

Background: Proteinase 3 activity is poorly controlled by physiological inhibitors, and its biological function is not well understood.
Results: We have designed irreversible phosphonate inhibitors based on structural differences between proteinase 3 and elastase.
Conclusion: They selectively inhibit proteinase 3 in biological fluids and can act as activity-based probes.
Significance: These inhibitors will help clarify proteinase 3 function.
Keywords: Inflammation, Neutrophil, Protease, Protease Inhibitor, Serine Protease, Activity-based Probes, Inflammatory Diseases, Proteinase 3 (Myeloblastin)
Abstract
The function of neutrophil protease 3 (PR3) is poorly understood despite of its role in autoimmune vasculitides and its possible involvement in cell apoptosis. This makes it different from its structural homologue neutrophil elastase (HNE). Endogenous inhibitors of human neutrophil serine proteases preferentially inhibit HNE and to a lesser extent, PR3. We constructed a single-residue mutant PR3 (I217R) to investigate the S4 subsite preferences of PR3 and HNE and used the best peptide substrate sequences to develop selective phosphonate inhibitors with the structure Ac-peptidylP(O-C6H4-4-Cl)2. The combination of a prolyl residue at P4 and an aspartyl residue at P2 was totally selective for PR3. We then synthesized N-terminally biotinylated peptidyl phosphonates to identify the PR3 in complex biological samples. These inhibitors resisted proteolytic degradation and rapidly inactivated PR3 in biological fluids such as inflammatory lung secretions and the urine of patients with bladder cancer. One of these inhibitors revealed intracellular PR3 in permeabilized neutrophils and on the surface of activated cells. They hardly inhibited PR3 bound to the surface of stimulated neutrophils despite their low molecular mass, suggesting that the conformation and reactivity of membrane-bound PR3 is altered. This finding is relevant for autoantibody binding and the subsequent activation of neutrophils in granulomatosis with polyangiitis (formerly Wegener disease). These are the first inhibitors that can be used as probes to monitor, detect, and control PR3 activity in a variety of inflammatory diseases.
Introduction
There are ∼100 active serine proteases with a trypsin/chymotrypsin-like fold that are encoded by distinct human genes, but the physiological substrates and in vivo function of most of them are still poorly characterized. Although they are potential therapeutic targets in a large number of diseases, only a few inhibitors, primarily those that interfere with the coagulation cascade (factor Xa, thrombin inhibitors), have been approved for clinical use (for review see Ref. 1). Human proteinase 3 (PR3)2 (EC 3.4.21.76) is a neutrophilic serine protease that shares many structural and functional characteristics with human neutrophil elastase (HNE) (EC 3.4.21.37) (2, 3). Large amounts of both proteases are stored intracellularly in so-called primary granules and contribute to the breakdown of extracellular matrix components in infectious and inflammatory diseases, especially those of the lung (4). PR3 has also been identified as the principal autoantigen in one clinical subtype of systemic autoimmune vasculitis, granulomatosis with polyangiitis (GPA) (formerly Wegener disease) (5–7). The PR3 in activated neutrophils with destabilized lysosomal membranes can induce apoptosis and hence accelerate their death in inflamed tissues (8). Unlike HNE, PR3 is also present in highly mobile secretory vesicles and is translocated to the outer plasma membrane under certain conditions of priming (9). Furthermore, very small amounts of PR3 are constitutively exposed on the outer surface of circulating neutrophils (10). This genetically determined constitutive distribution is a unique feature of human PR3 that may explain its function of autoantibody target in vasculitides (11). Naturally occurring inhibitors of PR3 in the extracellular compartment and blood plasma target HNE preferentially, which makes investigating and understanding its biological function particularly complex (12).
Peptidyldiphenyl phosphonate inhibitors are irreversible transition state inhibitors that form a tetrahedral adduct with the serine 195 residue (chymotrypsin numbering) of the catalytic triad (13, 14). They selectively inhibit serine proteases, are chemically stable in several buffers and in the plasma under acidic and neutral conditions, and are effective at low concentrations (15). They can also be used as activity-based probes for labeling serine proteases at the cell surface (16) and even within the cell when synthesized in a membrane-permeable form (17). These inhibitors, therefore, seem to be most appropriate for dissecting the intracellular and extracellular biological roles of enzymatically active PR3 whether free or membrane-bound. We and others have shown that the substrate binding site of PR3 extends on both side of the catalytic site and that the Asp residues at P2 and P2′ (nomenclature of Schechter and Berger (18)) are essential to obtain selectivity toward PR3 (19, 20). Our goal was to produce an inhibitor that was selective for PR3 and had a sequence that binds only to the nonprime subsites of the protease. Having an Asp at P2 is not sufficient to ensure a selective interaction with PR3; we therefore used the difference between the structures of the S4 subsites of PR3 and HNE to determine whether the cooperation between the S4 and the S2 subsites could provide inhibitors selective for PR3.
We designed a tetrapeptide to be the peptide moiety of a PR3-selective, irreversible, easy-to-handle chlorodiphenyl phosphonate inhibitor. This compound has proved to be a successful probe for detecting PR3 activity in biological samples or for visualizing and monitoring PR3 both inside cells and at the cell surface.
EXPERIMENTAL PROCEDURES
Production of proI217R
The proI217R mutant was produced in Sf9 insect cells using the pMT/BiP/proPR3/His vector as a matrix and two complementary primers (5′-ccaaggaatagactccttcgtgaggtggggatgtgcc-3′ and 5′-ggcacatccccacctcacgaaggagtctattccttgg-3′). The mutation was introduced using the QuikChange Lightning Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA), and its presence was checked by sequencing (MWG Biotech). We established a stable cell line using antibiotic selection, and the cells were cultured in Schneider medium supplemented with 10% fetal bovine serum. We used CuSO4 to induce synthesis of the protein, which was purified by affinity chromatography on a chelating Sepharose fast flow resin column (Amersham Biosciences), eluted with a gradient of imidazole. The proI217R was activated as described previously for recombinant wtPR3 and the K99L (19).
Synthesis of Peptidyl Phosphonate Inhibitors
The first step in the synthesis of the phosphonic analog of alanine was the preparation of tri(4-chlorophenyl)phosphite from 4-chlorophenol and phosphorus trichloride (21). Briefly, phosphorus trichloride (10 mmol) was added to 4-chlorophenol (30 mmol) dissolved in acetonitrile (50 ml), and the mixture was refluxed for 6 h. The volatile elements were removed in a vacuum, and the resulting crude phosphite, a yellow oil, was used directly in an amidoalkylation reaction. It was mixed with benzyl carbamate (12 mmol) and acetaldehyde (12 mmol) and refluxed in acetic acid for 3 h (Oleksyszyn and Powers (22)). The resulting carboxybenzyl-AlaP(O-C6H4-4-Cl)2 was a white solid, 56%; yield, purity after 56%; high resolution mass spectrometry calculated for (C22H20Cl2NO5P)H+, 480.0534; found, 480.0533; 1H NMR (CDCl3, ppm), δ 7.43-6.97 (m, 14H), 5.22-5.08 (m, 2H), 4.74-4.37 (m, 1H), 1.56 (dd, J = 18.2, 7.4 Hz, 3H); 31P NMR (CDCl3, ppm): δ 19.56 (s). The carboxybenzyl protecting group was then removed by incubation with 33% hydrobromic acid in acetic acid (2 h), and the volatile components were removed under reduced pressure. The product was crystallized from methanol/diethyl ether to give HBr*H2N-AlaP(OPh)2 as a white solid, 97%; high resolution mass spectrometry calculated for (C14H14Cl2NO3P)H+, 346.0167; found, 346.0172; 1H NMR (DMSO-d6, ppm): δ 8.85 (s, 3H), 7.57-7.44 (m, 4H), 7.36-7.16 (m, 4H), 4.45-4.24 (m, 1H), 1.55 (dd, J = 18.3, 7.2 Hz, 3H); 31P NMR (CDCl3, ppm): δ 16.49 (s).
The peptides were synthesized manually by the solid-phase method using Fmoc (fluoromethyloxycarbonyl chloride) chemistry. The following amino acid derivatives were used: Fmoc-Pro, Fmoc-Tyr(tBu), Fmoc-Asp(OtBu). The protected derivative of the C-terminal amino acid residue, Fmoc-Asp(OtBu), was attached to the 2-chlorotritylchloride resin (substitution of Cl− 1.46 meq/g) (Calbiochem) in the presence of an equimolar amount of diisopropylethylamine (based on the amino acid) under anhydrous conditions in dichloromethylene solution. Peptide chains were elongated in consecutive cycles of deprotection and coupling. They were deprotected with 20% piperidine in dimethylformamide (DMF)/n-methylpyrrolidone (1:1, v/v) plus 1% Triton X-100, whereas chain elongation used standard differential interference contrast/hydroxybenzotriazole chemistry; 3 eq of protected amino acid derivatives were used. The same method was used to couple biotin and fluorescent compounds (used as N-protected Fmoc derivatives). A 10-fold molar excess of N-acetylimidazole in DMF was used for N-terminal acetyl derivates. Bt-[PEG]66-Pro-Tyr-Asp-AlaP(O-C6H4-4-Cl)2 was synthesized by coupling the α-(9-fluorenylmethyloxycarbonyl)amino-ω-carboxyl poly(ethylene glycol) Fmoc-polyethylene glycol-66 (PEG66; Mr 3000 Da) to the amino group of Pro. The N-terminal biotin group was introduced stirring a 5-fold molar (1.8 mmol) excess of biotin and 1,3-diisopropylcarbidiimide in 2 ml of anhydrous DMSO for 6 h at 30 °C. The synthesized peptides were cleaved from the resin with trifluoroacetic acid/hexane/acetic acid (1:6:1, v/v/v).
Fully protected peptides were dissolved in DMF, and their carboxyl groups were activated with 1,3-diisopropylcarbidiimide to which were added HBr×H2N-AlaP(OPh)2 in DMF and diisopropylethylamine. The mixture was stirred for 6 h, and the DMF was evaporated off under reduced pressure. The resulting compound was suspended in trifluoroacetic acid/phenol/triisopropylsilane/H2O (88:5:2:5, v/v/v/v) for 2 h to remove side-chain protecting groups.
The crude peptides were purified by high performance liquid chromatography (HPLC) on a Beckman Gold System (Beckman) with an RP Kromasil-100, C8, 5 μm column (8 × 250 mm) (Knauer, Berlin, Germany). The solvent systems were 0.1% trifluoroacetic acid (A) and 80% acetonitrile in A (B). Either isocratic conditions or a linear gradient were applied (flow rate 3.0 ml/min, monitored at 226 nm). The purity of the synthesized peptides was checked on an RP Kromasil 100, C8, 5 μm column (4.6 × 250 mm) (Knauer). The peptides were eluted with a linear gradient of the above solvent system (10–90% B) for 30 min and a flow rate 1 ml/min, monitored at 226 nm. Mass spectrometric analysis was done on a MALDI MS (a Biflex III MALDI-TOF spectrometer, Bruker Daltonics, Ettlingen, Germany) using a CCA matrix.
Enzyme Studies
Free wtPR3, I217R, K99L, and HNE (Biocentrum, Krakow, Poland) were titrated with α-1-proteinase inhibitor (23). The specificity constants kcat/Km for fluorescence resonance energy transfer (FRET) substrates and para-nitroanilide (pNA) substrates synthesized by Genecust (Dudelange Luxembourg) were determined under first-order conditions (23). All assays were performed at 37 °C in 50 mm HEPES, 0.75 m NaCl, and 0.05% Nonidet P-40 (pH 7.4).
We measured the inactivation of proteases by substrate analog inhibitors in the presence of the substrate by competition for the enzyme binding site by the method of Tian and Tsou (24). Product formation in the presence of an irreversible inhibitor approaches an asymptote in this system, as described by: log([P∞] − [P]) = log[P∞] − 0.43A[Y]t, where [P∞] is the concentration of product formed at time approaching infinity, [P] is the concentration of product at time t, [Y] is the inhibitor concentration, and A is the apparent inhibition rate constant in the presence of the substrate. A is given by: A = k+o/(1 + K−1[S]), where k+o is the rate constant for association of the inhibitor with the enzyme, K−1 is the inverted Michaelis constant, and [S] is the substrate concentration. The apparent inhibition rate constant A is the slope of a plot of log([P∞] − [P]) against t, to give the second-order rate constant of inhibition k+o. The rates of inhibition of PR3 and HNE were measured in 50 mm HEPES, 0.75 m NaCl, and 0.05% Nonidet P-40 (pH = 7.4).
Identification of Cleavage Sites by Mass Spectrometry
HPLC-purified fragments of FRET substrates were diluted 20-fold in acetonitrile:water:formic acid (45:54.8:0.2) and analyzed by direct infusion in a 4-GHz MaXis ultra high resolution quadrupole-TOF mass spectrometer (Bruker Daltonics) equipped with an electrospray ion source. Spectra were acquired in positive ion MS mode over a 300–3000 m/z range with a nebulizer gas pressure of 0.3 bars. The drying gas flow was 4 liter/min, and the temperature was 180 °C. The acquisition rate was 1 Hz corresponding to spectra summations of 5494. External calibration was performed with the ESI-L Low Concentration Tuning Mix (Agilent Technologies). Electrospray ionization-high resolution mass spectrometry spectra were processed using DataAnalysis 3.1 software (Bruker Daltonics), and cleavage peptides were assigned using Paws Version 8.5.0.3 (ProteoMetrics, New York, NY).
Detection of PR3 in Biological Fluids
The volumes of cystic fibrosis (CF) sputa and 10× concentrated urines from patients with bladder cancer were adjusted to give a final PR3 concentration of 10–100 nm (4–40 ng). These samples were incubated with compound 5 (150 nm final) for 20 min at 37 °C in 50 mm HEPES, 0.75 m NaCl, and 0.05% Nonidet P-40 (pH 7.4). The reaction was stopped by adding 1 volume of 2× SDS reducing buffer and heating at 90 °C for 5 min. The components of the mixture were separated by SDS-PAGE, 12% SDS-polyacrylamide gel electrophoresis under denaturing conditions (4–40 ng of protein per lane). They were transferred to a nitrocellulose (Hybond)-ECL (Enhanced Chemiluminescence) membrane at 4 °C.
Extravidin-peroxidase Detection
Free sites on the membrane were blocked with 3% bovine serum albumin (BSA) in PBS (phosphate-buffered saline) or with 0.1% Tween in PBS for 90 min at room temperature (RT). Membranes were then given two quick washes with PBS, Tween 0.1% and incubated for 2 h at RT with extravidin horseradish peroxidase (HRP) (Sigma) (diluted 1/4000 in 3% BSA in PBS, Tween 0.1%). The extravidin-HRP-treated membrane was washed (3 × 10 min) with PBS, Tween 1% and then incubated with HRP substrate for 1 min. Reactive bands were identified by chemiluminescence (ECL kit).
Immunodetection
Free sites on the membranes were blocked by incubation with 5% nonfat dried milk in PBS, 0.1% Tween for 90 min at RT. They were washed twice with PBS, Tween 0.1% and incubated overnight with a rabbit primary anti-PR3 antibody (1:700) followed by a goat anti-rabbit IgG secondary antibody (1:7000). These membranes were then washed and processed as above.
ELISA
The wells of Nunc Maxisorp plates were coated with purified wtPR3 and HNE by incubating them with enzyme (0.05 μm in 100 μl PBS) at 37 °C for 1 h. The plates were washed 4 times with 200 μl of PBS, Tween 0.1%. Bt-[PEG]66-PYDAP(O-C6H4-4-Cl)2 (compound 9) (2.5 μm final) in 100 μl of HEPES buffer was added to each well, and the plates were incubated at 37 °C for 180 min. The wells were washed 4 times with 200 μl of PBS, Tween 0.1% and then saturated by adding 2% BSA in PBS (200 μl/well) and incubated for 1 h at 37 °C. The plates were washed again (4 × 200 μl PBS, Tween 0.1%) and incubated with extravidin-HRP (1/2000) or streptavidin-HRP (1/15000) in 0.1% Tween, 2% BSA in PBS for 2 h at RT. The plates were washed again and incubated with the chromogenic substrate ortho-phenylenediamine (Sigma). The reaction was stopped by adding H2SO4 (2 m), and the absorbance at 492 nm was measured.
Flow Cytometry and Confocal Microscopy
Neutrophils were isolated from heparinized peripheral blood samples from healthy volunteers who gave their informed consent (25). Purified quiescent neutrophils were suspended in PBS and fixed with 3.7% (v/v) paraformaldehyde (20 min at RT). They were washed twice with PBS and permeabilized with 0.5% (v/v) Triton X-100 (10 min at RT). Permeabilized cells were washed twice in PBS and then incubated for 20 min at RT with 10 μm Bt-[PEG]66-PYDAP(O-C6H4-4-Cl)2 (compound 9). These cells were then washed twice in PBS to remove unbound inhibitors, and free binding sites were blocked by incubation with 10% (w/v) BSA in PBS for 1 h. They were then incubated with Alexa Fluor® 488 streptavidin (Invitrogen, diluted 1/1000 in 3% (w/v) PBS) for 1 h at RT. Flow cytometry or confocal microscopy were performed immediately after this last wash. For flow cytometry, cells were suspended in 200 μl of PBS and examined in a flow cytometer equipped with a 488-nm argon laser. For microscopy, 200 μl of suspended cells were centrifuged (500 g, 5 min) on a glass slide, treated with DAPI (4′,6-diamidino-2-phenylindole), and examined under a Zeiss Axio Imager fluorescence microscope.
RESULTS
Design of the Selective PR3-recognizing Peptide Sequence
The charge distribution of the solvent-accessible surfaces in the vicinity of the substrate binding sites of PR3 and HNE differ significantly (20, 26). The S2 pocket is smaller and more hydrophilic in PR3 than in HNE mainly because the Leu at position 99 is replaced by a Lys in the vicinity of both the S2 and S4 pockets (26). Similarly, there is a charged arginyl residue at position 217 in HNE in the vicinity of the S4, whereas it is an Ile in PR3. We investigated the importance of the S4 subsite in PR3 by producing a new PR3 variant with an Arg at position 217 (I217R) (Fig. 1A). The activities of the wild type and the two PR3 mutants (I217R, K99L (19)) were then assayed with peptide-based FRET protease substrates derived from the PR3/NE substrate ABZ-QPMAV↓VQSVPQ-EDDnp (substrate 1) (19) bearing either a positively charged residue ABZ-QKMAV↓VQSVPQ-EDDnp (substrate 2) or a negatively charged residue QDMAV↓VQSVPQ-EDDnp (substrate 3) at P4. The wtPR3, the PR3 mutants, and HNE all cleaved substrate 1 at the V-V bond with similar specificity constants (Table 1). Although replacing P4 Pro by a positively charged residue in substrate 1 (substrate 2) had no significant influence on the rate of hydrolysis, an Asp at P4 (substrate 3) strongly impaired cleavage by wtPR3 and K99L but not by I217R, which cleaved at the V-V bond with a significantly larger kcat/Km (Table 1 and Fig. 1, A and B). Although replacing the Ala at P2 by Asp in substrate 1 (substrate 4) resulted in a large increase in the specificity constant with wtPR3 (19), I217R still cleaved at the same V-V bond with a similar specificity constant (Table 1). These observations strongly suggest that the S4 pocket greatly influences the substrate specificity of PR3. This was also demonstrated by replacing the Pro at P4 by Asp and the Ala at P2 by Asp. The resulting substrate (substrate 5) was not hydrolyzed by wtPR3 but was cleaved by I217R. As expected K99L did not cleave substrates with an Asp at either P2 or P4 (Table 1). HNE still cleaved substrate 5, but cleavage was one bond downstream from the V-V bond (Fig. 1, A and B), which is not a drawback when constructing inhibitors with no prime residues. We deduced from these data that the sequence Asp-X-Asp-Ala (DXDA) should be optimal for discriminating between PR3 and HNE in terms of selectivity, but that PR3 would have a greater affinity for Pro-X-Asp-Ala (PXDA).
FIGURE 1.

Hydrolysis of the polyvalent FRET substrate ABZ-QDMAVVQSVPQ-EDDnp (substrate 3) by wtPR3, the PR3 variant I217R, and HNE. A, HPLC C18 analysis of substrate fragments after extensive hydrolysis. Fragments were detected at 220 nm (blue) and 360 nm (green) as described (19). The insets show the relative positions of the key residues at positions 99 and 217 (chymotrypsin numbering) in the crystal structures of PR3 (PDB code 1FUJ) (26) and HNE (PDB code 1PPF) (49) and the molecular model of the PR3 variant I217R. The active site serine and Ile-217 are shown in yellow and orange, respectively. B, identification of the cleavage sites by mass spectrometry (top) and summary of the identified cleavage sites (bottom).
TABLE 1.
Kinetics of synthetic substrate cleavage by proteinase 3 and elastase
The arrows indicate the cleavage sites: ↓PR3, ↑I217R, ↕HNE. n.h., no hydrolysis.

* Values are the means ± S.D. of three experiments.
# Values were from Jégot et al., (19).
We previously observed that a wide range of amino acid residues could be accommodated at the PR3 P3 subsite, whereas Tyr was more appropriate for PR3 (12). We, therefore, constructed pNA substrates having the peptide sequences Ac-DYDA- and Ac-PYDA- from P4 to P1. The specificity constant for Ac-PYDA-pNA (substrate 6) was higher than for Ac-DYDA-pNA (substrate 7), but substrate 7 was also hydrolyzed at a significant rate (Table 1). These two sequences were selected to construct peptidyl di(chlorophenyl) phosphonate esters as inhibitors of PR3.
Synthesis and Inhibitory Properties of Ac-peptidylP(O-C6H4-4-Cl)2 Phosphonate Inhibitors and Their Biotinylated Derivatives
The phosphonate inhibitors and their biotinylated derivatives (Table 2) synthesized, purified by HPLC, and checked by mass spectrometry as described above were assayed for their capacities to inhibit the human neutrophil serine proteases PR3 and HNE by measuring residual activity after incubation with increasing amounts of each compound. We used the FRET substrates developed for each protease (ABZ-VADnVADYQ-EDDnp (27) (where nV stands for norvaline) and ABZ-APQQIMDDQ-EDDnp (28)). PR3 (1 nm final) was almost completely inhibited by incubation for 20 min with 0.5 μm Ac-PYDAP(O-C6H4-4-Cl)2 (compound 1), whereas HNE was not significantly inhibited. A far higher concentration of Ac-DYDAP(O-C6H4-4-Cl)2 (compound 2) was necessary to completely inhibit PR3, in keeping with the kinetic data reported above, whereas HNE was not inhibited. We confirmed these observations by measuring the second order rate constants kobs/[I] in competition experiments between the PR3 substrate and each inhibitor (24). The second order rate constant kobs/[I] for compound 1 was ∼80 times greater than that for compound 2 (Table 2). Replacing the charged Asp residue at P2 in compound 1 with an uncharged hydrophobic Phe residue (compound 3) reduced the kobs/[I] 80-fold (Table 2). Whereas compound 3 inhibited recombinant K99L, I217R, and wtPR3 to similar extents, compound 1 inhibited wtPR3 far better than the two PR3 variants, illustrating the close relationship between the S2 and S4 subsites (Fig. 2).
TABLE 2.
Rates of inhibition of wild type proteinase 3 and elastase by peptide phosphonates
n.s., not significant.

* Values are the means ± S.D. of three experiments.
FIGURE 2.
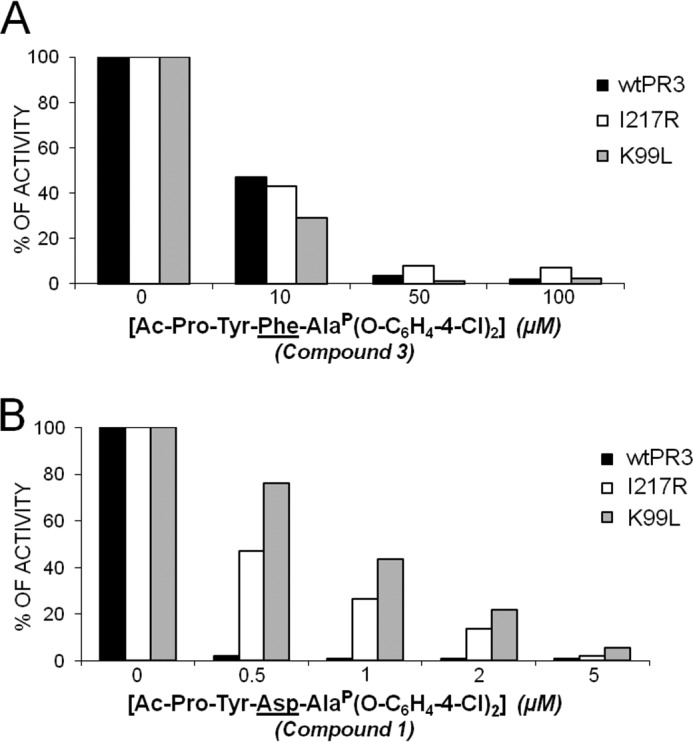
Inhibition of wtPR3 and its molecular variants I217R and K99L by increasing the amounts of Ac-PYFAP(O-C6H4-4-Cl)2 (compound 3) (A) and Ac-PYDAP(O-C6H4-4-Cl)2 (compound 1) (B) showing the relationship between the S4 and the S2 subsites of PR3.
We replaced the N-terminal acetyl group by biotin in compounds 1 and 2 to generate molecular probes for identifying PR3 activity in liquid biological samples, cells, or tissues (compounds 4 and 5). This resulted in significantly greater kobs/[I] values (Table 2). Thus incubating PR3 (1 nm final) for 20 min with a small molar excess of Bt-PYDAP(O-C6H4-4-Cl)2 (compound 5) completely inhibited the enzyme, whereas HNE was not inhibited when the inhibitor concentration was even 100-fold higher (Fig. 3, A and B). The biotinylated inhibitor also enabled us to identify PR3 by Western blotting under reducing and denaturing conditions but not HNE (Fig. 3C). Non-denaturing conditions prevented biotin being recognized by extravidin, suggesting that biotin helps the inhibitor interact with the binding site or stabilizes it. We constructed additional inhibitors with the same peptide sequence but bearing an aliphatic chain, a positive charge at their N terminus (compounds 6–9), or a hydrophobic residue (compounds 10 and 11) (Table 2 and Fig. 4) to examine the way biotin influences the interaction between phosphonate inhibitors and the active site of PR3. A five-carbon aliphatic chain improved the specificity constant, as much as biotin, which means that a hydrophobic moiety is enough to stabilize the inhibitor within the PR3 active site (Table 2). Introducing PEG66 as a spacer in compound 5 (compound 9) resulted in an inhibitor that was not fully selective for PR3 (Table 2) but still selectively visualized the PR3-bound biotinylated inhibitor despite the fact that biotin was not recognized by extravidin or streptavidin in the HNE-compound 9 complex under non-denaturing/non-reducing conditions (Fig. 5). We next examined the capacities of these phosphonate inhibitors to inhibit PR3 when it was in a complex biological environment and resisted degradation by other proteases.
FIGURE 3.

Selective inhibition of PR3 by Bt-PYDAP(O-C6H4-4-Cl)2 (compound 5). A, chemical structure of the biotinylated phosphonate inhibitor Bt-PYDAP(O-C6H4-4-Cl)2 (compound 5). B, residual activities of PR3 and HNE incubated with compound 5. PR3 (1 nm) and HNE (1 nm) were incubated for 20 min at 37 °C with various concentrations of compound 5. Inhibition was monitored by measuring the residual activities of PR3 on ABZ-VADnVADYQ-EDDnp (20 μm) and HNE on ABZ-APQQIMDDQ-EDDnp (20 μm). C, Western blot analysis of the irreversible PR3-compound 5 complex. PR3 (10 and 50 nm) and HNE (10 and 50 nm) were incubated for 20 min at 37 °C with compound 5 (50 nm final), and the mixtures were analyzed by Western blotting (4 ng of PR3 and 20 ng of HNE) and revealed using extravidin peroxidase.
FIGURE 4.

Chemical structures of +H2N-PYDAP(O-C6H4-4-Cl)2 (compound 8), Ac-PYDAP(O-C6H4-4-Cl)2 (compound 1), HX-PYDAP(O-C6H4-4-Cl)2 (compound 6), and Ac-AHX-PYDAP(O-C6H4-4-Cl)2 (compound 7). AHX, 6-amino hexanoic acid.
FIGURE 5.

Selective recognition of the PR3-compound 9 complex by extravidin/streptavidin under non-denaturing/non-reducing conditions. A, chemical structures of Bt-[PEG]66-PYDAP(O-C6H4-4-Cl)2 (compound 9). B, visualization by ELISA of the PR3-compound 9 (CP5) complex as revealed with streptavidin-HRP (1/2000). C, Western blot analysis of the PR3-compound 9 complex under non-denaturing/non-reducing conditions. PR3 and HNE (50 and 100 nm) were incubated for 20 or 180 min at 37 °C with compound 9 (150 nm), and the mixtures were analyzed by Western blotting using extravidin-peroxidase. D, selective interaction of biotinylated compound 9 with PR3 in a neutrophil lysate as shown by Western blotting under non-denaturing/non-reducing conditions and extravidin-peroxidase staining.
Selective Inhibition of PR3 by Bt-PYDAP(O-C6H4-4-Cl)2 (compound 5) in a Human Neutrophil Lysate and in Biological Samples
PR3 and its glycosylated forms present in a lysate of purified human neutrophils were totally inhibited by Bt-PYDAP(O-C6H4-4-Cl)2 when assayed under the same experimental conditions as those used for purified proteases (Fig. 6, A and B). Furthermore, the inhibitor suffered no loss of activity when it was incubated with the neutrophil lysate for 20 min before HPLC analysis, indicating that the inhibitor resisted degradation in an environment replete with proteases (Fig. 6C).
FIGURE 6.
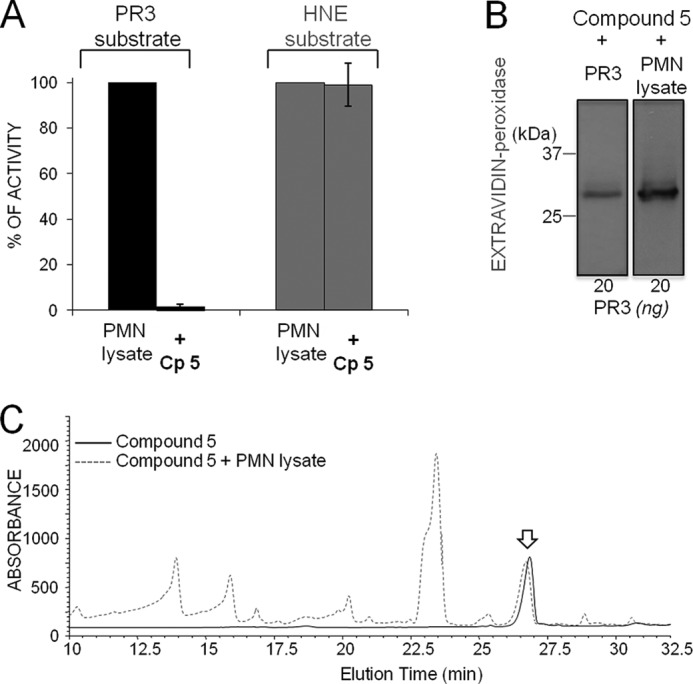
Selective inhibition of PR3 in a neutrophil lysate. A, PR3 (∼1 nm) and HNE (∼1 nm) were incubated for 20 min at 37 °C with 10 nm Bt-PYDAP(O-C6H4-4-Cl)2 (compound 5 (CP5)), and the inhibition was monitored by measuring the hydrolysis of 20 μm ABZ-VADnVADYQ-EDDnp (PR3) or 20 μm ABZ-APQQIMDDQ-EDDnp (HNE). B, detection of the irreversible complex of compound 5 with purified PR3 and PR3 in neutrophil lysate using extravidin-peroxidase: samples containing ∼50 nm PR3 were incubated for 20 min at 37 °C with compound 5 (50 nm final). The resulting products were separated by electrophoresis and transferred to a nitrocellulose membrane. C, stability of compound 5 (50 nm) (solid line) after incubation for 20 min at 37 °C with a neutrophil lysate (dotted line) and analysis by HPLC at 220 nm. The arrow indicates compound 5.
We then analyzed the supernatants of 10 sputa from CF patients for their PR3 activity before and after incubation with compound 5 (150 nm final). All the biological samples cleaved the selective PR3 substrate in the absence of inhibitor, and cleavage was always at the PR3-specific bond. The estimated PR3 concentration of these samples was in the 0.1–1 μm range based on the rate of substrate hydrolysis. Substrate hydrolysis was totally inhibited after incubation with compound 5 for 20 min, whereas HNE activity remained unchanged (Fig. 7A). Compound 5 was also used to selectively label PR3 activity in CF sputum, and the findings compared with those obtained with anti-PR3 antibodies (Fig. 7, B and C). The inhibitor was not broken down by crude supernatants, and the rate of PR3 inactivation was comparable to that of purified PR3. Thus, PR3 inhibition was not impaired by other soluble compounds in CF sputa. The same results were obtained with crude bronchoalveolar fluid samples (n = 5) from patients with idiopathic pulmonary fibrosis (not shown).
FIGURE 7.

Selective inhibition of PR3 in biological samples. A, inhibition of PR3 in a representative CF sputum supernatant. The volume of CF sputum was adjusted to give final concentrations of PR3 and HNE of ∼1 nm. Diluted samples were incubated with compound 5 (Cp5, 50 nm final) for 20 min at 37 °C. The residual activities of PR3 and HNE were measured as described in Fig. 3. Purified PR3 (1 nm) and HNE (1 nm) were used as controls. B, selective labeling of PR3 activity in CF sputum. CF volumes were adjusted to contain 4 or 20 ng PR3 (10 and 50 nm final) and incubated for 20 min at 37 °C with compound 5 (150 nm final), and the products were revealed with extravidin peroxidase. C, Western blot analysis of CF sputa with anti-PR3 antibodies showing active/inactive/degraded PR3 and additional bands due to antibodies interacting with unrelated CF sputum proteins. D, inhibition of PR3 in concentrated (10×) urine from three patients with bladder cancer. Proteolytically active PR3 in unconcentrated urine was ∼0.1–1 nm based on the rate of substrate hydrolysis. E, selective labeling of PR3 activity in concentrated (10×) urine (U) samples. F, Western blot analysis of concentrated urine samples (10×) using anti-PR3 antibodies.
We also used the urine of patients with bladder cancer as a model for detecting proteolytically active PR3 by compound 5. PR3 has been shown to be an antigenic marker of aggressive bladder cancer (29), but there was no indication as to whether PR3 was proteolytically active or whether phosphonate inhibitors resist degradation in this heterogeneous environment. Urine samples from control subjects and patients with a bladder cancer (n = 30) were concentrated 20-fold (controls) or 10-fold, PR3 activity was measured, and compound 5 was added before analysis by Western blotting. No activity was detected in the urines of controls in these experimental conditions. The active PR3 concentration in the cancer patient samples was estimated to be 0.1–1 nm based on the rate of substrate hydrolysis. Incubation for 20 min with compound 5 (50 nm final) completely inhibited the PR3 activity in the concentrated urine samples (Fig. 7D). Urinary PR3 activity was selectively labeled and compared well with the data from anti-PR3 antibodies (Fig. 7, E and F). Thus, the concentration of proteolytically active PR3 in the urine of patients with bladder cancer is elevated. It probably originates from cancer-associated neutrophils, as supported by our observation that other neutrophil-derived markers such as HNE and myeloperoxidase are also present in the urine of these patients (not shown).
Characterization of PR3 in Purified Neutrophils
Because the biotin in compound 9 still reacts with extravidin/streptavidin after the inhibitor has interacted with its target protease, we looked at the intracellular distribution of PR3 in purified permeabilized neutrophils. The whole cytoplasm of permeabilized neutrophils was intensely labeled but not the perinuclear environment, as previously observed for HNE (30) (Fig. 8 and supplemental Video 1). There was no PR3 labeling at the cell surface which confirms that the small variable fraction of PR3 constitutively exposed on the surface of quiescent neutrophils is inactive (31). Primed, activated neutrophils bear an “induced pool of active PR3” on their membranes, the binding of which clearly differs from that of HNE and affects the kinetic properties of the membrane-bound PR3 (PR3m) (32). This results in increased resistance to inhibition by α-1-proteinase inhibitor, its main natural inhibitor in plasma (31). The resistance has been attributed to steric hindrance of interactions with high molecular mass (50 kDa) serpin (33, 34). We examined this hypothesis using the cell-impermeable low Mr compound 1 to inhibit the PR3 on the surface of activated neutrophils purified from control subjects and patients with GPA. The phosphonate inhibitor behaved much like α-1-proteinase inhibitor as, despite its very small size (0.7 kDa), a large molar excess of inhibitor was needed to fully inhibit PR3m (Fig. 9A). We used flow cytometry and compound 9 to show that the PR3-inhibitor complexes remained at the surface of activated neutrophils (Fig. 9B). Because the membrane binding site on the PR3 surface involves a hydrophobic patch located near the S4 subsite (34, 35), we postulated that PR3m was in a resting state conformation that prevented small molecule inhibitors from rapidly interacting with PR3m (Fig. 9C). We checked this by comparing the Km values of the interactions between a peptide PR3 substrate, structurally similar to the phosphonate inhibitor, measured with the soluble and the membrane-bound proteases. The Km (determined by Hanes plot of the hydrolysis of PR3 substrate) for PR3m was ∼6 times higher than that for soluble PR3 indicating that the substrate binding region of PR3m is not freely accessible to substrates or inhibitors, unlike that of the soluble, active form. By contrast, the Km of an HNE peptide substrate (ABZ-APQQIMDDQ-EDDnp) was the same when it was measured in a suspension of activated neutrophils and with purified HNE (not shown).
FIGURE 8.

Selective labeling of intracellular PR3 in fixed, permeabilized quiescent neutrophils. A, residual activities of PR3 and HNE in permeabilized cells incubated for 90 min at RT with the polyvalent serine protease inhibitor Ala-Ala-Pro-Val-CH2Cl (AAPV-CH2Cl, 2 mm) for 20 min at RT with compound 9 (Cp9, 10 μm), or with the elastase inhibitor EPI-hNE-4 (1 μm). Inhibition was monitored using 100,000–150,000 quiescent cells. The residual activity of PR3 was measured using ABZ-VADnVADYQ-EDDnp (20 μm final) and that of HNE with ABZ-APQQIMDDQ-EDDnp (20 μm final). B, side (SS)- and forward (FS)-scatter of fixed, permeabilized purified neutrophils showing the homogeneity of the neutrophil population. C, flow cytometry analysis of permeabilized neutrophils that had been incubated with compound 9. The fluorescence of cells increased dramatically after incubation with compound 9 (green peak, mean fluorescence intensity = 67073), but it increased much less when the cells had been first treated with the polyvalent serine protease inhibitor Ala-Ala-Pro-Val-CH2Cl (blue peak, mean fluorescence intensity = 1900). D, fluorescence remained unchanged in cells preincubated with the elastase inhibitor EPI-hNE-4 (50) (magenta peak). The black peak shows the fluorescence of permeabilized neutrophils in the presence of streptavidin-Alexa Fluor® 488. E, confocal microscopy of permeabilized neutrophils treated first with EPI-hNE-4 and then incubated with compound 9 (right). Confocal microscope image stacks are as revealed with DAPI and streptavidin-Alexa Fluor® 488.
FIGURE 9.

Inhibition and labeling of PR3m. A, residual activity of soluble (gray bars) and PR3m from purified neutrophils of a healthy individual (black bars) and a GPA patient (white bars) in the presence of increased concentrations of compound 1. Purified neutrophils were activated with A23187 (1 μm) for 15 min at 37 °C (25). Purified PR3 (1 nm) and PR3m (∼1 nm based on the rate of substrate hydrolysis by ∼1 million cells) were incubated with a range of concentrations of compound 1 for 20 min at RT in PBS, and residual activities were measured fluorometrically. A large molar excess of compound 1 was required to inhibit PR3m on activated neutrophils. B, labeling of cell surface PR3m. Shown is a representative flow cytometry analysis of A23187-activated neutrophils before and after incubation with compound 9. Activated neutrophils were incubated for 20 min at RT with 50 μm compound 9 and visualized with streptavidin-Alexa Fluor® 488. The gray (mean fluorescence intensity = 562) peak corresponds to unspecific binding of streptavidin-Alexa Fluor® 488, and the black (mean fluorescence intensity = 1439) peak corresponds to labeled active PR3m. The 2.5 ± 0.4 (mean value of 4 independent experiments) -fold increase in mean fluorescence intensity shows that induced PR3m interacts with compound 9 and that the complex lies on the cell surface. C, proposed explanation for the low activity of induced PR3m and its stable exposure on activated neutrophils. Although constitutive PR3 is proteolytically inactive, induced PR3m, which may be co-localized with NB1 (CD177) in lipid drafts on the neutrophil surface (51), is in a resting state conformation that impairs the productive binding of substrates and inhibitors (left). An excess of substrate or inhibitor favors the conversion of latent PR3m to its fully active form (right).
DISCUSSION
PR3 and HNE are structurally very similar, and both are produced, stored and secreted at the same time by the same leukocytes. Both cleave protein substrates preferentially after small hydrophobic residues like Val/Ala and have long been regarded as proteases with almost indistinguishable substrate specificities. But evidence is slowly accumulating that each protease has a unique and non-redundant biological function that is mirrored by subtle differences in their overall structure. These affect their subcellular distributions and their substrate specificities (3, 36). For example, human PR3 has a hydrophobic patch on its surface that is responsible for its exposure on the surface of quiescent neutrophils, whereas HNE does not. This probably explains why PR3 is a disease-specific major autoantigen in GPA (35). The substrate binding specificities of PR3 and HNE extend to both sides of the cleaved peptide bond, so that subsites from S4 to S2′ influence the rate of substrate cleavage and sensitivity to inhibitors (20, 37).
Their stability, their lack of toxicity, and their selectivity make peptide derivatives of aminoalkyl phosphonate diphenyl esters convenient tools for in vitro and in vivo studies on the function and activity of serine proteases during the course of a variety of diseases (14). The phosphonate moiety of these irreversible inhibitors interacts with the S1′ site and forms a covalent bond with Ser-195. We have introduced an electron-withdrawing chlorine atom in the para position of the phenyl ester ring to increase the electrophilic potential of the phosphorous atom, thus making it more susceptible to the nucleophilic attack of the active site serine residue (38). The selectivity of phosphonate inhibitors depends only on the non-prime specificity of the target protease, whereas the C-terminal phosphonate group occupies the S1′ subsite of the protease. Selective PR3 substrates have exploited both the prime and non-prime specificity pockets (27). We wanted to produce PR3 inhibitors that consisted only of non-prime residues and discriminated between PR3 and HNE. We, therefore, investigated the non-prime subsite specificity of PR3 using two PR3 mutants that mimic the S4 and S2 subsites of HNE, which are the most critical non-prime sites for distinguishing between PR3 and HNE (26).
We used several FRET substrates that differed at the P4 and P2 positions to compare wtPR3, the two mutants of PR3 and HNE, followed by peptidyl-pNA substrates that have no prime side residues. These studies revealed a close cooperation between the S4 and the S2 subsites, which we exploited in our design of two short tetrapeptide sequences that are selectively recognized by PR3. DYDA and PYDA were chosen as the P4-P1 sequences to synthesize peptide derivatives of aminoalkyl phosphonate diphenyl esters. The two tetrapeptide derivatives were selective for PR3, but the second order inhibition constant kobs/[I] for the prolyl-containing inhibitor was 100 times larger, in keeping with the lesser ability of the PR3 S4 subsite to accommodate a negatively charged residue. Unexpectedly, biotinylation of the N terminus of Ac-PYDAP(O-C6H4-4-Cl)2 improved the kobs/[I] ∼25 times. This was probably because biotin improved the binding of the inhibitor to the protease. This idea is supported by the finding that biotin interacts poorly with extravidin/streptavidin once the inhibitor is bound to PR3. We also showed that attaching a short aliphatic sequence like hexanoic acid to the P4 prolyl residue of inhibitors improved inhibitor binding, whereas introducing a +NH2Pro at P4 greatly impaired the interaction. This apparently contradicts previous results showing that an +NH3-terminal residue poorly influenced the cleavage of FRET substrates bearing a prime extension and no Asp at P2 (20). Stabilizing the short tetrapeptide sequence, as was done here depends greatly on the Asp at P2. This stabilization is most certainly compromised by the presence of +NH2Pro in the environment of P2 = Asp. This further emphasizes the cooperation between the S4 and S2 subsites in PR3.
We also verified that phosphonate inhibitors are still active and resist degradation in a milieu replete with proteases, such as neutrophil lysates, bronchoalveolar fluids, and sputa from patients with inflammatory lung diseases or the urine of patients with bladder cancer. A spacer like polyethylene glycol placed between the biotin moiety and the inhibitor P4 residue preserved inhibitory properties and allowed the biotin moiety to be recognized by extravidin/streptavidin. Thus the biotinylated inhibitor can be used as a probe for visualizing PR3 activity in situ in permeabilized neutrophils by confocal microscopy. Unlike HNE, whose distribution is mainly perinuclear (30, 39), PR3 activity is present in punctate areas throughout the cytoplasm of neutrophils. This agrees with the results obtained by immunocytochemistry showing that the PR3 autoantigen, the target of cANCA (anti-neutrophil cytoplasmic antibodies), is distributed throughout the cytoplasm, whereas the HNE antigen (40, 41), the target of some pANCA (42, 43), is concentrated in the perinuclear region.
The most striking feature of PR3 is its constitutive expression as a proteolytically inactive form at the neutrophil surface and the peculiar binding of its induced form to the surface of activated neutrophils. This particular binding depends on a hydrophobic patch located beyond the S4 subsite of the protease (Phe-165, Phe-166, Phe-215, Ile-217, Trp-218, Phe-224) (26). The other neutrophilic serine proteases, HNE and cathepsin G, lack this hydrophobic patch (35, 44) as does rodent PR3. This difference between humans and rodents seriously complicates the use of rodents for studies on PR3 in vivo (45). We previously reported a weak PR3 activity in suspensions of activated human neutrophils (31). We also observed that induced PR3m was poorly inhibited by its main natural plasma inhibitor α-1-proteinase inhibitor and attributed this to steric hindrance at the cell surface. We now find that low molecular weight phosphonate inhibitors behave in the same way as α-1-proteinase inhibitor and that a large molar excess of inhibitor is need to fully inhibit PR3 in a suspension of activated neutrophils. This phenomenon cannot be readily accounted for by steric hindrance. The PR3 hydrophobic patch should not impair access of low Mr phosphonate inhibitors to the PR3 active site cleft. The weak PR3m activity and its resistance to inhibition might be due to its conformation being distorted; we recently used the monoclonal Ab MCPR3–7 to show that PR3 is in equilibrium between a highly favored active conformation and an inactive conformation (46). This antibody inhibits PR3 activity by an allosteric mechanism and competes with the neutrophil membrane for the binding of PR3. Interaction of the hydrophobic patch of PR3m with NB1 (CD177) or lipid rafts on the neutrophil surface might well employ the same mechanism to affect the substrate and inhibitor binding subsites. The conversion of the resting or latent conformation to the fully active form would be induced by an excess of substrate or inhibitor. This is consistent with our observation that the Km for cleavage of a peptide substrate by PR3 in a suspension of neutrophils is about six times greater than that of PR3 in solution. PR3m, therefore, seems to function much like other serine proteases, granzyme K, FIXa, DF, degP, and α-tryptase that have a resting state conformation due to one or more distorted loops surrounding the active site cleft that impair the productive binding of substrates and inhibitors (47).
The relative resistance of induced PR3m to inhibition and its apparently reduced enzymatic activity make it different from HNE, which remains fully active at the neutrophil surface and is easily removed from the surface by α-1-proteinase inhibitor (31, 48). This peculiar behavior of PR3m results in the prolonged exposure of a latent form of the protease at the cell surface during inflammatory episodes. This exposure of a latent activatable form plus the permanent, genetically determined, exposure of inactive PR3 at the cell surface could well explain why this protease is the major autoantigen mediating the binding of anti-PR3 antibodies and the subsequent activation of blood neutrophils in GPA.
Supplementary Material
Acknowledgments
We acknowledge the Association Vaincre La Mucoviscidose (AVLM). We thank Lise Vanderlynden (INSERM U-1100), Julien Burlaud-Gaillard (Département des Microscopies, Université Francois Rabelais), and Heike Reimann (Comprehensive Pneumology Center, Institute of Lung Biology and Disease) for technical assistance. The English text was edited by Dr. Owen Parkes.
This work was supported by the Ministère de l'Enseignement Supérieur et de la Recherche, the Région Centre, and the Fonds Européen de Développement Régional (project INFINHI) and Polish Ministry of Science and Higher Education (project IP2012 0596 72; to M. W.). This work was also supported by European Union Seventh Framework Programme (FP7/2007–2013; to D. E. J.) under Grant 261382 and by European Union Commission (FP7/ TransBioBC; to J. Z. and A. V.) under Grant 601933.

This article contains supplemental Video 1.
- PR3
- proteinase 3
- PR3m
- membrane-bound PR3
- HNE
- human neutrophil elastase
- GPA
- granulomatosis with polyangiitis
- pNA
- para-nitroaniline
- ABZ
- ortho-aminobenzoic acid
- EDDnp
- N-(2.4-dinitrophenyl)ethylenediamine
- Bt
- biotin
- CF
- cystic fibrosis
- Fmoc
- (fluoromethyloxycarbonyl chloride)
- DMF
- dimethylformamide
- RT
- room temperature.
REFERENCES
- 1. Turk B. (2006) Targeting proteases: successes, failures, and future prospects. Nat. Rev. Drug Discov. 5, 785–799 [DOI] [PubMed] [Google Scholar]
- 2. Korkmaz B., Moreau T., Gauthier F. (2008) Neutrophil elastase, proteinase 3, and cathepsin G: physicochemical properties, activity, and physiopathological functions. Biochimie 90, 227–242 [DOI] [PubMed] [Google Scholar]
- 3. Korkmaz B., Horwitz M. S., Jenne D. E., Gauthier F. (2010) Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol. Rev. 62, 726–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Owen C. A. (2008) Roles for proteinases in the pathogenesis of chronic obstructive pulmonary disease. Int. J. Chron. Obstruct. Pulmon. Dis. 3, 253–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jenne D. E., Tschopp J., Ludemann J., Utecht B., Gross W. L. (1990) Wegener's autoantigen decoded. Nature 346, 520. [DOI] [PubMed] [Google Scholar]
- 6. Kallenberg C. G. (2008) Pathogenesis of PR3-ANCA associated vasculitis. J. Autoimmun. 30, 29–36 [DOI] [PubMed] [Google Scholar]
- 7. Csernok E., Gross W. L. (2013) Current understanding of the pathogenesis of granulomatosis with polyangiitis (Wegener's). Expert Rev. Clin. Immunol. 9, 641–648 [DOI] [PubMed] [Google Scholar]
- 8. Gabillet J., Millet A., Pederzoli-Ribeil M., Tacnet-Delorme P., Guillevin L., Mouthon L., Frachet P., Witko-Sarsat V. (2012) Proteinase 3, the autoantigen in granulomatosis with polyangiitis, associates with calreticulin on apoptotic neutrophils, impairs macrophage phagocytosis, and promotes inflammation. J. Immunol. 189, 2574–2583 [DOI] [PubMed] [Google Scholar]
- 9. Witko-Sarsat V., Cramer E. M., Hieblot C., Guichard J., Nusbaum P., Lopez S., Lesavre P., Halbwachs-Mecarelli L. (1999) Presence of proteinase 3 in secretory vesicles: evidence of a novel, highly mobilizable intracellular pool distinct from azurophil granules. Blood 94, 2487–2496 [PubMed] [Google Scholar]
- 10. Halbwachs-Mecarelli L., Bessou G., Lesavre P., Lopez S., Witko-Sarsat V. (1995) Bimodal distribution of proteinase 3 (PR3) surface expression reflects a constitutive heterogeneity in the polymorphonuclear neutrophil pool. FEBS Lett. 374, 29–33 [DOI] [PubMed] [Google Scholar]
- 11. Schreiber A., Busjahn A., Luft F. C., Kettritz R. (2003) Membrane expression of proteinase 3 is genetically determined. J. Am. Soc. Nephrol. 14, 68–75 [DOI] [PubMed] [Google Scholar]
- 12. Korkmaz B., Kellenberger C., Viaud-Massuard M. C., Gauthier F. (2013) Selective inhibitors of human neutrophil proteinase 3. Curr. Pharm. Des. 19, 966–976 [DOI] [PubMed] [Google Scholar]
- 13. Sieńczyk M., Oleksyszyn J. (2009) Irreversible inhibition of serine proteases: design and in vivo activity of diaryl α-aminophosphonate derivatives. Curr. Med. Chem. 16, 1673–1687 [DOI] [PubMed] [Google Scholar]
- 14. Grzywa R., Sieńczyk M. (2013) Phosphonic esters and their application of protease control. Curr. Pharm. Des. 19, 1154–1178 [DOI] [PubMed] [Google Scholar]
- 15. Boduszek B., Brown A. D., Powers J. C. (1994) α-Aminoalkylphosphonate di(chlorophenyl) esters as inhibitors of serine proteases. J. Enzyme Inhib. 8, 147–158 [DOI] [PubMed] [Google Scholar]
- 16. Brown C. M., Ray M., Eroy-Reveles A. A., Egea P., Tajon C., Craik C. S. (2011) Peptide length and leaving-group sterics influence potency of peptide phosphonate protease inhibitors. Chem. Biol. 18, 48–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Woodard S. L., Jackson D. S., Abuelyaman A. S., Powers J. C., Winkler U., Hudig D. (1994) Chymase-directed serine protease inhibitor that reacts with a single 30-kDa granzyme and blocks NK-mediated cytotoxicity. J. Immunol. 153, 5016–5025 [PubMed] [Google Scholar]
- 18. Schechter I., Berger A. (1967) On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 27, 157–162 [DOI] [PubMed] [Google Scholar]
- 19. Jégot G., Derache C., Castella S., Lahouassa H., Pitois E., Jourdan M. L., Remold-O'Donnell E., Kellenberger C., Gauthier F., Korkmaz B. (2011) A substrate-based approach to convert SerpinB1 into a specific inhibitor of proteinase 3, the Wegener's granulomatosis autoantigen. FASEB J. 25, 3019–3031 [DOI] [PubMed] [Google Scholar]
- 20. Korkmaz B., Hajjar E., Kalupov T., Reuter N., Brillard-Bourdet M., Moreau T., Juliano L., Gauthier F. (2007) Influence of charge distribution at the active site surface on the substrate specificity of human neutrophil protease 3 and elastase: a kinetic and molecular modeling analysis. J. Biol. Chem. 282, 1989–1997 [DOI] [PubMed] [Google Scholar]
- 21. Sieńczyk M., Oleksyszyn J. (2006) Inhibition of trypsin and urokinase by Cbz-amino(4-guanidinophenyl)methanephosphonate aromatic ester derivatives: the influence of the ester group on their biological activity. Bioorg. Med. Chem. Lett. 16, 2886–2890 [DOI] [PubMed] [Google Scholar]
- 22. Oleksyszyn J., Powers J. C. (1989) Irreversible inhibition of serine proteases by peptidyl derivatives of α-aminoalkylphosphonate diphenyl esters. Biochem. Biophys. Res. Commun. 161, 143–149 [DOI] [PubMed] [Google Scholar]
- 23. Korkmaz B., Attucci S., Hazouard E., Ferrandiere M., Jourdan M. L., Brillard-Bourdet M., Juliano L., Gauthier F. (2002) Discriminating between the activities of human neutrophil elastase and proteinase 3 using serpin-derived fluorogenic substrates. J. Biol. Chem. 277, 39074–39081 [DOI] [PubMed] [Google Scholar]
- 24. Tian W. X., Tsou C. L. (1982) Determination of the rate constant of enzyme modification by measuring the substrate reaction in the presence of the modifier. Biochemistry 21, 1028–1032 [DOI] [PubMed] [Google Scholar]
- 25. Korkmaz B., Attucci S., Juliano M. A., Kalupov T., Jourdan M. L., Juliano L., Gauthier F. (2008) Measuring elastase, proteinase 3, and cathepsin G activities at the surface of human neutrophils with fluorescence resonance energy transfer substrates. Nat. Protoc. 3, 991–1000 [DOI] [PubMed] [Google Scholar]
- 26. Fujinaga M., Chernaia M. M., Halenbeck R., Koths K., James M. N. (1996) The crystal structure of PR3, a neutrophil serine proteinase antigen of Wegener's granulomatosis antibodies. J. Mol. Biol. 261, 267–278 [DOI] [PubMed] [Google Scholar]
- 27. Korkmaz B., Attucci S., Moreau T., Godat E., Juliano L., Gauthier F. (2004) Design and use of highly specific substrates of neutrophil elastase and proteinase 3. Am. J. Respir. Cell Mol. Biol. 30, 801–807 [DOI] [PubMed] [Google Scholar]
- 28. Derache C., Epinette C., Roussel A., Gabant G., Cadene M., Korkmaz B., Gauthier F., Kellenberger C. (2012) Crystal structure of greglin, a novel non-classical Kazal inhibitor, in complex with subtilisin. FEBS J. 279, 4466–4478 [DOI] [PubMed] [Google Scholar]
- 29. Zoidakis J., Makridakis M., Zerefos P. G., Bitsika V., Esteban S., Frantzi M., Stravodimos K., Anagnou N. P., Roubelakis M. G., Sanchez-Carbayo M., Vlahou A. (2012) Profilin 1 is a potential biomarker for bladder cancer aggressiveness. Mol. Cell Proteomics 10.1074/mcp.M111.009449 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kasperkiewicz P., Poreba M., Snipas S. J., Parker H., Winterbourn C. C., Salvesen G. S., Drag M. (2014) Design of ultrasensitive probes for human neutrophil elastase through hybrid combinatorial substrate library profiling. Proc. Natl. Acad. Sci. U.S.A. 111, 2518–2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Korkmaz B., Jaillet J., Jourdan M. L., Gauthier A., Gauthier F., Attucci S. (2009) Catalytic activity and inhibition of wegener antigen proteinase 3 on the cell surface of human polymorphonuclear neutrophils. J. Biol. Chem. 284, 19896–19902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Korkmaz B., Lesner A., Letast S., Mahdi Y. K., Jourdan M. L., Dallet-Choisy S., Marchand-Adam S., Kellenberger C., Viaud-Massuard M. C., Jenne D. E., Gauthier F. (2013) Neutrophil proteinase 3 and dipeptidyl peptidase I (cathepsin C) as pharmacological targets in granulomatosis with polyangiitis (Wegener granulomatosis). Semin. Immunopathol. 35, 411–421 [DOI] [PubMed] [Google Scholar]
- 33. Hajjar E., Mihajlovic M., Witko-Sarsat V., Lazaridis T., Reuter N. (2008) Computational prediction of the binding site of proteinase 3 to the plasma membrane. Proteins. 71, 1655–1669 [DOI] [PubMed] [Google Scholar]
- 34. Broemstrup T., Reuter N. (2010) How does proteinase 3 interact with lipid bilayers? Phys. Chem. Chem. Phys. 12, 7487–7496 [DOI] [PubMed] [Google Scholar]
- 35. Korkmaz B., Kuhl A., Bayat B., Santoso S., Jenne D. E. (2008) A hydrophobic patch on proteinase 3, the target of autoantibodies in Wegener granulomatosis, mediates membrane binding via NB1 receptors. J. Biol. Chem. 283, 35976–35982 [DOI] [PubMed] [Google Scholar]
- 36. Hajjar E., Broemstrup T., Kantari C., Witko-Sarsat V., Reuter N. (2010) Structures of human proteinase 3 and neutrophil elastase: so similar yet so different. FEBS J. 277, 2238–2254 [DOI] [PubMed] [Google Scholar]
- 37. Hajjar E., Korkmaz B., Reuter N. (2007) Differences in the substrate binding sites of murine and human proteinase 3 and neutrophil elastase. FEBS Lett. 581, 5685–5690 [DOI] [PubMed] [Google Scholar]
- 38. Sieńczyk M., Podgórski D., Bła¿ejewska A., Kulbacka J., Saczko J., Oleksyszyn J. (2011) Phosphonic pseudopeptides as human neutrophil elastase inhibitors: a combinatorial approach. Bioorg. Med. Chem. 19, 1277–1284 [DOI] [PubMed] [Google Scholar]
- 39. Clark J. M., Vaughan D. W., Aiken B. M., Kagan H. M. (1980) Elastase-like enzymes in human neutrophils localized by ultrastructural cytochemistry. J. Cell Biol. 84, 102–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Benson K. F., Li F. Q., Person R. E., Albani D., Duan Z., Wechsler J., Meade-White K., Williams K., Acland G. M., Niemeyer G., Lothrop C. D., Horwitz M. (2003) Mutations associated with neutropenia in dogs and humans disrupt intracellular transport of neutrophil elastase. Nat. Genet. 35, 90–96 [DOI] [PubMed] [Google Scholar]
- 41. Köllner I., Sodeik B., Schreek S., Heyn H., von Neuhoff N., Germeshausen M., Zeidler C., Krüger M., Schlegelberger B., Welte K., Beger C. (2006) Mutations in neutrophil elastase causing congenital neutropenia lead to cytoplasmic protein accumulation and induction of the unfolded protein response. Blood 108, 493–500 [DOI] [PubMed] [Google Scholar]
- 42. Csernok E., Lüdemann J., Gross W. L., Bainton D. F. (1990) Ultrastructural localization of proteinase 3, the target antigen of anti-cytoplasmic antibodies circulating in Wegener's granulomatosis. Am. J. Pathol. 137, 1113–1120 [PMC free article] [PubMed] [Google Scholar]
- 43. Tervaert J. W., Mulder L., Stegeman C., Elema J., Huitema M., The H., Kallenberg C. (1993) Occurrence of autoantibodies to human leucocyte elastase in Wegener's granulomatosis and other inflammatory disorders. Ann. Rheum. Dis. 52, 115–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kantari C., Millet A., Gabillet J., Hajjar E., Broemstrup T., Pluta P., Reuter N., Witko-Sarsat V. (2011) Molecular analysis of the membrane insertion domain of proteinase 3, the Wegener's autoantigen, in RBL cells: implication for its pathogenic activity. J. Leukoc. Biol. 90, 941–950 [DOI] [PubMed] [Google Scholar]
- 45. Korkmaz B., Jenne D. E., Gauthier F. (2013) Relevance of the mouse model as a therapeutic approach for neutrophil proteinase 3-associated human diseases. Int. Immunopharmacol. 17, 1198–1205 [DOI] [PubMed] [Google Scholar]
- 46. Hinkofer L. C., Seidel S. A., Korkmaz B., Silva F., Hummel A. M., Braun D., Jenne D. E., Specks U. (2013) A monoclonal antibody (MCPR3–7) interfering with the activity of proteinase 3 by an allosteric mechanism. J. Biol. Chem. 288, 26635–26648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hink-Schauer C., Estébanez-Perpiñá E., Wilharm E., Fuentes-Prior P., Klinkert W., Bode W., Jenne D. E. (2002) The 2.2-A crystal structure of human pro-granzyme K reveals a rigid zymogen with unusual features. J. Biol. Chem. 277, 50923–50933 [DOI] [PubMed] [Google Scholar]
- 48. Korkmaz B., Attucci S., Jourdan M. L., Juliano L., Gauthier F. (2005) Inhibition of neutrophil elastase by α1-protease inhibitor at the surface of human polymorphonuclear neutrophils. J. Immunol. 175, 3329–3338 [DOI] [PubMed] [Google Scholar]
- 49. Bode W., Wei A. Z., Huber R., Meyer E., Travis J., Neumann S. (1986) X-ray crystal structure of the complex of human leukocyte elastase (PMN elastase) and the third domain of the turkey ovomucoid inhibitor. EMBO J. 5, 2453–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Attucci S., Gauthier A., Korkmaz B., Delépine P., Martino M. F., Saudubray F., Diot P., Gauthier F. (2006) EPI-hNE4, a proteolysis-resistant inhibitor of human neutrophil elastase and potential anti-inflammatory drug for treating cystic fibrosis. J. Pharmacol. Exp. Ther. 318, 803–809 [DOI] [PubMed] [Google Scholar]
- 51. von Vietinghoff S., Tunnemann G., Eulenberg C., Wellner M., Cristina Cardoso M., Luft F. C., Kettritz R. (2007) NB1 mediates surface expression of the ANCA antigen proteinase 3 on human neutrophils. Blood 109, 4487–4493 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.