

Background: RNA processing abnormalities have been linked to amyotrophic lateral sclerosis (ALS).
Results: HuR promotes the expression of the ALS-associated TDP-43 and FUS/TLS RNA-binding proteins.
Conclusion: Through its regulation of TDP-43 and FUS/TLS, HuR potentially impacts a wide range of molecular and cellular phenotypes.
Significance: A network of proteins exists to maintain RNA processing, and ALS-associated abnormalities may stem from disruption of this network.
Keywords: Amyotrophic Lateral Sclerosis (ALS) (Lou Gehrig Disease), Astrocyte, Neuron, RNA Processing, RNA Splicing, 3′ Untranslated Region
Abstract
Posttranscriptional gene regulation is governed by a network of RNA-binding proteins (RBPs) that interact with regulatory elements in the mRNA to modulate multiple molecular processes, including splicing, RNA transport, RNA stability, and translation. Mounting evidence indicates that there is a hierarchy within this network whereby certain RBPs cross-regulate other RBPs to coordinate gene expression. HuR, an RNA-binding protein we linked previously to aberrant VEGF mRNA metabolism in models of SOD1-associated amyotrophic lateral sclerosis, has been identified as being high up in this hierarchy, serving as a regulator of RNA regulators. Here we investigated the role of HuR in regulating two RBPs, TDP-43 and FUS/TLS, that have been linked genetically to amyotrophic lateral sclerosis. We found that HuR promotes the expression of both RBPs in primary astrocytes and U251 cells under normal and stressed (hypoxic) conditions. For TDP-43, we found that HuR binds to the 3′ untranslated region (UTR) and regulates its expression through translational efficiency rather than RNA stability. With HuR knockdown, there was a shift of TDP-43 and FUS mRNAs away from polysomes, consistent with translational silencing. The TDP-43 splicing function was attenuated upon HuR knockdown and could be rescued by ectopic TDP-43 lacking the 3′ UTR regulatory elements. Finally, conditioned medium from astrocytes in which HuR or TDP-43 was knocked down produced significant motor neuron and cortical neuron toxicity in vitro. These findings indicate that HuR regulates TDP-43 and FUS/TLS expression and that loss of HuR-mediated RNA processing in astrocytes can alter the molecular and cellular landscape to produce a toxic phenotype.
Introduction
The genetic linkage of TAR DNA binding protein 43 (TDP-43) and fused in sarcoma/translocated in liposarcoma (FUS/TLS) with amyotrophic lateral sclerosis (ALS)3 has elevated RNA processing to the forefront as a potential mechanism of motor neuron degeneration in ALS (1). These RNA binding proteins contain one or more RNA recognition motifs (RRM) that bind to target mRNA ligands to direct splicing, RNA transport, stabilization, and other processes. They are subject to autoregulation to maintain tight control of expression levels (1–3). Loss of autoregulation with cytoplasmic protein accumulation is a leading hypothesis for cellular toxicity in some models of ALS. In addition to autoregulation, it is well established that many RBPs are subject to cross-regulation by other cellular factors that bind to their mRNAs and modulate splicing and stability (4–6). Although TDP-43 binds to FUS/TLS and positively regulates its mRNA and protein expression (7), little is known about cross-regulatory factors that may influence TDP-43 expression. Several studies have identified Hu antigen R (HuR) as a positive regulator of mRNAs encoding RRMs (5, 8, 9). HuR has three RRMs and regulates its target mRNA ligands through binding U- and AU-rich elements in the 5′ and 3′ UTRs. HuR plays an important role in stress response through a positive regulation of growth and cell survival factors at the level of RNA stability and translational efficiency (10). Similar to TDP-43 and FUS/TLS, HuR is predominantly nuclear in location (>90%) but shuttles to the cytoplasm as part of its function in regulating mRNA stability and translational efficiency (11, 12). This translocation can be augmented by cellular stressors such as heat shock or glucose deprivation (12, 13). We demonstrated previously that cellular toxicity related to ALS-associated mutant superoxide dismutase 1 (SOD1) could be reversed with HuR up-regulation (14). Here we show that HuR promotes TDP-43 and FUS/TLS expression by increasing translational efficiency. For TDP-43 mRNA, this regulation is mediated by the 3′ UTR to which HuR binds. Loss of HuR produces a splicing phenotype consistent with attenuated TDP-43 function. Suppression of HuR in primary astrocytes leads to the production of soluble factors that are toxic to cortical and motor neurons, similar to when TDP-43 is directly suppressed. These findings provide evidence that HuR plays a role in regulating ALS-associated RNA binding proteins and raise the possibility that HuR may contribute to motor neuron disease pathology by its unchecked positive regulation of ALS-associated TDP-43 and FUS/TLS.
EXPERIMENTAL PROCEDURES
Cell Culture, DNA Constructs, and Transfection
U251 MG, U251 Tet-On, U251 SOD1 clones, and doxycycline-inducible U251 HuR clones were maintained as described previously (14, 15). Cortical astrocytes were isolated as described previously (16) from G93A SOD1 mice (C57/Bl6 background, The Jackson Laboratory), littermate controls, and KSRP−/− mice (provided by Dr. Ching-Yi Chen, University of Alabama at Birmingham). The purity of astrocytes was confirmed by immunostaining for glial fibrillary acidic protein. Primary motor neurons were isolated and cultured on the basis of a protocol published previously (17). Motor neurons were plated on 15-mm coverslips (Assistant coverglasses, Carolina) coated with 0.5 mg/ml polyornithine (Sigma) and 0.5 mg/ml laminin (Sigma) and cultured in Neurobasal/B27 medium supplemented with 2% horse serum (Sigma) and 10 ng/ml each BDNF, ciliary neurotrophic factor, and glial cell line-derived neurotrophic factor (Peprotech). The small molecule inhibitor of HuR, MS-444, was provided by Novartis (Dr. Nicole Meisner) and was reconstituted in dimethyl sulfoxide. Cells were treated for 24 h. Rat primary cortical neurons were prepared with the assistance of the University of Alabama Tissue Culture Core Facility on the basis of a published protocol (18). The TDP-43 3′ UTR cDNA was provided by Dr. F. E. Baralle (International Centre for Genetic Engineering and Biotechnology). A 900-nt proximal segment, beginning at the stop codon, was cloned downstream from a luciferase open reading frame in PGL2 in the sense (900F) or antisense (900R) orientation using methods described previously (19). The following primers were used: upstream, 5′ TCAAGCTTCCCGGGACAGTGGGGTTGTGG TTGGT 3′; downstream, 5′ TAGACCTTCCCGGGCGCCAAGTTAAAAAAT 3′. A GFP-TDP-43 expression plasmid was provided by Dr. Zuoshang Xu (University of Massachusetts). FLAG-HuR, FLAG-KSRP, and the cystic fibrosis transmembrane conductance regulator (CFTR) hybrid minigene constructs are described elsewhere (19–21). All U251 cells and clones were transfected with Lipofectamine 2000 according to the instructions of the manufacturer (Invitrogen). Astrocytes were transfected by electroporation using a NeonTM transfection system (Invitrogen, MPK5000) following the protocol of the manufacturer.
Protein Preparation, Western Blotting, Antibodies, and Immunocytochemistry
Whole-cell lysates from cultured cells were prepared in the presence of protease inhibitors and sodium orthovanadate using the M-PER kit (Pierce) and quantitated using a BCA assay (Pierce Endogen). For Western blot analysis, 40 μg of extract were resolved by gel electrophoresis, blotted, and probed with the following antibodies: HuR 3A2 (Santa Cruz Biotechnology); TDP-43 (Cell Signaling Technology), FUS (Proteintech), HSP-70 (Santa Cruz Biotechnology), cleaved caspase 3 (Cell Signaling Technology), Actin (Sigma), luciferase (Millipore), GAPDH (Cell Signaling Technology), mouse IgG (Santa Cruz Biotechnology), FLAG (Sigma), and KSRP (provided by Dr. Ching-Yi Chen). Band densities were quantified using Quantity One software (Bio-Rad). For motor neuron immunofluorescence, cells were fixed on coverslips with 4% paraformaldehyde for 15 min. Slides were incubated with anti-cleaved caspase 3 (1:500) overnight at 4 °C. The next day, slides were incubated with Alexa Fluor 488 secondary antibody (1:1000, Invitrogen) for 1 h at room temperature. Images were visualized under an Olympus BX41 microscope equipped with a digital camera and processed using Adobe Photoshop (Adobe Systems). Cells were counted in a blinded manner.
RNA Isolation, Quantitative Real-time PCR, RNA Decay, and RNA Immunoprecipitation
Total RNA was isolated using a GE Healthcare Illustra RNAspin mini kit and quantitated with a Nanodrop2000 (Thermo Scientific). TDP-43 mRNA was quantitated by qRT-PCR with a ViiATM 7 real-time PCR system using optimized commercial primers and probes (Invitrogen). Primers and probes for luciferase and β-galactosidase are described elsewhere (16, 22). For RNA degradation analysis, cell cultures were treated with actinomycin D for up to 6 h, as described elsewhere (19). Degradation curves were generated with GraphPad (GraphPad software, San Diego, CA). For quantification of protein-RNA binding, 250 μg of total extracts from cultured cells were prepared, divided equally into three aliquots, and immunoprecipitated with HuR, TDP-43, or mouse IgG (negative control) using methods described previously (23). RNA was eluted from beads using the RNeasy kit (Qiagen) and then analyzed by qRT-PCR for TDP-43 and luciferase mRNA.
Polysome Analysis
U251 Cells were grown to 70% confluence and treated for 3 min at 37 °C and 5% CO2 with 0.1 mg/ml of cycloheximide in complete medium (1× Dulbecco's modified Eagle's medium/F12K 50/50, 7% FBS). Cells were washed twice with PBS supplemented with cycloheximide (0.1 mg/ml), scraped in 400 μl of polysome extraction buffer (15 mm Tris-Cl (pH 7.4), 15 mm MgCl2, 0.3 m NaCl, 1% Triton X-100, 0.1 mg/ml cycloheximide, and 1 mg/ml heparin) and then pelleted. Lysates of U251 cells were prepared and subjected to centrifugation through a linear 10–50% (w/v) sucrose gradient, and fractions were collected while being monitored for UV absorbance (254 nm) with a spectrophotometer. RNA isolation and analysis have been described previously (14).
shRNA and siRNA-mediated HuR Knockdown
For RNA interference, transfections were carried out using Lipofectamine 2000 (Invitrogen) and 30 nm of control scrambled small interfering RNA or gene-specific siRNA targeting TDP-43 or FUS (Invitrogen). The siRNA sequences for TDP-43 were as follows: set 1, 5′ GAACGAUGAACCCAUUGAATT 3′ (sense); set 2, 5′ CCAAUGCUGAACCUAAGCATT 3′ (sense). The sequences for control scrambled siRNA were as follows: set 1, 5′ GAAUCAGAUGCACAUGAGUTT 3′; set 2, 5′ ACGGCCUAAUCUAACAGACTT 3′ (24). Both sets of primers were pooled prior to transfection. SMARTpool siRNAs against HuR and GFP were purchased from Thermo Scientific. shRNA-mediated HuR knockdown has been described previously (14, 25).
Luciferase Assay
Luciferase and β-galactosidase measurements were performed as described previously (19). Briefly, TDP-43 3′UTR reporters, siRNAs, and a pSV40-β-galactosidase plasmid were cotransfected into U251 MG cells in triplicate. Cells were harvested 72 h after transfection, and lysates were prepared for further analysis. Luciferase activity was measured using a kit (Promega) and a Synergy 2 multimode microplate reader (Bio-Tek) and normalized to β-galactosidase activity. For translational efficiency, luciferase and β-galactosidase activity and respective mRNA levels were measured by qRT-PCR. Translational efficiency was calculated on the basis of the following equation: [Luciferase activity/Luciferase mRNA]/[β-galactosidase activity/β-galactosidase mRNA] as described previously (22).
RNA Binding Assay
The TDP-43 3′ UTR probe template was derived from the 900-nt proximal portion (900F) and used to transcribe a biotin-labeled probe with T7 RNA polymerase, as described previously (26). Templates and synthesis of RNA probes for TNF-α 3′ UTR and pBSK (negative control) have been described elsewhere (27). An ELISA-based RNA binding assay was used to assess RNA binding according to a protocol published previously (26). Briefly, recombinant GST-HuR protein (27) was affixed to the ELISA well and incubated with a biotinylated TDP-43, TNF-α 3′ UTR, or control riboprobe in RNA binding buffer. For cold probe competition, labeled TDP-43 probe (0.02 pmol) was added to the ELISA well in the presence of excess molar amounts of unlabeled TDP-43 3′ UTR or pBSK probes. Binding curves were calculated using GraphPad Prism Software v. 5.0 (GraphPad Software).
CFTR Minigene Assay
A CFTR minigene (21) was cotransfected with TDP-43, HuR, or control GFP siRNA into U251 cells using Lipofectamine 2000 (Invitrogen). The mRNA was assessed by RT-PCR for CFTR exon 9 splicing using the following primers: 5′-CAACTTCAAGCTCCTAAGCCACTGC-3′ and 5′-TAGGATCCGGTCACCAGGAAGTTGGTTAAATCA-3′.
Cell Viability and Neuronal Toxicity Assays
Cell viability/proliferation of astrocytes was assessed by Vialight assay (Lonza). For motor neuron and cortical neuron toxicity, cells were treated with 10% conditioned medium from HuR, TDP-43, or control GFP knockdown astrocytes in duplicate at 37 °C for 24 h. A total of three independent experiments were done with different motor neuron cultures. In treatment groups and controls, the yield of purified motor neurons was equivalent. Cleaved caspase 3-positive cells were counted in ten high-power fields in a blinded manner and expressed as a percentage of total cells. For cortical neurons, caspase 3/7 activity was measured using a SensoLyte AMC kit (AnaSpec).
Statistics
All statistical analyses were performed with GraphPad Prism V. 5 software using two-tailed Student's t test.
RESULTS
HuR Regulates TDP-43 and FUS/TLS
We used shRNA to knock down HuR in U251 clones expressing mutant or wild-type SOD1 and in primary astrocytes (14, 28). These clones have been used previously by our laboratory to assess dysregulation of VEGF mRNA processing in the presence of mutant SOD1 (14, 28). We achieved excellent knockdown of HuR compared with control shRNA, as determined by Western blot analysis (Fig. 1, A and B). In the U251 clones, there was a concomitant 3- to 4-fold attenuation of TDP-43 and FUS/TLS. Hsp70, another RNA binding protein, was not affected, indicating a specificity of regulation (29). In astrocytes, the attenuation of FUS/TLS was ∼3-fold in WT and G93A SOD1 astrocytes, whereas TDP-43 was suppressed more than 5-fold compared with control shRNA. We then assessed the effect of HuR overexpression using doxycycline-inducible HuR U251 clones described previously (14). We observed up-regulation of TDP-43 (2-fold) and FUS/TLS (3- to 6-fold) with induction of HuR (Fig. 1C). Again, there was no change in Hsp70 expression, indicating a specificity of the effect. As another method of HuR suppression, we used the small molecule inhibitor MS-444, which blocks HuR function through inhibition of its dimerization and cytoplasmic translocation (30). Astrocytes or U251 clones were treated for 24 h and then harvested. We observed more than 50% attenuation of TDP-43 and FUS/TLS in the U251 clones and primary astrocytes, without an obvious difference between mutant SOD1-expressing cells and wild-type controls (Fig. 2). For primary astrocytes, we checked hsp70 and did not see any attenuation with HuR knockdown. Overall, the effects seen in U251 clones and primary astrocytes were similar, which is consistent with our previous observations that these cell models have substantial overlap (15). To examine the effects of HuR knockdown on the stress response, we subjected wild-type and G93A SOD1 mouse astrocytes to hypoxia (1% O2) for 48 h (Fig. 3). We observed an ∼3- to 5-fold induction of TDP-43 and FUS/TLS by Western blot analysis, but not of Hsp70, in both cell types (Fig. 3A). HuR showed up-regulation as well, greater in wild-type than mutant astrocytes (1.5- versus 3-fold). With HuR silencing, we observed no induction of TDP-43 and FUS/TLS. Rather, there was an ∼60% attenuation of TDP-43 and ∼90% with FUS/TLS compared with control siRNA (Fig. 3B). With the control siRNA, we observed the induction of each RBP. Hsp70 did not show any appreciable differences. Therefore, depletion of HuR blunted the physiological up-regulation of TDP-43 and FUS/TLS in response to hypoxia in wild-type and mutant SOD1 astrocytes. To determine whether there was any reciprocal effect on HuR expression, we silenced TDP-43 or FUS/TLS in primary astrocytes (Fig. 4, A and B) but did not observe any change in HuR expression. Interestingly, we did not see a change in FUS/TLS with TDP-43 knockdown as reported elsewhere (7). As an additional control, we assessed primary astrocytes from a mouse in which the RNA binding protein KSRP (which contains KH-type RNA binding domains) was knocked out (16, 31). We observed no change in TDP-43 or FUS/TLS expression compared with astrocytes from littermate controls (Fig. 4C). These findings suggest that the cross-regulation is specific but not bidirectional and that HuR is an upstream regulator of TDP-43 and FUS/TLS.
FIGURE 1.
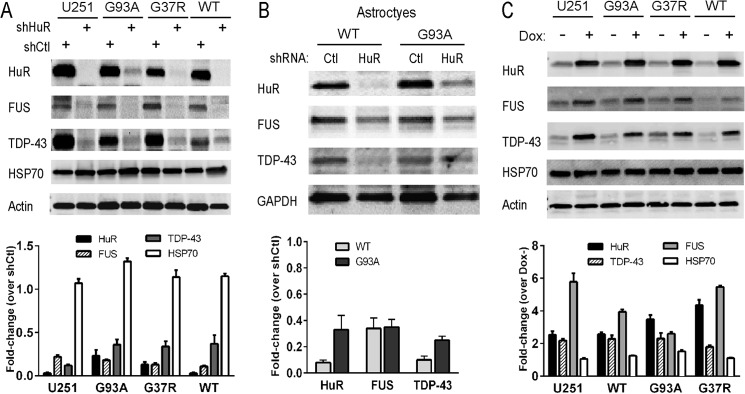
HuR promotes TDP-43 and FUS/TLS expression. A and B, Western blot analysis of cell extracts following shRNA-mediated silencing of HuR in U251 parent cells and U251 SOD1 clones (A) and primary astrocytes from WT or G93A SOD1 mutant mice (B). Ctl, control. C, Western blot of U251 clones (and parent cell line) constitutively expressing forms of SOD1 in which HuR is induced with doxycycline treatment. Antibodies are shown to the left of each blot. Quantitative densitometry is shown below each blot. Bands for the proteins were normalized to GAPDH or actin and then expressed as a fold change over the control (either siCtl for knockdown or doxycycline (Dox) for overexpression of HuR). Results are representative of three independent experiments.
FIGURE 2.

The HuR inhibitor MS-444 attenuates TDP-43 and FUS/TLS expression. A and B, Western blot analysis of wild-type and mutant SOD1 U251 clones (A) or cortical astrocytes (B) after treatment with MS-444 for 24 h (50 μm for astrocytes and 100 μm for U251 clones). Antibodies are shown to the left. Bottom panels, densitometric quantification of bands shown in the blots. Values for each protein were adjusted to the loading controls (GAPDH or actin) and are shown as a fold change over dimethyl sulfoxide (DMSO) vehicle control. The results are representative of three independent experiments.
FIGURE 3.

Hypoxia induction of TDP-43 and FUS/TLS is blocked with HuR silencing. A, primary astrocytes from WT or G93A mice were subjected to 1% O2 or normoxic conditions for 48 h and assessed by Western blot analysis. Antibodies are shown to the left of the blots. B, primary astrocytes were treated with siHuR or siGFP (control) RNAs and subjected to hypoxic conditions as in A. Bottom panels, quantitative densitometry was calculated as described in Fig. 1. Results are representative of three independent experiments.
FIGURE 4.
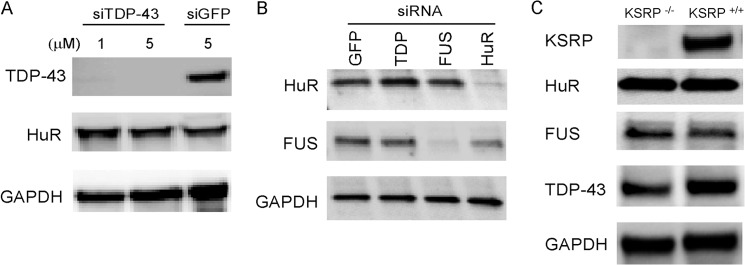
Knockdown of TDP-43, FUS/TLS, or KSRP does not affect HuR or other RBP expression in astrocytes. A, Western blot analysis of primary astrocytes after transfection with TDP-43 or control (siGFP) siRNAs. B, astrocytes were knocked with the siRNA shown, and extracts were assessed for HuR or FUS/TLS expression. C, extracts were prepared from KSRP−/− and KSRP+/+ (littermate) mouse astrocytes and assessed by Western blot analysis. Antibodies are shown to the left of the blots.
HuR Modulates Gene Expression via the 3′ UTR of TDP-43
The 3′ UTR of TDP-43 is lengthy (∼3.0 kb) and displays alternative splicing with different polyadenylation sites that produce short and long transcripts (7, 32). The proximal portion (initial 900 nt) of the 3′ UTR contains a number of cis elements that represent potential binding sites for HuR (Fig. 5A) (33, 34). We subcloned this part of the 3′ UTR (900F) downstream from the luciferase open reading frame to determine its impact on reporter expression. The reverse complement of this sequence was used as a control (900R). Plasmids were transfected into U251 cells along with a β-galactosidase control plasmid. Luciferase values were normalized to β-galactosidase activity. We knocked down TDP-43 in U251 cells and transfected the reporter plasmids (Fig. 5B, top panel). With 900R, we observed no difference in luciferase activity between siTDP-43 and control siRNA. The 900F reporter, however, showed a significant increase in luciferase activity when TDP-43 was knocked down (∼2.5-fold, p < 0.001), consistent with the negative autoregulatory effect observed previously (2). Interestingly, 900F contains a portion of the TDP-43-binding region that includes the major 34-nt sequence identified by cross-linking immunoprecipitation with a TDP-43 antibody (35). To confirm that the increased luciferase activity was related to changes in protein expression, we assessed luciferase protein by Western blot analysis and found a marked enhancement of expression in TDP-43-silenced cells (Fig. 5B, bottom panel). After HuR knockdown, there was a 3-fold attenuation (p < 0.001) of luciferase activity with 900F compared with control siRNA (Fig. 5C), with no effect on the control 3′ UTR. MS-444 treatment of U251 cells transfected with the 900F construct produced a dose-dependent reduction in luciferase activity (Fig. 5D). On the other hand, ectopic expression of HuR produced a nearly 4-fold increase (p < 0.001) in luciferase activity compared with the control (Fig. 5E). In summary, HuR modulates gene expression via the TDP-43 3′ UTR.
FIGURE 5.

HuR Regulates TDP-43 via the 3′ UTR. A, schematic of the luciferase reporter constructs used to assess the TDP-43 3′ UTR. The portion of the 3′UTR (900F) used in these experiments was cloned downstream of the luciferase coding region at the PflM1 site. Potential HuR binding sites are shown below (33, 34). A fragment representing the reverse complement (900R) was used as a control. TDPBR, TDP-43 binding region as defined by Ayala et al. (35). B, top panel, luciferase constructs were transfected into U251 cells along with TDP-43 or control siRNA. Luciferase activity was normalized to an internal transfection control (β-galactosidase). Bottom panel, Western blot analysis of protein extract from the same transfection experiment. C, the same as B, except HuR was knocked down with siRNA. Some cells (control) were not treated with any siRNA. D, U251 cells were transfected with the 900F construct and treated with vehicle (dimethyl sulfoxide (DMSO)) or MS-444 at the doses shown for 24 h. E, luciferase constructs were transfected into a U251 clone that can be induced to express HuR with doxycycline treatment (shaded boxes). Luciferase activity was normalized to the internal transfection control. All results are mean ± S.E. of at least three independent tests. **, p < 0.005; ***, p < 0.001.
Knockdown of HuR Affects the Translational Efficiency of TDP-43 mRNA
We next sought to determine how HuR regulates the expression of TDP-43. We first assessed the impact of HuR silencing or transgenic up-regulation on TDP-43 mRNA in the U251 clones. With HuR knockdown, we saw no significant changes in mRNA levels compared with shControl (Fig. 6A). Likewise, in astrocytes, there was no significant change in mRNA levels (data not shown). With ectopic HuR expression (after doxycycline induction), the mRNA levels remained unchanged (Fig. 6B). Assessment of TDP-43 mRNA decay showed no difference in the decay curves following actinomycin D treatment, indicating no change in RNA half-life up to the 6-h time frame (Fig. 6C). These data suggest that HuR was affecting translation. To address this possibility, we assessed translational efficiency using a method initially described by Vasudevan and Steitz (36) and, more recently, used in our laboratory with modification (22). In HuR-silenced U251 cells, we cotransfected the luciferase reporter containing the TDP-43 3′ UTR (900F) with a β-galactosidase reporter without regulatory elements in the 3′ UTR. The RNA levels of the reporters were measured by qRT-PCR in parallel with reporter activity. Translational efficiency, calculated as described previously (22), declined ∼5-fold compared with siGFP control or non-silenced parental cells (p < 0.0005, Fig. 6D). To further determine the possibility that HuR knockdown affects translational efficiency, we quantitated TDP-43 and FUS mRNAs in polysome fractions after HuR knockdown (Fig. 7). For polysome preparation, we used U251 cells treated with shHuR or shGFP control. We achieved excellent knockdown of HuR and, again, no differences in total mRNA levels compared with control (Fig. 7, A and B). Fractions were collected and analyzed by qRT-PCR (Fig. 7, C and D). We observed a prominent shift of TDP-43 and FUS/TLS mRNAs away from the polysome fractions in shHuR-treated cells versus the control. GAPDH mRNA was unaffected and localized predominantly to the polysome fractions. These data are consistent with the luciferase reporter results and support the conclusion that HuR promotes translational efficiency of TDP-43 and FUS/TLS.
FIGURE 6.

HuR does not affect TDP-43 mRNA expression or stability but suppresses translational efficiency. A and B, HuR was knocked down (A) or overexpressed (B) in U251 clones as indicated, and TDP-43 mRNA was measured by qRT-PCR and expressed as a percentage of the housekeeping gene S9. C, analysis of TDP-43 mRNA decay in U251 cells after transfection with siRNA to TDP-43, HuR, or GFP. Cells were treated with actinomycin D for the time interval indicated, followed by measurement of TDP-43 mRNA levels. RNA values are expressed as a percentage of the baseline value prior to actinomycin D treatment (time 0). D, the 900F luciferase construct (Fig. 5A) and a β-galactosidase control were transfected into U251 cells along with siRNA to HuR, GFP, or no siRNA (−). Luciferase and β-galactosidase activity and mRNA levels were measured, and translational efficiency was calculated by the following equation: [Luciferase activity / Luciferase mRNA] / [β-galactosidase activity / β-galactosidase mRNA] (see “Experimental Procedures”). All data points represent the mean ± S.E. of at least three independent tests. ***, p < 0.0005.
FIGURE 7.

HuR knockdown leads to shift of TDP-43 and FUS/TLS away from polysomes. A, Western blot analysis showing knockdown of HuR in U251 cells versus shControl (Ctl) prior to polysome fractionation. B, qRT-PCR showing total mRNA levels of TDP-43 and FUS/TLS after HuR knockdown. C, representative absorbance profile of cytoplasmic extract fractionated by sucrose gradient. D, percent distribution of TDP-43, FUS/TLS, and GAPDH mRNA in the fractions. RNA was quantitated by qRT-PCR. The experiment was repeated one time with similar results. RQ, relative quantity.
HuR Binds to the TDP-43 3′ UTR
The data so far indicate that HuR can modulate TDP-43 expression via the portion of the 3′ UTR that contains AU-rich elements. We next determined whether HuR could directly bind the TDP-43 3′ UTR. We performed RNA immunoprecipitation (RNA-IP) of U251 cells (Fig. 8A). We observed a pull-down of TDP-43 mRNA with a HuR antibody compared with the IgG control. RNA-IP with an anti-TDP-43 antibody also pulled down TDP-43 mRNA, but 5-fold less. To confirm that the pull-down was related to the 3′ UTR, cells were transfected with the 900F luciferase reporter plasmid and subjected to RNA-IP. The HuR antibody yielded ∼10-fold more luciferase mRNA than the TDP-43 antibody. Although these results suggest increased HuR binding to the 3′ UTR compared with TDP-43, we cannot exclude the possibility of different immunoprecipitation efficiencies between the antibodies. We next incubated biotin-labeled TDP-43 3′ UTR (900F) with lysates from cells transiently transfected with FLAG-HuR or FLAG-KSRP (control). The lysates were then immunoprecipitated with streptavidin-agarose beads. A Western blot analysis identified FLAG-HuR in the precipitate with anti-FLAG antibodies (Fig. 8B). A control biotinylated probe (pBSK) was negative for HuR, whereas the control with a known HuR target (TNF-α 3′ UTR) was positive. FLAG-KSRP was not detected with the TDP-43 3′ UTR pull-down. Because 900F contains a portion of the TDP-43-binding region, we wanted to determine whether HuR binding overlapped this region. We assessed HuR binding to this region (250F) versus the proximal 650 nt (650F) using biotinylated probes and the U251 doxycycline-inducible HuR cell line (Fig. 8C). With doxycycline induction, we observed pull-down of FLAG-HuR with 650F but not 250F, suggesting that HuR binding does not overlap with the TDP-43-binding region. We next assessed in vitro binding features with the TDP-43 3′ UTR using an ELISA-based RNA binding assay and recombinant HuR protein (26, 27, 37). We observed strong binding of HuR to TDP-43 3′ UTR, similar to that of TNF-α 3′ UTR, whereas no binding was observed with the control 3′ UTR (Fig. 9A). Likewise, HuR binding to TDP-43 3′ UTR showed a dose-dependent increase (Fig. 9B). Finally, unlabeled TDP-43 3′ UTR could compete for HuR binding, whereas unlabeled control 3′ UTR could not (Fig. 9C). In summary, HuR can specifically bind to the TDP-43 3′ UTR with a pattern similar to RNA ligands associated previously with HuR (38, 39).
FIGURE 8.
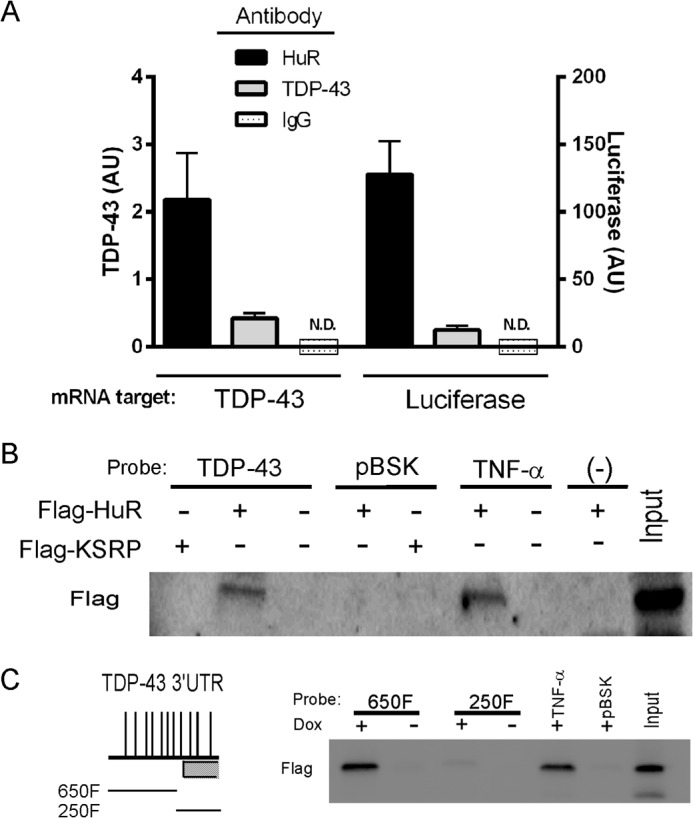
HuR binds to the TDP-43 3′UTR. A, U251MG cell extracts were subjected to RNA immunoprecipitation (RNA-IP) with the antibodies shown, and TDP-43 mRNA in the precipitate was quantified by qRT-PCR. U251 cells were also transfected with the 900F TDP-43 3′ UTR luciferase construct (Fig. 5) and immunoprecipitated with the same antibodies, and luciferase mRNA was quantified in the precipitate. AU, arbitrary units; ND, not detected. B, U251 MG cells were transiently transfected with FLAG-HuR or FLAG-KSRP expression plasmids, and lysates were incubated with biotin-labeled 3′UTR RNA probes for TDP-43, TNF-α, pBSK (control), or no probe (−) as shown. The extracts were precipitated with streptavidin beads and assessed by Western blot analysis using the antibodies shown on the left. C, the U251 doxycycline (Dox)-inducible FLAG-HuR clone was induced with doxycycline, and lysates were incubated with biotinylated TDP-43 3′ UTR probes derived from 900F (schematically shown to the left, see Fig. 5) and immunoprecipitated as in B.
FIGURE 9.
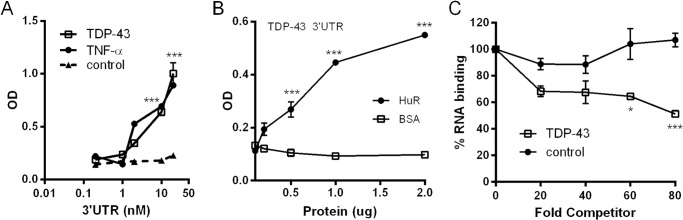
Recombinant HuR binds to the TDP-43 3′UTR. An ELISA-based RNA-binding assay was used to assess RNA binding (see “Experimental Procedures” and Ref. 26). With HuR affixed to the ELISA well, the binding reaction was initiated by addition of a biotinylated TDP-43, TNF-α 3′UTR, or control riboprobe. A, HuR binds to TDP-43 3′ UTR similarly as to the TNF-α 3′ UTR. B, HuR binding is protein concentration-dependent. Varying amounts of HuR or control protein (BSA) was adsorbed to the ELISA well prior to the RNA binding reaction. C, addition of unlabeled TDP43 3′ UTR, but not a control riboprobe, inhibits binding. All data represent the mean ± S.E. of three independent measurements. *, p < 0.01; ***, p < 0.001.
Knockdown of HuR Alters a Downstream Function of TDP-43
Because we established that TDP-43 expression is dependent on HuR, we next determined whether a downstream function of TDP-43 would be attenuated with HuR knockdown. A major role of TDP-43 is regulating alternative splicing, and exon 9 exclusion in the CFTR gene was the initial splicing function linked to TDP-43 (40, 41). We used a CFTR minigene (Fig. 10A) to assess splicing after HuR knockdown. U251 cells were treated with siRNA for HuR, TDP-43, or control and then transfected with the minigene. We assessed exon 9 inclusion/exclusion by RT-PCR as described previously (42). We observed a significant increase in exon 9 inclusion when either TDP-43 or HuR was knocked down compared with control siRNA (Fig. 10B). This molecular phenotype could be reversed by cotransfecting a GFP-TDP-43 expression plasmid that lacks the 3′ UTR regulatory elements (Fig. 10C). Therefore, knockdown of HuR reduced a downstream splicing function of TDP-43.
FIGURE 10.

Knockdown of HuR induces CFTR exon 9 skipping. A, schematic of the hybrid minigene containing CFTR exon 9 and its flanking introns. FP, forward primer; RP, reverse primer. B, the CFTR hybrid minigene was cotransfected with TDP-43, HuR, or control GFP siRNA into U251 cells, and the mRNA was assessed by RT-PCR for exon 9 splicing. A representative image is shown. The arrow depicts an aberrant splicing product from a cryptic 3′ splice site as previously described (40). Bottom panel, densitometric quantification of bands shown as a ratio of exon 9 inclusion (+) to exclusion (−). C, rescue study showing that transfection of a TDP-43 expression plasmid lacking regulatory elements in the 3′ UTR can reverse the splicing defect caused by HuR knockdown. Data points represent the mean ± S.E. of five independent tests. **, p < 0.01; ***, p < 0.001. The efficiency of HuR or TDP-43 knockdown was monitored by Western blot analysis.
Knockdown of HuR in Astrocytes Produces Motor Neuron and Cortical Neuron Toxicity
We have shown previously that posttranscriptional gene regulation can inhibit secretion of soluble factors by astrocytes, including inflammatory cytokines (16). The regulation of TDP-43 and FUS/TLS by HuR, as shown here, would have an additional impact because they can modulate (positively or negatively) a number of mRNA targets encoding secreted proteins (7, 43). Therefore, we investigated whether HuR knockdown in astrocytes could impact motor neuron viability. We collected astrocyte-conditioned medium (ACM) following knockdown with HuR, TDP-43, or control siRNA and added it to primary motor neurons in culture. The purity of motor neurons was determined by staining with anti-choline acetyltransferase antibodies (Fig. 11A). After 24 h, apoptosis was assessed by staining motor neurons with cleaved caspase 3 antibody (Fig. 11, B and C). We observed a significant 4-fold increase in apoptosis with siHuR ACM versus the control (p < 0.001). A similar toxic pattern was seen with TDP-43 knockdown. We also tested primary cortical neurons and found a 3- to 5-fold increase in apoptotic cells with siTDP-43 and siHuR ACM versus the control (Fig. 12, A and B). This was accompanied by a significant increase in caspase 3 and 7 activity, as measured directly in the culture well (Fig. 12C). To determine whether HuR or TDP-43 knockdown was inducing astrocyte cell death (and releasing toxic factors), we assessed cell viability by an ATP-based viability assay (Fig. 12D). We found no significant effect on astrocyte viability with any of the siRNAs compared with untreated cells, indicating that the toxic effect of the conditioned media was neural-specific. In summary, attenuation of HuR or TDP-43 shifted the astrocytes to a “toxic” phenotype.
FIGURE 11.
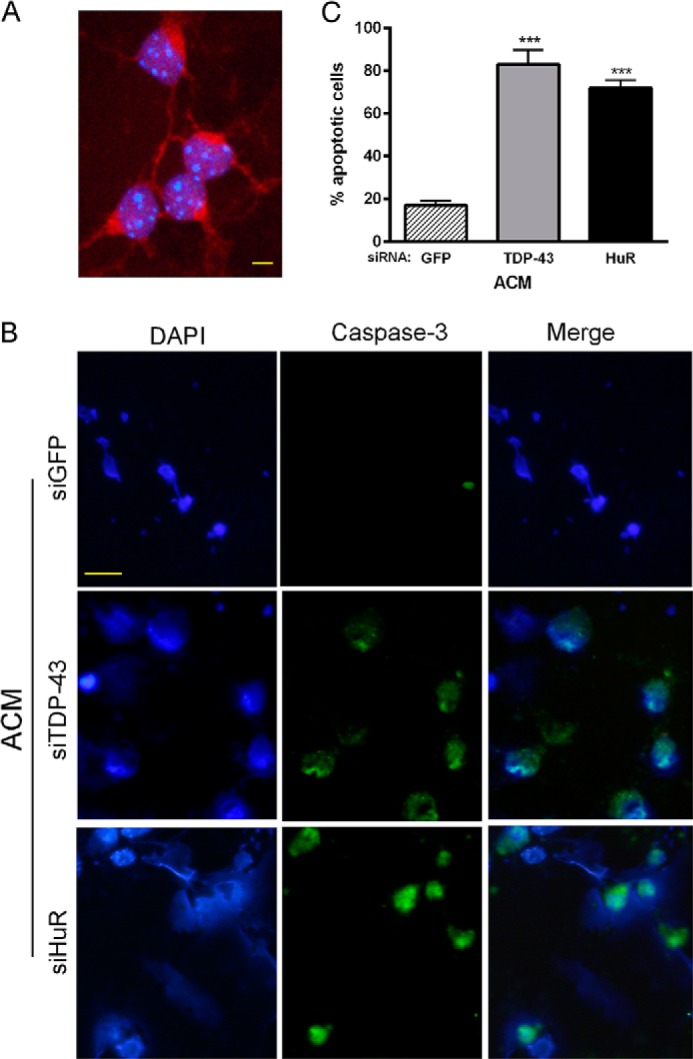
Conditioned medium from HuR and TDP-43 knockdown astrocytes is toxic to motor neurons. A, cultured motor neurons used in the assay were stained with choline acetyltransferase antibody (red) to verify purity. DAPI staining (blue) was used to identify nuclei. Scale bar = 20 μm. B, motor neurons were incubated with conditioned medium from astrocytes (ACM) in which TDP-43 or HuR was knocked down or transfected with the GFP control siRNA. Cells were fixed and stained for cleaved caspase 3 (green) and DAPI (blue). Scale bar = 50 μm. C, cells positive for cleaved caspase 3 staining were scored by a blinded observer and expressed as a percentage of total cells. Data points are the mean ± S.E. of 10 high-power fields from three independent experiments. ***, p < 0.001 compared with the siGFP control.
FIGURE 12.

Conditioned medium from HuR and TDP-43 knockdown astrocytes is toxic to cortical neurons but not astrocytes. A, cortical neurons were incubated with conditioned medium from astrocytes (ACM) in which TDP-43 or HuR was knocked down or were transfected with the GFP control siRNA. Cells were fixed and stained for cleaved caspase 3 (red) and DAPI (blue). STS, staurosporine. Scale bar = 50 μm. B, cells positive for cleaved caspase 3 staining were scored by a blinded observer and expressed as a percentage of total cells. Data points are the mean ± S.E. of 10 high-power fields from three independent experiments. C, caspase 3 and 7 activity in cortical neurons was measured directly in the tissue culture well after incubation with the ACM (siGFP was set at 1). D, astrocyte viability after siRNA transfection was assessed by an ATP-based assay. ***, p < 0.001 compared with siGFP control.
DISCUSSION
HuR plays a major role in regulating gene programs that govern a range of cellular functions, including stress response, inflammation, proliferation, and senescence (11, 44). The far-reaching effects of HuR on RNA metabolism stem, in part, from its regulation of other RNA regulators (5, 8, 9). Here we show that HuR regulates two RNA binding proteins, TDP-43 and FUS/TLS, which together regulate thousands of splicing events in coding and non-coding RNA targets (7, 43, 45). Indeed, following HuR knockdown, there was a significant attenuation of exon 9 exclusion in the CFTR gene, which is the first splicing function linked to TDP-43 (Fig. 10) (40). The cross-regulation observed here was unidirectional because neither TDP-43 nor FUS/TLS knockdown affected HuR expression. The hierarchy of RNA regulators is further underscored by a recent observation that TDP-43 binds to and regulates FUS/TLS mRNA in the brain (7). Interestingly, hnRNPA1, another RBP recently linked to ALS, was also identified as a possible mRNA target of HuR, although the effect on its posttranscriptional regulation has not been investigated (8, 43). These findings play into the concept of the ribonome in which RNA regulators are interconnected and coordinate the dynamic expression of a broad set of functionally related genes (46, 47). The auto- and cross-regulation of RBPs is necessary to maintain the proper balance for these coordinated responses.
Many of the downstream targets of TDP-43 and FUS/TLS, typically characterized by long introns, play vital roles in synaptic transmission, neurodevelopment, and neurological disease (7, 43, 45). The biological consequences of HuR knockdown in astrocytes, which would also affect non-TDP-43 and FUS/TLS mRNA targets, are likely complex. We did not observe any alterations in astrocyte viability, but HuR-silenced astrocytes produced soluble factors that were toxic to motor neurons (Fig. 11). Astrocytes play an important role in CNS homeostasis, including synaptic maintenance, blood-brain barrier and blood flow, neuroprotection, and energy production (48). The broad range of functions is attributable to a versatile molecular phenotype, including the array of factors capable of being secreted, which vary depending on the microenvironment and signals therein (e.g. hypoxia, inflammation, and tissue injury) (49, 50). The astrocyte may also contribute to disease pathology (49). In ALS, the astrocyte has been implicated in the propagation of motor neuron death (and, therefore, clinical disease), possibly through the secretion of toxic factors (51–56). Our data suggest that perturbation of posttranscriptional regulation by silencing HuR shifts the secretome of the astrocyte to a toxic one for motor neurons but not astrocytes themselves (Figs. 11 and 12). Because TDP-43 knockdown produced a similar toxic effect, it is possible that the phenotype of HuR knockdown resulted from concomitant TDP-43 attenuation. Indeed, a recent report by Yang et al. (55) found that partial in vivo knockdown of TDP-43 in glial cells in the spinal cord, without loss of expression in motor neurons, led to significant motor neuron cell loss. Another report, however, observed no motor neuron toxicity following TDP43 knockout in astrocytes, although a different model was used (on the basis of in vitro differentiated astrocytes from glial restricted precursor cells) (57). We also observed cortical neuron toxicity from these knockdowns, which potentially extends the ramifications of our findings to other neurodegenerative diseases (Fig. 12). A similar impact of altered posttranscriptional regulation in astrocytes has been described recently in our laboratory, where deletion of the RNA destabilizer KSRP led to toxicity of cortical neurons (16). Taken together, disruption of RNA homeostasis through alterations of RBPs can have a significant impact on astrocyte phenotype and neuronal maintenance. The nature of the toxic phenotype observed in this study has not yet been characterized.
Our data show that HuR positively regulates gene expression through the TDP-43 3′ UTR and that negative autoregulation could be overrun with increasing HuR expression (Fig. 5). Because of the proximity of AU-rich elements with the TDP-43 binding region, it is possible that the two proteins may compete in some way, although our findings indicate that HuR binding does not overlap a major binding locus for TDP-43 (Fig. 8) (35). Our data imply that there is a balance between the positive effects of HuR and the negative autoregulatory effects of TDP-43 to control expression. This balance could be perturbed with HuR overexpression, knockdown, or inhibition, resulting in the up- or down-regulation of TDP-43 (Figs. 1–3 and 5). Likewise, FUS/TLS expression levels could be altered with changes in HuR expression. This finding is relevant to neurodegenerative disease, where cytoplasmic mislocalization of mutant TDP-43 or FUS/TLS can circumvent the normal autoregulatory pathways that curtail expression (3, 58). Therefore, a possibly unfettered positive regulation by HuR in these pathological states could contribute to the accumulation of cytosolic TDP-43 of FUS/TLS.
Interestingly, knockdown of HuR did not alter the TDP-43 mRNA level or half-life. Rather, there was a loss of translational efficiency (Figs. 6 and 7). RNA stability and translational efficiency, although closely connected, are distinct levels of regulation, and HuR can modulate one process without affecting the other for a given RNA target (10, 59). A similar dissociation of RNA stability from translational efficiency was observed with cytochrome c, where HuR augmented protein expression but did not affect mRNA levels (60). The role of HuR in regulating translation is complex, with evidence of both positive and negative effects either through the 5′ or 3′ UTR (11, 60–63). Enhanced translation is often linked to HuR localization to the cytosol, its association with polysomes, and/or competition with silencing miRNAs (10, 12, 64). Although we did not test the role of the 3′ UTR in HuR regulation of FUS/TLS, the pattern of polysome dissociation with HuR knockdown was similar to TDP-43 mRNA, suggesting that HuR regulates its translational efficiency. The 3′ UTR of FUS/TLS contains many putative AU-rich elements and was identified in a recent analysis of 3′ UTRs as a possible binding target of HuR (8).
There was no obvious impact of mutant SOD1 on HuR regulation of TDP-43 or FUS/TLS in U251 cells or astrocytes, unlike with endothelial growth factor (VEGF), where mutant SOD1 suppressed RNA and protein expression and shortened the RNA half-life (14, 15). Therefore, the biochemical effect of mutant SOD1 is target-specific despite its capacity to bind different AU-rich 3′ UTRs (37).
HuR plays an important role in the cellular responses to stress, including heat shock, UV irradiation, oxidative stress, glucose deprivation, and hypoxia (10, 12, 13, 65). With hypoxia, we saw an up-regulation of HuR in astrocytes and a concomitant increase in TDP-43 and FUS/TLS (Fig. 3), indicating a physiological stressor in which this cross-regulation comes into play. With hypoxic exposure, HuR promotes the expression of key response genes, such as VEGF and Hif-1α, through posttranscriptional pathways (65). Both TDP-43 and FUS/TLS are also stress response proteins that can translocate to the cytoplasm and assemble into stress granules with different triggers, including heat shock, oxidants, and osmotic shifts (66–69). These granules are considered part of a survival mechanism whereby the translational apparatus is broken down for certain mRNAs in favor of those that promote damage repair and stress response (70). The downstream impact of hypoxia-induced TDP-43 and FUS/TLS up-regulation via HuR in astrocytes remains to be determined.
In summary, we show that HuR is a regulator of ALS-associated TDP-43 and FUS/TLS and, as a consequence, their downstream functions. Our findings reinforce the concept that there is a balance within the RNA world that is maintained by tightly regulated networks of RNA binding proteins. Disruption of this balance can have devastating consequences, possibly aggravating the accumulation of toxic proteins or, as in this report, creating a toxic astrocyte phenotype.
Acknowledgments
We thank Dr. Laurent Corcos (Faculty of Medicine, Brest Cedex 3, France) for advice regarding the cftr minigene experiments and Drs. Claudia Fallini and Gary Bassell (Emory University, Atlanta, GA), for teaching us how to isolate and culture motor neurons. We also acknowledge the University of Alabama Tissue Core Facility for assistance with neuronal cultures.
This work was supported, in whole or in part, by NINDS, National Intitutes of Health Grants NS064133, R21NS081743, and R21NS085497 (to P. H. K.) and NS057664 (to L. L.). This work was also supported by a Merit Review award from the Department of Veterans Affairs (to P. H. K.).
- ALS
- amyotrophic lateral sclerosis
- ChAT
- choline acetyltransferase
- CFTR
- cystic fibrosis transmembrane conductance regulator
- FUS/TLS
- fused in sarcoma/translocated in liposarcoma
- HuR
- Hu antigen R
- KSRP
- KH-type splicing regulatory protein
- RRM
- RNA recognition motif
- RBP
- RNA binding protein
- RNA-IP
- RNA immunoprecipitation
- TDP-43
- TAR DNA-binding protein 43
- TDPBR
- TDP-43 binding region
- TIAR
- TIA-1 related protein
- TNF-α
- tumor necrosis factor α
- UTR
- untranslated region
- VEGF
- vascular endothelial growth factor
- qRT-PCR
- quantitative real-time PCR
- ACM
- astrocyte-conditioned medium
- nt
- nucleotide.
REFERENCES
- 1. Robberecht W., Philips T. (2013) The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 14, 248–264 [DOI] [PubMed] [Google Scholar]
- 2. Buratti E., Baralle F. E. (2011) TDP-43: new aspects of autoregulation mechanisms in RNA binding proteins and their connection with human disease. FEBS J. 278, 3530–3538 [DOI] [PubMed] [Google Scholar]
- 3. Zhou Y., Liu S., Liu G., Oztürk A., Hicks G. G. (2013) ALS-associated FUS mutations result in compromised FUS alternative splicing and autoregulation. PLoS Genet. 9, e1003895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huelga S. C., Vu A. Q., Arnold J. D., Liang T. Y., Liu P. P., Yan B. Y., Donohue J. P., Shiue L., Hoon S., Brenner S., Ares M., Jr., Yeo G. W. (2012) Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep. 1, 167–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pullmann R., Jr., Kim H. H., Abdelmohsen K., Lal A., Martindale J. L., Yang X., Gorospe M. (2007) Analysis of turnover and translation regulatory RNA-binding protein expression through binding to cognate mRNAs. Mol. Cell Biol. 27, 6265–6278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ni J. Z., Grate L., Donohue J. P., Preston C., Nobida N., O'Brien G., Shiue L., Clark T. A., Blume J. E., Ares M., Jr. (2007) Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev. 21, 708–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Polymenidou M., Lagier-Tourenne C., Hutt K. R., Huelga S. C., Moran J., Liang T. Y., Ling S. C., Sun E., Wancewicz E., Mazur C., Kordasiewicz H., Sedaghat Y., Donohue J. P., Shiue L., Bennett C. F., Yeo G. W., Cleveland D. W. (2011) Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 14, 459–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dassi E., Zuccotti P., Leo S., Provenzani A., Assfalg M., D'Onofrio M., Riva P., Quattrone A. (2013) Hyper conserved elements in vertebrate mRNA 3′-UTRs reveal a translational network of RNA-binding proteins controlled by HuR. Nucleic Acids Res. 41, 3201–3216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mukherjee N., Corcoran D. L., Nusbaum J. D., Reid D. W., Georgiev S., Hafner M., Ascano M., Jr., Tuschl T., Ohler U., Keene J. D. (2011) Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol. Cell 43, 327–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Abdelmohsen K., Kuwano Y., Kim H. H., Gorospe M. (2008) Posttranscriptional gene regulation by RNA-binding proteins during oxidative stress: implications for cellular senescence. Biol. Chem. 389, 243–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Srikantan S., Gorospe M. (2012) HuR function in disease. Front. Biosci. (Landmark Ed) 17, 189–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brennan C. M., Steitz J. A. (2001) HuR and mRNA stability. Cell. Mol. Life Sci. 58, 266–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Burkhart R. A., Pineda D. M., Chand S. N., Romeo C., Londin E. R., Karoly E. D., Cozzitorto J. A., Rigoutsos I., Yeo C. J., Brody J. R., Winter J. M. (2013) HuR is a post-transcriptional regulator of core metabolic enzymes in pancreatic cancer. RNA Biol. 10, 1312–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu L., Wang S., Zheng L., Li X., Suswam E. A., Zhang X., Wheeler C. G., Nabors L. B., Filippova N., King P. H. (2009) Amyotrophic lateral sclerosis-linked mutant SOD1 sequesters Hu antigen R (HuR) and TIA-1 related protein (TIAR): implications for impaired posttranscriptional regulation of vascular endothelial growth factor. J. Biol. Chem. 284, 33989–33998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lu L., Zheng L., Viera L., Suswam E., Li Y., Li X., Estévez A. G., King P. H. (2007) Mutant Cu/Zn-superoxide dismutase associated with amyotrophic lateral sclerosis destabilizes vascular endothelial growth factor mRNA and downregulates its expression. J. Neurosci. 27, 7929–7938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li X., Lin W. J., Chen C. Y., Si Y., Zhang X., Lu L., Suswam E., Zheng L., King P. H. (2012) KSRP: A checkpoint for inflammatory cytokine production in astrocytes. Glia 60, 1773–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fallini C., Bassell G. J., Rossoll W. (2010) High-efficiency transfection of cultured primary motor neurons to study protein localization, trafficking, and function. Mol. Neurodegener. 5, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kaech S., Banker G. (2006) Culturing hippocampal neurons. Nat. Protoc. 1, 2406–2415 [DOI] [PubMed] [Google Scholar]
- 19. Nabors L. B., Suswam E., Huang Y., Yang X., Johnson M. J., King P. H. (2003) Tumor necrosis factor-α induces angiogenic factor up-regulation in malignant glioma cells: a role for RNA stabilization and HuR. Cancer Res. 63, 4181–4187 [PubMed] [Google Scholar]
- 20. Lin W.-J., Duffy A., Chen C.-Y. (2007) Localization of AU-rich element-containing mRNA in cytoplasmic granules containing exosome subunits. J. Biol. Chem. 282, 19958–19968 [DOI] [PubMed] [Google Scholar]
- 21. Dujardin G., Buratti E., Charlet-Berguerand N., Martins de Araujo M., Mbopda A., Le Jossic-Corcos C., Pagani F., Ferec C., Corcos L. (2010) CELF proteins regulate CFTR pre-mRNA splicing: essential role of the divergent domain of ETR-3. Nucleic Acids Res. 38, 7273–7285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Suswam E. A., Shacka J. J., Walker K., Lu L., Li X., Si Y., Zhang X., Zheng L., Nabors L. B., Cao H., King P. H. (2013) Mutant tristetraprolin: a potent inhibitor of malignant glioma cell growth. J. Neurooncol. 113, 195–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Suswam E. A., Nabors L. B., Huang Y., Yang X., King P. H. (2005) IL-1β induces stabilization of IL-8 mRNA in malignant breast cancer cells via the 3′ untranslated region: involvement of divergent RNA-binding factors HuR, KSRP and TIAR. Int. J. Cancer 113, 911–919 [DOI] [PubMed] [Google Scholar]
- 24. Iguchi Y., Katsuno M., Niwa J., Yamada S., Sone J., Waza M., Adachi H., Tanaka F., Nagata K., Arimura N., Watanabe T., Kaibuchi K., Sobue G. (2009) TDP-43 depletion induces neuronal cell damage through dysregulation of Rho family GTPases. J. Biol. Chem. 284, 22059–22066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Filippova N., Yang X., Wang Y., Gillespie G. Y., Langford C., King P. H., Wheeler C., Nabors L. B. (2011) The RNA-binding protein HuR promotes glioma growth and treatment resistance. Mol. Cancer Res. 9, 648–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. King P. H. (2000) RNA-binding analyses of HuC and HuD with the VEGF and c-myc 3′-untranslated regions using a novel ELISA-based assay. Nucleic Acids Res. 28, E20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nabors L. B., Gillespie G. Y., Harkins L., King P. H. (2001) HuR, an RNA stability factor, is expressed in malignant brain tumors and binds to adenine and uridine-rich elements within the 3′ untranslated regions of cytokine and angiogenic factor mRNAs. Cancer Res. 61, 2154–2161 [PubMed] [Google Scholar]
- 28. Filippova N., Yang X., King P., Nabors L. B. (2012) Phosphoregulation of the RNA-binding protein Hu antigen R (HuR) by Cdk5 affects centrosome function. J. Biol. Chem. 287, 32277–32287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Henics T., Nagy E., Oh H. J., Csermely P., von Gabain A., Subjeck J. R. (1999) Mammalian Hsp70 and Hsp110 proteins bind to RNA motifs involved in mRNA stability. J. Biol. Chem. 274, 17318–17324 [DOI] [PubMed] [Google Scholar]
- 30. Meisner N.-C., Hintersteiner M., Mueller K., Bauer R., Seifert J.-M., Naegeli H.-U., Ottl J., Oberer L., Guenat C., Moss S., Harrer N., Woisetschlaeger M., Buehler C., Uhl V., Auer M. (2007) Identification and mechanistic characterization of low-molecular-weight inhibitors for HuR. Nat. Chem. Biol. 3, 508–515 [DOI] [PubMed] [Google Scholar]
- 31. Lin W. J., Zheng X., Lin C. C., Tsao J., Zhu X., Cody J. J., Coleman J. M., Gherzi R., Luo M., Townes T. M., Parker J. N., Chen C. Y. (2011) Posttranscriptional control of type I interferon genes by KSRP in the innate immune response against viral infection. Mol. Cell Biol. 31, 3196–3207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Avendaño-Vázquez S. E., Dhir A., Bembich S., Buratti E., Proudfoot N., Baralle F. E. (2012) Autoregulation of TDP-43 mRNA levels involves interplay between transcription, splicing, and alternative polyA site selection. Genes Dev. 26, 1679–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ray D., Kazan H., Chan E. T., Peña Castillo L., Chaudhry S., Talukder S., Blencowe B. J., Morris Q., Hughes T. R. (2009) Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nat. Biotechnol. 27, 667–670 [DOI] [PubMed] [Google Scholar]
- 34. Lebedeva S., Jens M., Theil K., Schwanhäusser B., Selbach M., Landthaler M., Rajewsky N. (2011) Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol. Cell 43, 340–352 [DOI] [PubMed] [Google Scholar]
- 35. Ayala Y. M., De Conti L., Avendaño-Vázquez S. E., Dhir A., Romano M., D'Ambrogio A., Tollervey J., Ule J., Baralle M., Buratti E., Baralle F. E. (2011) TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 30, 277–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vasudevan S., Steitz J. A. (2007) AU-rich-element-mediated upregulation of translation by FXR1 and Argonaute 2. Cell 128, 1105–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li X., Lu L., Bush D. J., Zhang X., Zheng L., Suswam E. A., King P. H. (2009) Mutant copper-zinc superoxide dismutase associated with amyotrophic lateral sclerosis binds to adenine/uridine-rich stability elements in the vascular endothelial growth factor 3′-untranslated region. J. Neurochem. 108, 1032–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fialcowitz E. J., Brewer B. Y., Keenan B. P., Wilson G. M. (2005) A Hairpin-like structure within an AU-rich mRNA-destabilizing element regulates trans-factor binding selectivity and mRNA decay kinetics. J. Biol. Chem. 280, 22406–22417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ma W.-J., Cheng S., Campbell C., Wright A., Furneaux H. (1996) Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. J. Biol. Chem. 271, 8144–8151 [DOI] [PubMed] [Google Scholar]
- 40. Buratti E., Dörk T., Zuccato E., Pagani F., Romano M., Baralle F. E. (2001) Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 20, 1774–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Buratti E., Baralle F. E. (2001) Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J. Biol. Chem. 276, 36337–36343 [DOI] [PubMed] [Google Scholar]
- 42. Pagani F., Buratti E., Stuani C., Romano M., Zuccato E., Niksic M., Giglio L., Faraguna D., Baralle F. E. (2000) Splicing factors induce cystic fibrosis transmembrane regulator exon 9 skipping through a nonevolutionary conserved intronic element. J. Biol. Chem. 275, 21041–21047 [DOI] [PubMed] [Google Scholar]
- 43. Lagier-Tourenne C., Polymenidou M., Hutt K. R., Vu A. Q., Baughn M., Huelga S. C., Clutario K. M., Ling S. C., Liang T. Y., Mazur C., Wancewicz E., Kim A. S., Watt A., Freier S., Hicks G. G., Donohue J. P., Shiue L., Bennett C. F., Ravits J., Cleveland D. W., Yeo G. W. (2012) Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 15, 1488–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Simone L. E., Keene J. D. (2013) Mechanisms coordinating ELAV/Hu mRNA regulons. Curr. Opin. Genet. Dev. 23, 35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tollervey J. R., Curk T., Rogelj B., Briese M., Cereda M., Kayikci M., König J., Hortobágyi T., Nishimura A. L., Zupunski V., Patani R., Chandran S., Rot G., Zupan B., Shaw C. E., Ule J. (2011) Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 14, 452–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Keene J. D., Tenenbaum S. A. (2002) Eukaryotic mRNPs may represent posttranscriptional operons. Mol. Cell 9, 1161–1167 [DOI] [PubMed] [Google Scholar]
- 47. Mansfield K. D., Keene J. D. (2009) The ribonome: a dominant force in co-ordinating gene expression. Biol. Cell 101, 169–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Blackburn D., Sargsyan S., Monk P. N., Shaw P. J. (2009) Astrocyte function and role in motor neuron disease: a future therapeutic target? Glia 57, 1251–1264 [DOI] [PubMed] [Google Scholar]
- 49. Sofroniew M. V., Vinters H. V. (2010) Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dong Y., Benveniste E. N. (2001) Immune function of astrocytes. Glia 36, 180–190 [DOI] [PubMed] [Google Scholar]
- 51. Haidet-Phillips A. M., Hester M. E., Miranda C. J., Meyer K., Braun L., Frakes A., Song S., Likhite S., Murtha M. J., Foust K. D., Rao M., Eagle A., Kammesheidt A., Christensen A., Mendell J. R., Burghes A. H., Kaspar B. K. (2011) Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 29, 824–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nagai M., Re D. B., Nagata T., Chalazonitis A., Jessell T. M., Wichterle H., Przedborski S. (2007) Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yamanaka K., Chun S. J., Boillee S., Fujimori-Tonou N., Yamashita H., Gutmann D. H., Takahashi R., Misawa H., Cleveland D. W. (2008) Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 11, 251–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bi F., Huang C., Tong J., Qiu G., Huang B., Wu Q., Li F., Xu Z., Bowser R., Xia X. G., Zhou H. (2013) Reactive astrocytes secrete lcn2 to promote neuron death. Proc. Natl. Acad. Sci. U.S.A. 110, 4069–4074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yang C., Wang H., Qiao T., Yang B., Aliaga L., Qiu L., Tan W., Salameh J., McKenna-Yasek D. M., Smith T., Peng L., Moore M. J., Brown R. H., Jr., Cai H., Xu Z. (2014) Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 111, E1121–E1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Di Giorgio F. P., Carrasco M. A., Siao M. C., Maniatis T., Eggan K. (2007) Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat. Neurosci. 10, 608–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Haidet-Phillips A. M., Gross S. K., Williams T., Tuteja A., Sherman A., Ko M., Jeong Y. H., Wong P. C., Maragakis N. J. (2013) Altered astrocytic expression of TDP-43 does not influence motor neuron survival. Exp. Neurol. 250, 250–259 [DOI] [PubMed] [Google Scholar]
- 58. Buratti E., Baralle F. E. (2012) TDP-43: gumming up neurons through protein-protein and protein-RNA interactions. Trends Biochem. Sci. 37, 237–247 [DOI] [PubMed] [Google Scholar]
- 59. Barreau C., Paillard L., Osborne H. B. (2005) AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 33, 7138–7150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kawai T., Lal A., Yang X., Galban S., Mazan-Mamczarz K., Gorospe M. (2006) Translational control of cytochrome c by RNA-binding proteins TIA-1 and HuR. Mol. Cell Biol. 26, 3295–3307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kullmann M., Göpfert U., Siewe B., Hengst L. (2002) ELAV/Hu proteins inhibit p27 translation via an IRES element in the p27 5′UTR. Genes Dev. 16, 3087–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Meng Z., Jackson N. L., Choi H., King P. H., Emanuel P. D., Blume S. W. (2008) Alterations in RNA-binding activities of IRES-regulatory proteins as a mechanism for physiological variability and pathological dysregulation of IGF-IR translational control in human breast tumor cells. J. Cell Physiol. 217, 172–183 [DOI] [PubMed] [Google Scholar]
- 63. Durie D., Lewis S. M., Liwak U., Kisilewicz M., Gorospe M., Holcik M. (2011) RNA-binding protein HuR mediates cytoprotection through stimulation of XIAP translation. Oncogene 30, 1460–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Meisner N.-C., Filipowicz W. (2011) Properties of the regulatory RNA-binding protein HuR and its role in controlling miRNA repression (Großhans H. ed.) pp. 106–123, Springer, New York: [DOI] [PubMed] [Google Scholar]
- 65. Gorospe M., Tominaga K., Wu X., Fähling M., Ivan M. (2011) Post-transcriptional control of the hypoxic response by RNA-binding proteins and microRNAs. Front. Mol. Neurosci. 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sama R. R., Ward C. L., Kaushansky L. J., Lemay N., Ishigaki S., Urano F., Bosco D. A. (2013) FUS/TLS assembles into stress granules and is a prosurvival factor during hyperosmolar stress. J. Cell Physiol. 228, 2222–2231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bosco D. A., Lemay N., Ko H. K., Zhou H., Burke C., Kwiatkowski T. J., Jr., Sapp P., McKenna-Yasek D., Brown R. H., Jr., Hayward L. J. (2010) Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 19, 4160–4175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Colombrita C., Zennaro E., Fallini C., Weber M., Sommacal A., Buratti E., Silani V., Ratti A. (2009) TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 111, 1051–1061 [DOI] [PubMed] [Google Scholar]
- 69. Vaccaro A., Tauffenberger A., Ash P. E., Carlomagno Y., Petrucelli L., Parker J. A. (2012) TDP-1/TDP-43 regulates stress signaling and age-dependent proteotoxicity in Caenorhabditis elegans. PLoS Genet. 8, e1002806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Anderson P., Kedersha N. (2006) RNA granules. J. Cell Biol. 172, 803–808 [DOI] [PMC free article] [PubMed] [Google Scholar]