

Abstract
The centromere is a chromatin region that is required for accurate inheritance of eukaryotic chromosomes during cell divisions. Among the different centromere-associated proteins (CENP) identified, CENP-B has been independently domesticated from a pogo-like transposase twice: Once in mammals and once in fission yeast. Recently, a third independent domestication restricted to holocentric lepidoptera has been described. In this work, we take advantage of the high-quality genome sequence and the wealth of functional information available for Drosophila melanogaster to further investigate the possibility of additional independent domestications of pogo-like transposases into host CENP-B related proteins. Our results showed that CENP-B related genes are not restricted to holocentric insects. Furthermore, we showed that at least three independent domestications of pogo-like transposases have occurred in metazoans. Our results highlight the importance of transposable elements as raw material for the recurrent evolution of important cellular functions.
Keywords: pogo, Drosophila, exaptation, functional domain, holocentric chromosomes
Centromere-Associated Protein B Homologs Are Present in Mammals, Fission Yeast, and Holocentric Lepidoptera
CENP-B is one of the earliest described cases of transposable element (TE) exaptations in the human genome (Tudor et al. 1992; Smit 1996). Human CENP-B has extensive sequence and domain similarity to transposases encoded by the pogo superfamily of TEs. It is widespread and highly conserved in mammals, whereas it is undetectable in other metazoans (Casola et al. 2008). Other than in mammals, three CENP-B homologs have been described in fission yeast: Abp1 (Autonomous replicating sequence-binding protein 1), Cbh1 (CENP-B homolog 1), and Cbh2 (CENP-B homolog 2). Fission yeast and human CENP-B proteins are functionally related. Fission yeast CENP-B homologs show partially redundant function in the formation of centromeric heterochromatin and in chomosome segregation (Irelan et al. 2001). They also play a role in the silencing of TEs and TE-associated genes (Cam et al. 2008; Lorenz et al. 2012) and in DNA replication (Zaratiegui et al. 2011). In humans, although the role of CENP-B has been controversial (Marshall and Choo 2012), it has been recently shown that CENP-B provides an alternative redundant pathway for kinetochore formation in vivo (Fachinetti et al. 2013). Sequence and functional relationship between mammal and fission yeast CENP-B homologs is the result of convergent domestication: Different pogo-like transposases have been exapted independently in the two lineages to give rise to host proteins with centromere-binding activity (Casola et al. 2008).
Recently, a CENP-B homolog has been described in the holocentric lepidoptera Spodoptera frugiperda (d’Alençon et al. 2011). Although in most eukaryotes the kinetochore protein complex, connecting chromosomes to spindle microtubules during cell division, usually binds to a single locus called the centromere, in holocentric chromosomes kinetochore proteins bind along the entire length of the chromosomes. Spodoptera frugiperda CENP-B ability to bind in vivo to a retrotransposon derived sequence and its nuclear localization suggest that this protein is functionally related to other CENP-B homologs (d’Alençon et al. 2011). Orthologs of S. frugiperda CENP-B have been identified in other holocentric lepidoptera, Bombyx mori and Helicoverpa armigera, but not in other invertebrates. These findings suggest that there has been a third convergent domestication of a transposase into a CENP-B-related (CR) protein that appears to be restricted to holocentric lepidopteran species (d’Alençon et al. 2011). These results prompted us to further investigate whether CR proteins can be identified in the Drosophila melanogaster genome. Drosophila melanogaster has one of the highest quality genomes in terms of sequence and functional annotation (St Pierre et al. 2013), and its DNA is organized in nonholocentric chromosomes: Two metacentric and two telocentric ones.
CAG Is the Closest CR Protein in D. melanogaster
To identify CR proteins in D. melanogaster, we used the protein sequences of the previously identified CENP-B homologs and D. melanogaster pogo transposase as queries in BLASTp searches against the D. melanogaster protein database. As expected, we found that the pogo transposase was the best hit in all searches (20–30% identity, e values 4e−34–3e−12). We also found that CAG (CG12346) was the only host protein showing local significant sequence similarity with human CENP-B (34% identity, e value 4e−17), fission yeast Cbh1 (26% identity, e value 2e−05), and Drosophila pogo transposase (26% identity, e value 9e−10). Reciprocal BLAST searches using CAG as a query confirmed that the closest sequence in fission yeast is Cbh1 (25% identity, e value 7e−06). On the other hand, CAG shows significant sequence homology with 40 blast hits in human, being the host genes TIGD6 (34% identity, e value 3e−18) and CENP-B (34% identity, e value 5e−18) the highest scoring hits. Although four other proteins containing CENP-B domains have been described in D. melanogaster, we could not detect them in an exhaustive search using BLASTp, tBLASTN, and HMMER, suggesting that they are not closely related to CAG and previously described CR proteins (Benchabane et al. 2011).
To further determine that CAG is a transposon-derived gene and not a transposon remnant, we followed the conservative approach proposed by Feschotte and Pritham (2007). CAG fulfills the six criteria proposed by these authors. First, we did not find evidence of transposon hallmarks, that is, Terminal Inverted Repeats (TIRs), in CAG flanking regions, suggesting that CAG is not a transposon. Second, CAG shows significant sequence and domain architecture similarities (see below) with pogo transposase and other transposase-derived genes suggesting that it has a transposon origin. Third, the coding capacity of CAG is intact and it is evolving under functional constrain contrary to TE-coding regions of nonautonomous transposons that typically evolve neutrally (dN/dS = 0.08312 estimated using a cDNA alignment with D. yakuba). Fourth, synteny around CAG is conserved in most species of the Drosophila genus (see below) as opposed to TEs that are not expected to be maintained at orthologous positions. Fifth, CAG expresses two alternative transcripts and shows peaks of expression in different developmental stages (Marygold et al. 2013) in contrast to TE genes that are often not expressed (Deloger et al. 2009). And sixth, there are seven reported alleles for this gene, and some of them are lethal, suggesting that CAG has a critical biological function in vivo (St Pierre et al. 2013).
Thus, we can conclude that CAG has a transposon origin and it is the closest D. melanogaster host gene encoding a CR protein.
CAG Domain Architecture Is Similar to pogo Transposase and Other CR Proteins
We checked whether, besides sequence conservation, the domain architecture of CAG is also conserved when compared with previously identified CENP-B homologs and D. melanogaster pogo transposase (fig. 1A). Using hmmscan (Finn et al. 2011), we found that CAG shows the same composite DNA binding domain (DBD) structure found in human and S. frugiperda CR proteins (fig. 1A). The majority of the highly conserved amino acids shared by proteins having this composite DBD are also conserved in CAG suggesting that this domain is functional (supplementary fig. S1, Supplementary Material online). Furthermore, a 3D model of CAG DBD build using human CENP-B as a template, shows that the fold of this domain is similar in both proteins (fig. 1B).
Fig. 1.—
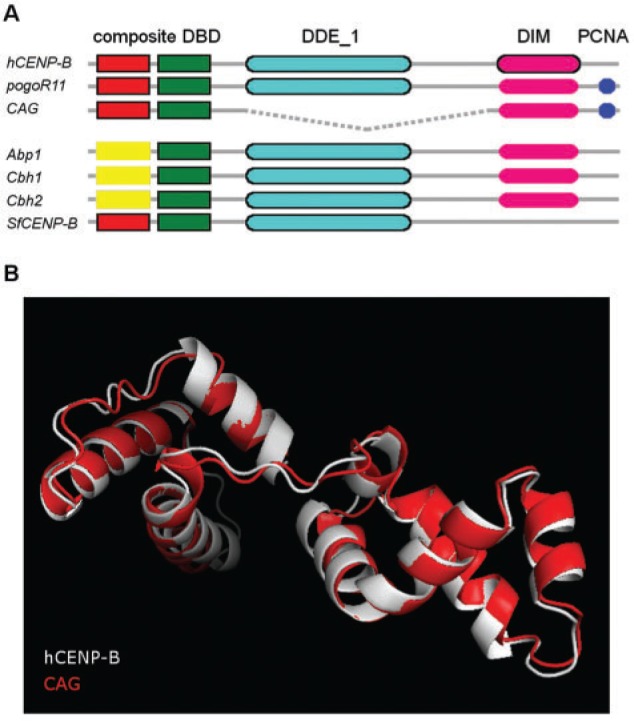
Domain structure of pogo transposase and CR proteins. (A) Human CENP-B (hCENP-B), D. melanogaster pogo transposase (pogoR11), D. melanogaster CAG (CAG), yeast CENP-B homologs (Abp1, Cbh1, Cbh2), and Spodoptera frugiperda CENP-B homolog (SfCENP-B). CENP-B_N domain is shown in red, d1iufa1 in yellow, HTH_Tnp_Tc5 in green, DDE_1 in light blue, DIM in pink, and PCNA in dark blue. Domains predicted by hmmscan are shown as black-lined boxes, the other domains were inferred from experimental evidence. The discontinuous line indicates the deleted region. (B) 3D structure prediction of D. melanogaster CAG DBD using human CENP-B as a template. Z-score = −6.66 and −6.7 for CENP-B and CAG, respectively.
CAG is the only of the seven proteins being compared that does not have a DDE_1 endonuclease domain (DDE) next to the DBD domain (fig. 1A). Indeed, CAG is shorter than the other proteins probably due to an internal deletion, which is a common feature in this class of transposons (Negoua et al. 2013). In fact, out of the 44 pogo copies in the D. melanogaster genome, 35 showed internal deletions and one of them, FBti0020096, has a similar deletion as CAG.
A dimerization domain (DIM) near the C-terminal region is present in human CENP-B (Tawaramoto et al. 2003). hmmscan does not detect any C-terminal DIM domain in the other six proteins. However, it has been described that the pogo transposase, CAG and the three yeast CENP-B homologs self-dimerize (Wang et al. 1999; Irelan et al. 2001; Giot et al. 2003; Tawaramoto et al. 2003; Cam et al. 2008; Lorenz et al. 2012). Given their common evolutionary origin, we hypothesized that the DIM domain might be located in the C-terminal region in these proteins as well. Finally, the pogo transposase has a PCNA (proliferating cell nuclear antigen) binding domain in the C-terminal end (Warbrick et al. 1998). Although this domain has not been identified in CAG at the sequence level, there is experimental evidence for CAG binding to PCNA suggesting that besides CAG other pogo-related proteins might have also conserved this function (fig. 1A).
Taken together, these results indicate that CAG is similar to the pogo transposase both at sequence and domain architecture levels, further confirming that CAG has a transposon origin. The lack of the DDE domain is explained by an internal deletion that is common in this family of transposons. Except for the absence of the DDE domain, CAG sequence and domain architecture are also similar to other described CENP-B homologs.
Protein–Protein Interaction Network Suggests CAG Is Functionally Related to Other CR Proteins
Because CAG is a protein of unknown function, we searched for proteins directly interacting with CAG to shed light on the biological processes in which this protein might be involved. Although data from protein-protein interaction (PPI) networks is still noisy and partial, functional annotation based on interaction networks provides reliable insight into the biology of proteins with unknown function (Titz et al. 2004; Sharan et al. 2007). There is experimental evidence for the interaction between CAG and 15 other proteins, and six of them have nucleic acid binding capacity (supplementary table S1, Supplementary Material online). CAG directly interacts with Mi-2, which is a component of the nucleosome remodeling and histone deacetylation (NuRD) complex with chromatin binding and remodeling activity (Bouazoune and Brehm 2005). As mentioned above, CAG has preserved the pogo transposase capacity to interact with PCNA (Warbrick et al. 1998). Besides playing a crucial role in DNA replication and repair (Warbrick et al. 1998), cell cycle control and sister chromatin cohesion (Maga and Hubscher 2003), PCNA is also a component of the microtubule associated complex (Hughes et al. 2008). CAG also interacts with Cdc5 and snama, which are involved in cell cycle control (supplementary table S1, Supplementary Material online).
To further investigate the functional annotation of CAG, we expanded the network of proteins that directly interact with CAG by incorporating their respective interaction partners (Chua et al. 2006). The resulting list of 842 proteins in the neighborhood-2 of CAG showed a significant enrichement for 72 biological process Gene Ontology (GO) terms related to cell cycle and mitotic spindle organization and nucleic acid metabolism among others (fig. 2). Fifty-eight out of the 72 CAG enriched GO terms are also enriched in the human CENP-B neighborhood-2, further suggesting that CAG and CENP-B interacting partners are involved in similar biological processes.
Fig. 2.—

Biological processes overrepresented in the CAG 2-neighbourhood PPI. Hierarchical representation of the 72 biological process GO terms enriched in the neighbourhood-2 of CAG PPI network. Node colors indicate the level of significance. The overrepresented GO terms were categorized into four groups related to the neigbourhood-1 genes of CAG PPI: Cell cycle and spindle organization, response to stimulus and regulation of metabolic process, nucleic acids metabolism, and protein metabolism. GO terms enriched also in the neighborhood-2 of human CENP-B are represented inside gray boxes.
CR Genes Are Present in Holocentric and Nonholocentric Insecta
To determine whether CAG is present in species other than D. melanogaster, we searched for CAG orthologs using a BLASTp search against ensembl metazoa protein database (see Materials and Methods). CAG has nine 1-to-1 orthologs in the Drosophila genus that showed a sequence identity from 97.2% to 39.8% (table 1). The DBD architecture is conserved in all of them and the length of the protein is highly similar except in D. simulans, where only the first DBD domain is present (table 1). Other than sequence identity and protein length, synteny is also conserved in the six closest Drosophila species except in D. simulans. Furthermore, CAG is evolving under functional constrain in these ten Drosophila species (average difference of synonymous and nonsynonymous substitutions per site over all nine sequence pairs is 17.99, P value ≪ 0.001) suggesting that CAG orthologs are functional genes.
Table 1.
CR Genes Identified in Holocentric and Nonholocentric Insecta
| Class | Order | Species | Protein Identifiera | Protein Sequence Identityb (%) | Protein Length | Conserved Protein Domains |
|
|---|---|---|---|---|---|---|---|
| HTH_CENP-B_N | HTH_Tnp_Tc5 | ||||||
| Insecta | Diptera | Drosophila melanogaster | CAG (CG12346) | 100 | 225 | X | X |
| D. simulans | GD15259 | 97.22 | 111 | X | — | ||
| D. sechelia | GM20484 | 94.67 | 225 | X | X | ||
| D. yakuba | GE13064 | 92.44 | 225 | X | X | ||
| D. erecta | GG22708 | 93.24 | 207 | X | X | ||
| D. ananassae | GF13390 | 59.11 | 222 | X | X | ||
| D. pseudoobscura | GA11571 | 57.46 | 228 | X | X | ||
| D. persimilis | GL17090 | 58.77 | 228 | X | X | ||
| D. willistoni | GK19073 | 40.29 | 222 | X | X | ||
| D. virilis | GJ16124 | 39.81 | 227 | X | X | ||
| Insecta | Lepidoptera | Bombyx mori | BGIBMGA013031 | 32.09 | 278 | X | X |
| BGIBMGA008012 | 26.35 | 501 | X | X | |||
| BGIBMGA007903 | 25 | 468 | X | X | |||
| BGIBMGA013624 | 29.63 | 722 | X | X | |||
| Insecta | Lepidoptera | Heliconius melpomene | HMEL009793 | 35.38 | 255 | X | X |
| HMEL010729 | 33.8 | 295 | X | X | |||
| HMEL014790 | 31.55 | 533 | X | X | |||
| HMEL007960 | 50 | 533 | X | X | |||
| HMEL011593 | 35.38 | 192 | X | X | |||
| Insecta | Lepidoptera | Helicoverpa armigera | 94B11_25* | 22.22# | 488 | X | X |
| Insecta | Lepidoptera | Spodoptera frugiperda | 72F01* | 23.61# | 488 | X | X |
| Insecta | Coleoptera | Tribolium castaneum | TC003750 | 26.39 | 1175 | X | X |
| TC001653 | 30 | 486 | X | — | |||
| TC005011 | 51.49 | 212 | — | X | |||
aAll sequences can be downloaded from Ensembl Metazoa except those with an “*” that can be downloaded from LepidoDB.
bProtein sequence identity estimated using BLASTp except for those with an “#” estimated using ClustalW (see Materials and Methods).
Other than in the Drosophila genus, CAG has homologs in four Lepidoptera species and in one Coleoptera with sequence identities ranging from 51.5% to 22.2% (table 1). We could only detect TIRs flanking Heliconius melpomene HMEL010729 suggesting that the other identified homologs are not transposons but transposon-derived genes (see Materials and Methods).
Overall, our results show that CR genes are present both in holocentric, Lepidoptera, and in nonholocentric, Diptera and Coleoptera, species.
CAG Belongs to the CR Clade
We constructed a phylogenetic tree to find out where insect CENP-B homologs are located in the previously published phylogeny containing a representative set of pogo transposases and pogo-derived genes (Casola et al. 2008). Phylogenetic trees of the full sequence set containing nonmetazoan transposases and transposase-derived genes can be found in supplementary figures S2 and S3, Supplementary Material online (see Materials and Methods). Our tree recovers the two monophyletic clades in metazoans: CR and Jerky related (JR) (fig. 3). CAG is located in the CR clade, and as expected, its closest transposase is the D. melanogaster pogo. The closest non-Drosophila CAG homolog is Tribolium castaneum TC005011. Most of the other insect CENP-B homolog genes, including the already described S. frugiperda and H. armigera CENP-B homologs, also fell in the CR clade. Insect and mammalian CR proteins form subclades inside the CR clade (fig. 3). Other than between D. melanogaster_CAG and D. ananassae_GF13390 transposase-derived genes, synteny is also conserved among Sfru_72F01, Harmi_94B11_25, and Bombyx_ BGIBMGA013624 suggesting that at least two additional independent exaptations, besides the mammal and fission yeast exaptations reported by Casola et al (2008), have occurred.
Fig. 3.—

Phylogenetic distribution of pogo-related transposases and transposase-derived genes in metazoans. JR and CR indicate that the sequences belong to the JR clade and the CR clades, respectively. Filled-boxes depict pogo-related transposases and empty boxes depict transposase-derived genes. Numbers in the nodes show posterior probabilities (black) and bootstrap values (red). Shaded branches correspond to new CR proteins identified in this work and in d'Alençon et al 2011 (table 1) that have been incorporated to the previously published phylogeny (Casola et al 2008). Dotted lines represent branches not drawn to scale. Trees including nonmetazoans pogo-related transposases and transposase-derived genes are depicted in supplementary figures S2 and S3, Supplementary Material online.
Note that Helicon. melpomene homologs form two clusters, one in the JR clade and one in the CR clade, showing extensive sequence identity indicating that they are either recent duplications or miss-annotated transposons (fig. 3).
Pogo-like Transposases Have Been Recurrently Exapted into CR Proteins in Metazoans
In this work, we have identified CAG as the closest CR protein in the D. melanogaster genome. Similar to other CR proteins, CAG has originated from the domestication of a pogo transposase and might be functionally related to other CENP-B homologs as suggested by the conservation of three out of the four functional domains (fig. 1) and the GO enrichment analyses of CAG PPI network (fig. 2). Knowledge about the contribution of each particular domain to the overall functions of CR proteins is scarce (Okada et al. 2007; Lorenz et al. 2012). However, conservation of DBD domain appears to be particularly important because it has been demonstrated that binding of this domain is sufficient to promote chromatin assembly in humans (Okada et al. 2007). Both sequence identity and 3D structure prediction show that CAG has a highly conserved DBD domain (fig. 1).
Other than in D. melanogaster, we were also able to identify CR proteins in T. castaneum, which is also a nonholocentric insect, indicating that CR proteins are not restricted to holocentric insecta (table 1) (d’Alençon et al 2011). Insect CENP-B homologs do not form a single monophyletic clade: Most sequences are part of the CR clade and a few belong to the JR clade. Furthermore, insect and mammalian CR proteins form moderately supported subclades inside the CR clade (fig. 3). These results suggest that at least three independent domestications of pogo-like transposases into CR proteins have occurred in metazoans (fig. 3).
Pogo-like transposases might have a predisposition to be recruited as centromeric proteins because 1) their DBD might provide them with the intrinsic ability to interact with centromeric DNA, and/or 2) interaction with the centromere might be indirect through their interaction with other host proteins with this ability (Feschotte and Pritham 2007; Casola et al. 2008). Our results further support both hypotheses. All CR proteins described so far conserved their DBD suggesting that they all probably have the ability to directly bind to DNA (fig. 1A, table 1). In the case of CAG, indirect capacity to interact with DNA is also provided through its interaction with PCNA (Warbrick et al. 1998; Maga and Hubscher 2003) and other DNA binding proteins (supplementary table S1, Supplementary Material online).
Overall, our results suggest that the numerous TE exaptations already described might just be the tip of the iceberg, and highlight the role of TEs as raw material for the recurrent evolution of important cellular functions (Bowen and Jordan 2007; Sinzelle et al. 2009).
Materials and Methods
CAG Identification
Human CENP-B (P07199), fission yeast Abp1 (NP_596460), Cbh1 (CAB16408), Cbh2 (CAA19330.1), S. frugiperda CENP-B (72F01) (d’Alençon et al. 2011), and pogo transposase (S20478) were used as BLASTp queries against D. melanogaster nr protein database with BLOSUM45 scoring matrix, low-complexity region filter, and an e value cutoff of 1e-04. Significant hits were used in a reciprocal BLASTp search using these same parameters.
Domain Architecture Analysis
Hmmscan software implemented at HMMER 3.1b1 (Finn et al. 2011) was used to search for occurrences of the domains deposited in Pfam-A database in the protein sequences under study. This information was complemented with the structural data available for human CENP-B (PDBIDs: 1HLV, 1BW6, 1UFI) and fission yeast Abp-1 (PDBID: 1IUF). The experimentally determined molecular interactions reported by Warbrick et al. (1998), Giot et al. (2003), Guruharsha et al. (2011), and Irelan et al. (2001) that were accessed through the PSICQUIC web server (Aranda et al. 2011) and FlyBase (Marygold et al. 2013) were also taken into account.
CAG 3D Modeling
The crystal structure of human CENP-B DBD (PDB ID: 1HLV) was used as a template for the modeling of D. melanogaster CAG DBD. The first 145 aminoacids (aa) of CAG were aligned to the 131 aa contained in the crystalized human CENP-B DBD using a combination of a global alignment built with ClustalW and two local alignments of the HTH regions built with hmmalign software (Finn et al. 2011) and the pfam profiles CENP-B_N and HTH_Tnp_Tc5. The resulting alignment was manually refined taking into account the predicted secondary structure of CAG and the description of the secondary structure of the crystal. Several alignments were used as an input to build CAG DBD models using the automodel class in Modeller 9.7 (Eswar et al. 2007). The resulting models were evaluated taking into account their stereochemical properties calculated by PROCHECK (Laskowski et al. 1993) and their pseudoenergetic profiles and z-scores calculated by PROSA-II (Wiederstein and Sippl 2007). STAMP was used to visualize the superimposition of the models with human CENP-B (Russell and Barton 1992).
CAG PPI Network
A list of proteins with experimental evidence of direct interaction with CAG (Q7JR24) and human CENP-B (P07199) were retrieved from Drosophila Protein Interaction Map and the PSICQUIC web server (Aranda et al. 2011). Only those binary interactions obtained using experimental detection methods and interaction types “association”, “physical association,” or “direct interaction” were kept.
The PPI interaction network at neighborhood-1 of CAG and CENP-B was expanded to neighborhood-2 using both experimentally determined and predicted interactions deposited at Interolog Finder web server (Wiles et al. 2010). We retrieved a list of 1,413 interactions involving 842 proteins for CAG and a list of 1,665 interactions involving 1,174 proteins for CENP-B. Cytoscape was used to visualize the interactions and BinGO to assess the overrepresentation of GO terms (Maere et al. 2005). Those GO terms showing a Benjamin and Hochberg False Discovery Rate corrected P value <1e−10 in a hypogeometric statistical test were represented, together with their parent terms, in a hierachical layout. The intersection of the GO terms that were enriched in both CAG and CENP-B neighborhood was performed by comparing the generated output files.
Codon-Based Test of Purifying Selection
Coding sequences for nine CAG orthologus in Drosophila species were aligned by ClustalW (Thompson et al. 1994). Codon-based tests of selection analyses were conducted in MEGA5 (Hall 2013) using the Nei–Gojobori method (Nei and Gojobori 1986). All ambiguous positions were removed for each sequence pair. The average difference of synonymous and nonsynonymous substitutions per site was calculated. The variance of the difference was computed using the bootstrap method (500 replicates).
Identification of CAG Orthologs and CR Genes
BLASTp searches using CAG sequence as a query were performed against ensembl metazoa protein databases. Protein sequences of those hits showing an E value smaller than 10−4 and a protein sequence identity greater than 25% along at least 100 aa, were retrieved and used for further analyses. We then checked whether the identified proteins were reported as orthologs in ensembl metazoa compara, Genomicus, and OrthoDB Arthropods, and kept only the ones that were reported as orthologs in at least one of the three databases. The two previously described CR genes in S. frugiperda and H. armigera were also included in the analyses (d’Alençon et al 2011). For these two sequences percentage of protein sequence identity was estimated using ClustalW. We then confirmed that the sequences had not been annotated as TEs during their respective genome annotation projects (Tribolium Genome Sequencing Consortium 2008; Duan et al. 2010; Heliconius Genome Consortium 2012). To further confirm that these sequences correspond to host proteins and not to transposases, we searched for Terminal Inverted Repeats in the 5′ and 3′ 600 base-pair regions flanking the CDS. To this end, we performed local sequence alignments using the Smith–Waterman algorithm for all possible sliding windows of 24 bp in the 5′-flanking region and the reverse sequence of the 3′-flanking region. Drosophila melanogaster PogoR11 was used as a positive control.
Phylogenetic Analysis of pogo-Related Sequences
Global multiple sequence alignments were performed using MAFFT (L-INS-I algorithm) (Katoh and Standley 2013). Local alignment of the DBD was performed using hmmalign and the hidden markov models HTH_CENP-B_N and HTH_Tnp_Tc5. Both alignments were combined and manually curated to obtain a final multiple sequence alignment of 449 residues with a proportion of gaps of 15.90%. This alignment was used to reconstruct the phylogeny of the pogo-related transposases and transposase-derived genes. We estimated the maximum-likelihood (ML) tree using RAxML (Stamatakis 2014). We used the best-fit amino acid substitution matrix (LG) estimated by ProtTest 3 (Darriba et al. 2011) with a GAMMA model of rate heterogeneity and the ML estimate of alpha-parameter. The best tree out of 100 inferences was optimized and 100 bootstrap replicates were performed (supplementary fig. S2, Supplementary Material online). Because bootstrap support in most of the branches in the metazoan sequences were smaller than 70, we decided to perform an independent inference using a Bayesian approach. We constructed the phylogenetic tree with PhyloBayes using the LG empirical mixture model with a discrete gamma distribution with four categories where constant sites were removed. Two Markov chains were run in parallel with a subsampling frequency of 100 until convergence was reached (population effective size of 242, maximum difference of 0.105175, mean difference of 0.00381158) (supplementary fig. S3, Supplementary Material online). ETE Toolkit (Huerta-Cepas et al. 2010) was used to annotate and visualize phylogenetic trees.
Genomicus (Louis et al. 2013) was used to check for synteny conservation in the different subclades identified.
Supplementary Material
Supplementary table S1 and figures S1–S3 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank Barbara Negre and members of the González lab for critically reading the manuscript, Alex de Mendoza and Iñaki Ruiz-Trillo for advice on phylogenetic analyses, and Claudio Casola for sharing his data set of pogo transposases and pogo-derived protein sequences. J.G. is a Ramón y Cajal fellow (RYC-2010-07306). This work was supported by grants from the European Commission (Marie Curie CIG PCIG-GA-2011-293860) and from the Spanish Government (Fundamental Research Projects Grant BFU-2011-24397) awarded to J.G.
Literature Cited
- Aranda B, et al. PSICQUIC and PSISCORE: accessing and scoring molecular interactions. Nat Methods. 2011;8:528–529. doi: 10.1038/nmeth.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benchabane H, et al. Jerky/Earthbound facilitates cell-specific Wnt/Wingless signalling by modulating β-catenin-TCF activity. EMBO J. 2011;30:1444–1458. doi: 10.1038/emboj.2011.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouazoune K, Brehm A. dMi-2 chromatin binding and remodeling activities are regulated by dCK2 phosphorylation. J Biol Chem. 2005;280:41912–41920. doi: 10.1074/jbc.M507084200. [DOI] [PubMed] [Google Scholar]
- Bowen NJ, Jordan IK. Exaptation of protein coding sequences from transposable elements. Genome Dyn. 2007;3:147–162. doi: 10.1159/000107609. [DOI] [PubMed] [Google Scholar]
- Cam HP, Noma K, Ebina H, Levin HL, Grewal SIS. Host genome surveillance for retrotransposons by transposon-derived proteins. Nature. 2008;451:431–436. doi: 10.1038/nature06499. [DOI] [PubMed] [Google Scholar]
- Casola C, Hucks D, Feschotte C. Convergent domestication of pogo-like transposases into centromere-binding proteins in fission yeast and mammals. Mol Biol Evol. 2008;25:29–41. doi: 10.1093/molbev/msm221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua HN, Sung W-K, Wong L. Exploiting indirect neighbours and topological weight to predict protein function from protein-protein interactions. Bioinformatics. 2006;22:1623–1630. doi: 10.1093/bioinformatics/btl145. [DOI] [PubMed] [Google Scholar]
- d’Alençon E, et al. Characterization of a CENP-B homolog in the holocentric Lepidoptera Spodoptera frugiperda. Gene. 2011;485:91–101. doi: 10.1016/j.gene.2011.06.007. [DOI] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27:1164–1165. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deloger M, et al. Identification of expressed transposable element insertions in the sequenced genome of Drosophila melanogaster. Gene. 2009;439:55–62. doi: 10.1016/j.gene.2009.03.015. [DOI] [PubMed] [Google Scholar]
- Duan J, et al. SilkDB v2.0: a platform for silkworm (Bombyx mori) genome biology. Nucleic Acids Res. 2010;38:D453–D456. doi: 10.1093/nar/gkp801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N, et al. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2007;2:2.9.1–2.9.31. doi: 10.1002/0471140864.ps0209s50. [DOI] [PubMed] [Google Scholar]
- Fachinetti D, et al. A two-step mechanism for epigenetic specification of centromere identity and function. Nat Cell Biol. 2013;15:1056–1066. doi: 10.1038/ncb2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 2007;41:331–368. doi: 10.1146/annurev.genet.40.110405.090448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giot L, et al. A protein interaction map of Drosophila melanogaster. Science. 2003;302(5651):1727–1736. doi: 10.1126/science.1090289. [DOI] [PubMed] [Google Scholar]
- Guruharsha KG, et al. A protein complex network of Drosophila melanogaster. Cell. 2011;147(3):690–703. doi: 10.1016/j.cell.2011.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BG. Building phylogenetic trees from molecular data with MEGA. Mol Biol Evol. 2013;30:1229–1235. doi: 10.1093/molbev/mst012. [DOI] [PubMed] [Google Scholar]
- Heliconius Genome Consortium. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature. 2012;487:94–98. doi: 10.1038/nature11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J, Dopazo J, Gabaldón T. ETE: a python environment for tree exploration. BMC Bioinformatics. 2010;11:24. doi: 10.1186/1471-2105-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JR, et al. A microtubule interactome: complexes with roles in cell cycle and mitosis. PLoS Biol. 2008;6:e98. doi: 10.1371/journal.pbio.0060098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irelan JT, Gutkin GI, Clarke L. Functional redundancies, distinct localizations and interactions among three fission yeast homologs of centromere protein-B. Genetics. 2001;157:1191–1203. doi: 10.1093/genetics/157.3.1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DB, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- Lorenz DR, et al. CENP-B cooperates with Set1 in bidirectional transcriptional silencing and genome organization of retrotransposons. Mol Cell Biol. 2012;32:4215–4225. doi: 10.1128/MCB.00395-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis A, Muffato M, Roest Crollius H. Genomicus: five genome browsers for comparative genomics in eukaryota. Nucleic Acids Res. 2013;41:D700–D705. doi: 10.1093/nar/gks1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S, Heymans K, Kuiper M. BiNGO: a cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- Maga G, Hubscher U. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J Cell Sci. 2003;116:3051–3060. doi: 10.1242/jcs.00653. [DOI] [PubMed] [Google Scholar]
- Marshall OJ, Choo KHA. Putative CENP-B paralogues are not present at mammalian centromeres. Chromosoma. 2012;121:169–179. doi: 10.1007/s00412-011-0348-3. [DOI] [PubMed] [Google Scholar]
- Marygold SJ, et al. FlyBase: improvements to the bibliography. Nucleic Acids Res. 2013;41:D751–D757. doi: 10.1093/nar/gks1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negoua A, Rouault J-D, Chakir M, Capy P. Internal deletions of transposable elements: the case of Lemi elements. Genetica. 2013;141:369–379. doi: 10.1007/s10709-013-9736-3. [DOI] [PubMed] [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Okada T, et al. CENP-B controls centromere formation depending on the chromatin context. Cell. 2007;131:1287–1300. doi: 10.1016/j.cell.2007.10.045. [DOI] [PubMed] [Google Scholar]
- Russell RB, Barton GJ. Multiple protein sequence alignment from tertiary structure comparison: assignment of global and residue confidence levels. Proteins. 1992;14:309–323. doi: 10.1002/prot.340140216. [DOI] [PubMed] [Google Scholar]
- Sharan R, Ulitsky I, Shamir R. Network-based prediction of protein function. Mol Syst Biol. 2007;3:88. doi: 10.1038/msb4100129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinzelle L, Izsvák Z, Ivics Z. Molecular domestication of transposable elements: from detrimental parasites to useful host genes. Cell Mol Life Sci. 2009;66:1073–1093. doi: 10.1007/s00018-009-8376-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA. Tiggers and other DNA transposon fossils in the human genome. Proc Natl Acad Sci U S A. 1996;93:1443–1448. doi: 10.1073/pnas.93.4.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Pierre SE, Ponting L, Stefancsik R, McQuilton P, FlyBase Consortium FlyBase 102ő advanced approaches to interrogating FlyBase. Nucleic Acids Res. 2013;42:D780–D788. doi: 10.1093/nar/gkt1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanyon CA, et al. A Drosophila protein-interaction map centered on cell-cycle regulators. Genome Biol. 2004;5:R96. doi: 10.1186/gb-2004-5-12-r96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawaramoto MS, et al. Crystal structure of the human centromere protein B (CENP-B) dimerization domain at 1.65-A resolution. J Biol Chem. 2003;278:51454–1461. doi: 10.1074/jbc.M310388200. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titz B, Schlesner M, Uetz P. What do we learn from high-throughput protein interaction data? Expert Rev Proteomics. 2004;1:111–121. doi: 10.1586/14789450.1.1.111. [DOI] [PubMed] [Google Scholar]
- Tribolium Genome Sequencing Consortium. The genome of the model beetle and pest Tribolium castaneum. Nature. 2008;452:949–955. doi: 10.1038/nature06784. [DOI] [PubMed] [Google Scholar]
- Tudor M, Lobocka M, Goodell M, Pettitt J, O’Hare K. The pogo transposable element family of Drosophila melanogaster. Mol Gen Genet. 1992;232:126–134. doi: 10.1007/BF00299145. [DOI] [PubMed] [Google Scholar]
- Wang H, Hartswood E, Finnegan DJ. Pogo transposase contains a putative helix-turn-helix DNA binding domain that recognises a 12 bp sequence within the terminal inverted repeats. Nucleic Acids Res. 1999;27:455–461. doi: 10.1093/nar/27.2.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warbrick E, Heatherington W, Lane DP, Glover DM. PCNA binding proteins in Drosophila melanogaster: the analysis of a conserved PCNA binding domain. Nucleic Acids Res. 1998;26:3925–3932. doi: 10.1093/nar/26.17.3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiles AM, et al. Building and analyzing protein interactome networks by cross-species comparisons. BMC Syst Biol. 2010;4:36. doi: 10.1186/1752-0509-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaratiegui M, et al. CENP-B preserves genome integrity at replication forks paused by retrotransposon LTR. Nature. 2011;469:112–115. doi: 10.1038/nature09608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.