

Background: The selectivity profiles of the closely related chemokine-binding proteins Evasin-1 and -4 differ.
Results: Using phage display, we identified the N-terminal region of Evasin-4 as key for the interaction with CC chemokines.
Conclusion: Evasin-1 and -4 use different domains for target binding.
Significance: Phage display allowed rapid insight into their different selectivities, which could aid rational design of inhibitory proteins.
Keywords: Cell Migration, Chemokine, Chemotaxis, Epitope Mapping, Phage Display, Chemokine Binding Protein, Pharmacophore
Abstract
To elucidate the ligand-binding surface of the CC chemokine-binding proteins Evasin-1 and Evasin-4, produced by the tick Rhipicephalus sanguineus, we sought to identify the key determinants responsible for their different chemokine selectivities by expressing Evasin mutants using phage display. We first designed alanine mutants based on the Evasin-1·CCL3 complex structure and an in silico model of Evasin-4 bound to CCL3. The mutants were displayed on M13 phage particles, and binding to chemokine was assessed by ELISA. Selected variants were then produced as purified proteins and characterized by surface plasmon resonance analysis and inhibition of chemotaxis. The method was validated by confirming the importance of Phe-14 and Trp-89 to the inhibitory properties of Evasin-1 and led to the identification of a third crucial residue, Asn-88. Two amino acids, Glu-16 and Tyr-19, were identified as key residues for binding and inhibition of Evasin-4. In a parallel approach, we identified one clone (Y28Q/N60D) that showed a clear reduction in binding to CCL3, CCL5, and CCL8. It therefore appears that Evasin-1 and -4 use different pharmacophores to bind CC chemokines, with the principal binding occurring through the C terminus of Evasin-1, but through the N-terminal region of Evasin-4. However, both proteins appear to target chemokine N termini, presumably because these domains are key to receptor signaling. The results also suggest that phage display may offer a useful approach for rapid investigation of the pharmacophores of small inhibitory binding proteins.
Introduction
The recruitment of leukocytes to sites of inflammation is highly dependent on the activity of chemokines. These proteins are small chemoattractant cytokines that are classified into four subfamilies (CC, CXC, C, and CX3C chemokines) based on the conservation of two cysteines at their N termini. Following immobilization on cell surface glycosaminoglycans, chemokines interact with specific seven-transmembrane G protein-coupled receptors, leading to intracellular signaling and migration of leukocytes to the inflammatory sites, a process that is crucial to the immune response (1).
To avoid detection, parasites and pathogens have developed several strategies to subvert the immune system of infected hosts, including the secretion of cytokine inhibitors, analgesic molecules, and evasion from complement-mediated killing (2). One particular cytokine inhibitor strategy is the production of chemokine-binding proteins (CKBP),2 which have been identified in several organisms, including viruses, worms, and arthropods (3). These CKBPs inhibit the activity of chemokines by blocking their interaction with specific receptor(s) or by inhibiting their interaction with glycosaminoglycans, and some CKBPs are able to simultaneously prevent both interactions (4). CKBPs exhibit differences in terms of their selectivity for chemokines: proteins from the 35-kDa virally encoded family only bind and neutralize CC chemokines (5); the poxvirus CKBP M-T7 binds interferon-γ as well as all chemokine classes; M3 isolated from an herpesvirus binds chemokines from all four subfamilies (6); the viral CKBPs that contain the SECRET (smallpox virus-encoded chemokine receptor) domain recognize a limited number of chemokines from the CC, CXC, and CX3C subfamilies in addition to binding TNFα (7). We have recently described the cloning and the characterization of Evasin-4, the third member of a family of tick-derived CKBPs that we have collectively termed Evasins (8). In contrast to Evasin-1 and -3, which are highly selective (9, 10), Evasin-4 displays a broad selectivity and binds at least 20 CC chemokines although it does not recognize members of the CXC, XC, and CX3C subfamilies.
Several studies aimed at identifying amino acids that are involved in the interaction between antagonistic binding proteins and their targets have revealed a number of critical residues on the surface of chemokines (11–15). The crystal structure of a complex between CCL4 and the viral CC chemokine inhibitor (vCCI), a 35-kDa CKBP, as well as mutagenesis studies of the interaction between vCCI and CCL5 have highlighted the importance of vCCI residues Asp-141, Glu-143, and Tyr-217 (16, 17). In particular, the interaction between Glu-143 and a conserved positively charged residue (an arginine or a lysine) at position 18 in the mature chemokine sequence is thought to play an essential role in forming and/or stabilizing the complex. These results were confirmed by the analysis of the vCCI·CCL11 complex by NMR (15). The subsequent solution of A41 and CrmD structures, two CKBPs with different specificities, reveals an overall β-sandwich fold similar to that of vCCI, despite a sequence conservation of <20% (18, 19). However, these three CKBPs do not use the same inhibition strategy: vCCI and A41 bind chemokines mainly via their β-sheet II, whereas CrmD exhibits a negatively charged β-sheet I interaction with the N and 40s loops of the chemokine. Finally, the structure of the broad spectrum CKBP M3 in complex with CCL2 reveals a 2:2 stoichiometry, with a homodimer of M3 bound to two independent CCL2 monomers (20). Importantly, M3 binds to the N-terminal region of CCL2, which, in addition to being important for signaling, contains a proline at position 8 that is responsible for chemokine dimerization. This interaction essentially mimics the chemokine dimerization interface; it also blocks residues involved in glycosaminoglycan binding. Because dimerization, glycosaminoglycan binding, and the N-terminal signaling domain are essential for CCL2 activity, M3 seems to be a particularly well designed inhibitor (21, 22). It should be noted that in all chemokine·CKBP complexes observed so far, patches of residues rather than distinct single amino acids are involved in the interaction. Moreover, the amino acids identified as critical for the interaction are frequently not conserved between chemokines bound by the CKBP and may also be present in chemokines that are not targets of the CKBP (17–20).
The Evasins are strikingly different from other CKBPs in that they are small, with molecular masses ranging from 7–11 kDa. The structures of Evasin-1 alone and in complex with CCL3, and the structure of Evasin-3 have been solved, demonstrating novel folds. Furthermore, the Evasin-1·CCL3 complex reveals a unique mechanism of binding compared with other CKBP structures solved to date (23). Sequence analysis predicts that Evasin-4 will have the same fold as Evasin-1 because the positions of the cysteines, predicted to form disulfide bridges, are conserved in both proteins, and the secondary structure of Evasin-4 is predicted to be similar to that of Evasin-1. The structure of the complex of Evasin-1 with CCL3 revealed two prominent π-π interactions between the chemokine and the CKBP, involving amino acids Phe-14 and Trp-89, respectively. However, mutation of these two residues did not completely abolish binding, which motivated the current investigation to: (i) define the precise binding interactions that confer strict chemokine selectivity to Evasin-1 and (ii) understand the interactions that enable many members of the CC chemokine class to bind Evasin-4. To this end, we have explored the potential of phage display as a tool for the preliminary mapping of these interactions.
Here we show, for the first time, the successful display of functional CKBPs from the Evasin family on the surface of filamentous phage. Many immunoglobulins and related cysteine-containing eukaryotic proteins, in addition to enzymes, short peptides, and various artificial scaffolds, have been functionally displayed as fusions to filamentous phage coat proteins on the surface of phage (3, 24–26). Phage display allows a direct link between the nucleotide sequence encoded by the phage(mid) and the phenotype of the displayed protein variant or protein variant mutant library and is a powerful approach for studying ligand binding without the need for individually expressing and chromatographically purifying large numbers of individual protein variants. Using this technique, we report progress toward identifying sequence determinants that confer a broad yet selective binding profile of Evasin-4 for CC chemokines and confirm the importance of Phe-14 and Trp-89 for the interaction of Evasin-1 with CCL3. Phage-displayed alanine mutants of Evasin-4 led to the identification of Glu-16 and Tyr-19 as residues crucial for its interaction with several chemokines, and phage-displayed Evasin-1 mutants identified Phe-14, Asn-88, and Trp-89 as key residues for its inhibitory activity. The data also highlight the differential mode of chemokine recognition by these CKBPs, with the carboxyl terminus of Evasin-1 and amino terminus of Evasin-4 being important. Lastly, using a series of mutants of one of the Evasin-4 ligands, CCL5, we showed that this CKBP binds to the N terminus of the chemokine, as was observed in the crystal structure of the complex of Evasin-1 and CCL3. Thus, despite having different binding hotspots, both Evasins block the key signaling domain of chemokines.
EXPERIMENTAL PROCEDURES
Phage ELISA
TG1 bacteria (Lucigen, Middleton, WI) were transformed with the pMS101C vector containing a mutated open reading frame of Evasin-1 or -4. Briefly, this vector comprises a repressible Lac promotor, the 5′-UTR and modified secretion signal coding sequence derived from the SPY periplasmic stress protein of Escherichia coli, a variant c-Myc tag, a His7 purification tag and a junctional amber codon to allow translational read-through of the full-length M13 gIII phage coat protein gene in suppressor strains of E. coli. Additionally, to support the periplasmic folding of certain eukaryotic protein domains, the vector incorporates an expressible molecular chaperone. Ampicillin-resistant colonies were incubated overnight at 30 °C in 2% glucose medium. The following day, the overnight culture was inoculated into fresh medium and grown to turbidity. To produce phage, the culture was then infected with M13KO7 phage (Invitrogen) and incubated stationary at 37 °C for 2 h. Finally, the infected culture was inoculated into ampicillin+ kanamycin+ medium and incubated overnight at 30 °C.
To perform the ELISA, biotinylated chemokines (Almac Group, Craigavon, UK) at 1 μg/ml were added to wells coated previously overnight with 10 μg/ml neutravidin (Thermo Fisher Scientific). Following a 1 h incubation to allow chemokine capture, wells were blocked with 5% milk in phosphate-buffered saline (PBS) for an additional hour. The bacterial cultures containing the rescued phage were also blocked with the same solution for 30 min. After the washing steps, the blocked phage were transferred to the chemokine-coated wells and incubated for 1 h. Bound phage were revealed using a rabbit anti-fd bacteriophage primary antibody (Sigma-Aldrich), a HRP-coupled anti-rabbit IgG secondary antibody (Jackson ImmunoResearch Laboratories), and the QuantaBlu Fluorogenic Substrate Solution (Thermo Fisher Scientific) according to the manufacturer's instructions.
Expression and Purification of Evasin Mutants
Evasin-1 mutants were transiently expressed in human embryonic kidney 293 cells (HEK 293) as described previously (10). Evasin-4 mutated sequences were introduced into a plasmid containing the sequence of IgG1 Fc domain, and proteins were purified on a Protein A column as described in Ref. 8.
Analysis of Selectivity
The selectivity profiles of Evasin-4 mutants were analyzed using SPR on a BIAcore 2000 system (GE Healthcare Life Sciences). The analyses were performed by immobilizing the Evasin-4-Fc protein on a CM5 chip coated previously with an anti-human Fc using the human antibody capture kit (GE Healthcare) according to the manufacturer's instructions, under the conditions determined previously for the Evasin-4-Fc fusion protein (8). A coating level of 4150 relative units was obtained. Wild type or mutated Evasin-4-Fc was suspended at 50 μg/ml in HBS-EP running buffer (0.01 m Hepes, pH 7.4, 0.15 m NaCl, 3 mm EDTA, 0.005% surfactant P20) and injected for 2 min at 30 μl/min. The baseline was stabilized by 14 min of continuous running buffer flow. Each binding experiment was performed twice. For kinetic characterization, five dilutions of the NusA chemokine fusion protein (NusA-CCL3, NusA-CCL5, or NusA-CCL8) produced as described previously (27) were prepared. Chemokine dilutions were injected for 3 min followed by a dissociation time of 15 min. The CM5 chip was then regenerated using 3 m MgCl2 for 30 s. Data were analyzed using BIAevaluation software (version 4.1.1, GE Healthcare), and curves were fitted using a 1:1 Langmuir binding model.
Relative binding of the purified Evasin-4-Fc mutants was also investigated by ELISA. Briefly, biotinylated chemokines (Almac Group) were coated on streptavidin microplates (Greiner Bio-One, Frickenhausen, Germany), and serial dilutions of Evasin-4 mutants were then added. Following washes, bound Evasin-4 was detected using a polyclonal anti-Evasin-4 antibody raised in rabbits (Eurogentec, Seraing, Belgium) and an HRP-coupled anti-rabbit IgG (Jackson ImmunoResearch Europe, Newmarket, United Kingdom). Plates were developed by the addition of 3,3′,5,5′-tetramethylbenzidine (Sigma-Aldrich) as enzyme substrate. The reaction was terminated by the addition of 2 m H2SO4, and absorbance at 450 nm was read on a Synergy HT spectrophotometer (BioTek Instruments, Luzern, Switzerland).
Neutralization Assay
The ability of Evasin variants to inhibit chemokine-induced in vitro chemotaxis was assessed using ChemoTx System chemotaxis plates with a pore size of 5 μm (NeuroProbe, Inc.). Assays were performed in the presence of increasing concentrations of antibodies using semi-stable L1.2/chemokine receptor transfectants obtained as described previously (28). An agonist chemokine concentration corresponding to the EC80 determined beforehand was used. Briefly, 105 cells were added to the top of the filter, and 32 μl of chemokine/Evasin solution were placed in the wells of the lower plate. Plates were incubated at 37 °C and 5% CO2 for 4 h, and FMAT was used to evaluate the migration of the cells as described previously (27).
CCL5 Mutants
Mutations H23A, E26A, G32P, G32K, E66A, 44AANA47, and 55AAWVA59 were introduced in CCL5 DNA by site-directed mutagenesis, as described previously (29). Chemokines were expressed in E. coli and purified using standard protocols for chemokines (30). Binding of CCL5 mutants to coated Evasin-4 by SPR was performed on a BIAcore 3000 system as described elsewhere (8).
RESULTS
Putative Binding Interactions of Evasin-4 and CCL3 from an in Silico Model
Despite several attempts, we were unable to obtain crystals of Evasin-4, either alone or in complex with chemokine. We therefore opted for a different approach to delineate the binding properties of Evasin-4. Because the cysteine residues of Evasin-1 and Evasin-4 are well conserved, it strongly suggests that they share the same disulfide arrangement. Moreover, as shown in Fig. 1, the secondary structure of Evasin-4 is predicted to be similar to that of Evasin-1, supporting the hypothesis that Evasin-1 and Evasin-4 share the same fold. We therefore constructed an in silico model of the structure of Evasin-4 in complex with CCL3 based on the crystal structure of the Evasin-1·CCL3 complex using Maestro software (Schrödinger). Evasin-1 and -4 sequences were initially aligned using Geneious software (Biomatters, Ltd.), and the alignment was manually modified to avoid gaps in the β-sheets and α-helix of Evasin-1. Amino acids 1–13 of Evasin-4 were not modeled into the structure of the complex due to the absence of an equivalent N terminus in Evasin-1. The other main difference between Evasin-1 and Evasin-4 is the presence of a long C terminus enriched in basic residues in Evasin-1. Using the alignment shown in Fig. 1, a model of the Evasin-4·CCL3 structure was obtained (Fig. 2B). The structure of four loops (Ala-21–Thr-22; Leu-34–Leu-36; Thr-56–Asn-59; Gln-76–Asn-78) could not be modeled, as gaps were present at these positions in the sequence alignment.
FIGURE 1.
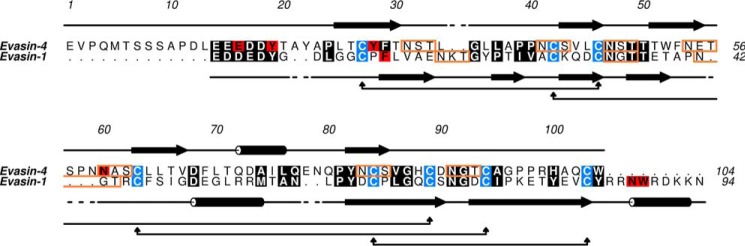
Alignment of Evasin-1 and Evasin-4 sequences. Conserved amino acids are highlighted in black, amino acids mentioned in the text are in red, and cysteines are in blue. The secondary structure of each protein is indicated (Evasin-1: based on Protein Data Bank code 3FPU; Evasin-4, predicted): cylinders stand for α-helixes, and arrows indicate β-sheets. Predicted glycosylation sites are highlighted by orange boxes.
FIGURE 2.

Modeling of Evasin-4 onto the structure of the Evasin-1·CCL3 complex. A, structure of Evasin-1·CCL3 complex (Protein Data Bank code 3FPU). The structure has been rotated to highlight the π-π interaction between Trp-89 of Evasin-1 and Phe-29 of CCL3 in the enlarged region. B, homology model of the Evasin-4·CCL3 complex, based on Evasin-1. When gaps were present in the Evasin-1/Evasin-4 sequence alignment, the structure of the correspondent loop could not be predicted, and these areas are therefore colored in red. The enlarged region shows the proximity of Glu-16 and Tyr-19 of Evasin-4 to Ser-16 and Arg-17 of CCL3. C, electrostatic surface of CCL3 (left) and Evasin-4 (right) generated in PyMOL. Positively charged regions are highlighted in blue, and negatively charged regions are colored in red. Compared with the complex displayed in B, CCL3 is rotated 90 degrees around the y axis to show the negative cluster of the chemokine putatively interacting with the acidic N terminus of Evasin-4 (double arrow).
Following inspection of the modeled interface between the chemokine and Evasin-4, we identified 14 amino acids as potentially important candidates contributing to binding (Fig. 3B). The results obtained by this manual analysis concurred well with the protein-protein interactions predicted using the Protein Interactions Calculator webserver (31). The majority of these amino acids were in the first 30 residues of Evasin-4, a domain predicted to be the major contributor to the interface surface by the ProFACE server (32), suggesting that the N-terminal region of Evasin-4 plays an important role in the chemokine·Evasin complex. This was further supported by the analysis of the electrostatic map generated from Evasin-4 and CCL3 structures in PyMOL software: the strongly negatively charged EEEDD motif in the N-terminal part of Evasin-4 may interact with a basic cluster on the surface of CCL3 formed by Arg-17, Lys-44, Arg-45, and Arg-47 of the chemokine (Fig. 2C).
FIGURE 3.

Alanine-scanning mutagenesis of Evasin-1 and -4. A, validation of Evasin display on phage particles. Evasin-4 was cloned into pMS101C vector and expressed on M13 phage fused to the gIII protein. Phage particles were used in ELISA against the indicated biotinylated protein. Tie-2 was used as an irrelevant protein (negative control). As expected from previous studies (8, 9), Evasin-4 binds to CCL5 but not to CCL2 or CXCL8. B, phage ELISA of mutated Evasin-1 (left panel) or Evasin-4 (right panel) against biotinylated CCL3 or CCL5, respectively. Fluorescence was normalized to wild type Evasin, which is designated as 100% of binding. Data are presented as the mean ± S.E. of two to three phage display experiments, with three measurements performed for each experiment and analyzed using an unpaired two-tailed t test. ns, non significant; *, p < 0.05; **, p < 0.01; ****, p < 0.0001 versus wild type Evasin. C, inhibition of 1 nm CCL3L1-mediated chemotaxis of L1.2/CCR5 cells with Evasin-1 and its derivatives. D, inhibition of 10 nm CCL5-mediated chemotaxis of L1.2/CCR3 cells with Evasin-4-Fc and its derivatives. Chemotaxis data are presented as the mean of three measurements ± S.E. and are representative of at least two independent experiments.
Phage ELISA Profiling to Identify Evasin Mutants with Reduced Binding
To experimentally determine residues involved in the interaction between the Evasins and chemokine, we developed a phage display approach. The small size of the Evasins suggested that they might be able to be expressed and correctly folded on the surface of M13 filamentous phage, which would allow us to screen a number of Evasin mutants without having to purify each one. Indeed, preliminary experiments confirmed that Evasins displayed on phage retain functional binding to their known ligands (Fig. 3A). We took advantage of these properties to study the potential role of the 14 residues of Evasin-4 identified in our model to be involved in the interaction with the chemokine. These amino acids were mutated to alanine, and the resultant individually mutated clones were screened for binding to chemokine ligands by a phage enzyme-linked immunosorbent assay (ELISA). In parallel, we performed the same study with Evasin-1: examining the structure of the Evasin-1·CCL3 complex, we selected 10 Evasin-1 amino acids present at the interface as having a potential involvement in ligand binding. We also included two residues thought to form π-π interactions with aromatic residues from CCL3, Phe-14 and Trp-89 (23). We mutated these positions to alanine, anticipating that reduced binding should be observed at least with F14A and W89A mutations, which would allow us to validate our approach.
As expected, for Evasin-1 displayed on phage, binding to CCL3 was shown to be substantially reduced for the F14A and W89A mutants (Fig. 3B). Interestingly, we also identified a third mutation, N88A, that showed a decreased signal similar to that of F14A and W89A. In the case of Evasin-4, the Y19A mutation dramatically decreased the binding of Evasin-4 to CCL5 by >80%, whereas E16A induced a 50% reduction in binding (Fig. 3B). The other mutations had no significant impact on Evasin-4 binding to the chemokine, and we therefore decided to focus on these two mutations. In the course of preliminary evaluations in a parallel study aimed at altering the potency of Evasin-4 for certain chemokines, we identified another Evasin-4 mutant containing the double mutation Y28Q/N60D that also showed reduced relative binding when displayed on phage particles, and we therefore decided to include this variant in our subsequent analyses.
Characterization of Recombinant Evasin Mutants
We next expressed and purified the key Evasin mutants to confirm the results obtained by phage display and to further investigate the functional role of the amino acids identified as potentially crucial for binding. To facilitate purification, Evasins were expressed as C-terminally tagged proteins, with a His6 tag fused to Evasin-1 and an Fc to Evasin-4 as described previously (8, 10).
The inhibitory properties of Evasin-1 single mutants as well as those of the double mutant F14A/W89A were assessed by chemotaxis assays using L1.2 cells stably transfected with specific chemokine receptors. To increase the sensitivity of our assay, we used CCL3L1, an isoform that has 95% sequence identity with CCL3 and similar affinity for CCR5 but is 100-fold more potent in chemotaxis (33). W89A and N88A mutations induced a 10-fold decrease in the ability of Evasin-1 to inhibit CCL3L1, whereas the effect of the F14A mutation was even more pronounced, showing a 200-fold increase of the half-maximal inhibitory concentration (IC50). As expected, the double mutant F14A/W89A was unable to inhibit chemotaxis mediated by 1 nm CCL3L1 at physiologically relevant concentrations of the Evasin (Fig. 3C).
The three Evasin-4 mutants were characterized by SPR and chemotaxis assays, using CCL3, CCL5, and CCL8 as binding partners. In the SPR experiments, we used the previously described NusA chemokine fusion proteins (27) to increase the molecular weight of the analyte and hence the sensitivity of the response signal on the BIAcore 2000 instrument, whereas the Evasins were immobilized via their Fc domain on an anti-human Fc-coated chip. The results obtained by SPR correlate well with the relative levels of inhibition observed for the chemotaxis assay. E16A showed a slight but reproducible reduction in binding capacity by SPR and potency in inhibition of chemotaxis, whereas the reduction shown by Y19A was more pronounced (Fig. 3D and Table 1). Because the single mutations did not lead to complete abrogation of Evasin-4 binding to CC chemokines, we created a double mutant Evasin-4-Fc E16A/Y19A. The binding of this protein to CCL3, CCL5, and CCL8 was almost undetectable by SPR, and we were therefore unable to fit the data (Fig. 4). To confirm these results, the binding of the E16A/Y19A mutant to CCL3 and CCL5 was investigated by ELISA. Again, the double mutant showed poorer relative binding than the single mutants (data not shown). As expected, the reduced binding correlates with a loss of potency in the inhibition of chemotaxis induced by the three chemokines tested (Table 1).
TABLE 1.
Characterization of Evasin-4-Fc mutants
Inhibitory properties were determined by inhibition of L1.2/CCR5 (40 nm CCL3 or 40 nm CCL5) or L1.2/CCR2 (10 nm CCL8) chemotaxis and are characterized by the associated IC50. Dissociation constants (KD) were measured by SPR. −, no inhibition of chemotaxis or no binding; n.d., not determined as the fitting was not good enough to determine an accurate KD due to low signal and chemokine oligomerization.
| ka × 103 | kd × 10−4 | KD | IC50 | |
|---|---|---|---|---|
| m−1 s−1 | s−1 | nm | nm | |
| CCL3 | ||||
| Evasin-4-Fc | 5 | 4 | 71 | 11 |
| E16A | 4 | 4 | 81 | 17 |
| Y19A | 5 | 15 | 283 | 37 |
| E16A/Y19A | − | − | − | 36 |
| Y28Q/N60D | 29 | 89 | 310 | 133 |
| CCL5 | ||||
| Evasin-4-Fc | 31 | 84 | 274 | 97 |
| E16A | 36 | 134 | 377 | 176 |
| Y19A | n.d | n.d. | n.d. | 7598 |
| E16A/Y19A | − | − | − | − |
| Y28Q/N60D | 34 | 248 | 726 | 380 |
| CCL8 | ||||
| Evasin-4-Fc | 17 | 8 | 47 | 0.8 |
| E16A | 13 | 15 | 112 | 1.1 |
| Y19A | 26 | 55 | 209 | 1.7 |
| E16A/Y19A | − | − | − | 17.3 |
| Y28Q/N60D | 31 | 78 | 248 | 2.0 |
FIGURE 4.

SPR analysis of Evasin-4-Fc alanine mutants. CCL3, CCL5, and CCL8 suspended at 800 nm in running buffer were injected on Evasin-4-Fc coated on an anti-human Fc chip. To improve detection, chemokine NusA fusion proteins were used. Evasin-4-Fc wild type (black solid line) displayed a strong binding to the three chemokines tested whereas Evasin-4-Fc E16A (black dotted line) and Evasin-4-Fc Y19A (black dashed line) showed reduced binding, especially in the case of CCL5. The double mutant Evasin-4-Fc E16A Y19A (dashed and dotted line) has almost no detectable interaction with any of the chemokines tested. R.U., response units.
For the three ligands tested, Evasin-4 Y28Q/N60D displayed considerably reduced binding, mainly due to a faster dissociation (Table 1). The mutant showed a reduced potency to inhibit chemotaxis of transfected L1.2 cells compared with wild type Evasin-4, confirming the results obtained by SPR (Fig. 3D).
Residues of CCL5 Involved in Binding to Evasin-4
Finally, to determine the site of the chemokine targeted by Evasin-4, we took advantage of our collection of CCL5 mutants. To this end, we first screened the ability of Evasin-4 to inhibit the chemotactic response of L1.2/CCR5 transfectants induced by CCL5 mutants and observed that only one mutant, T8P, showed reduced inhibition by Evasin-4 (Fig. 5A). Evasin-4 inhibited the mutants in the 40s and 50s loops with equal potency (Fig. 5A), as well as the mutants in the 20s and 30s loops, and E26A and E66A, mutants deficient in oligomerization (34) (data not shown). We then determined the affinity of CCL5 T8P as well as that of CCL5 F12A for Evasin-4 by SPR (Fig. 5B). CCL5 F12A was not used in the chemotaxis assay as the mutation of Phe-12, a pivotal residue involved in receptor activation (35), abrogates the chemotactic activity of the chemokine. Binding to both the mutants was significantly reduced: CCL5 has an affinity of 12.6 nm (ka 1.03 × 105 (m−1 s−1), kd 1.29 × 10−3 (s−1)) for Evasin-4, whereas the KD of CCL5 T8P and CCL5 F12A were, respectively, 290.2 nm (ka of 1.95 × 104 (m−1 s−1), kd of 5.65 × 10−3 (s−1)), and 130.5 nm (ka of 3.21 × 104 (m−1 s−1), kd of 4.19 × 10−3 (s−1)).
FIGURE 5.
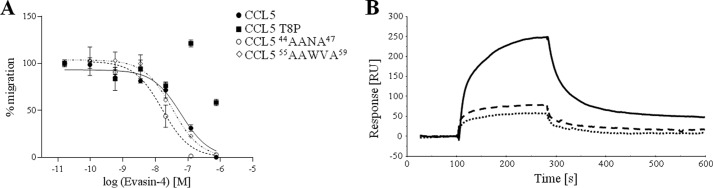
Interaction between Evasin-4 and CCL5 mutants. A. Inhibition of chemokine-mediated chemotaxis of L1.2/CCR5 cells with Evasin-4. CCL5 or the indicated mutant was used at 10 nm to induce cell migration. Evasin-4 inhibits the activity of all tested mutants (44AANA47, 55AAWVA59, H23A, E26A, G32P, G32K, and E66A) except CCL5 T8P. Data are presented as the mean of three measurements ± S.E. B, CCL5 mutants suspended at 200 (CCL5 E26A) or 1000 nm (CCL5 T8P and CCL5 F12A, respectively) in running buffer were injected on coated Evasin-4. As expected from previous studies, the obligate tetramer CCL5 E26A (black solid line) displayed a strong binding to Evasin-4. By comparison, CCL5 T8P (black dotted line) and CCL5 F12A (black dashed line) showed a significantly reduced binding to the CKBP, even at high concentrations. RU, response units.
DISCUSSION
Chemokines are key players in both the innate and the adaptive immune response. Proof of their crucial role is revealed by the strategies taken by pathogens to inhibit them: viruses secrete chemokine receptor homologues and chemokine mimics as well as CKBPs to confuse their host immune system (36), and eukaryotic parasites such as worms and ticks produce CKBPs (8–10, 37). The majority of CKBPs have fairly broad binding selectivities for chemokines; however, the tick-derived CKBPs Evasin-1 and Evasin-3 are highly selective as Evasin-1 only recognizes CCL3, CCL4, and CCL18, and the Evasin-3 only ELR CXC chemokines. However, Evasin-4, which is predicted to share a conserved topology with Evasin-1, has been shown to bind at least 20 CC chemokines (10). We were intrigued by the distinct ligand recognition characteristics of these small but related proteins and therefore sought to identify the amino acids responsible for chemokine recognition by Evasin-1 and -4.
The elucidation and mapping of residues essential for the interaction between proteins typically requires extensive cloning, expression and purification steps to produce the necessary useful range of protein mutants. To circumvent this, we reasoned that the small size of the Evasins might facilitate their expression and display on filamentous phage particles. This technology, linking genotype and phenotype, is well suited to screen large polypeptide libraries for particular ligand or antigen binding properties and is now increasingly employed by the pharmaceutical industry as a platform for the isolation of potential therapeutic and diagnostic candidates from synthetic or naturally diversified molecular repertoires (38). By successfully applying phage display to the study of Evasin family members, we have extended the spectrum of amenable polypeptide classes reported for this technology and simultaneously have explored the potential of the technique for rapidly assessing the role of candidate residues in ligand binding. Validation of the approach was obtained with Evasin-1 by confirming the importance of residues Phe-14 and Trp-89, which had been identified previously (23), whereas Asn-88 was identified from a set of 11 residues found at the interface of the CKBP and CCL3 in the crystal structure of the complex.
To identify residues in Evasin-4 responsible for binding chemokines, we constructed a model of the complex with CCL3. We chose this chemokine rather than with CCL5 for which we had a series of mutants to test experimentally because the structure of CCL3 in complex with the other tick-derived CKBP Evasin-1 had been solved. The model of the putative Evasin-4·CCL3 interface suggested a potential set of amino acids as candidates for mutagenesis. Mutation of these residues to alanine led to the identification of Glu-16 and Tyr-19 as crucial for the affinity of Evasin-4, at least for binding to CCL3, CCL5, and CCL8. Upon inspection of the in silico Evasin-4·CCL3 model, it appeared that Tyr-19 of Evasin-4 may interact with Arg-17 in CCL3 via a cation-π interaction, which is well described in protein-protein interaction literature (39). Most of the chemokines that Evasin-4 inhibits have an arginine or a lysine at the equivalent position, supporting the hypothesis that this amino acid may be involved in stabilizing the complex (Fig. 6). Certain chemokines such as CCL18 or CCL22 have an aromatic amino acid at this position (either a tryptophan or a tyrosine), allowing the formation of a π-π interaction between the Evasin and the chemokine. The only CC chemokines inhibited by Evasin-4 that do not have a positively charged or aromatic amino acid at the equivalent position are CCL15 and CCL17. CCL15 has a longer N terminus than other CC chemokines, which it is unstructured in the NMR structure (40) but could potentially provide alternative binding contacts. In CCL17, the positional equivalent of Arg-17 in CCL3 is occupied by a glycine. However, the preceding amino acid is a lysine, which may be close enough to the Tyr-19 of Evasin-4 to allow interaction. We confirmed this hypothesis by showing that the single mutant, Y19A, and the double mutant, E16A/Y19A, were both unable to inhibit CCL17-mediated chemotaxis of L1.2/CCR4 transfectants (data not shown). With regard to Glu-16, this residue may act through a stabilizing interaction by forming hydrogen bond(s) with the polar amino acid present at position 16 in CC chemokines (a serine in CCL3).
FIGURE 6.

Alignment of CC chemokines inhibited by Evasin-4. Conserved positions are shaded in gray, whereas positions mentioned in the text are highlighted in red (Phe-12 and equivalent amino acid), blue (positively charged or aromatic residue at position 17), or orange (hydrophobic residue at position 40).
In a parallel study, we identified a clone, Evasin-4 Y28Q/N60D, which showed reduced binding to CCL3, CCL5, and CCL8. In trying to rationalize the structural mechanism(s) by which the Y28Q and N60D mutations lead to impaired binding, we observed from our in silico model of the Evasin-4·CCL3 complex that both amino acids are predicted to be part of the interface between Evasin-4 and CCL3 (ProFACE server (32)). Tyr-28 is predicted to form hydrophobic interactions with Ile-40 of CCL3 (31), one of the most conserved positions in CC chemokines (Fig. 6). The substitution of the tyrosine with a hydrophilic glutamine may destabilize this interaction leading to a faster dissociation of the complex. Interestingly, we mutated Tyr-28 into an alanine in our first approach, but this mutant retains binding to CCL5 when displayed on phage particles. It therefore appears that this amino acid could contribute to the off-rate stabilization of the complex but is not necessary for the initial formation of the complex. Regarding Asn-60, this amino acid is not predicted to interact specifically with any CCL3 residue. However, as it is part of an NXS motif, Asn-60 is predicted to be glycosylated post-transcriptionally in mammalian cells (41). The absence of glycosylation induced by the N60D mutation may impact the conformation of the protein, leading to a less stable complex between the chemokine and Evasin-4, although glycosylation has not been shown to affect biological activity for Evasin-1 and -3 (9, 10).
Our initial analysis of the in silico model of Evasin-4·CCL3 revealed the presence of two Evasin-4 phenylalanines in the vicinity of the CCL3 Phe-12 residue, which could potentially allow a stabilization mediated by π-π interactions. However, the mutation of each of these phenylalanines to alanines (F29A and F53A) did not impair chemokine binding by phage-displayed Evasin-4. Therefore, these amino acids do not appear to be essential for the interaction between the two proteins. These results are supported by the fact that an aromatic residue at the position equivalent to Phe-12 in CCL3 is present in only half of the chemokines inhibited by Evasin-4 (Fig. 6).
From our results, it appears that Evasin-1 and Evasin-4 do not use the same binding modality to target CC chemokines. Evasin-1 requires both N and C termini to interact stably with CCL3 through Phe-14 and Trp-89 (23) and Asn-88 identified in this study. Conversely, only the N-terminal region of Evasin-4 appears to have a crucial role in the interaction with the chemokine as suggested initially by the modeled complex structure and confirmed by the alanine mutants identified by phage display. This is confirmed by the high number of predicted hydrogen bonds in this area of the complex. These differences may be further supported by the presence of a longer N terminus in Evasin-4, whereas Evasin-1 has a highly charged extended C terminus. However, we initially thought from the model of the complex that Evasin-4 would interact with the basic residues in the 40s loop of the chemokine, but this was disproved as this CKBP had unaltered activity on 44AANA47-CCL5.
Chemokines have a highly conserved monomeric fold despite different primary amino acid sequences, which can result in identity as low as 20%. Therefore, it is likely that the individual interactions between the CKBP and the chemokine may differ, which is reflected by the differences in affinity, but the composite of the interactions allows binding and neutralization. Thus, the amino acids identified in this study that play a role in the binding of Evasin-4 do not impact to the same extent on the three chemokines tested, but contribute to its activity for these chemokines. For example, the double mutation Y28Q/N60D reduced the inhibitory activity of Evasin-4 on CCL3 10-fold, whereas it had only a 2-fold reduction for the inhibition of CCL8. Similarly, the drastic effect of the Y19A mutation on the interaction with CCL5 was less pronounced on CCL3 and CCL8.
It is well established that the N terminus of chemokines is required for optimal binding and signaling through specific G protein-coupled receptors (42). The conformational change induced in the N terminus of CCL3 following Evasin-1 binding was therefore suggested to be important in explaining the inhibition (23). Based on the involvement of the Evasin-4 residues Glu-16 and Tyr-19 in our model, we can postulate that binding may also involve certain N-terminal residues of its ligands. This hypothesis is supported by our finding that among a collection of CCL5 mutants, only the two N-terminal mutants T8P and F12A lost interaction with Evasin-4.
Binding the N-terminal region of chemokines appears to be a common feature of several CKBPs. For example, vCCI from several viruses, as well as M3 and CrmD bind the 20s loop of their respective ligands (11, 12, 16, 19, 20). In particular, the vCCIs bind to the conserved Arg or Lys equivalent to Arg-17 in CCL3. With the exception of A41 (18), all viral CKBPs as well as the eukaryotic smCKBP isolated from Schistosoma mansoni inhibit chemotaxis as demonstrated for the Evasins. Moreover, most CKBPs have broad specificities similar to Evasin-4 but with the exception of A41, which is more selective as has been shown for Evasin-1 and -3. These observations suggest that viral and eukaryotic organisms convergently evolved to inhibit host chemokines and that the targeting of positively charged amino acids present in the 20s loop may be a common mechanism.
Lastly, this study highlights the use of phage display for the rapid identification of residues responsible for the interaction between two proteins. Protein-protein interactions have been studied by phage display before (43), and here, we show that small soluble proteins such as these tick-derived CKBPs may be displayed on phage particles. We suggest that this technique could be extended to other proteins such as cytokines.
Acknowledgments
We thank A. Garin, A. Hermant, and S. Krohn for help with chemokine experiments and biology, N. Barillat, F. Bollin, F. Borlat, J.-P. Gaudry, and L. Glez for advice in protein expression and purification, N. Bosson and M. De Tiani for help with phage display experiments, P. Malinge for advice in SPR, A. Mantovani for L1.2 receptor transfectants and C. Power, M. Kosco-Vilbois, and N. Fischer for fruitful scientific discussions.
This work was supported by the European Union Seventh Framework Programme (FP7–2007-2013) Grant HEALTH-F4–2011-281608 TIMER (to P. B. and A. E. I. P.) and NIAID, National Institutes of Health Grant RO1AI37113 (to T. M. H.).
- CKBP
- chemokine-binding protein
- SPR
- surface plasmon resonance
- vCCI
- viral CC chemokine inhibitor.
REFERENCES
- 1. Salanga C. L., Handel T. M. (2011) Chemokine oligomerization and interactions with receptors and glycosaminoglycans: the role of structural dynamics in function. Exp. Cell Res. 317, 590–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zipfel P. F., Hallström T., Riesbeck K. (2013) Human complement control and complement evasion by pathogenic microbes–tipping the balance. Mol. Immunol. 56, 152–160 [DOI] [PubMed] [Google Scholar]
- 3. Alcami A., Saraiva M. (2009) Chemokine binding proteins encoded by pathogens. Adv. Exp. Med. Biol. 666, 167–179 [DOI] [PubMed] [Google Scholar]
- 4. Alexander-Brett J. M., Fremont D. H. (2007) Dual GPCR and GAG mimicry by the M3 chemokine decoy receptor. J. Exp. Med. 204, 3157–3172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lalani A. S., Ness T. L., Singh R., Harrison J. K., Seet B. T., Kelvin D. J., McFadden G.., Moyer R. W. (1998) Functional comparisons among members of the poxvirus T1/35kDa family of soluble CC-chemokine inhibitor glycoproteins. Virology 250, 173–184 [DOI] [PubMed] [Google Scholar]
- 6. van Berkel V., Barrett J., Tiffany H. L., Fremont D. H., Murphy P. M., McFadden G., Speck S. H., Virgin H. W., IV (2000) Identification of a gammaherpesvirus selective chemokine binding protein that inhibits chemokine action. J. Virol. 74, 6741–6747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alejo A., Ruiz-Argüello M. B., Ho Y., Smith V. P., Saraiva M., Alcami A. (2006) A chemokine-binding domain in the tumor necrosis factor receptor from variola (smallpox) virus. Proc. Natl. Acad. Sci. U.S.A. 103, 5995–6000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Déruaz M., Bonvin P., Severin I. C., Johnson Z., Krohn S., Power C. A., Proudfoot A. E. (2013) Evasin-4, a tick-derived chemokine-binding protein with broad selectivity can be modified for use in preclinical disease models. FEBS J. 280, 4876–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Déruaz M., Frauenschuh A., Alessandri A. L., Dias J. M., Coelho F. M., Russo R. C., Ferreira B. R., Graham G. J., Shaw J. P., Wells T. N., Teixeira M. M., Power C. A., Proudfoot A. E. (2008) Ticks produce highly selective chemokine binding proteins with antiinflammatory activity. J. Exp. Med. 205, 2019–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Frauenschuh A., Power C. A., Déruaz M., Ferreira B. R., Silva J. S., Teixeira M. M., Dias J. M., Martin T., Wells T. N., Proudfoot A. E. (2007) Molecular cloning and characterization of a highly selective chemokine-binding protein from the tick Rhipicephalus sanguineus. J. Biol. Chem. 282, 27250–27258 [DOI] [PubMed] [Google Scholar]
- 11. Beck C. G., Studer C., Zuber J. F., Demange B. J., Manning U., Urfer R. (2001) The viral CC chemokine-binding protein vCCI inhibits monocyte chemoattractant protein-1 activity by masking its CCR2B-binding site. J. Biol. Chem. 276, 43270–43276 [DOI] [PubMed] [Google Scholar]
- 12. Ruiz-Argüello M. B., Smith V. P., Campanella G. S., Baleux F., Arenzana-Seisdedos F., Luster A. D., Alcami A. (2008) An ectromelia virus protein that interacts with chemokines through their glycosaminoglycan binding domain. J. Virol. 82, 917–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seet B. T., Singh R., Paavola C., Lau E. K., Handel T. M., McFadden G. (2001) Molecular determinants for CC-chemokine recognition by a poxvirus CC-chemokine inhibitor. Proc. Natl. Acad. Sci. U.S.A. 98, 9008–9013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Webb L. M., Smith V. P., Alcami A. (2004) The gammaherpesvirus chemokine binding protein can inhibit the interaction of chemokines with glycosaminoglycans. FASEB J. 18, 571–573 [DOI] [PubMed] [Google Scholar]
- 15. Kuo N. W., Gao Y. G., Schill M. S., Isern N., Dupureur C. M., Liwang P. J. (2014) Structural insights into the interaction between a potent anti-inflammatory protein, vCCI, and the human CC chemokine, Eotaxin-1. J. Biol. Chem. 289, 6592–6603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. White G. E., McNeill E., Christou I., Channon K. M., Greaves D. R. (2011) Site-directed mutagenesis of the CC chemokine binding protein 35K-Fc reveals residues essential for activity and mutations that increase the potency of CC chemokine blockade. Mol. Pharmacol. 80, 328–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang L., Derider M., McCornack M. A., Jao S. C., Isern N., Ness T., Moyer R., LiWang P. J. (2006) Solution structure of the complex between poxvirus-encoded CC chemokine inhibitor vCCI and human MIP-1β. Proc. Natl. Acad. Sci. U.S.A. 103, 13985–13990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bahar M. W., Kenyon J. C., Putz M. M., Abrescia N. G., Pease J. E., Wise E. L., Stuart D. I., Smith G. L., Grimes J. M. (2008) Structure and function of A41, a vaccinia virus chemokine binding protein. PLoS Pathog. 4, e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xue X., Lu Q., Wei H., Wang D., Chen D., He G., Huang L., Wang H., Wang X. (2011) Structural basis of chemokine sequestration by CrmD, a poxvirus-encoded tumor necrosis factor receptor. PLoS Pathog. 7, e1002162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alexander J. M., Nelson C. A., van Berkel V., Lau E. K., Studts J. M., Brett T. J., Speck S. H., Handel T. M., Virgin H. W., Fremont D. H. (2002) Structural basis of chemokine sequestration by a herpesvirus decoy receptor. Cell 111, 343–356 [DOI] [PubMed] [Google Scholar]
- 21. Paavola C. D., Hemmerich S., Grunberger D., Polsky I., Bloom A., Freedman R., Mulkins M., Bhakta S., McCarley D., Wiesent L., Wong B., Jarnagin K., Handel T. M. (1998) Monomeric monocyte chemoattractant protein-1 (MCP-1) binds and activates the MCP-1 receptor CCR2B. J. Biol. Chem. 273, 33157–33165 [DOI] [PubMed] [Google Scholar]
- 22. Proudfoot A. E., Handel T. M., Johnson Z., Lau E. K., LiWang P., Clark-Lewis I., Borlat F., Wells T. N., Kosco-Vilbois M. H. (2003) Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. U.S.A. 100, 1885–1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dias J. M., Losberger C., Déruaz M., Power C. A., Proudfoot A. E., Shaw J. P. (2009) Structural basis of chemokine sequestration by a tick chemokine binding protein: the crystal structure of the complex between Evasin-1 and CCL3. PLoS One 4, e8514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dey B., Thukral S., Krishnan S., Chakrobarty M., Gupta S., Manghani C., Rani V. (2012) DNA-protein interactions: methods for detection and analysis. Mol. Cell Biochem. 365, 279–299 [DOI] [PubMed] [Google Scholar]
- 25. Hammers C. M., Stanley J. R. (2014) Antibody phage display: technique and applications. J. Invest. Dermatol. 134, e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rentero Rebollo I., Heinis C. (2013) Phage selection of bicyclic peptides. Methods 60, 46–54 [DOI] [PubMed] [Google Scholar]
- 27. Magistrelli G., Gueneau F., Muslmani M., Ravn U., Kosco-Vilbois M., Fischer N. (2005) Chemokines derived from soluble fusion proteins expressed in Escherichia coli are biologically active. Biochem. Biophys. Res. Commun. 334, 370–375 [DOI] [PubMed] [Google Scholar]
- 28. Fra A. M., Locati M., Otero K., Sironi M., Signorelli P., Massardi M. L., Gobbi M., Vecchi A., Sozzani S., Mantovani A. (2003) Cutting edge: scavenging of inflammatory CC chemokines by the promiscuous putatively silent chemokine receptor D6. J. Immunol. 170, 2279–2282 [DOI] [PubMed] [Google Scholar]
- 29. Proudfoot A. E., Fritchley S., Borlat F., Shaw J. P., Vilbois F., Zwahlen C., Trkola A., Marchant D., Clapham P. R., Wells T. N. (2001) The BBXB motif of RANTES is the principal site for heparin binding and controls receptor selectivity. J. Biol. Chem. 276, 10620–10626 [DOI] [PubMed] [Google Scholar]
- 30. Proudfoot A. E., Borlat F. (2000) Purification of recombinant chemokines from E. coli in Chemokines Protocols (Proudfoot A. E., Wells T. N., Power C. A., eds) pp. 75–88 Humana Press, Totowa, NJ: [DOI] [PubMed] [Google Scholar]
- 31. Tina K. G., Bhadra R., Srinivasan N. (2007) PIC: protein interactions calculator. Nucleic Acids Res. 35, W473–W476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saha R. P., Bahadur R. P., Pal A., Mandal S., Chakrabarti P. (2006) ProFace: a server for the analysis of the physicochemical features of protein-protein interfaces. BMC Struct. Biol. 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Menten P., Struyf S., Schutyser E., Wuyts A., De Clercq E., Schols D., Proost P., Van Damme J. (1999) The LD78β isoform of MIP-1α is the most potent CCR5 agonist and HIV-1-inhibiting chemokine. J. Clin. Invest. 104, R1–R5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Czaplewski L. G., McKeating J., Craven C. J., Higgins L. D., Appay V., Brown A., Dudgeon T., Howard L. A., Meyers T., Owen J., Palan S. R., Tan P., Wilson G., Woods N. R., Heyworth C. M., Lord B. I., Brotherton D., Christison R., Craig S., Cribbes S., Edwards R. M., Evans S. J., Gilbert R., Morgan P., Randle E., Schofield N., Varley P. G., Fisher J., Waltho J. P., Hunter M. G. (1999) Identification of amino acid residues critical for aggregation of human CC chemokines macrophage inflammatory protein (MIP)-1α, MIP-1β, and RANTES: characterization of active disaggregated chemokine variants. J. Biol. Chem. 274, 16077–16084 [DOI] [PubMed] [Google Scholar]
- 35. Pakianathan D. R., Kuta E. G., Artis D. R., Skelton N. J., Hébert C. A. (1997) Distinct but overlapping epitopes for the interaction of a CC-chemokine with CCR1, CCR3 and CCR5. Biochemistry 36, 9642–9648 [DOI] [PubMed] [Google Scholar]
- 36. Finlay B. B., McFadden G. (2006) Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell 124, 767–782 [DOI] [PubMed] [Google Scholar]
- 37. Smith P., Fallon R. E., Mangan N. E., Walsh C. M., Saraiva M., Sayers J. R., McKenzie A. N., Alcami A., Fallon P. G. (2005) Schistosoma mansoni secretes a chemokine binding protein with antiinflammatory activity. J. Exp. Med. 202, 1319–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pande J., Szewczyk M. M., Grover A. K. (2010) Phage display: concept, innovations, applications and future. Biotechnol. Adv. 28, 849–858 [DOI] [PubMed] [Google Scholar]
- 39. Crowley P. B., Golovin A. (2005) Cation-π interactions in protein-protein interfaces. Proteins 59, 231–239 [DOI] [PubMed] [Google Scholar]
- 40. Sticht H., Escher S. E., Schweimer K., Forssmann W. G., Rösch P., Adermann K. (1999) Solution structure of the human CC chemokine 2: a monomeric representative of the CC chemokine subtype. Biochemistry 38, 5995–6002 [DOI] [PubMed] [Google Scholar]
- 41. Chauhan J. S., Rao A., Raghava G. P. (2013) In silico platform for prediction of N-, O- and C-glycosites in eukaryotic protein sequences. PLoS One 8, e67008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chevigné A., Fievez V., Schmit J. C., Deroo S. (2011) Engineering and screening the N-terminus of chemokines for drug discovery. Biochem. Pharmacol. 82, 1438–1456 [DOI] [PubMed] [Google Scholar]
- 43. Sidhu S. S., Koide S. (2007) Phage display for engineering and analyzing protein interaction interfaces. Curr. Opin. Struct. Biol. 17, 481–487 [DOI] [PubMed] [Google Scholar]