14. doi: 10.1016/0027-5107(69)90135-3. [DOI] [PubMed] [Google Scholar]
- 251.Witkin EM. The radiation sensitivity of Escherichia coli B: a hypothesis relating filament formation and prophage induction. Proc Natl Acad Sci U S A. 1967;57:1275–1279. doi: 10.1073/pnas.57.5.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Witkin EM. Ultraviolet mutagenesis and inducible DNA repair in Escherichia coli. Bacteriol.Rev. 1976;40:869–907. doi: 10.1128/br.40.4.869-907.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Witkin EM. Ultraviolet-induced mutation and DNA repair. Annu Rev Microbiol. 1969;23:487–514. doi: 10.1146/annurev.mi.23.100169.002415. [DOI] [PubMed] [Google Scholar]
- 254.Wolff E, Kim M, Hu K, Yang H, Miller JH. Polymerases leave fingerprints: analysis of the mutational spectrum in Escherichia coli rpoB to assess the role of polymerase IV in spontaneous mutation. J Bacteriol. 2004;186:2900–2905. doi: 10.1128/JB.186.9.2900-2905.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Wong JH, Brown JA, Suo Z, Blum P, Nohmi T, Ling H. Structural insight into dynamic bypass of the major cisplatin-DNA adduct by Y-family polymerase Dpo4. EMBO J. 2010;29:2059–2069. doi: 10.1038/emboj.2010.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Woodgate R. Construction of a umuDC operon substitution mutation in Escherichia coli. Mutat.Res. 1992;281:221–225. doi: 10.1016/0165-7992(92)90012-7. [DOI] [PubMed] [Google Scholar]
- 257.Woodgate R. Evolution of the two-step model for UV-mutagenesis. Mutat.Res. 2001;485:83–92. doi: 10.1016/s0921-8777(00)00076-8. [DOI] [PubMed] [Google Scholar]
- 258.Woodgate R, Ennis DG. Levels of chromosomally encoded Umu proteins and requirements for in vivo UmuD cleavage. Mol.Gen.Genet. 1991;229:10–16. doi: 10.1007/BF00264207. [DOI] [PubMed] [Google Scholar]
- 259.Woodgate R, Levine AS, Koch WH, Cebula TA, Eisenstadt E. Induction and cleavage of Salmonella typhimurium UmuD protein. Mol.Gen.Genet. 1991;229:81–85. doi: 10.1007/BF00264216. [DOI] [PubMed] [Google Scholar]
- 260.Woodgate R, Rajagopalan M, Lu C, Echols H. UmuC mutagenesis protein of Escherichia coli: purification and interaction with UmuD and UmuD'. Proc.Natl.Acad.Sci.USA. 1989;86:7301–7305. doi: 10.1073/pnas.86.19.7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Xing G, Kirouac K, Shin YJ, Bell SD, Ling H. Structural insight into recruitment of translesion DNA polymerase Dpo4 to sliding clamp PCNA. Mol.Microbiol. 2009;71:678–691. doi: 10.1111/j.1365-2958.2008.06553.x. [DOI] [PubMed] [Google Scholar]
- 262.Yamada M, Nunoshiba T, Shimizu M, Gruz P, Kamiya H, Harashima H, Nohmi T. Involvement of Y-family DNA polymerases in mutagenesis caused by oxidized nucleotides in Escherichia coli. J Bacteriol. 2006;188:4992–4995. doi: 10.1128/JB.00281-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Yeiser B, Pepper ED, Goodman MF, Finkel SE. SOS-induced DNA polymerases enhance long-term survival and evolutionary fitness. Proc.Natl.Acad.Sci.USA. 2002;99:8737–8741. doi: 10.1073/pnas.092269199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Yin J, Seo KY, Loechler EL. A role for DNA polymerase V in G --> T mutations from the major benzo[a]pyrene N2-dG adduct when studied in a 5'-TGT sequence in E. coli. DNA Repair (Amst) 2004;3:323–334. doi: 10.1016/j.dnarep.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 265.Yuan B, Cao H, Jiang Y, Hong H, Wang Y. Efficient and accurate bypass of N2-(1-carboxyethyl)-2’-deoxyguanosine by DinB DNA polymerase in vitro and in vivo. Proc Natl Acad Sci U S A. 2008;105:8679–8684. doi: 10.1073/pnas.0711546105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Zang H, Goodenough AK, Choi JY, Irimia A, Loukachevitch LV, Kozekov ID, Angel KC, Rizzo CJ, Egli M, Guengerich FP. DNA adduct bypass polymerization by Sulfolobus solfataricus DNA polymerase Dpo4: analysis and crystal structures of multiple base pair substitution and frameshift products with the adduct 1,N2-ethenoguanine. J Biol Chem. 2005;280:29750–29764. doi: 10.1074/jbc.M504756200. [DOI] [PubMed] [Google Scholar]
- 267.Zang H, Irimia A, Choi JY, Angel KC, Loukachevitch LV, Egli M, Guengerich FP. Efficient and high fidelity incorporation of dCTP opposite 7,8-dihydro-8-oxodeoxyguanosine by Sulfolobus solfataricus DNA polymerase Dpo4. J Biol Chem. 2006;281:2358–2372. doi: 10.1074/jbc.M510889200. [DOI] [PubMed] [Google Scholar]
- 268.Zhang H, Eoff RL, Kozekov ID, Rizzo CJ, Egli M, Guengerich FP. Structure-function relationships in miscoding by Sulfolobus solfataricus DNA polymerase Dpo4: guanine N2,N2-dimethyl substitution produces inactive and miscoding polymerase complexes. J Biol Chem. 2009;284:17687–17699. doi: 10.1074/jbc.M109014274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Zhang H, Eoff RL, Kozekov ID, Rizzo CJ, Egli M, Guengerich FP. Versatility of Y-family Sulfolobus solfataricus DNA polymerase Dpo4 in translesion synthesis past bulky N2-alkylguanine adducts. J Biol Chem. 2009;284:3563–3576. doi: 10.1074/jbc.M807778200. [DOI] [PMC free article] [PubMed] [Google Scholar]