

Background: Canonical histones are regulated by stem-loop-binding protein (SLBP) and are expressed during S phase.
Results: Arsenic decreases SLBP levels via epigenetic mechanisms, and histone mRNAs acquire a poly(A) tail.
Conclusion: Poly(A) tails allow for the presence of canonical histones outside of S phase.
Significance: These mechanisms may be involved in arsenic-induced carcinogenesis.
Keywords: Cell Cycle, Epigenetics, Histone, Metal, Polyadenylation, Arsenic, SLBP, Canonical Histone mRNA
Abstract
The replication-dependent histone genes are the only metazoan genes whose messenger RNA (mRNA) does not terminate with a poly(A) tail at the 3′-end. Instead, the histone mRNAs display a stem-loop structure at their 3′-end. Stem-loop-binding protein (SLBP) binds the stem-loop and regulates canonical histone mRNA metabolism. Here we report that exposure to arsenic, a carcinogenic metal, decreased cellular levels of SLBP by inducing its proteasomal degradation and inhibiting SLBP transcription via epigenetic mechanisms. Notably, arsenic exposure dramatically increased polyadenylation of canonical histone H3.1 mRNA possibly through down-regulation of SLBP expression. The polyadenylated H3.1 mRNA induced by arsenic was not susceptible to normal degradation that occurs at the end of S phase, resulting in continued presence into mitosis, increased total H3.1 mRNA, and increased H3 protein levels. Excess expression of canonical histones have been shown to increase sensitivity to DNA damage as well as increase the frequency of missing chromosomes and induce genomic instability. Thus, polyadenylation of canonical histone mRNA following arsenic exposure may contribute to arsenic-induced carcinogenesis.
Introduction
Arsenic is a well established human carcinogen, and even today millions of people are exposed to dangerous levels of naturally occurring arsenic in drinking water throughout the world (1). Exposure to a carcinogenic metal, such as arsenic, has been associated with altered gene expression patterns both in vitro and in vivo, and it has been demonstrated that the arsenic-induced changes in gene expression may be epigenetically regulated (2–9). Although arsenic is capable of inducing changes in gene expression by altering the levels of post-translational histone modifications in the promoters of genes, alterations in the expression of the histone genes themselves by arsenic has not been studied. Addressing this question may provide insight into arsenic-induced carcinogenesis because alterations in histone stoichiometry contribute to genomic instability. Histone genes are highly conserved (10) and can be classified into three distinct groups: 1) replication-dependent genes, which encode the canonical histones, whose expression is limited to the S phase of the cell cycle, such as the histone H3.1; 2) replication-independent genes, which encode the so-called “replacement histones” that are expressed at low but constant levels throughout the cell cycle and in non-dividing differentiated cells, such as the histone variant H3.3; and 3) genes encoding tissue-specific isotypes, such as the exclusively testicularly expressed mammalian H1t and H3t (11).
Interestingly, the canonical histone genes are the only metazoan genes whose messenger RNA (mRNA) does not terminate at the 3′-end with a poly(A) tail. Instead of a poly(A) tail, canonical histones contain a conserved 26-nucleotide sequence that can form a stem-loop structure, consisting of a four-nucleotide loop and a six-base pair stem. These 26 highly conserved nucleotides are the binding site for the stem-loop-binding protein (SLBP)4 (10). SLBP is critical for canonical histone pre-mRNA processing and histone mRNA translation (12) and is a major mediator of histone mRNA stability (13, 14). During the normal transcription of canonical histone genes, the stem-loop structure at the 3′-UTR end of mRNAs and numerous associated proteins, i.e. SLBP, U7 RNA of the U7 small nuclear ribonucleoprotein, will form a stable complex, which will subsequently assemble with other additional factors including heat-labile factor and hypothetical cleavage factor, resulting in the cleavage of the pre-mRNA (10). The SLBP accompanies the mature histone mRNA into the cytoplasm as a component of the histone messenger ribonucleoprotein particle where it participates in efficient translation of histone mRNA (15). Mutations or depletion of SLBP can result in misprocessing of the canonical histone mRNA, leading to the aberrant expression of polyadenylated mRNA from each of the five histone genes (16–18).
SLBP is dynamic and cell cycle-regulated. SLBP accumulates and is depleted in parallel to histone mRNAs; they rapidly increase at the G1/S phase transition, remain high throughout S phase, and begin to fall dramatically at the end of S phase (19). Translation regulation and proteasomal degradation function cooperatively to maintain very low SLBP levels in G1 phase, and these events are independent of the proteasomal degradation pathway mediating SLBP degradation at the S/G2 transition (20). Although several studies have shown the post-translational mechanisms of SLBP depletion, the mechanism underlying transcriptional regulation of SLBP expression is not clear.
Although arsenic is considered a weak mutagen, many studies demonstrate that arsenic exerts its toxicity and carcinogenicity primarily through epigenetic mechanisms. Arsenic has been demonstrated to silence genes by increasing DNA methylation and altering histone modifications at the promoter of genes (21, 22). The mechanisms underlying arsenic-induced epigenetic alterations are vague, but various investigations have reported on the effect of arsenic on a number of epigenetic enzymes, such as EZH2 (23, 24) and G9a (25). In this study, we identified a new response to arsenic exposure causing the depletion of SLBP via epigenetic regulation at its promoter and enhanced proteasomal degradation. This effect subsequently led to a dramatic increase in polyadenylated H3.1 mRNA, resulting in increased total H3.1 mRNA and H3 protein levels and the presence of H3.1 transcripts in M phase of the cell cycle. To our knowledge, this is the first study to report the aberrant polyadenylation of canonical histone mRNA by a human carcinogen.
EXPERIMENTAL PROCEDURES
Peripheral Blood Mononuclear Cell (PBMC) Sample Collection
Blood samples for PBMCs were obtained by venipuncture from a volunteer. Blood was collected into heparin-containing Vacutainer tubes, and PBMCs were isolated using a standard Ficoll-Hypaque separation technique. After isolation, PBMCs were plated at a density of 5 × 105/ml in 10-cm2 culture dishes and allowed to incubate overnight prior to treatment.
Cell Culture and Transformation
BEAS-2 B (human bronchial epithelial cells) and A549 (adenocarcinomic human alveolar basal epithelial cells) were cultured in DMEM containing 10% inactivated FBS and 1% penicillin-streptomycin. PBMCs and BL41(Burkitt lymphoma cells) were cultured in RPMI 1640 medium containing 10% inactivated FBS, 1% penicillin-streptomycin, and 1% glutamate. All cells were cultured at 37 °C in an incubator with a humidified atmosphere containing 5% CO2, 95% air. BL41 and PBMCs were plated at 5 × 105/ml in 10-cm2 culture dishes; BEAS-2B and A549 cells were plated at 2.5 × 105/ml in 10-cm2 culture dishes. For arsenic exposure, cells were treated with sodium meta-arsenite (NaAsO2) with doses ranging from 0 to 1 μm for 48 h. BEAS-2B, arsenic-transformed BEAS-2B, and Cr(VI)-transformed clones (26, 27), respectively, were grown in DMEM containing 10% FBS and 1% penicillin-streptomycin. Transformation assays were conducted as described previously (28, 29). BEAS-2B cells obtained from transformed and non-transformed arsenic clones were plated and cultured for 24 h without arsenic. To inhibit proteasome activity, BL41 and BEAS-2B cells were co-cultured with MG-132 (Calbiochem/EMD) (0.1 μm) and 0–1 μm sodium arsenite for 48 h. To inhibit DNA methyltransferase I activity, cells were treated with10 μm o5-aza-2′-deoxycytidine for 48 h after an initial 48 h exposure of 1 μm arsenic. To inhibit histone deacetylase activity, cells were treated with 5 mm sodium butyrate for 16 h after an initial 48-h exposure to 1 μm arsenic.
RNA Isolation, Amplification, and Hybridization for Gene Expression
Total RNA was extracted from each sample using TRIzol (Invitrogen) according to the manufacturer's protocol. cRNA probes were synthesized and labeled using the GeneChip Whole Transcript Terminal Labeling Expression kit (Affymetrix) and subjected to hybridization with the GeneChip Human Gene 1.0 ST Array (Affymetrix), which contains 28,869 well annotated genes. Hybridization and scanning of the arrays were performed using a standard procedure.
Data Analysis to Identify Differentially Expressed Genes
Microarray data analysis was performed as described previously (28). Briefly, the data analysis was performed using GeneSpring v12.0 (Agilent Technologies). The expression value of each probe set was determined after quantile normalization using the robust multiarray average algorithm and baseline transformation to the median levels of control samples. Differentially expressed genes were identified using one-way analysis of variance (p < 0.05). Functional annotation was analyzed with the gene ontology classification system using DAVID software.
Real Time Quantitative PCR
Total RNA was extracted from each cell line using TRIzol (Invitrogen) and converted to single-stranded cDNA using the SuperScript III First-Strand Synthesis System (Invitrogen) according to the manufacturer's instruction with oligo(dT) primers used to examine the polyadenylation status of the histone genes. Quantitative real time PCR analysis was performed using Power SYBR Green PCR Master Mix (Qiagen) on the ABI PRISM 7900HT system. All PCRs were performed in triplicate. Relative gene expression levels were normalized to GAPDH or β-actin expression. The results are presented as -fold change to the level expressed in control cells. The SLBP primers used are as follows: forward, 5′-CAGTCTTGCCACAACTTCAATC-3′; reverse, 5′-ATGGAGCCGATTATGAGAACAC-3′.
Northern Blotting
BEAS-2B cells were treated with 1 μm arsenic for 48 h. Total RNA was extracted using TRIzol reagent (Ambion) following the manufacturer's protocol. Polyadenylated (poly(A)+) mRNA was purified from 75 μg of total RNA using the Dynabeads mRNA Purification kit (Ambion). The unbound flow-through fraction was also collected as poly(A)− mRNA. All obtained poly(A)+ mRNA, 10 μg of total RNA, and 10 μg of poly(A)− mRNA were separated on an agarose gel and transferred to a nylon membrane for Northern blotting. H3.1 cDNA was used as the probe.
The H3.1 cDNA was obtained by double digestion and gel extraction from pcDNA3.1-FLAG-H3.1 plasmid, which was previously constructed by our laboratory. 25 ng of H3.1 cDNA was then labeled with [α-32P]dCTP to generate DNA probe using the Random Primers DNA Labeling System (Invitrogen). The labeled DNA probe was purified using a Probe Quant G-50 microcolumn (Amersham Biosciences) and added to the membrane for hybridization using rapid hybridization buffer (Amersham Biosciences). The membrane was washed, UV cross-linked, and then exposed to a phosphorimaging screen. The phosphorimaging screen was finally scanned by Typhoon FLA 1000 (GE Healthcare), and the data were analyzed and quantitated by ImageQuant TL 1D v8.1 (GE Healthcare) and ImageJ software. All procedures were carried out according to the manufacturer's protocol.
Whole-cell Protein Extraction
Whole-cell lysates were extracted by incubating with ice-cold radioimmunoprecipitation assay buffer (50 mm Tris-HCl, pH 7.4, 1% Nonidet P-40, 0.25% sodium deoxycholate, 150 mm NaCl, 1 mm EDTA) supplemented with a protease inhibitor mixture for 20 min on ice followed by centrifugation at 14,000 × g for 15 min. The supernatant was collected as whole-cell lysate.
Western Blotting
The protein concentration was determined using Bio-Rad DC (detergent-compatible) protein assay, and 15 μg of total protein was separated by 15% SDS-PAGE and transferred to PVDF membranes. Immunoblotting was performed with anti-stem-loop-binding protein antibody, anti-histone 3 rabbit polyclonal antibody, anti-mouse monoclonal actin, secondary anti-rabbit HRP-conjugated antibody, and secondary anti-mouse HRP-conjugated antibody (Abcam). The detection was accomplished by chemical fluorescence following an ECL Western blotting protocol with Pierce ECL Western Blotting Substrate.
Sodium Butyrate and 5-Aza-2′-deoxycytidine Treatment
To test whether the reduction of SLBP mRNA level by arsenic treatment is regulated via epigenetic mechanisms, we treated BEAS-2B cells with an inhibitor of histone acetylation (sodium butyrate) or DNA methylation (5-aza-2′-deoxycytidine) after arsenic exposure. The BEAS-2B cells were first treated with 0 or 1 μm arsenic for 48 h and then treated with 5 mm sodium butyrate for 24 h or treated with 10 μm 5-aza-2′-deoxycytidine for a total of 48 h with a medium change after 24 h, respectively. The cells were collected, and total RNA was extracted for quantitative real time PCR (RT-qPCR) to monitor change in the SLBP mRNA level. The SLBP primers are listed above. γ-Tubulin was used as internal control, and the primers used are as follows: forward, 5′-CGGCTGAATGACAGGTATCCTA-3′; reverse, 5′-CTCGTCCTGGTTGGGAAACA-3′.
Chromatin Immunoprecipitation
Mono- and dinucleosomes were purified by sucrose gradient from arsenic-treated and untreated BEAS-2B cells as described (29). ChIP was performed using H3K4 trimethylation (H3K4me3) antibody (Millipore 07-473) and H3K27 trimethylation (H3K27me3) antibody (Millipore 07-449). The primers used for qPCR are as follows: SLBP forward, 5′-GGGATGGTGCGGATCTACAG-3′; SLBP reverse, 5′-GTTGACTGGGTTTGTATCCTGAAGA-3′; γ-globin forward, 5′-TCTACCCATGGACCCAGAGGT-3′; γ-globin reverse, 5′-CCACATGCAGCTTGTCACAGT-3′.
SLBP Overexpression
Human embryonic kidney 293 (HEK293) cells were transfected with either an empty pcDNA-FLAG mammalian expression vector or a pcDNA-FLAG-SLBP vector using Lipofectamine reagent (Invitrogen). 24 h later, transfected cells were treated with NaAsO2 (Sigma) at the indicated concentrations for 48 h. At the end of the treatment, total proteins and total RNAs were collected from the cell samples for Western blotting and RT-PCR analyses, respectively.
Cell Synchronization
BEAS-2B cell synchronization was achieved by double thymidine block or nocodazole treatment. Cells were treated with 2 mm thymidine for 24 h, released in fresh DMEM for 14 h, and treated with 2 mm thymidine for another 24 h. The cells were then collected at G1/early S phase, released in fresh medium for 2 h at mid-S or released for 5 h at late S. To get mitosis-arrested cells, BEAS-2B cells were treated with 2 mm thymidine for 24 h, released in fresh DMEM for 3 h, and treated with 200 ng/ml nocodazole for another 16 h. The M phase-arrested cells were harvested by shake-off with gentle pipetting. Arsenic was added during the synchronization process to make sure the total treatment time was 48 h.
Statistical Procedures
Statistical significance was calculated using an unpaired, two-tailed t test with * indicating a p value less than 0.05 and ** indicating a p value less than 0.01.
RESULTS
Arsenic Increases Histone Cluster Gene Expression at Transcriptional and Protein Levels
Exposure to arsenic has been associated with altered gene expression in both in vitro and in vivo models (3, 4, 30, 31), and various studies have demonstrated that PMBCs collected from individuals exposed to arsenic exhibit marked changes in their gene expression (3, 4). Therefore, to mimic environmentally relevant doses of exposure and identify gene expression alterations, we exposed PBMCs to low doses of arsenic (0–1 μm) for 48 h and analyzed their gene expression profiles using 1.0 ST Affymetrix gene chips. 1 μm arsenic is equivalent to 163 ppb arsenic, and many individuals are exposed to much higher levels in Bangladesh, e.g. 500 ppb (32). A study by Arain et al. (33) shows that individuals exposed to drinking water contaminated with 53–175 ppb arsenic have blood arsenic levels between 2.39 and 7.66 μg/liter. The Agency for Toxic Substances and Disease Registry states that normal blood arsenic levels should be under 1 μg/liter. Unexpectedly, we observed that 90% of all histone genes were up-regulated by arsenic. Of all the up-regulated genes, 61 were histone genes, amounting to 6, 7, and 8% of the up-regulated genes at 0.1, 0.5, and 1 μm treatments, respectively (supplemental Table S1), that encode canonical histones. These histone genes were not only the most abundantly up-regulated set of genes, but they were also among the genes with the highest -fold increases (Fig. 1A).
FIGURE 1.

Canonical histone gene transcription and H3 protein levels are increased by arsenic. A, total RNA was extracted from PBMCs after a 48-h treatment with 0, 0.1, 0.5, or 1 μm arsenic. Representative Affymetrix microarray -fold changes in the expression of canonical, replication-dependent histone genes are shown. B and D–F, mRNA levels of H3.1 (B), MTG1 (D), HMOX1 (E), and granzyme K (GMZK) (F) were analyzed by quantitative RT-qPCR. Relative mRNA levels were normalized to 18 S rRNA expression and are presented as -fold change to the level expressed in PBMCs. Data are mean ± S.D. (n = 3). C, BL41 cells were treated with 0, 0.1, 0.5, or 1 μm of arsenic for 48 h. Cells were lysed with radioimmune precipitation assay buffer, and whole-cell lysate was run on 15% SDS acrylamide gels. Upper panel, representative Western blot of H3 protein in BL41 cells. Lower panel, quantification of H3 protein levels in BL41. Relative protein levels were calculated based on band intensity measured with ImageJ software. Statistical significance was calculated using an unpaired, two-tailed t test with * indicating a p value less than 0.05 and ** indicating a p value less than 0.01. Error bars represent S.D.
To validate the results obtained from our microarray study, RT-qPCR was performed on a subset of four genes, including the H3.1 gene, which exhibited at least a 2-fold change in gene expression. Random primers were used to detect total mRNA. Three up-regulated genes (H3.1, MTG1, and HMOX1) and one down-regulated gene (GMZK) were chosen based on their levels of expression in the microarray study. Differential expression of these genes in either control or arsenic-treated samples as measured by RT-qPCR was in agreement with the microarray results (Fig. 1, B and D–F). Note the dose-dependent increase in H3.1 mRNA (Fig. 1B).
We next utilized a Burkitt lymphoma cell line, BL41, to further examine whether arsenic also changes histone protein levels. The BL41 cell line has widely been used in arsenic studies because of their increased sensitivity to arsenic treatment (34–43). Western blotting results show that H3 protein levels were increased by 1 μm arsenic treatment in BL41 cells (Fig. 1C). Moreover, an investigation in immortalized human bronchial epithelial BEAS-2B cells supported these findings by demonstrating that 0.5 and 1.0 μm arsenic treatments increased H3 protein levels in a dose-dependent manner (see Fig. 4A). We conclude that arsenic exposure increases histone mRNA and protein levels.
FIGURE 4.

SLBP protein levels are decreased by arsenic treatment. BEAS-2B and BL41 cells were treated with 0, 0.1, 0.5, or 1 μMNaAsO2 for 48 h, and BEAS-2B clones were transformed with NaAsO2. Cells were lysed with radioimmune precipitation assay buffer, and whole-cell lysate was run on 12% SDS acrylamide gels. A, representative Western blot of SLBP and H3 protein levels in BEAS-2B cells. Band intensities were quantified using Image J software. B, representative Western blot of SLBP protein levels in arsenic-transformed BEAS-2B clones and spontaneous clones. Band intensities were quantified using ImageJ software. Band intensities were pooled for control clones and arsenic-transformed clones to better visualize the dramatic decrease of SLBP levels in arsenic-transformed clones. C, representative Western blot of SLBP protein levels in BL41 cells. Band intensities were quantified using ImageJ software. Statistical significance was calculated using an unpaired, two-tailed t test with * indicating a p value less than 0.05 and ** indicating a p value less than 0.01. Error bars represent S.D. Contr., control.
Arsenic Causes an Increase in Polyadenylation of H3.1 mRNA
The increase in histone mRNA could be due to increased gene transcription or increased mRNA stability. Histone mRNAs do not contain a poly(A) tail. Poly(A) tails are a feature of mRNAs that increases the half-life and stability of the mRNA. The aberrant pre-mRNA processing results in an accumulation of polyadenylated histone mRNA, which may contribute to the observed increase in histone mRNA levels following arsenic exposure. To test the possibility, we used Northern blotting to measure the percentage of polyadenylated H3.1 mRNA in total H3.1 mRNA before and after arsenic exposure. BEAS-2B cells were utilized because of the known association of arsenic exposure and lung cancer (44) as well as the ability of this cell line to undergo malignant transformation after metal exposure (21, 22, 27, 45). Poly(A)+ mRNAs were purified from total RNA using poly(dT) beads, and the unbound fraction was designated as the poly(A)− fraction. Northern blotting was carried out using H3.1 cDNA as the probe. As shown in Fig. 2A, three bands were detected from total RNA substrate (lanes 2 and 5). The top band appears to be nonspecific because it was absent in both poly(A)+ (lanes 4 and 7) and poly(A)− fractions (lanes 3 and 6). The middle band and the bottom band represent H3.1 mRNA with and without a poly(A) tail, respectively, because they were detectable in poly(A)+ (lanes 4 and 7) and poly(A)− fractions (lanes 3 and 6), respectively. About 25% of H3.1 mRNA was already polyadenylated even in the absence of arsenic (lane 2). Notably, the amount of polyadenylated H3.1 mRNA was doubled following arsenic exposure (Fig. 2B).
FIGURE 2.

Polyadenylated H3.1 mRNA is doubled by arsenic treatment. A, Northern blot. Total RNA was prepared from BEAS-2B cells treated or untreated with 1 μm arsenic for 48 h. 75 μg was used to purify poly(A)+ mRNA using poly(dT) beads, and all purified polyadenylated mRNA was separated on the agarose gel (lanes 4 and 7). The unbound flow-through fraction was designated as poly(A)−. 10 μg of total mRNA and poly(A)+ fractions were also separated on the gel. Northern blot was carried out using H3.1 cDNA as the probe. The poly(A)− fractions in treated cells are decreased by arsenic exposure, whereas poly(A)+ fractions are increased (compare lanes 2 and 5). B, ImageJ quantification of the bands in lanes 2 and 5 of Fig. 2A. The bar graph shows relative quantification of polyadenylated H3.1 mRNA. The band intensities from lanes 2 and 5 in A were quantified using ImageJ software. The sum of poly(A)+ and poly(A)− in each lane was designated as “total” and set to 1, respectively. The percentage of the poly(A)+ fraction was calculated by dividing the intensity of the poly(A)+ band by each total. C, total mRNA and poly(A)+ mRNA were purified by using random and poly(dT) primers in a reverse transcription reaction, respectively. The levels of total H3.1 mRNA and poly(A)+ H3.1 mRNA were measured by qPCR and normalized by GAPDH. The total mRNA before and after the treatment was set to 1, respectively. D and E, total RNA was extracted from each cell line after a 48-h treatment with 0, 01, 0.5, or 1 μm NaAsO2. mRNA was then converted to cDNA using oligo(dT) primers. H3.1 polyadenylation was then measured by quantitative RT-PCR. Polyadenylation levels of H3.1 in PBMCs (D) and BL41 cells (E) are shown. The relative gene expression level was normalized to 18 S rRNA expression and is presented as -fold change to the level expressed in each cell line. Error bars represent S.D.
Next, we applied RT-qPCR to confirm the arsenic-induced increase of poly(A) H3.1 mRNA (Fig. 2C). During reverse transcription, random hexamers prime randomly along the RNAs, whereas oligo(dT) primers only prime at poly(A) tails of mRNA. Thus, utilizing PCR amplifications using cDNAs resulting from random hexamers and oligo(dT) primers, we were able to measure the relative amount of total mRNA and poly(A)+ mRNA, respectively. In agreement with the Northern blotting analysis, polyadenylated H3.1 mRNA was increased by arsenic exposure (Fig. 2C). Arsenic-induced up-regulation of polyadenylated H3.1 mRNA was confirmed in two other cell lines, BL41 (human lung carcinoma cells) and PBMCs (Fig. 2, D and E).
We next examined whether polyadenylation of histone mRNA is arsenic-specific. Whereas the marked increase in polyadenylation of H3.1 mRNA was observed in arsenic-transformed BEAS-2B clones, it was absent in Cr(VI)-transformed BEAS-2B clones, indicating that the aberrant polyadenylation may be specific to arsenic and not other metals (Fig. 3).
FIGURE 3.

Polyadenylation of H3.1 mRNA is specific for arsenic exposure. Total RNA was extracted from arsenic- and Cr(VI)-transformed BEAS-2B clones, clones spontaneously grown in agar, and parental BEAS-2B cells. mRNA was then converted to cDNA using oligo(dT) primers. H3.1 polyadenylation was then measured by quantitative RT-PCR. The relative gene expression level was normalized to 18 S rRNA expression and is presented as -fold change to the level expressed in BEAS-2B clones and parental cells or spontaneous control clones. A, polyadenylation of H3.1 in control and arsenic-transformed clones. C1–C3 are spontaneous control clones, and As1–As4 are arsenic-transformed clones. B, polyadenylation of H3.1 in control and Cr(VI)-transformed clones. P is a parental BEAS-2B cell, C1 is a spontaneous control clone, and Cr1 and Cr2 are Cr(VI)-transformed BEAS-2B clones. Statistical significance was calculated using an unpaired, two-tailed t test with * indicating a p value less than 0.05 and ** indicating a p value less than 0.01. Error bars represent S.D.
Arsenic Down-regulates SLBP Expression by Enhancing Its Proteasomal Degradation
Because SLBP is essential for canonical histone mRNA processing (47) and its depletion or mutation can result in an increase of aberrantly polyadenylated histone mRNA (16), we decided to investigate whether protein levels of SLBP are affected by arsenic exposure. A 48-h treatment with arsenic in BEAS-2B decreased SLBP (Fig. 4A), and this depletion was confirmed in arsenic-transformed BEAS-2B clones and BL41 cells (Fig. 4, B and C). SLBP can undergo proteasomal degradation when it becomes phosphorylated at Thr-60 and Thr-61 (48). To investigate whether increased proteasomal degradation of SLBP contributed to arsenic-induced depletion of SLBP, we treated BEAS-2B cells with arsenic in the presence of the proteasomal inhibitor MG-132. As expected the exposure to arsenic alone resulted in a decrease of SLBP protein levels at 1 μm. However, when BEAS-2B cells were treated with arsenic in the presence of 0.1 μm MG-132, the depletion of SLBP was markedly decreased at that same concentration of arsenic, indicating that arsenic treatment depletes SLBP by inducing its proteasomal degradation (Fig. 5).
FIGURE 5.

Proteasomal inhibitor restores arsenic-induced reduction of SLBP protein levels. BEAS-2B cells were treated with 0, 0.1, 0.5, and 1 μm arsenic and with 0.1 μm MG-132 (M) for 48 h. Cells were lysed with radioimmune precipitation assay buffer, and whole-cell lysate was run on 12% SDS acrylamide gels. SLBP protein levels were then measured with anti-SLBP antibody. A, representative Western blot of SLBP levels. B, quantification of SLBP protein levels. Relative protein levels were calculated based on band intensity measured with ImageJ software. Statistical significance was calculated using an unpaired, two-tailed t test with * indicating a p value less than 0.05, ** indicating a p value less than 0.01 for arsenic versus control (C), and # indicating a p value less than 0.5 for arsenic versus arsenic + MG-132. Error bars represent S.D.
Arsenic Depletes SLBP by Epigenetically Regulating SLBP mRNA Expression
To examine whether other mechanisms contributed to arsenic-induced reduction of SLBP, we investigated the effects of arsenic on SLBP mRNA. Interestingly, we found that the expression of mRNA for SLBP was decreased by 40% in BEAS-2B (Fig. 6, A and B).
FIGURE 6.
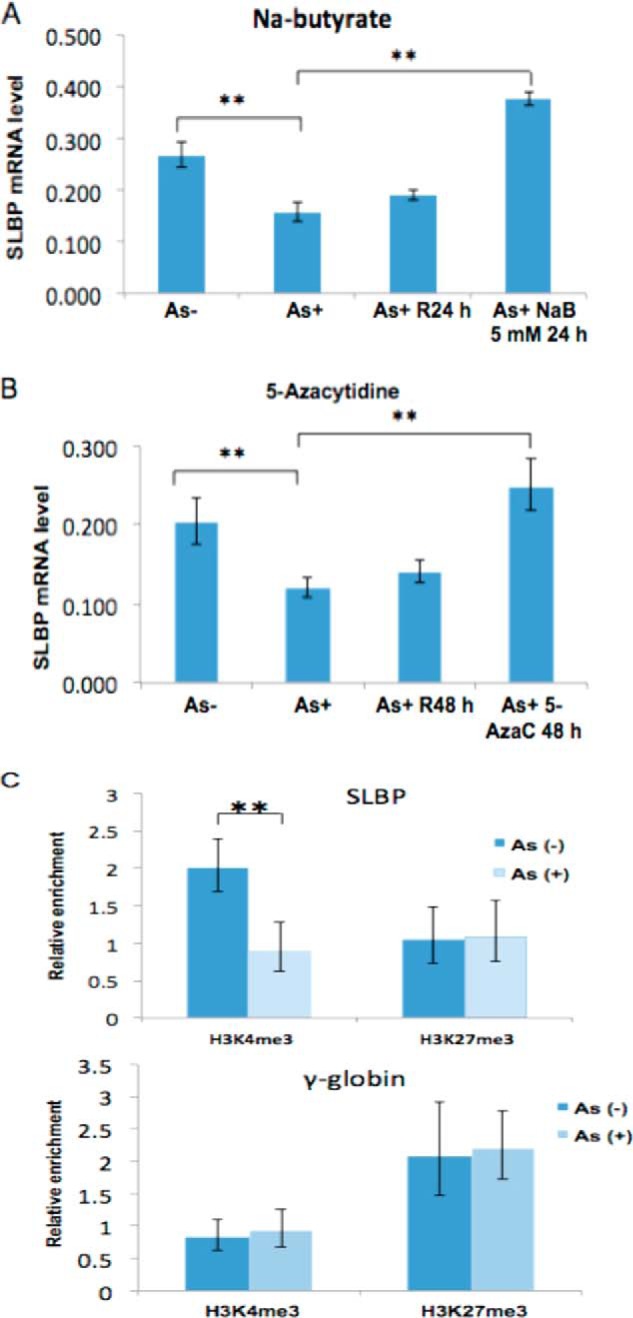
Arsenic down-regulates SLBP mRNA via epigenetic mechanisms. A, arsenic-induced reduction of the SLBP mRNA level was rescued by inhibitors of histone acetylation. After treating BEAS-2B cells with 1 μm arsenic for 48 h, the cells were recovered in arsenic-free medium, treated with or without 5 mm sodium butyrate (NaB) for 24 h, and subjected to RT-qPCR using SLBP primers. γ-Tubulin was used as an internal control. R24 h, cells cultured in regular medium without sodium butyrate. B, arsenic-induced reduction of the SLBP mRNA level was rescued by inhibitors of DNA methylation. After treating BEAS-2B cells with 1 μm arsenic for 48 h, the cells were recovered in arsenic-free medium treated with or without 10 μm 5-aza-2′-deoxycytidine for 48 h and subjected to RT-qPCR using SLBP primers. γ-Tubulin was used as an internal control. R48 h, cells cultured in regular medium without 5-aza-2′-deoxycytidine (AzaC). C, ChIP analysis of the SLBP promoter region. Mono- and dinucleosomes were purified by sucrose gradient from arsenic-treated and untreated BEAS-2B cells, and ChIP assays were performed. Levels of the active histone mark H3K4me3 at the SLBP promoter region were decreased by more than 50% after arsenic treatment, whereas H3K27me3 remained unchanged. γ-Globin was used as a negative control. Statistical significance was calculated using an unpaired, two-tailed t test with * indicating a p value less than 0.05 and ** indicating a p value less than 0.01. Error bars represent S.D.
Given that previous studies have shown that arsenic can alter histone modifications and DNA methylation at the promoters of genes, leading to gene silencing (22, 25, 49), we decided to investigate whether the arsenic-induced down-regulation of SLBP mRNA was epigenetically regulated. Fig. 6B shows that the arsenic-induced reduction of SLBP mRNA levels can be rescued by an inhibitor of histone deacetylases (sodium butyrate) or DNA methyltransferases (5-azacytidine), suggesting that the effect of arsenic on SLBP transcription is regulated by epigenetic mechanisms. This was further supported by ChIP assays. In the presence of arsenic, H3K4me3, an active transcriptional mark, was decreased by more than 50% around the SLBP promoter region (Fig. 6C). γ-Tubulin was used as internal control. No significant change in H3K27me3 levels, a mark associated with repressive transcription, was observed before and after arsenic treatment (Fig. 6C).
Overexpression of SLBP Prevents Arsenic-induced Polyadenylation of H3.1 mRNA
To further address the role of SLBP in arsenic-induced misprocessing of canonical histone mRNA, we investigated whether overexpression of SLBP could suppress the increase in polyadenylated H3.1 mRNA induced by arsenic exposure. Given their increased ability for transfection efficiency, HEK293 cells were utilized. Interestingly, overexpression of FLAG-tagged SLBP decreased the level of polyadenylated H3.1 mRNA even in the absence of arsenic treatment (Fig. 7A). In addition, treatment of cells with 0.5 μm arsenic resulted in the suppression of endogenous SLBP protein and an increase in H3.1 polyadenylation (Fig. 7). Notably, in the cells overexpressing FLAG-tagged SLBP, arsenic exposure failed to increase polyadenylated H3.1 mRNA (Fig. 7A), suggesting that SLBP plays a key role in the arsenic-induced polyadenylation of histone mRNA.
FIGURE 7.
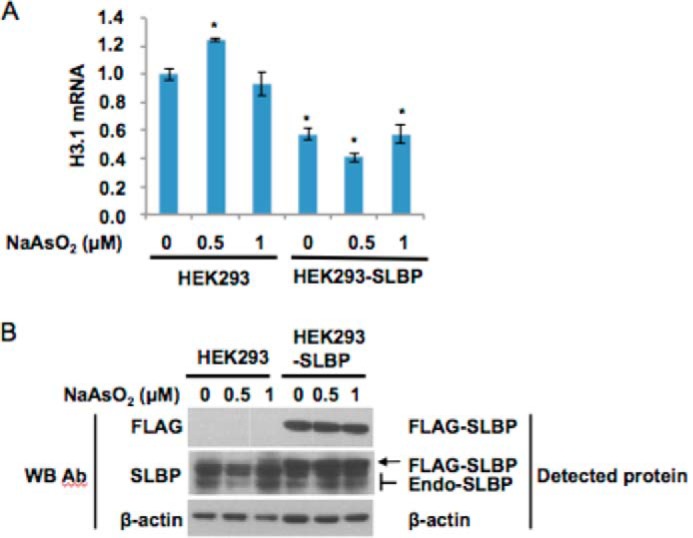
Overexpression of SLBP decreases H3.1 mRNA levels. HEK293 cells were transfected with the indicated plasmid vectors and treated as described under “Experimental Procedures.” A, real time RT-PCR was performed, and relative histone H3.1 poly(A) mRNA levels were calculated as described under “Experimental Procedures” and plotted as mean ± S.E. Statistical significance (compared with the untransfected and untreated sample; p < 0.05, Student's t test) are indicated with an asterisk. Error bars represent S.D. B, Western blot (WB) showing SLBP levels in transfected cells versus controls. Western blotting was conducted as described under “Experimental Procedures.” Endo-SLBP, endogenous SLBP; Ab, antibody.
Polyadenylated H3.1 mRNA Exists to a Greater Extent Outside of S Phase
The polyadenylated canonical histone mRNA is not susceptible to normal degradation that occurs at the end of S phase, which may result in the presence of a greater amount of canonical histone mRNA outside of S phase. This may have a significant impact on cellular processes because a failure to repress histone expression following DNA replication is highly toxic due to abnormal chromosome segregation in mitosis (50). To test the possibility that arsenic-induced polyadenylation of H3.1 mRNA allows for its presence outside of S phase, we synchronized BEAS-2B cells in the presence or absence of 1 μm arsenic for 48 h. Cell cycle phase was analyzed by propidium iodide staining using flow cytometry (not shown). RT-qPCR was used to measure the levels of polyadenylated mRNA. Arsenic exposure decreased mRNA for SLBP in the G1/early S and mid-S phases (Fig. 8A), and this was accompanied by increased polyadenylated H3.1 mRNA in the mid- and later parts of S phase (Fig. 8B). Importantly, the levels of polyadenylated H3.1 mRNA were increased almost 3-fold during mitosis by arsenic exposure as compared with the control (Fig. 8B).
FIGURE 8.
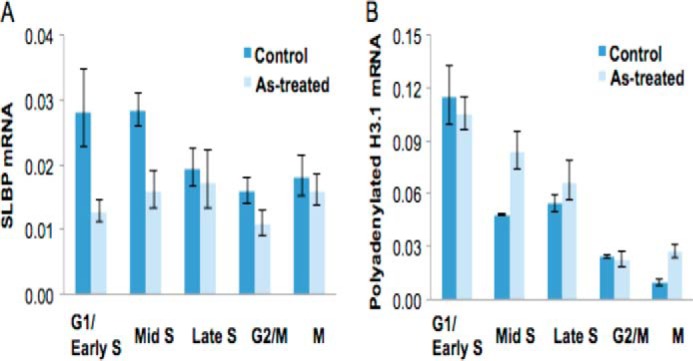
Polyadenylated H3.1 mRNA exists outside of S phase. BEAS-2B cells were treated with (light blue) or without (blue) 1 μm arsenic for 48 h, and cells were synchronized by double thymidine block at G1/early S, mid-S, and late S phases; thymidine-nocodazole treatment was used for arrest in mitosis (M). Total RNAs were isolated and reverse transcribed into cDNA by poly(dT) primers followed by qPCR to measure mRNA levels for SLBP and H3.1. Tubulin expression was used as an internal control. A, SLBP mRNA levels in synchronized BEAS-2B cells treated with arsenic. B, polyadenylated H3.1 mRNA levels in synchronized BEAS-2B cells treated with arsenic. Error bars represent S.D.
DISCUSSION
This is the first study to describe aberrant polyadenylation of H3.1 mRNA as a result of a loss of SLBP induced by arsenic exposure. Arsenic ultimately works to deplete SLBP at the mRNA and protein levels by inducing epigenetic changes at the SLBP promoter and promoting its proteasomal degradation. These events subsequently lead to an increase in H3.1 polyadenylated mRNA and H3 protein levels and the presence of H3.1 mRNA and probably other canonical histone mRNAs outside of S phase.
Arsenic-induced Increase in Histone Gene Expression and Polyadenylation of Canonical Histone mRNA
Microarray analysis of arsenic-treated PBMCs displayed a remarkable increase in 61 histone genes. Arsenic has been demonstrated to affect the global landscape of histone modifications in the cell (1) as well as alter the expression of histone modifying enzymes such as EZH2 (23); however, this is the first time arsenic has been reported to increase the expression of the histone genes themselves and remarkably on a large scale.
We discovered that the observed increase in canonical histone gene expression was at least in part due to the aberrant polyadenylation of canonical histone mRNAs, and this was confirmed in a number of cell lines, such as BEAS-2B, arsenic-transformed BEAS-2B, BL41, and PBMC (Fig. 2). Although the arsenic-induced increase of H3.1 poly(A) mRNA was evident, there still appears to be a great deal of H3.1 mRNA without a poly(A) tail after arsenic treatment. Moreover, 1 μm arsenic only partially depletes SLBP protein levels in BEAS-2B (Fig. 4A), indicating that H3.1 mRNA stability and translation are still being facilitated by SLBP to some extent after arsenic exposure. Lanzotti et al. (16) found that Drosophila embryos containing a mutation in the SLBP gene displayed alternative processing of only a portion of canonical histone transcripts, whereas some transcripts were still processed via a stem-loop structure. In this study, we observed a dose effect of arsenic on H3.1 poly(A) mRNA and SLBP protein levels in several cell lines; perhaps higher arsenic doses could completely abolish SLBP processing of H3.1 mRNA. Although many H3.1 transcripts are still processed via SLBP after arsenic exposure, the arsenic-induced increase in H3.1 mRNA is still likely to have a large impact on the cell given that the poly(A) tail increases translation and stability of transcripts. Also, the poly(A) tail will allow the mRNA to bypass normal degradation processes for canonical histone mRNAs that occur at the end of S phase, which will provide for its presence outside of S phase as seen in Fig. 8.
Polyadenylated H3.1 mRNA was detected at significantly high levels after a 48-h arsenic treatment in the cancer cell line BL41, but a smaller increase was observed in the non-cancerous human PBMCs (Fig. 2D). Given the tendency of cancer cells to quickly adapt to perturbations in its environment, the cancer cell lines are likely to be better poised than normal PBMCs to promptly respond to loss of a critical protein, such as SLBP, which facilitates cellular division. Polyadenylated H3.1 mRNA (Fig. 2D) was not as high as total H3.1 mRNA (transcripts with or without a poly(A) tail) (Fig. 1B) in arsenic-treated PBMCs. This alludes to the possibility that arsenic may be affecting H3.1 on the transcriptional level as well as the post-transcriptional level. Further studies are needed to verify what kind of mechanisms other than the addition of poly(A) tails are involved in the observed increase in canonical histone mRNAs in PBMCs.
Arsenic-transformed BEAS-2B clones contained high levels of polyadenylated H3.1 mRNA compared with control clones (Fig. 3A). Cr(VI)-transformed BEAS-2B clones did not display an increase in H3.1 poly(A) mRNA (Fig. 3B). The observance that Cr(VI)-transformed clones did not display poly(A) H3.1 mRNA indicates that Cr(VI) exposure is unlikely to induce depletion of SLBP. Thus, we postulate that the mode of action of arsenic to decrease SLBP levels and induce aberrant polyadenylation of H3.1 mRNA is likely due to a toxic effect specific to arsenic that is not observed in Cr(VI) toxicity.
Arsenic exposure also increases H3 protein levels in BL41 cells (Fig. 1C) as well as BEAS-2B cells (Fig. 4A). The increase likely occurred because of the aberrant polyadenylation of H3.1 mRNA. The highest dose of arsenic, 1 μm, resulted in the highest increase in H3 protein levels for both cell lines, and this dose also induced the most dramatic increase in polyadenylated H3.1 mRNA (Fig. 2, D and E). BEAS-2B cells treated with 0.5 μm arsenic also displayed a statistically significant increase in polyadenylated H3.1 mRNA. Polyadenylation of mRNA affords increased stability and translation compared with normal canonical histone processing. Because untreated BL41 cells maintained expression of SLBP (Fig. 4C) and contained only basal levels of polyadenylated H3.1 mRNA (Fig. 2E), H3.1 mRNA stability and translation were regulated by SLBP in control cells. Therefore, Figs. 1C, 2E, and 4C allow comparison of SLBP versus poly(A) regulation of H3.1 mRNA on the outcome of protein expression. The poly(A) tail increases the half-life of the mRNA by acting as a wall protecting the mRNA from degradation, whereas enzymes in the cytosol gradually degrade it until they reach the mRNA (51). The poly(A) tail also functions to enhance translation of the transcript by guiding the mRNA to the ribosome and promoting initiation of translation (51). Similar to a poly(A) tail, SLBP in conjunction with other RNA-binding proteins stabilize the transcript and facilitate translation (47); however, our results demonstrate that 3′ polyadenylation performs these functions more efficiently.
Possible Mechanisms of Polyadenylation of Canonical Histone mRNA
The mechanisms underlying poly(A) tail formation following the depletion of SLBP are unclear and require further investigation. After nuclear levels of SLBP have been depleted, the stem-loop structure is unbound and via an unknown mechanism is degraded, allowing the formation of the poly(A) tail. Normally, there are two proteins attached directly to the stem-loop structure, SLBP and 3′hExo, a 3′–5′ exoribonuclease, both of which bind cooperatively (52, 53). When one binds, the stem-loop conformation is changed, and there is an increased affinity for the other to bind. There are large conformational differences between the stem-loop in the complex versus that in free solution (52, 53). Future studies should investigate the possible role of 3′hExo in the degradation of the stem-loop structure prior to polyadenylation. Given the role of 3′hExo in the degradation of canonical histone mRNA at the end of S phase (52, 54), it is possible that this enzyme is being stimulated by the altered stem-loop conformation due to the absence of SLBP to degrade the stem-loop structure prior to polyadenylation.
Other hypotheses point toward the notion that polyadenylation occurs on the transcript due to downstream poly(A) signal sequences in the canonical histone genes. At least one canonical (AAUAAA) poly(A) signal sequence is present in the region 3′ of all five histone genes (16). It is unclear whether transcription through the poly(A) signal sequence also includes the stem-loop structure or whether the stem-loop sequence is skipped over by the RNA polymerase. Alternatively, the stem-loop structure could be spliced out of the mRNA post-transcriptionally.
Arsenic-induced Depletion of SLBP
Several studies have reported that polyadenylated canonical histone mRNAs occur in cells that have a mutation in the SLBP gene or have lost SLBP protein expression (10, 12, 16). Our study is in agreement with these observations by demonstrating that cell lines expressing polyadenylated H3.1 mRNA also display depleted SLBP protein levels. BEAS-2B, BL41, and arsenic-transformed BEAS-2B clones contained lower SLBP levels than their control counterparts (Fig. 4).
A decrease in SLBP mRNA occurred after arsenic exposure; however, the decrease in mRNA was not as dramatic as the decrease in SLBP protein levels, indicating that arsenic affects SLBP at both mRNA and protein levels. Our study demonstrates that arsenic-induced depletion of SLBP is mediated by proteasomal degradation (Fig. 5).
The mechanisms underlying arsenic-induced SLBP degradation are still unclear. Tang et al. (55) reported that arsenic inhibits CK2 activity, whereas an investigation by McNeely et al. (56) demonstrated that arsenic enhanced the activity of Cdk1. Degradation of promyelocytic leukemia protein is dependent on phosphorylation by CK2 (57); however, arsenic-induced degradation of promyelocytic leukemia protein occurs without CK2 phosphorylation (58). Based on these findings, it is possible that arsenic-induced SLBP degradation may be mediated by alternative mechanisms to CK2 that occur at an early stage of S phase either by phosphorylating Thr-60 prematurely or modifying another site on SLBP that signals degradation. The studies cited above indicate that arsenic influences the activity of the kinases that control SLBP degradation; whether the machinery is repressed or enhanced by arsenic may be different among cell types. It is worth noting here that phosphorylation may not be the only modification that influences SLBP degradation. Other modifications such as methylation, acetylation, and sumoylation have not been investigated, and future studies should investigate the possibility that these post-translational modifications play a role in SLBP degradation. In addition, arsenic has a propensity to affect methyl and acetyl groups on proteins, and this tendency may mediate arsenic-induced SLBP degradation.
Many studies have demonstrated that arsenic exerts its toxicity and carcinogenicity primarily through epigenetic mechanisms. In line with these reports, our study revealed that arsenic affects SLBP mRNA expression via epigenetic mechanisms (Fig. 6). Inhibition of histone deacetylase and DNA methyltransferase activity led to recovery of SLBP mRNA levels after arsenic treatment. Inhibition of these enzymes promotes an active epigenetic landscape at the promoter, such as decreased DNA methylation and the presence of acetylated histones. H3K4me3, an active transcriptional mark, was decreased at the SLBP promoter after arsenic treatment (Fig. 6C). Given the tendency of arsenic to affect epigenetic enzymes, future investigations should focus on the effect of arsenic on the epigenetic machinery that writes (KMT2A) and erases (KDM5b) H3K4me3. Our data also indicated that arsenic-induced hypermethylation at the SLBP gene is likely to have contributed to its silencing, which is not surprising considering the cross-talk between histone modifications and DNA methylation. Many reports have described promoter hypermethylation after arsenic treatment (21, 22), but the mechanisms are unclear. Regardless of the mechanism, we have demonstrated that the effects of arsenic on SLBP transcription are reversible, and protein levels can be restored.
An assay in which SLBP was overexpressed further supported the notion that SLBP plays a key role in arsenic-induced polyadenylation of histone mRNA. Interestingly, overexpression of FLAG-tagged SLBP decreased the level of polyadenylated H3.1 mRNA even in the absence of arsenic treatment (Fig. 7). The data are in agreement with the idea that in normal cells the amount of SLBP is not sufficient for its binding to every stem-loop region on histone mRNA, allowing cells to generate a small number of polyadenylated H3.1 mRNAs. This explains why we were always able to detect a certain amount of polyadenylated H3.1 mRNA without arsenic exposure.
Significance of the Presence of Polyadenylated Canonical Histone mRNA Outside of S Phase
Unlike normally processed canonical histone mRNAs, polyadenylated mRNAs are not degraded at the end of S phase (59, 60); thus, it was not surprising that we found polyadenylated H3.1 mRNA outside of S phase in arsenic-exposed BEAS-2B cells that were cell cycle-synchronized. The levels of polyadenylated H3.1 mRNA were increased almost 3-fold during mitosis by arsenic exposure as compared with the control. Although double thymidine block generally arrests cells at the G1/S border and makes it hard to obtain data about whether polyadenylated H3.1mRNA also exists in G1 phase, it is clear that polyadenylated H3.1 mRNA was present to a greater extent during mitosis. The continued presence of polyadenylated H3.1 transcripts after S phase is likely due to the association of the mRNA with different translation initiation factors. SLBP associates with CBP80/20-dependent translation complex, which allows for the rapid degradation of histone mRNA following inhibition of DNA replication, whereas poly(A) tails associate with eukaryotic translation initiation factor 4E, which does not couple degradation with cessation of DNA replication (61).
The presence of H3.1 outside of S phase may compete with variant histones for replication-independent nucleosome assembly. The histone variant H3.3 is constitutively expressed throughout the cell cycle and in quiescent cells. H3.3-containing nucleosomes perform specialized functions depending on their location (62). Its presence at promoters facilitates active chromatin complexes, allowing transcriptional machinery to bind to the promoter and activate other epigenetic changes associated with transcription (63–65). H3.3 seems to have an opposite effect when it is present at telomeres; it was shown that H3.3 is required for silencing of telomeric repeats (66). The primary structures of H3.3 and H3.1 are very similar with only five amino acid differences at positions 31, 87, 89, 90, and 96 (67). Given the structural similarities between H3.1 and H3.3 and the specialized roles H3.3 plays in the nucleosome, the presence of H3.1 outside of S phase may interfere with nucleosome remodeling involving H3.3 and consequently interfere with gene expression, cell cycle control, and genomic stability. Alterations in histone stoichiometry due to increased H3 protein levels may also contribute to the genomic instability (50). In fact, loss of SLBP in Drosophila causes genomic instability and impaired cellular proliferation (69). Future studies investigating mechanisms underlying arsenic-induced carcinogenesis should consider these possibilities. Chromatin remodeling pathways involving H3.3 are dysregulated in human cancers (70), and dysregulation of other histone variants may contribute to carcinogenesis (46). It should be noted here that other canonical histones besides H3.1 may be present outside of S phase due to arsenic exposure because other studies have reported that SLBP depletion leads to the aberrant expression of polyadenylated mRNA from each of the histone genes (16–18), and this study demonstrated an increase in expression of 61 histone genes following arsenic treatment in PBMCs. The presence of other canonical histone proteins along with H3.1 outside of S phase may further interfere with nucleosome assembly and chromatin remodeling and contribute to arsenic-induced carcinogenesis.
In conclusion, our work has identified a new response to arsenic exposure causing the depletion of SLBP via epigenetic regulation at the SLBP promoter and enhanced proteasomal degradation. This effect subsequently leads to polyadenylation of H3.1 mRNA, resulting in increased H3 protein levels and the presence of H3.1 transcripts outside of S phase. Our hypothesis is that H3.1 genes are initially transcribed with a normal stem-loop construct at the 3′-end of the mRNA. Arsenic depletes nuclear levels of SLBP, resulting in H3.1 transcripts not bound to SLBP. These unbound transcripts lose their stem-loop structure at the 3′-end and acquire a poly(A) tail, which increases the half-life and facilitates translation of the mRNA, two factors that provide for increased H3 protein levels. The poly(A) H3.1 mRNA is not susceptible to normal degradation that occurs at the end of S phase, allowing its presence in other phases of the cell cycle. Excess expression of canonical histones and/or their expression outside of S phase can increase sensitivity to DNA damage and increase the frequency of missing chromosomes, which lead to genomic instability (68). Although arsenic is classified as a class 1 human carcinogen, the mechanics behind arsenic-induced cancers are not fully understood. Future research should determine the roles polyadenylated canonical histone mRNAs play in arsenic-induced carcinogenesis.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants ES010344, ES014454, ES000260, ES022935, ES023174, and ES005512.

This article contains supplemental Table S1.
- SLBP
- stem-loop-binding protein
- PBMC
- peripheral blood mononuclear cell
- RT-qPCR
- quantitative real time PCR
- H3K4me3
- H3K4 trimethylation
- H3K27me3
- H3K27 trimethylation.
REFERENCES
- 1. Chervona Y., Hall M. N., Arita A., Wu F., Sun H., Tseng H. C., Ali E., Uddin M. N., Liu X., Zoroddu M. A., Gamble M. V., Costa M. (2012) Associations between arsenic exposure and global posttranslational histone modifications among adults in Bangladesh. Cancer Epidemiol. Biomarkers Prev. 21, 2252–2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Medeiros M., Zheng X., Novak P., Wnek S. M., Chyan V., Escudero-Lourdes C., Gandolfi A. J. (2012) Global gene expression changes in human urothelial cells exposed to low-level monomethylarsonous acid. Toxicology 291, 102–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andrew A. S., Jewell D. A., Mason R. A., Whitfield M. L., Moore J. H., Karagas M. R. (2008) Drinking-water arsenic exposure modulates gene expression in human lymphocytes from a U.S. population. Environ. Health Perspect. 116, 524–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Argos M., Kibriya M. G., Parvez F., Jasmine F., Rakibuz-Zaman M., Ahsan H. (2006) Gene expression profiles in peripheral lymphocytes by arsenic exposure and skin lesion status in a Bangladeshi population. Cancer Epidemiol. Biomarkers Prev. 15, 1367–1375 [DOI] [PubMed] [Google Scholar]
- 5. Stueckle T. A., Lu Y., Davis M. E., Wang L., Jiang B. H., Holaskova I., Schafer R., Barnett J. B., Rojanasakul Y. (2012) Chronic occupational exposure to arsenic induces carcinogenic gene signaling networks and neoplastic transformation in human lung epithelial cells. Toxicol. Appl. Pharmacol. 261, 204–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ahlborn G. J., Nelson G. M., Ward W. O., Knapp G., Allen J. W., Ouyang M., Roop B. C., Chen Y., O'Brien T., Kitchin K. T., Delker D. A. (2008) Dose response evaluation of gene expression profiles in the skin of K6/ODC mice exposed to sodium arsenite. Toxicol. Appl. Pharmacol. 227, 400–416 [DOI] [PubMed] [Google Scholar]
- 7. Jensen T. J., Wozniak R. J., Eblin K. E., Wnek S. M., Gandolfi A. J., Futscher B. W. (2009) Epigenetic mediated transcriptional activation of WNT5A participates in arsenical-associated malignant transformation. Toxicol. Appl. Pharmacol. 235, 39–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li J., Gorospe M., Barnes J., Liu Y. (2003) Tumor promoter arsenite stimulates histone H3 phosphoacetylation of proto-oncogenes c-fos and c-jun chromatin in human diploid fibroblasts. J. Biol. Chem. 278, 13183–13191 [DOI] [PubMed] [Google Scholar]
- 9. Barr F. D., Krohmer L. J., Hamilton J. W., Sheldon L. A. (2009) Disruption of histone modification and CARM1 recruitment by arsenic represses transcription at glucocorticoid receptor-regulated promoters. PLoS One 4, e6766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dominski Z., Marzluff W. F. (1999) Formation of the 3′ end of histone mRNA. Gene 239, 1–14 [DOI] [PubMed] [Google Scholar]
- 11. Albig W., Doenecke D. (1997) The human histone gene cluster at the D6S105 locus. Hum. Genet. 101, 284–294 [DOI] [PubMed] [Google Scholar]
- 12. Marzluff W. F. (2005) Metazoan replication-dependent histone mRNAs: a distinct set of RNA polymerase II transcripts. Curr. Opin. Cell Biol. 17, 274–280 [DOI] [PubMed] [Google Scholar]
- 13. Graves R. A., Pandey N. B., Chodchoy N., Marzluff W. F. (1987) Translation is required for regulation of histone mRNA degradation. Cell 48, 615–626 [DOI] [PubMed] [Google Scholar]
- 14. Kaygun H., Marzluff W. F. (2005) Translation termination is involved in histone mRNA degradation when DNA replication is inhibited. Mol. Cell. Biol. 25, 6879–6888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Whitfield M. L., Kaygun H., Erkmann J. A., Townley-Tilson W. H., Dominski Z., Marzluff W. F. (2004) SLBP is associated with histone mRNA on polyribosomes as a component of the histone mRNP. Nucleic Acids Res. 32, 4833–4842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lanzotti D. J., Kaygun H., Yang X., Duronio R. J., Marzluff W. F. (2002) Developmental control of histone mRNA and dSLBP synthesis during Drosophila embryogenesis and the role of dSLBP in histone mRNA 3′ end processing in vivo. Mol. Cell. Biol. 22, 2267–2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sullivan E., Santiago C., Parker E. D., Dominski Z., Yang X., Lanzotti D. J., Ingledue T. C., Marzluff W. F., Duronio R. J. (2001) Drosophila stem loop binding protein coordinates accumulation of mature histone mRNA with cell cycle progression. Genes Dev. 15, 173–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sullivan K. D., Steiniger M., Marzluff W. F. (2009) A core complex of CPSF73, CPSF100, and Symplekin may form two different cleavage factors for processing of poly(A) and histone mRNAs. Mol. Cell 34, 322–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rattray A. M., Nicholson P., Müller B. (2013) Replication stress-induced alternative mRNA splicing alters properties of the histone RNA-binding protein HBP/SLBP: a key factor in the control of histone gene expression. Biosci. Rep. 33, e00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Djakbarova U., Marzluff W. F., Köseoğlu M. M. (2014) Translation regulation and proteasome mediated degradation cooperate to keep stem-loop binding protein low in G1-phase. J. Cell. Biochem. 115, 523–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Arita A., Costa M. (2009) Epigenetics in metal carcinogenesis: nickel, arsenic, chromium and cadmium. Metallomics 1, 222–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brocato J., Costa M. (2013) Basic mechanics of DNA methylation and the unique landscape of the DNA methylome in metal-induced carcinogenesis. Crit. Rev. Toxicol. 43, 493–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hong G. M., Bain L. J. (2012) Sodium arsenite represses the expression of myogenin in C2C12 mouse myoblast cells through histone modifications and altered expression of Ezh2, Glp, and Igf-1. Toxicol. Appl. Pharmacol. 260, 250–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li L., Qiu P., Chen B., Lu Y., Wu K., Thakur C., Chang Q., Sun J., Chen F. (2014) Reactive oxygen species contribute to arsenic-induced EZH2 phosphorylation in human bronchial epithelial cells and lung cancer cells. Toxicol. Appl. Pharmacol. 276, 165–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhou X., Sun H., Ellen T. P., Chen H., Costa M. (2008) Arsenite alters global histone H3 methylation. Carcinogenesis 29, 1831–1836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Clancy H. A., Sun H., Passantino L., Kluz T., Muñoz A., Zavadil J., Costa M. (2012) Gene expression changes in human lung cells exposed to arsenic, chromium, nickel or vanadium indicate the first steps in cancer. Metallomics 4, 784–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sun H., Clancy H. A., Kluz T., Zavadil J., Costa M. (2011) Comparison of gene expression profiles in chromate transformed BEAS-2B cells. PLoS One 6, e17982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sun H., Shamy M., Kluz T., Muñoz A. B., Zhong M., Laulicht F., Alghamdi M. A., Khoder M. I., Chen L. C., Costa M. (2012) Gene expression profiling and pathway analysis of human bronchial epithelial cells exposed to airborne particulate matter collected from Saudi Arabia. Toxicol. Appl. Pharmacol. 265, 147–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen D., Fang L., Li H., Tang M. S., Jin C. (2013) Cigarette smoke component acrolein modulates chromatin assembly by inhibiting histone acetylation. J. Biol. Chem. 288, 21678–21687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Davey J. C., Bodwell J. E., Gosse J. A., Hamilton J. W. (2007) Arsenic as an endocrine disruptor: effects of arsenic on estrogen receptor-mediated gene expression in vivo and in cell culture. Toxicol. Sci. 98, 75–86 [DOI] [PubMed] [Google Scholar]
- 31. Martin-Chouly C., Morzadec C., Bonvalet M., Galibert M. D., Fardel O., Vernhet L. (2011) Inorganic arsenic alters expression of immune and stress response genes in activated primary human T lymphocytes. Mol. Immunol. 48, 956–965 [DOI] [PubMed] [Google Scholar]
- 32. Kile M. L., Houseman E. A., Baccarelli A. A., Quamruzzaman Q., Rahman M., Mostofa G., Cardenas A., Wright R. O., Christiani D. C. (2014) Effect of prenatal arsenic exposure on DNA methylation and leukocyte subpopulations in cord blood. Epigenetics 9, 774–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Arain M. B., Kazi T. G., Baig J. A., Afridi H. I., Sarajuddin, Brehman K. D., Panhwar H., Arain S. S. (2014) Co-exposure of arsenic and cadmium through drinking water and tobacco smoking: risk assessment on kidney dysfunction. Environ. Sci. Pollut. Res. Int. 10.1007/s11356-014-3339-0 [DOI] [PubMed] [Google Scholar]
- 34. Chen Y. J., Li H. M., Lu W., Qing C. (2013) Blocking effect of arsenic trioxide on the proliferation and cell cycle of human Burkitt lymphoma cells and its related mechanism. Zhongguo Shi Yan Xue Ye Xue Za Zhi 21, 1454–1459 [DOI] [PubMed] [Google Scholar]
- 35. He Y., Yang J. M., Wang J. M., Zhou H., Lü S. Q., Hu X. X. (2008) Synergistic effects of arsenic trioxide and proteasome inhibitor bortezomib on apoptosis induction in Raji cell line. Zhongguo Shi Yan Xue Ye Xue Za Zhi 16, 794–798 [PubMed] [Google Scholar]
- 36. Li H. M., Long Y., Qing C., Yu M., Li Z. H., Zhang X. M., Li X. J., Chen Y. J., Zhang Y. L., Liang Y. (2011) Arsenic trioxide induces apoptosis of Burkitt lymphoma cell lines through multiple apoptotic pathways and triggers antiangiogenesis. Oncol. Res. 19, 149–163 [DOI] [PubMed] [Google Scholar]
- 37. Lombardo T., Cavaliere V., Costantino S. N., Kornblihtt L., Alvarez E. M., Blanco G. A. (2012) Synergism between arsenite and proteasome inhibitor MG132 over cell death in myeloid leukaemic cells U937 and the induction of low levels of intracellular superoxide anion. Toxicol. Appl. Pharmacol. 258, 351–366 [DOI] [PubMed] [Google Scholar]
- 38. Lu D., Bai X. C., Gui L., Li M., Zheng W. S., Han X. Q., Luo S. Q. (2003) Arsenic trioxide-induced apoptosis of human malignant lymphoma cell lines and its mechanisms. Di Yi Jun Yi Da Xue Xue Bao 23, 997–1001 [PubMed] [Google Scholar]
- 39. Lu D., Bai X. C., Gui L., Su Y. C., Deng F., Liu B., Li X. M., Zeng W. S., Cheng B. L., Luo S. Q. (2004) Hydrogen peroxide in the Burkitt's lymphoma cell line Raji provides protection against arsenic trioxide-induced apoptosis via the phosphoinositide-3 kinase signalling pathway. Br. J. Haematol. 125, 512–520 [DOI] [PubMed] [Google Scholar]
- 40. Lu D., Bai X. C., Liu B., Li X. M., Deng F., Li M., Cheng B. L., Luo S. Q. (2004) Hydrogen peroxide inhibits arsenic trioxide-induced apoptosis of Burkitt lymphoma cells. Di Yi Jun Yi Da Xue Xue Bao 24, 375–378 [PubMed] [Google Scholar]
- 41. Muscarella D. E., Bloom S. E. (2002) Differential activation of the c-Jun N-terminal kinase pathway in arsenite-induced apoptosis and sensitization of chemically resistant compared to susceptible B-lymphoma cell lines. Toxicol. Sci. 68, 82–92 [DOI] [PubMed] [Google Scholar]
- 42. Shen L., Chen T. X., Wang Y. P., Lin Z., Zhao H. J., Zu Y. Z., Wu G., Ying D. M. (2000) As2O3 induces apoptosis of the human B lymphoma cell line MBC-1. J. Biol. Regul. Homeost. Agents 14, 116–119 [PubMed] [Google Scholar]
- 43. Zhu Q., Chen G., Huang Y. (2000) The relationship between sensitivity to arsenic trioxide and antioxidative capacity of malignant hematopoietic cells. Zhonghua Zhong Liu Za Zhi 22, 359–361 [PubMed] [Google Scholar]
- 44. Smith A. H., Ercumen A., Yuan Y., Steinmaus C. M. (2009) Increased lung cancer risks are similar whether arsenic is ingested or inhaled. J. Expo. Sci. Environ. Epidemiol. 19, 343–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Passantino L., Muñoz A. B., Costa M. (2013) Sodium metavanadate exhibits carcinogenic tendencies in vitro in immortalized human bronchial epithelial cells. Metallomics 5, 1357–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hake S. B., Xiao A., Allis C. D. (2007) Linking the epigenetic ‘language’ of covalent histone modifications to cancer. Br. J. Cancer 96, (suppl.) R31–R39 [PubMed] [Google Scholar]
- 47. Marzluff W. F., Wagner E. J., Duronio R. J. (2008) Metabolism and regulation of canonical histone mRNAs: life without a poly(A) tail. Nat. Rev. Genet. 9, 843–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zheng L., Dominski Z., Yang X. C., Elms P., Raska C. S., Borchers C. H., Marzluff W. F. (2003) Phosphorylation of stem-loop binding protein (SLBP) on two threonines triggers degradation of SLBP, the sole cell cycle-regulated factor required for regulation of histone mRNA processing, at the end of S phase. Mol. Cell. Biol. 23, 1590–1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Boellmann F., Zhang L., Clewell H. J., Schroth G. P., Kenyon E. M., Andersen M. E., Thomas R. S. (2010) Genome-wide analysis of DNA methylation and gene expression changes in the mouse lung following subchronic arsenate exposure. Toxicol. Sci. 117, 404–417 [DOI] [PubMed] [Google Scholar]
- 50. Meeks-Wagner D., Hartwell L. H. (1986) Normal stoichiometry of histone dimer sets is necessary for high fidelity of mitotic chromosome transmission. Cell 44, 43–52 [DOI] [PubMed] [Google Scholar]
- 51. Preiss T., Hentze M. W. (1998) Dual function of the messenger RNA cap structure in poly(A)-tail-promoted translation in yeast. Nature 392, 516–520 [DOI] [PubMed] [Google Scholar]
- 52. Jurado A. R., Tan D., Jiao X., Kiledjian M., Tong L. (2014) Structure and function of pre-mRNA 5′-end capping quality control and 3′-end processing. Biochemistry 53, 1882–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tan D., Marzluff W. F., Dominski Z., Tong L. (2013) Structure of histone mRNA stem-loop, human stem-loop binding protein, and 3′hExo ternary complex. Science 339, 318–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hoefig K. P., Rath N., Heinz G. A., Wolf C., Dameris J., Schepers A., Kremmer E., Ansel K. M., Heissmeyer V. (2013) Eri1 degrades the stem-loop of oligouridylated histone mRNAs to induce replication-dependent decay. Nat. Struct. Mol. Biol. 20, 73–81 [DOI] [PubMed] [Google Scholar]
- 55. Tang F., Liu G., He Z., Ma W. Y., Bode A. M., Dong Z. (2006) Arsenite inhibits p53 phosphorylation, DNA binding activity, and p53 target gene p21 expression in mouse epidermal JB6 cells. Mol. Carcinog. 45, 861–870 [DOI] [PubMed] [Google Scholar]
- 56. McNeely S. C., Taylor B. F., States J. C. (2008) Mitotic arrest-associated apoptosis induced by sodium arsenite in A375 melanoma cells is BUBR1-dependent. Toxicol. Appl. Pharmacol. 231, 61–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Scaglioni P. P., Yung T. M., Cai L. F., Erdjument-Bromage H., Kaufman A. J., Singh B., Teruya-Feldstein J., Tempst P., Pandolfi P. P. (2006) A CK2-dependent mechanism for degradation of the PML tumor suppressor. Cell 126, 269–283 [DOI] [PubMed] [Google Scholar]
- 58. Percherancier Y., Germain-Desprez D., Galisson F., Mascle X. H., Dianoux L., Estephan P., Chelbi-Alix M. K., Aubry M. (2009) Role of SUMO in RNF4-mediated promyelocytic leukemia protein (PML) degradation: sumoylation of PML and phospho-switch control of its SUMO binding domain dissected in living cells. J. Biol. Chem. 284, 16595–16608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Harris M. E., Böhni R., Schneiderman M. H., Ramamurthy L., Schümperli D., Marzluff W. F. (1991) Regulation of histone mRNA in the unperturbed cell cycle: evidence suggesting control at two posttranscriptional steps. Mol. Cell. Biol. 11, 2416–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Whitfield M. L., Zheng L. X., Baldwin A., Ohta T., Hurt M. M., Marzluff W. F. (2000) Stem-loop binding protein, the protein that binds the 3′ end of histone mRNA, is cell cycle regulated by both translational and posttranslational mechanisms. Mol. Cell. Biol. 20, 4188–4198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Choe J., Kim K. M., Park S., Lee Y. K., Song O. K., Kim M. K., Lee B. G., Song H. K., Kim Y. K. (2013) Rapid degradation of replication-dependent histone mRNAs largely occurs on mRNAs bound by nuclear cap-binding proteins 80 and 20. Nucleic Acids Res. 41, 1307–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Szenker E., Ray-Gallet D., Almouzni G. (2011) The double face of the histone variant H3.3. Cell Res. 21, 421–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chow C. M., Georgiou A., Szutorisz H., Maia e Silva A., Pombo A., Barahona I., Dargelos E., Canzonetta C., Dillon N. (2005) Variant histone H3.3 marks promoters of transcriptionally active genes during mammalian cell division. EMBO Rep. 6, 354–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Delbarre E., Jacobsen B. M., Reiner A. H., Sørensen A. L., Küntziger T., Collas P. (2010) Chromatin environment of histone variant H3.3 revealed by quantitative imaging and genome-scale chromatin and DNA immunoprecipitation. Mol. Biol. Cell 21, 1872–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schwartz B. E., Ahmad K. (2005) Transcriptional activation triggers deposition and removal of the histone variant H3.3. Genes Dev. 19, 804–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Goldberg A. D., Banaszynski L. A., Noh K. M., Lewis P. W., Elsaesser S. J., Stadler S., Dewell S., Law M., Guo X., Li X., Wen D., Chapgier A., DeKelver R. C., Miller J. C., Lee Y. L., Boydston E. A., Holmes M. C., Gregory P. D., Greally J. M., Rafii S., Yang C., Scambler P. J., Garrick D., Gibbons R. J., Higgs D. R., Cristea I. M., Urnov F. D., Zheng D., Allis C. D. (2010) Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140, 678–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hake S. B., Garcia B. A., Kauer M., Baker S. P., Shabanowitz J., Hunt D. F., Allis C. D. (2005) Serine 31 phosphorylation of histone variant H3.3 is specific to regions bordering centromeres in metaphase chromosomes. Proc. Natl. Acad. Sci. U.S.A. 102, 6344–6349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kurat C. F., Recht J., Radovani E., Durbic T., Andrews B., Fillingham J. (2014) Regulation of histone gene transcription in yeast. Cell. Mol. Life Sci. 71, 599–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Salzler H. R., Davidson J. M., Montgomery N. D., Duronio R. J. (2009) Loss of the histone pre-mRNA processing factor stem-loop binding protein in Drosophila causes genomic instability and impaired cellular proliferation. PLoS One 4, e8168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Schwartzentruber J., Korshunov A., Liu X. Y., Jones D. T., Pfaff E., Jacob K., Sturm D., Fontebasso A. M., Quang D. A., Tönjes M., Hovestadt V., Albrecht S., Kool M., Nantel A., Konermann C., Lindroth A., Jäger N., Rausch T., Ryzhova M., Korbel J. O., Hielscher T., Hauser P., Garami M., Klekner A., Bognar L., Ebinger M., Schuhmann M. U., Scheurlen W., Pekrun A., Frühwald M. C., Roggendorf W., Kramm C., Dürken M., Atkinson J., Lepage P., Montpetit A., Zakrzewska M., Zakrzewski K., Liberski P. P., Dong Z., Siegel P., Kulozik A. E., Zapatka M., Guha A., Malkin D., Felsberg J., Reifenberger G., von Deimling A., Ichimura K., Collins V. P., Witt H., Milde T., Witt O., Zhang C., Castelo-Branco P., Lichter P., Faury D., Tabori U., Plass C., Majewski J., Pfister S. M., Jabado N. (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.