

Background: TNF-α-induced NF-κB pathway is associated with the progression of several cancers and abrogation of TNF signaling a potential target for cancer treatment.
Results: Novel biscoumarin inhibits TNF signaling in vitro and in vivo in IBD model.
Conclusion: The lead compound interrupts the trimeric structure of TNF to achieve this effect.
Significance: This study introduces a novel TNF inhibitor with the potential to target pro-inflammatory diseases.
Keywords: Cancer Therapy, Cell Invasion, Hepatocellular Carcinoma, Inflammatory Bowel Disease (IBD), NF-kappa B (NF-KB), Tumor Necrosis Factor (TNF)
Abstract
TNF is a pleotropic cytokine known to be involved in the progression of several pro-inflammatory disorders. Many therapeutic agents have been designed to counteract the effect of TNF in rheumatoid arthritis as well as a number of cancers. In the present study we have synthesized and evaluated the anti-cancer activity of novel biscoumarins in vitro and in vivo. Among new compounds, BIHC was found to be the most cytotoxic agent against the HepG2 cell line while exhibiting less toxicity toward normal hepatocytes. Furthermore, BIHC inhibited the proliferation of various hepatocellular carcinoma (HCC) cells in a dose- and time-dependent manner. Subsequently, using in silico target prediction, BIHC was predicted as a TNF blocker. Experimental validation was able to confirm this hypothesis, where BIHC could significantly inhibit the recombinant mouse TNF-α binding to its antibody with an IC50 of 16.5 μm. Furthermore, in silico docking suggested a binding mode of BIHC similar to a ligand known to disrupt the native, trimeric structure of TNF, and also validated with molecular dynamics simulations. Moreover, we have demonstrated the down-regulation of p65 phosphorylation and other NF-κB-regulated gene products upon BIHC treatment, and on the phenotypic level the compound shows inhibition of CXCL12-induced invasion of HepG2 cells. Also, we demonstrate that BIHC inhibits infiltration of macrophages to the peritoneal cavity and suppresses the activity of TNF-α in vivo in mice primed with thioglycollate broth and lipopolysaccharide. We comprehensively validated the TNF-α inhibitory efficacy of BIHC in an inflammatory bowel disease mice model.
Introduction
Tumor necrosis factor (TNF, also called TNF-α) is a proinflammatory cytokine involved in multiple biological processes including immune system regulation, cell proliferation, and survival (1). TNF was originally identified as an anti-tumor protein produced by macrophages in 1975 (2); however, later studies have shown that it can be produced by several types of cells including natural killer cells, B and T lymphocytes, mast cells, neutrophils, endothelial cells, muscle cells, etc. (3). Functionally, TNF is a cell-derived inflammatory mediator produced in response to bacterial antigens, such as lipopolysaccharide (LPS) (4). During chronic inflammation, however, an exaggerated immune response involving the production of TNF is able to dysregulate a variety of signaling cascades, and thereby contribute to oncogenesis (5). TNF is functional in its trimeric form and is known for its specific and efficient activation of nuclear factor κ B (NF-κB) signaling pathway that has been closely associated with carcinogenesis (6, 7). Researchers have hence identified TNF as a “master switch” that plays a pivotal role in the link between inflammation and cancer.
NF-κB is a transcription factor known to transcribe more than 200 genes involved in inflammation, cell proliferation, and survival, which include the Bcl-2 family proteins, Mcl-1, cyclin D1, survivin, cyclooxygenase-2 (COX-2), matrix metalloproteinases (MMPs), c-Myc, interleukins, growth factors, and cell adhesion molecules (8). It is present in the cytoplasm, either in its homodimeric or in its heterodimeric form, and associated with an inhibitory peptide, inhibitory κB (IκBα) (9, 10). A ubiquitin-mediated degradation of inhibitory IκBα protein and phosphorylation of various serine residues can then lead to nuclear translocation with subsequent effects on transcription (11). NF-κB1 (p50), NF-κB2 (p52), RelA (p65), RelB, and c-Rel are the five types of NF-κB subunits that have been identified until the current stage, out of which the p50-p65 heterodimer is the most common heterodimer associated with IκBα (12). Given that small regulatory changes to NF-κB signaling may contribute to tumorigenesis (13) a manipulation of TNF-mediated NF-κB signaling by small molecules could be a potent therapeutic strategy to combat inflammatory diseases, as well as to combat associated consequences of cancer development.
Currently, several monoclonal antibodies as well as synthetic and natural chemical moieties have been identified as TNF blockers (14). 2,5-Dimethoxy-4-iodoamphetamine, xanthine derivatives, and curcumin are some of the known small molecules with anti-inflammatory effect that act via inhibiting TNF expression and signaling (15–18). In addition, certolizumab, infliximab, golimumab, and adalimumab are the monoclonal antibodies used for this very purpose (19–22). On the disease level, lactone and coumarins have also previously been reported to interfere with NF-κB signaling in several type of cancers (23).
In the context of the present study we have now synthesized several novel biscoumarin derivatives and evaluated their anti-proliferative efficacy against human hepatocellular carcinoma (HCC)8 cells, and as a result, we were able to identify BIHC as a potent cytotoxic agent. Furthermore, we predicted the putative target of BIHC as TNF-α using an in silico cheminformatics approach, and this target was confirmed experimentally. We subsequently evaluated whether BIHC can abrogate TNF-induced NF-κB signaling cascade in HCC cells and found that as expected it caused down-regulation of p65 and its regulated gene products. Additionally, ELISA-based TNF binding studies revealed clear dose-response relationships between the concentration of BIHC and antibody binding of TNF-α. Also, we evaluated the effect of BIHC on the release of TNF by macrophages stimulated with lipopolysaccharide (LPS) in an in vivo mouse model and observed a significant reduction in the levels of TNF in the intraperitoneal fluid. Besides evaluating the role of BIHC on TNF-α-induced NF-κB signaling and in vivo anti-inflammatory activities, we also found that the suppression of dextran sodium sulfate (DSS) induced colitis in an inflammatory bowel disease mouse model through the regulation of various cytokines. Hence, we are able to report overall the synthesis, mode-of-action analysis, and comprehensive in vitro and in vivo evaluation of novel biscoumarins as TNF-α inhibitors.
EXPERIMENTAL PROCEDURES
Suppliers and Physical Measurements
Materials and reagents were purchased from commercial suppliers and used as received. Melting points were determined through open capillary method using Sigma melting point apparatus and are uncorrected. IR spectra were recorded on a Shimadzu IR spectrophotometer. 1H NMR and 13C NMR spectra were recorded on an Agilent Varian spectrometer in dimethyl sulfoxide (DMSO) at 400 MHz using TMS as internal standard, and chemical shifts are in δ. Mass spectroscopic analysis was done on a Shimadzu LC-MS. Analytical TLCs were performed on precoated Merck 0.25-mm silica gel 60 F254 plates using 40% ethyl acetate in n-hexane as eluent, and the spots were detected under UV light. Biological reagents such as Hoechst 33342, MTT, Tris, glycine, NaCl, SDS, BSA, and β-actin antibody were purchased from Sigma-Aldrich. DMEM, FBS, 0.4% trypan blue vital stain, and antibiotic-antimycotic mixture were obtained from Invitrogen. Antibodies against cyclin D1, Bcl-2, survivin, Mcl-1, lamin B1, and PARP were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies to phospho-specific p65 (Ser-536) and p65 were purchased from Cell Signaling Technology (Beverly, MA). Goat anti-rabbit-HRP conjugate and goat anti-mouse HRP were purchased from Sigma-Aldrich. CXCL12 was purchased from ProSpec-Tany TechnoGene Ltd. (Rehovot, Israel). Dextran sulfate sodium salt was obtained from MP Biomedicals. Etacept was obtained from CIPLA, Mumbai, India. Sulfasalazine was from Cadila Healthcare Ltd., Ahmedabad, India. Murine mini ELISA development kits for TNF-α, IFN-γ, IL-6, IL-1β, and IL-10 were purchased from PeproTech. All other chemicals were of analytical grade and were purchased from Sisco Research Laboratories (SRL, Mumbai, India).
Synthesis of Substituted Biscoumarins
General Procedure for One-pot Synthesis of bis-(4-Hydroxycoumarin)methanes
A mixture of (a) aromatic/heteroaromatic aldehydes (1 mmol), (b) 4-hydroxycoumarin (1.5 mmol) and (c) thiamine hydrochloride (0.2 mmol) in 4–5 ml of water was refluxed. The completion of reaction was monitored by thin layer chromatography with the solvent system ethyl acetate:hexane (4:6). After completion of the reaction, the reaction mixture was brought to room temperature. The solid crude product was separated by filtration, washed with water, and dried to give pure products. Spectral properties are consistent with the assigned structures.
Cell Lines
Human hepatocellular carcinoma cell lines (HepG2 and Hep3B) were obtained from American Type Culture Collection (Manassas, VA). HCCLM3 cells were kindly provided by Prof. Kam Man Hui, National Cancer Centre, Singapore. All HCC cell lines were cultured in DMEM containing 1× antibiotic-antimycotic solution with 10% FBS.
MTT Assay
The anti-proliferative effect of biscoumarins against HCC cells was determined by the MTT dye uptake method as described previously (24). Briefly, the cells (5 × 103/ml) were incubated in triplicate in a 96-well plate in the presence or absence of the indicated concentrations of compounds in a final volume of 0.2 ml for different time intervals at 37 °C. Thereafter, 20 μl of MTT solution (5 mg/ml in PBS) was added to each well. After a 2-h incubation at 37 °C, 0.1 ml of lysis buffer (20% SDS, 50% dimethylformamide) was added, incubation was done for 1 h at 37 °C, and subsequently the optical density at 570 nm was measured by a Tecan plate reader.
In Vitro TNF-α Binding Assay
The binding of TNF-α antibody binding was measured using a colorimetric-based Quantikine ELISA kit (R&D Systems). Different concentrations of BIHC (5–40 μm) were incubated with recombinant mouse TNF-α for 30 min at room temperature, and then 100 μl of the reaction mixture was transferred to a 96-well plate coated with a polyclonal antibody specific for mouse TNF-α followed by incubation for 2 h at room temperature. After washing, 100 μl of horseradish peroxidase-conjugated anti-TNF-α antibody was added to each well. After 30 min at room temperature, 100 μl of a stop solution (diluted HCl) was added, and the optical density of the streptavidin-POD enzyme conjugate was determined using a model 680 microplate reader (Bio-Rad) at 450 nm. Heparin (50 μg/ml) was used as a positive control.
SPR Analysis
Interaction of TNF-α with BIHC was examined using a Biacore-100 system. The TNF-α-immobilized sensor chip was prepared as described previously (25, 26). HBS-EP running buffer (10 mm HEPES, 0.15 m NaCl, 3 mm EDTA, and 0.005% (w/v) Tween 20, pH 7.4) having different concentrations of BIHC (10, 20, 30, 40, and 50 μm) was injected onto the surface of the sensor chip coated with TNF-α. Using the instrument default protocol, the flow rate was kept at a moderate speed of 30 μl/min. BIHC was allowed to interact with TNF-α for 2 min. The kinetic parameters for the binding of BIHC with TNF-α were obtained using a 1:1 binding model with mass transfer of BIA evaluation software.
In Silico Molecular Docking Studies
The co-crystal structure of TNF dimer with its inhibitor was used for structure-based molecular docking studies (27). A small molecule chromone-based compound has been reported to disrupt the native trimer of TNF-α binding to a dimeric complex, thereby exposing the hydrophobic protein-protein interface (27). We used the co-crystal structure of this compound and TNF-α as basis for our molecular docking studies (see Fig. 2A). We docked the full series of 23 biscoumarins to the TNF-α dimer using the default settings in MOE as described earlier (7, 28).
FIGURE 2.

In silico and in vitro analysis of the lead compound on TNF structure and functionality. A, the x-ray structure of TNF-α (yellow and blue graphic) and a small molecule inhibitor (all small molecules shown as sticks in element-wise coloring, carbon atoms in green) is shown. Six aromatic residues are exposed in the interface and are shown in stick representation. B, predicted binding mode of BIHC occupying a similar region in the hydrophobic binding site. C, ensemble of 10 snapshots from a molecular dynamics simulation of BIHC in the binding site highlighting flexibility of the ligand in the hydrophobic environment. D, the inhibitory effects of BIHC on the binding of TNF-α to anti-TNF-α antibody. Recombinant mouse TNF-α was incubated with the indicated concentrations of BIHC (5–40 μm). The bound TNF-α was quantified using the Quantikine kit. The percentage of inhibition of the binding of TNF-α to anti-TNF-α antibody against the concentration of BIHC is shown. Heparin (50 μg/ml) was used as a positive control. Data are represented as the mean ± S.D. for four replicates. * indicates p value < 0.05, and ** indicates p value < 0.01.
We further evaluated the predicted binding mode of BIHC by performing a molecular dynamics simulation. Thus, we parameterized the ligand in the Generalized Amber Force Field (29) and calculated point charges using Gaussian 03 (30) at HF/6-31G* level and RESP fitting (31). We prepared the TNF-α complex using MOE, capped termini using acetyl and N-methyl groups, and added a box of TIP3P water molecules (32) with minimum wall distance of 10 Å. Simulations were performed using the Amber force field 99SB-ILDN (33) using the GPU implementation of pmemd in AMBER12 (34). After careful equilibration including several minimization, heating, and cooling steps (35) we performed 10 ns of unrestrained molecular dynamics simulation.
Western Blotting
For detection of various proteins, BIHC-treated whole-cell extracts were lysed in lysis buffer (20 mm Tris (pH 7.4), 250 mm NaCl, 2 mm EDTA (pH 8.0), 0.1% Triton X-100, 0.01 mg/ml aprotinin, 0.005 mg/ml leupeptin, 0.4 mm PMSF, and 4 mm NaVO4). Tissue lysate was prepared using modified radioimmunoprecipitation assay buffer. Lysates were then spun at 14,000 rpm for 10 min to remove insoluble material and resolved on a 10% SDS gel. After electrophoresis, the proteins were electro-transferred to a nitrocellulose membrane, blocked with 5% nonfat milk, and probed with various antibodies (1:1000) overnight at 4 °C. The blot was washed, exposed to HRP-conjugated secondary antibodies for 1 h, and finally examined by chemiluminescence (ECL; GE Healthcare, Little Chalfont, Buckinghamshire, UK).
Invasion Assay
The in vitro invasion assay was performed using the Bio-Coat Matrigel invasion assay system (BD Biosciences), according to the manufacturer's instructions (36). 1 × 105 HepG2 cells were suspended in serum-free DMEM and seeded into the Matrigel Transwell chambers consisting of polycarbonate membranes with 8-μm pores. After preincubation with or without BIHC (25 μm) for 8 h, the Transwell chambers were then placed into appropriate wells of a 24-well plate, in which either the basal medium only or basal medium containing CXCL12 had been added. After incubation for 24 h, the upper surfaces of the Transwell chambers were wiped with cotton swabs, and the invading cells were fixed and stained with crystal violet solution. The invading cells were then counted in five randomly selected areas under microscopic observation.
In Vivo Anti-inflammatory Studies
Adult Swiss albino mice (20–25 g) were injected intraperitoneally with 2 ml of 3% thioglycolate (TG) broth (Sigma) or sterile saline as described earlier (37). After 10 min, BIHC (0.5, 1.5 or 5 mg/kg) suspended in saline was injected through a lateral tail vein. After 24 h, LPS (1.0 μg) was injected intraperitoneally, and 1 h later, the peritoneal cavities were lavaged with 4 ml of PBS containing 3 mm EDTA. After the total number of inflammatory cells was counted, the lavage fluid (2 ml) was centrifuged at 1,500 rpm for 5 min, and the supernatant was stored at −20 °C for the detection of TNF-α. The amount of TNF-α was determined by using a Quantikine ELISA kit (R&D Systems). Heparin (10 mg/kg) was used as a positive control.
Inflammatory Bowel Disease Model
6–8-week-old Swiss albino mice weighing about 30–36 grams were kept in a 12-h light and 12-h dark cycle and fed with standard chow formula and reverse osmosis (RO) water and were acclimatized for 1 week. Colitis was induced by administering 5% DSS (w/v) in RO water for 4 days to all the mice except RO control, Etacept alone, Sulfasalazine alone, and BIHC alone. The mice were randomly divided into nine groups (n = 6), and each group received the treatment from day 5 to day 9 as follows: Group 1, RO water control; Group 2, DSS-treated; Group 3, DSS- and Etacept-treated (5 mg/kg/day, subcutaneously injected); Group 4, DSS- and Sulfasalazine-treated (500 mg/kg/day, oral gavage); Group 5, DSS- and BIHC-treated (2.5 mg/kg/day, intraperitoneal injection); Group 6, DSS- and BIHC-treated (5 mg/kg/day, intraperitoneal injection); Group 7, BIHC alone (5 mg/kg/day, subcutaneous injection); Group 8, Etacept alone (5 mg/kg/day, subcutaneous injection); and Group 9, Sulfasalazine alone (500 mg/kg/day, oral gavage). All the animals were given free access to RO water after day 4 until the completion of the experiment. Animals were euthanized at day 10 through cardiac puncture, and blood was collected to assess the levels of various pro- and anti-inflammatory cytokines in serum. The colon was excised from caecum to anus, flushed with ice-cold phosphate-buffered saline (10 mm, pH 7.4), and further processed for histological analysis. The efficacy of BIHC against colitis was compared with Etacept (standard TNF-α inhibitor) and Sulfasalazine (standard therapeutic agent for colitis).
Determination of Disease Activity Index (DAI)
DAI was scored as described earlier by Cooper et al. (38). In brief, weight loss, stool consistency, and gross bleeding are the three individual parameters considered to assign the score to determine DAI. Stool consistency scored 0 for normal stool; 2 for loose stool; and 4 for diarrhea. Rectal bleeding scored 0 for normal; 2 for occult bleeding; and 4 for gross bleeding. Lastly, severity of colitis in the colons were analyzed by measuring the length of the colon, which is an indirect evidence of colonic inflammation.
Myeloperoxidase (MPO) Activity
The excised colonic tissue from all the groups were homogenized in 50 mm potassium phosphate buffer pH 6.0 containing 0.5% hexadecyltrimethyl ammonium bromide. Tissue debris were removed by centrifuging the homogenates at 3,000 rpm for 4 min at 4 °C. Supernatant (10 μl) was taken in 96-well plate in triplicates, and 200 μl of ODA-H2O2 reagent (0.167 mg/ml o-dianisidine dihydrochloride and 0.05% H2O2) was added to each well including the well containing 10 μl of buffer alone, which served as blank. Absorbance was measured at 450 nm using multimode plate reader (Thermo Scientific) at 0, 30, and 60 S. The difference between two time points was taken, and the MPO activity was calculated using the formula (MPO constant: 1.13×10−2).
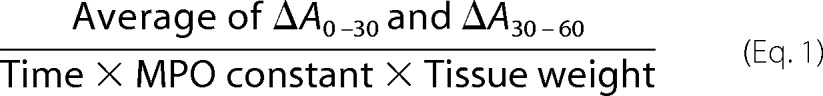 |
Estimation of Serum Cytokine Levels
The modulatory effect of BIHC on the levels of pro- and anti-inflammatory cytokines (TNF-α, IFN-γ, IL-1β, IL-6, and IL-10) were estimated using ELISA kits according to the manufacturer's protocol (PeproTech).
Histological Evaluation
The colonic tissues were fixed overnight in 10% phosphate-buffered formalin and dehydrated by processing with alcohol and chloroform mixture. The processed tissues were embedded in paraffin wax, and sections of 5-μm thickness were prepared. Further sections were stained with hematoxylin-eosin dye (H&E) and photographed under an Axio Imager.A2 microscope (Carl Zeiss AG, Oberkochen, Germany). The method of Gonzalez-Rey et al. (39) was followed for histological scoring of inflammation of colon section on a 0–3 graded scale: 0 for no sign of inflammation; 1 for low leukocyte infiltration; 2 for moderate leukocyte infiltration, thickening of the colon, moderate goblet cell loss, and focal loss of crypts; and 3 for transmural infiltration, massive loss of goblet cells, and diffuse loss of crypts. Irrespective of the treatments each slide was scored from five random spots.
Statistical Analysis
The statistical analysis was done using a software Origin 8 (OriginLab). The Mann-Whitney U test was used to determine p values.
RESULTS
Chemistry
7,7′-Dihydroxy-6,6′-dimethoxy-3,3′-biscoumarin was isolated from Erycibe obtusifolia (40). Additionally, the biscoumarin was synthesized by the condensation of 4-hydroxycoumarin and aldehydes using Et2AlCl (41) or tetra-n-butylammonium bromide (TBAB) (42) as catalysts. Previously, microwave-assisted or sonochemically (43) mediated catalyst-free condensation reactions were reported. In the current work we employed the thiamine hydrochloride-catalyzed condensation reaction of 4-hydroxycoumarins with aromatic, heteroaromatic, and aliphatic aldehydes for the synthesis of biscoumarin compounds. Details of the condensation reaction between 4-hydroxycoumarin and various aldehydes are summarized in Scheme 1 in Fig. 1A. Upon treatment of 4-hydroxycoumarin and various aldehydes reaction in the presence of thiamine hydrochloride in refluxing water, took place within 4–8 h, giving white solid products.
FIGURE 1.
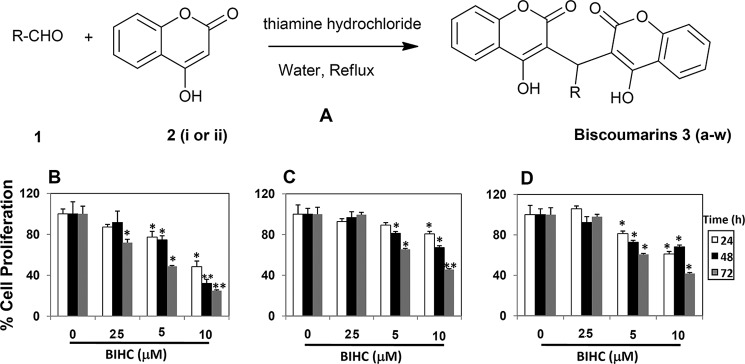
Synthesis (A) and cytotoxicity evaluation (B–D) of novel biscoumarins toward HCC cells. A, scheme represents the synthesis of biscoumarins (R-aromatic/heteroaromatic group). Twenty-three new biscoumarin derivatives were designated as 3(a-w). The anti-proliferative effects of BIHC against HepG2 (B), HCCLM3 (C), and Hep3B (D) cells were determined by MTT assay and reported as the percentage of viable cells relative to the control. Values are means ± S.E. of three independent experiments. * indicates p value < 0.05.
Biology
Biscoumarin Analogs Suppress Proliferation of HCC Cells in a Dose- and Time-dependent Manner
We evaluated the cytotoxic effects of the newly synthesized series of compounds on HepG2 cells using the MTT assay. Among the tested compounds, BIHC was found to be most effective with an IC50 value of 6.4 μm. Furthermore, we investigated whether BIHC can inhibit the proliferation of various HCC cell lines, namely HepG2, Hep3B, and HCCLM3, by using the MTT assay. BIHC significantly inhibited the proliferation of all the three HCC cell lines in a dose- and time-dependent manner (Fig. 1, B–D).
In Silico Mode-of-action Analysis of BIHC
Human protein targets were predicted for BIHC, which was shown to have the highest anti-proliferative effect in the HCC cell line HepG2 (IC50 = 6.4 μm). For this purpose, we employed in silico target prediction algorithms, which are trained on known bioactivity data and which have been successfully applied in different situations before (44, 45). We employed a Laplacian-modified Naïve Bayes classifier as published previously (46). Five targets were predicted with a log-odds score exceeding the cut-off of 5. Given the well established role of TNF in cancer as described in previous studies (47, 48) this target was chosen for further computational as well as experimental analysis.
In Silico Interaction Studies of Biscoumarins with TNF
A small molecule inhibitor of TNF-α, namely 6,7-dimethyl-3-[(methyl{2-[methyl({1-[3-(trifluoromethyl)phenyl]-1H-indol-3-yl}methyl)amino]ethyl}amino)methyl]-4H-chromen-4-one (DIC), which promotes subunit disassembly of the trimeric cytokine family member, has been reported previously (27) (Fig. 2A). Using in silico docking (as described under “Experimental Procedures”) we found that all 23 biscoumarins seem to occupy similar regions of the binding pocket (see Fig. 2C for the predicted binding pose of BIHC showing highest binding affinity). Docking scores correlated to a good extent with molecular weight (r2 = 0.62), as expected in a hydrophobic environment, which is lacking distinct anchor points for small molecule binding. Therefore, we did not attempt to rationalize binding affinities and particular ligand-protein interactions in more detail. Strikingly, we found the most active compound BIHC to show the lowest strain energy in comparison with all other compounds in the series. Hence, we hypothesize that conformational aspects seem to be crucial for small molecule binding to TNF-α. We furthermore performed an unrestrained molecular dynamics simulation to investigate the predicted binding mode from molecular docking. We found the complex to be very stable with a root mean square deviation of Cα positions smaller than 1.5 Å. Although the ligand is highly flexible in the hydrophobic binding interface, it remains close to the predicted binding mode over the full trajectory length of 10 ns (Fig. 2B). Therefore, the predicted binding mode of BIHC in the interface of TNF-α is overall found to be plausible and appears to be dominated by hydrophobic (and overall rather unspecific) interactions.
Inhibitory Effect of BIHC on TNF-α/anti-TNF-α Antibody Binding
To determine whether BIHC can block the binding of TNF-α to its antibody, and hence lend support to the target predicted in silico, BIHC was incubated in vitro with recombinant TNF-α and its inhibitory effect was quantified. It was found across a dose range from 5 to 40 μm that BIHC inhibited the binding of TNF-α to its antibody in a dose-dependent manner, resulting in an IC50 value of 16.5 μm (Fig. 2D).
SPR Analysis of BIHC with TNF
We analyzed the interaction between BIHC and TNF-α via SPR analysis using the Biacore system. The N terminus of TNF-α was immobilized on the streptavidin-coated sensor chip. The injection of different concentrations of BIHC onto the surface of a sensor chip coated with TNF-α resulted in the sensorgrams as shown in Fig. 3A. 8.5 ± 1.5 × 103 m−1 s−1 as association and 3.2 ± 0.7 × 10−3 s−1 as dissociation rate constants were obtained for the BIHC interaction with TNF-α. The dissociation equilibrium constant (Kd) was found to be 21.4 μm, signifying the affinity of BIHC toward TNF-α.
FIGURE 3.
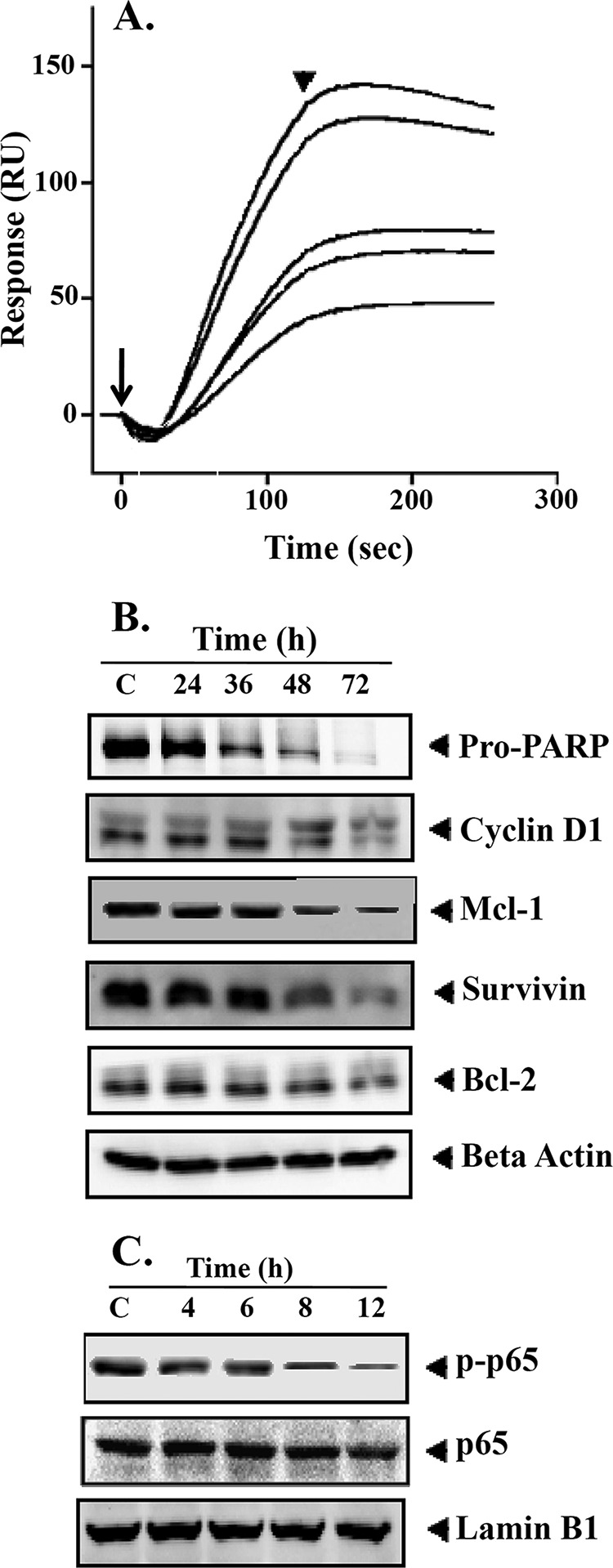
A, interaction of BIHC toward TNF-α. Various concentrations of BIHC (10–50 μm) were injected onto the surface of a TNF-α-immobilized sensor chip. Obtained sensorgrams were overlaid using the BIA evaluation software. RU, resonance units. Long and short arrows represent the beginning of the association and dissociation phases, respectively. B, BIHC suppresses NF-κB-regulated gene products involved in proliferation, survival, and angiogenesis. HepG2 cells (2 × 106/ml) were treated with 25 μm BIHC for the indicated time intervals, after which whole-cell extracts were prepared, 30 μg of proteins of those extracts was resolved on 10% SDS-PAGE, and membrane was sliced according to molecular weight and probed against PARP, cyclin D1, Mcl-1, survivin, and Bcl-2 antibodies. The same blots were stripped and reprobed with β-actin antibody to verify equal protein loading. The results of cyclin D1, Survivin, and Bcl-2 shown are representative of two independent experiments. C, HepG2 cells (2 × 106/ml) were treated with 25 μm BIHC for the indicated time intervals, after which whole-cell extracts were prepared and 30 μg of proteins of those extracts was resolved on 10% SDS-PAGE, and membrane was sliced according to molecular weight and probed against phospho-p65 antibody. The same blots were stripped and reprobed with p65 and lamin B1 antibodies.
BIHC Down-regulates the Expression of Cyclin D1, Mcl-1, Survivin, and Bcl-2 in HCC Cells
NF-κB activation has been shown to regulate the expression of various gene products involved in cell survival, proliferation, angiogenesis, and chemoresistance (10). We found that the cell cycle regulator, cyclin D1, and the anti-apoptotic proteins, Mcl-1, survivin, and Bcl-2, were constitutively expressed in HCC cells. However, their expression decreased in a time-dependent manner upon BIHC treatment, with maximum suppression observed at around 72 h (Fig. 3B). This also supports our hypothesis of TNF-α as a target of this compound.
BIHC Causes PARP Cleavage in HCC Cells
Whether suppression of constitutively active NF-κB in HepG2 cells by BIHC leads to apoptosis was investigated in the next step so that functional conclusions with regard to compound action could also be drawn. It was found that BIHC treatment of HepG2 cells resulted in time-dependent impact on cleavage of a 116-kDa PARP protein into an 85-kDa fragment (Fig. 3B). Given the biological role of PARP these results clearly suggest that BIHC induces substantial apoptosis in HepG2 cells.
BIHC Inhibits Phosphorylation and Translocation of p65 in HCC Cell Lines
In the next step we investigated the effect of BIHC on the constitutive phosphorylation of p65, given that phosphorylation is required for its transcriptional activity (9, 10). HepG2 cells were preincubated with different concentrations of BIHC, and nuclear extracts were prepared and tested for p65 phosphorylation and translocation using Western blot analysis. As shown in Fig. 3C, BIHC treatment abrogated both p65 phosphorylation and translocation in a time-dependent manner, with maximum inhibition seen at 12 h.
BIHC Inhibits CXCL12-induced HCC Cell Invasion
We also determined the effect of BIHC on HCC cell invasion, a key hallmark of cancer. Using an in vitro Boyden chamber assay, we found that HepG2 cells invaded faster under the influence of CXCL12 (Fig. 4). To elucidate further the effect on BIHC on CXCL12-induced cell invasion, we used BD BioCoat Matrigel invasion assay system and found that the treatment with BIHC significantly suppressed CXCL12-induced invasion of HepG2 cells (Fig. 4).
FIGURE 4.
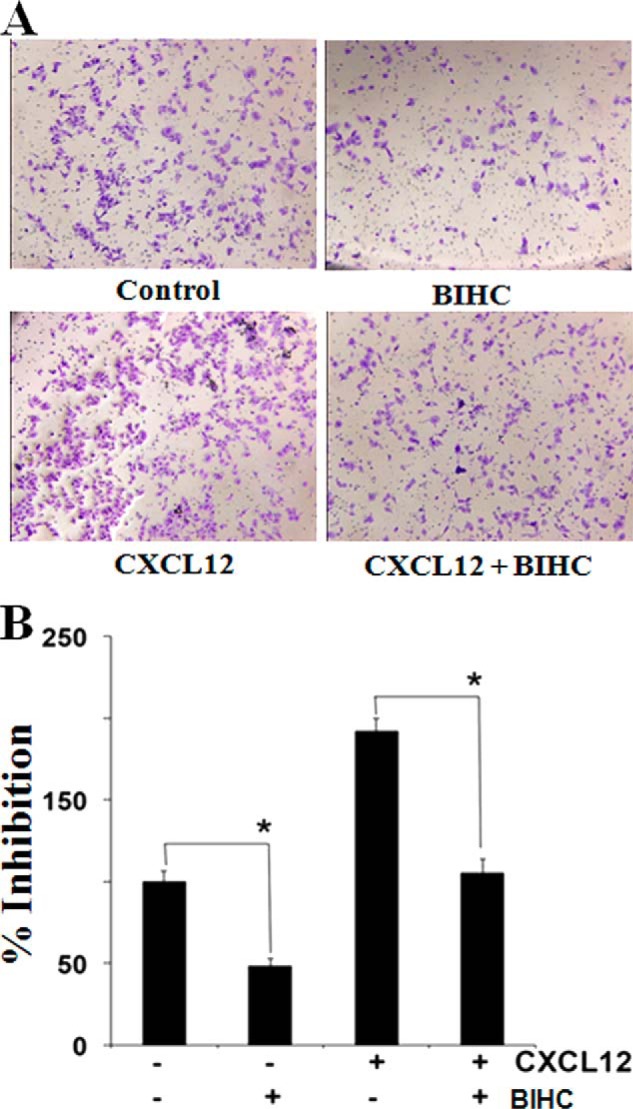
A, cell invasion assay for evaluating the inhibitory effect of BIHC on HepG2 cell invasion. HepG2 cells were pretreated with CXCL12 (20 μm for 2 h) followed by incubation with BIHC for 8 h. The cells were also treated with 25 μm BIHC alone for 8 h or CXCL12 (20 μm for 2 h) alone. The cells were fixed with 4% paraformaldehyde before staining with 0.5% crystal violet as described under “Experimental Procedures.” B, the percentage of the migratory cells of the treated group was normalized against the untreated group. The values are the means ± S.E. of three independent experiments. *, p < 0.05.
Effect of BIHC on the Secretion of TNF-α in Vivo
The effects of BIHC on the local production of TNF-α in the peritoneal cavities of TG-treated mice were examined to confirm the anti-inflammatory role of BIHC. BIHC effectively inhibited the infiltration of inflammatory cells in the lavage fluid by 47, 55, and 64% at 0.5, 1.5, and 5 mg/kg, respectively, as compared with the control (Fig. 5A). Heparin (10 mg/kg), which was used as a positive control, inhibited the infiltration of inflammatory cells in the lavage fluid by 81%. In addition, higher levels of TNF-α were detected in the mice primed with TG and LPS as compared with saline-injected control mice. TNF-α release was reduced from 42 (control) to 26, 22, or 19 × 10−2 ng/ml, corresponding to 38, 46, and 55% reduction as compared with the control at 0.5, 1.5, and 5 mg/kg of BIHC, respectively (Fig. 5B). Heparin, the positive control, inhibited TNF-α release from 42 to 12 × 10−2 ng/ml, corresponding to 70% reduction at 10 mg/kg of dose. These results support the effects of the direct binding of BIHC to TNF-α.
FIGURE 5.
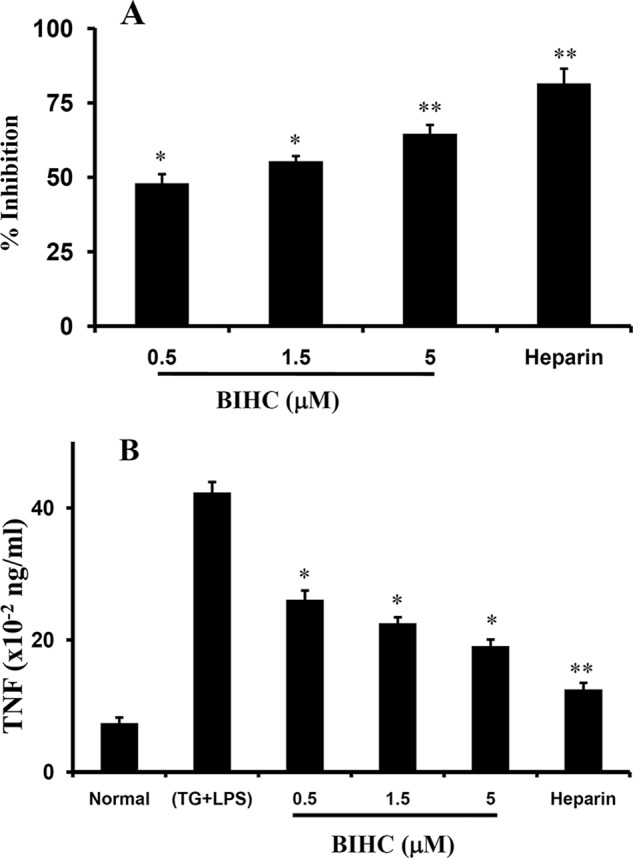
In vivo anti-inflammatory activity of BIHC. A, the anti-inflammatory effects of BIHC on TG + LPS-induced inflammation in mice. Swiss albino mice were injected intraperitoneally with 2 ml of 3% TG and LPS (1 μg). Intravenous administration of BIHC (0.5, 1.5, or 5 mg/kg) and heparin (10 mg/kg) effectively suppressed the inflammation. The bar graph represents the percentage of inhibition of the infiltration of inflammatory cells into the peritoneal fluid by BIHC and heparin. B, the effects of BIHC (0.5, 1.5, or 5 mg/kg) and heparin (10 mg/kg) on the local production of TNF-α in the peritoneal cavities of the mice primed with TG and LPS. Data from the quantification of TNF-α are presented as the mean ± S.D. for three independent experiments, and each experiment was conducted with six mice per group. *, p < 0.05, and **, p < 0.01 versus control.
Effect of BIHC on DSS-induced Disease Activity Index, Colon Length, and MPO Activity
The severity of DSS-induced colitis was monitored by assessing the DAI throughout the experimental period. We observed the increase in DAI in the animals administered with DSS as compared with control group (Fig. 6A). After day 6, DAI was significantly decreased in the groups treated with BIHC (p < 0.01), Etacept (p < 0.01), and Sulfasalazine (p < 0.01) in comparison with the DSS control group. Animals treated with a higher dose of BIHC (5 mg/kg of body weight) showed better recovery than its counterpart (2.5 mg/kg of body weight). We also observed the significant reduction in length of the colon in DSS-induced animals (Fig. 6B). On the other hand, treatment with BIHC, Etacept, and Sulfasalazine showed a substantial increase in length of the colon as compared with DSS control group. As an index of colonic inflammation, neutrophil infiltration was assessed via MPO activity from the colons of the control and DSS-induced experimental animals. The results revealed a significant (p < 0.001) increase in the activity of MPO in the colons of DSS-induced mice in comparison with RO control (Fig. 6C), signifying the severe intestinal inflammation. In contrast, treatment with BIHC, Etacept, and Sulfasalazine showed a substantial (p < 0.001) reduction in MPO activity in the colons as compared with animals treated with DSS alone, indicating the anti-inflammatory potential of BIHC.
FIGURE 6.
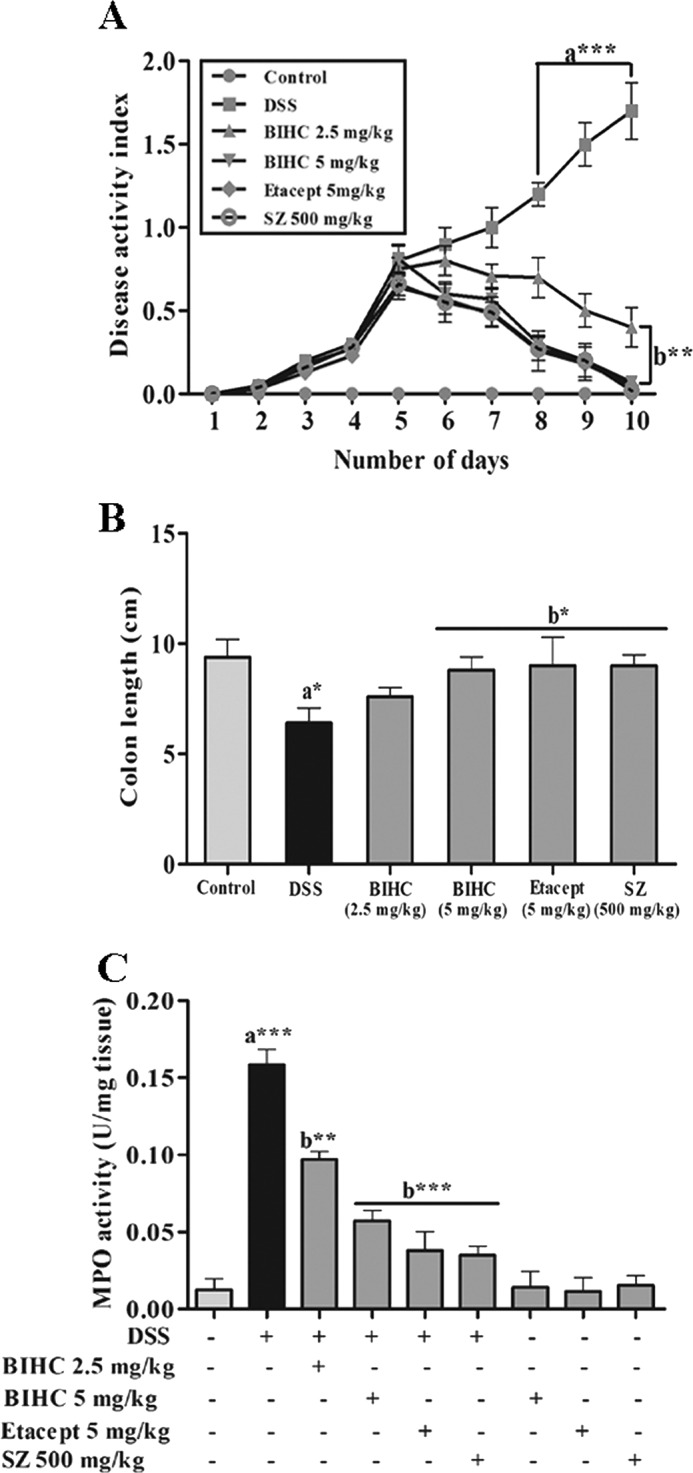
Effect of BIHC on DSS-induced DAI, colon length, and MPO activity. A, the severity of DSS-induced colitis was monitored daily by assessing the DAI throughout the experimental period. From day 4 onward DSS induced a significant increase in DAI. SZ: Sulfasalazine. B, severity of the colonic inflammation was assessed indirectly by measuring the colon length of RO control and experimental mice. C, severity of colonic inflammation was assessed by colonic MPO activity from RO control and experimental mice. Data are presented as mean ± S.E. *, p < 0.05, **, p < 0.01; ***, p < 0.001; n = 6; a: significant in comparison with RO control; b: significant in comparison with DSS control.
Effect of BIHC on Histopathology
We carried out the histopathological examination of colons using H&E staining (Fig. 7A) and assigned the scores to present in the form of a bar graph (Fig. 7B). We observed the distortion of the cryptic epithelium, as well as mucosal and submucosal infiltration of inflammatory leukocytes in the colon of DSS-induced animals (Fig. 7A, panel ii), whereas the animals treated with BIHC (5 mg/kg of body weight) (Fig. 7A, panel iii) displayed normal architecture with epithelial crypt structures and lesser infiltration of inflammatory leukocytes, which was highly comparable with Etacept (Fig. 7A, panel iv)- and Sulfasalazine (Fig. 7A, panel v)-treated animals.
FIGURE 7.

A, histopathological images (100×) of H&E-stained colon cross sections of RO control mice and DSS-induced mice treated with BIHC, Etacept, and Sulfasalazine. Panel i, colon section of the RO control mice showing normal crypt structures with intact mucosal and submucosal epithelium. Panel ii, colon section of the DSS-treated mice showing severe transmural inflammation and distorted crypt epithelium along with mucosal and submucosal infiltration of inflammatory cells. Panels iii–v, representative colon sections of the DSS-induced mice treated with BIHC, Etacept, and Sulfasalazine, respectively, showing restoration of crypt epithelial degeneration along with lesser infiltration of inflammatory cells. B, DSS treatment severely increased the histopathological score in comparison with RO control. Treatment with BIHC, Etacept, and Sulfasalazine (SZ) significantly reduced the DSS-induced histopathological scores. Scores are presented as mean ± S.E. of five independent observations of colon sections. ***, p < 0.001; a: significant in comparison with RO control; b: significant in comparison with DSS control.
Effect of BIHC on DSS-induced Secretion of pro- and anti-inflammatory Cytokines
Cytokines play a critical role in the progression of colitis, and essentially an imbalance in the levels of pro- and anti-inflammatory cytokines may prevent the prognosis of colitis. The levels of pro- and anti-inflammatory cytokines from serum of all the groups were estimated using ELISA kits. It was apparent that the levels of pro-inflammatory cytokines (TNF-α, IFN-γ, IL-1β, and IL-6) were notably increased (p < 0.01) and the anti-inflammatory cytokine (IL-10) was decreased (p < 0.01) in DSS-induced animals (Fig. 8, A–D). In contrast, animals treated with BIHC (5 mg/kg of body weight), Etacept, and Sulfasalazine significantly (p < 0.01) reverted back the DSS-induced pro- and anti-inflammatory cytokines. These data were further supported by immunoblotting of colon tissue homogenate against TNF-α antibody. Colon tissue homogenate of the mice treated with DSS alone exhibited elevated levels of TNF-α, whereas colon homogenate of the mice treated with BIHC (5 mg/kg of body weight), Etacept, and Sulfasalazine exhibited substantially decreased elevated levels of TNF-α (Fig. 8E).
FIGURE 8.

Effect of BIHC on DSS-induced secretion of pro- and anti-inflammatory cytokines. Serum cytokines levels of experimental animals were estimated using murine mini ELISA development kits according to the manufacturer's protocol. A–E, levels of serum cytokines: TNF-α (A); IFN-γ (B); IL-1β (C); IL-6 (D); and IL-10 (E). SZ, Sulfasalazine. F, Western blot of TNF-α expression in colon tissues. Data are presented as mean ± S.E. of five independent experiments. *, p < 0.05, **, p < 0.01; a: significant in comparison with control; b: significant in comparison with DSS control.
DISCUSSION
TNF is reported to be elevated in several types of cancer and one of the leading causes of inflammatory diseases, which renders it a potential therapeutic target in this context (49–51). The current study focuses on the synthesis of a library of novel biscoumarins, the identification of its most potent member, which was BIHC, the computational prediction of its putative mode of action and its validation, as well as the comprehensive biological characterization of the compound.
Our cheminformatics approach presented as a predicted target TNF-α, and further structure-based analysis was able to develop a hypothesis for the interaction of BIHC with dimeric TNF. To validate the predicted target we evaluated the inhibitory activity of BIHC by an ELISA-based in vitro TNF-α binding assay. Results showed a dose-dependent inhibition of antibody binding, comparable with heparin, with an IC50 of 16.5 μm. Furthermore, the SPR analysis revealed interaction of BIHC with TNF-α, signifying the preference of biscoumarins over the inflammatory marker. Given that TNF signaling activates a series of cytoplasmic kinases, which finally phosphorylate subunits of NF-κB (10), we then evaluated phosphorylation levels of p65 in HepG2 cells treated with BIHC and found that they decrease in a time-dependent manner. NF-κB is a transcription factor that transcribes the cluster of genes coding various proteins involved in cell survival, angiogenesis, anti-apoptosis, and metastasis (7, 52), and to assess the functional impact we then profiled the expression pattern of NF-κB-regulated gene products (Mcl-1, survivin, Bcl-2, cyclin D1) upon BIHC treatment, where we observed a significant decline of anti-apoptotic proteins. Furthermore, we confirmed the apoptosis-inducing effect of BIHC by detecting cleavage of pro-PARP. Given that NF-κB also transcribes genes encoding proteins with migratory function we have furthermore confirmed that BIHC substantially reduced cell migration upon CXCL12 stimulation.
Furthermore, we were also able to gather in vivo supports for the effects described above in vitro. For this, we injected TG broth intraperitoneally to adult Swiss albino mice to induce the infiltration of immune cells. Upon administration of LPS to TG-injected mice high levels of TNF in the lavage fluid collected from peritoneal cavity were observed. In the mouse group treated with BIHC, the levels of TNF have been significantly reduced, in a dose-dependent manner, comparable at high doses to heparin treatment.
In addition to monoclonal antibodies, several synthetic small molecules and phytochemicals have been studied extensively as TNF-α inhibitors in developing therapeutic agents against various cancers and inflammatory diseases (13, 53, 54). A wide array of phytochemicals including flavonoids (butein, quercetin), polyphenols (curcumin, capsaicin), alkaloids (piperine, berberine), and terpenoids (lactones) have been implemented in this direction (54). Although these natural compounds interfere with TNF signaling, they largely exhibit non-specificity and induce their effect in a multifactorial way. For instance, curcumin has been demonstrated to induce its inhibitory effect on TNF-α, JAK-STAT, JNK, NF-κB, and MAPK signaling pathways, suggesting the diverse targets of natural compounds (55–57). On the other hand, BIHC specifically inhibits the TNF signaling, thereby contributing to suppression of cancer cell proliferation.
Finally, to provide the direct evidence on structural distortion of TNF-α, we demonstrated the effect of BIHC on TNF-α-induced inflammation in an inflammatory bowel disease mouse model and compared it with anti-inflammatory drugs such as Etacept (Etanercept) and Sulfasalazine. Early treatment strategies in ulcerative colitis employ the use of corticosteroids and 5-aminosalicylic acid derivatives (Sulfasalazine drugs), and the disease at the advanced stage demands the use of high potential therapeutics such as monoclonal antibodies (Etanercept, Adalimumab) (58, 59). Despite the fact that Sulfasalazine drugs and monoclonal antibodies against TNF are the plausible remedy for ulcerative colitis, the long term use of the former may result in nausea, flatulence, and diarrhea, and the latter may induce neoplastic transformation and severe immunosuppression. Therefore, designing and development of new selective TNF-α inhibitors is critically essential to override the harmful side effects of existing therapeutics. In the present study, intraperitoneal injection of BIHC at a dose of 5 mg/kg of body weight (approximately equivalent to a 300-mg dose in a 60-kg human) into colitis-induced mouse resulted in a substantial decrease in disruption of mucosal barrier, crypt architecture, disease activity index, and myeloperoxidase activity, as well as a substantial increase in colon length, which is highly significant and comparable with Etacept and Sulfasalazine. Furthermore, the reversal of pro- and anti-inflammatory cytokines in the BIHC-treated group is an additive evidence to demonstrate the mode of action of BIHC. In summary, herein we describe the synthesis, mode-of-action analysis, and in vitro and in vivo activity of a novel biscoumarin small molecule that targets TNF-α.
Acknowledgments
We thank Gajanan D. Katkar, Paul M, Naveen Kumar SK, and Jagadish S for kind help during the study.
This work was supported by grants from National University of Singapore (NUS) Academic Research Fund (Grant R-184-000-207-112) (to G. S.). This research was also supported by the University Grants Commission (41-257-2012-SR), Vision Group Science and Technology, and Department of Science and Technology (Grant SR/FT/LS-142/2012) (to Basappa).
- HCC
- hepatocellular carcinoma
- BIHC
- 3,3′-((2-butyl-5-chloro-1H-imidazol-4-yl)methylene)bis(4-hydroxy-2H-chromen-2-one)
- DSS
- dextran sulfate sodium
- PARP
- poly(ADP-ribose) polymerase
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- TG
- thioglycolate
- DAI
- disease activity index
- RO
- reverse osmosis
- MPO
- myeloperoxidase.
REFERENCES
- 1. Hehlgans T., Pfeffer K. (2005) The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: players, rules and the games. Immunology 115, 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carswell E. A., Old L. J., Kassel R. L., Green S., Fiore N., Williamson B. (1975) An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. U.S.A. 72, 3666–3670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bradley J. R. (2008) TNF-mediated inflammatory disease. J. Pathol. 214, 149–160 [DOI] [PubMed] [Google Scholar]
- 4. Hawes A. S., Fischer E., Marano M. A., Van Zee K. J., Rock C. S., Lowry S. F., Calvano S. E., Moldawer L. L. (1993) Comparison of peripheral blood leukocyte kinetics after live Escherichia coli, endotoxin, or interleukin-1 α administration: studies using a novel interleukin-1 receptor antagonist. Ann. Surg. 218, 79–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Locksley R. M., Killeen N., Lenardo M. J. (2001) The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 104, 487–501 [DOI] [PubMed] [Google Scholar]
- 6. Wu Y., Zhou B. P. (2010) TNF-α/NF-κB/Snail pathway in cancer cell migration and invasion. Br. J. Cancer 102, 639–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chemical Computing Group Inc. (2013) Molecular Operating Environment (MOE), Chemical Computing Group Inc., Montreal, Quebec H3A 2R7, Canada [Google Scholar]
- 8. Nemchenko A., Hill J. A. (2010) NEMO nuances NF-κB. Circ. Res. 106, 10–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sethi G., Tergaonkar V. (2009) Potential pharmacological control of the NF-κB pathway. Trends Pharmacol. Sci. 30, 313–321 [DOI] [PubMed] [Google Scholar]
- 10. Li F., Sethi G. (2010) Targeting transcription factor NF-κB to overcome chemoresistance and radioresistance in cancer therapy. Biochim. Biophys. Acta 1805, 167–180 [DOI] [PubMed] [Google Scholar]
- 11. Sasaki C. Y., Barberi T. J., Ghosh P., Longo D. L. (2005) Phosphorylation of RelA/p65 on serine 536 defines an IκBα-independent NF-κB pathway. J. Biol. Chem. 280, 34538–34547 [DOI] [PubMed] [Google Scholar]
- 12. Nabel G. J., Verma I. M. (1993) Proposed NF-κB/IκB family nomenclature. Genes Dev. 7, 2063. [DOI] [PubMed] [Google Scholar]
- 13. Shrimali D., Shanmugam M. K., Kumar A. P., Zhang J., Tan B. K., Ahn K. S., Sethi G. (2013) Targeted abrogation of diverse signal transduction cascades by emodin for the treatment of inflammatory disorders and cancer. Cancer Lett. 341, 139–149 [DOI] [PubMed] [Google Scholar]
- 14. Sethi G., Sung B., Kunnumakkara A. B., Aggarwal B. B. (2009) Targeting TNF for treatment of cancer and autoimmunity. Adv. Exp. Med. Biol. 647, 37–51 [DOI] [PubMed] [Google Scholar]
- 15. Siddiqui A. M., Cui X., Wu R., Dong W., Zhou M., Hu M., Simms H. H., Wang P. (2006) The anti-inflammatory effect of curcumin in an experimental model of sepsis is mediated by up-regulation of peroxisome proliferator-activated receptor-γ. Crit. Care Med. 34, 1874–1882 [DOI] [PubMed] [Google Scholar]
- 16. Essayan D. M. (2001) Cyclic nucleotide phosphodiesterases. J. Allergy Clin. Immunol. 108, 671–680 [DOI] [PubMed] [Google Scholar]
- 17. Snir O., Hesselberg E., Amoudruz P., Klareskog L., Zarea-Ganji I., Catrina A. I., Padyukov L., Malmström V., Seddighzadeh M. (2013) Genetic variation in the serotonin receptor gene affects immune responses in rheumatoid arthritis. Genes Immun. 14, 83–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Basappa, Murugan S., Kavitha C. V., Purushothaman A., Nevin K. G., Sugahara K., Rangappa K. S. (2010) A small oxazine compound as an anti-tumor agent: a novel pyranoside mimetic that binds to VEGF, HB-EGF, and TNF-α. Cancer Lett. 297, 231–243 [DOI] [PubMed] [Google Scholar]
- 19. Tanaka T., Hishitani Y., Ogata A. (2014) Monoclonal antibodies in rheumatoid arthritis: comparative effectiveness of tocilizumab with tumor necrosis factor inhibitors. Biologics 8, 141–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alli-Akintade L., Pruthvi P., Hadi N., Sachar D. (2014) Race and fistulizing perianal Crohn's disease. J. Clin. Gastroenterol. 10.1097/MCG.0000000000000117 [DOI] [PubMed] [Google Scholar]
- 21. Dasgupta B., Combe B., Louw I., Wollenhaupt J., Zerbini C. A., Beaulieu A., Schulze-Koops H., Durez P., Wolff V., Yao R., Weng H. H., Govoni M., Vastesaeger N. (2014) Patient and physician expectations of add-on treatment with golimumab for rheumatoid arthritis: relationships between expectations and clinical and quality of life outcomes. Arthritis Care Res. (Hoboken) 10.1002/acr.22371 [DOI] [PubMed] [Google Scholar]
- 22. Kobayashi T., Yokoyama T., Ito S., Kobayashi D., Yamagata A., Okada M., Oofusa K., Narita I., Murasawa A., Nakazono K., Yoshie H. (2014) Periodontal and serum protein profiles in patients with rheumatoid arthritis treated with tumor necrosis factor inhibitor Adalimumab. J. Periodontol. 10.1902/jop.2014.140194 [DOI] [PubMed] [Google Scholar]
- 23. Rüngeler P., Castro V., Mora G., Gören N., Vichnewski W., Pahl H. L., Merfort I., Schmidt T. J. (1999) Inhibition of transcription factor NF-κB by sesquiterpene lactones: a proposed molecular mechanism of action. Bioorg. Med. Chem. 7, 2343–2352 [DOI] [PubMed] [Google Scholar]
- 24. Kumar B., Paricharak S., Dinesh K. R., Sivaraman Siveen K., Fuchs J., Rangappa S., Mohan C. D., Mohandas N., Kumar A. P., Sethi G., Bender A., Basappa S., Rangappa K. S. (2014) Synthesis, biological evaluation and in silico and in vitro mode-of-action analysis of novel dihydropyrimidones targeting PPAR-γ. RSC Adv. 4, 45143–45146 [Google Scholar]
- 25. Basappa, Sugahara K., Thimmaiah K. N., Bid H. K., Houghton P. J., Rangappa K. S. (2012) Anti-tumor activity of a novel hS-mimetic-vascular endothelial growth factor binding small molecule. PLoS One 7, e39444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fongmoon D., Shetty A. K., Basappa, Yamada S., Sugiura M., Kongtawelert P., Sugahara K. (2007) Chondroitinase-mediated degradation of rare 3-O-sulfated glucuronic acid in functional oversulfated chondroitin sulfate K and E. J. Biol. Chem. 282, 36895–36904 [DOI] [PubMed] [Google Scholar]
- 27. He M. M., Smith A. S., Oslob J. D., Flanagan W. M., Braisted A. C., Whitty A., Cancilla M. T., Wang J., Lugovskoy A. A., Yoburn J. C., Fung A. D., Farrington G., Eldredge J. K., Day E. S., Cruz L. A., Cachero T. G., Miller S. K., Friedman J. E., Choong I. C., Cunningham B. C. (2005) Small-molecule inhibition of TNF-α. Science 310, 1022–1025 [DOI] [PubMed] [Google Scholar]
- 28. Revanna C., SivaramanáSiveen K., NanjundaáSwamy S. (2014) Synthesis and biological evaluation of tetrahydropyridinepyrazoles (‘PFPs’) as inhibitors of STAT3 phosphorylation. MedChemComm 5, 32–40 [Google Scholar]
- 29. Wang J., Wolf R. M., Caldwell J. W., Kollman P. A., Case D. A. (2004) Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 [DOI] [PubMed] [Google Scholar]
- 30. Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Montgomery J., J. A., Vreven T., Kudin K. N., Burant J. C., Millam J. M., Iyengar S. S., Tomasi J., Barone V., Mennucci B., Cossi M., Scalmani G., Rega N., Petersson G. A., Nakatsuji H., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., shida M., Nakajima T., Honda Y., Kitao O., Nakai H., Klene M., Li X., Knox J. E., Hratchian H. P., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Ayala P. Y., Morokuma K., Voth G. A., Salvador P., Dannenberg J. J., Zakrzewski V. G., Dapprich S., Daniels A. D., Strain M. C., Farkas O., Malick D. K., Rabuck A. D., Raghavachari K., Foresman J. B., Ortiz J. V., Cui Q., Baboul A. G., Clifford S., Cioslowski J., Stefanov B. B., Liu G., Liashenko A., Piskorz P., Komaromi I., Martin R. L., Fox D. J., Keith T., Al-Laham M. A., Peng C. Y., Nanayakkara A., Challacombe M., Gill P. M. W., Johnson B., Chen W., Wong M. W., Gonzalez C., Pople J. A. (2004) Gaussian 03, Revision C, 02 Ed., Gaussian, Inc., Wallingford, CT [Google Scholar]
- 31. Bayly C. I., Cieplak P., Cornell W., Kollman P. A. (1993) A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 97, 10269–10280 [Google Scholar]
- 32. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 [Google Scholar]
- 33. Lindorff-Larsen K., Piana S., Palmo K., Maragakis P., Klepeis J. L., Dror R. O., Shaw D. E. (2010) Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Le Grand S., Götz A. W., Walker R. C. (2013) SPFP: Speed without compromise: a mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Phys. Commun. 184, 374–380 [Google Scholar]
- 35. Fuchs J. E., Huber R. G., von Grafenstein S., Wallnoefer H. G., Spitzer G. M., Fuchs D., Liedl K. R. (2012) Dynamic regulation of phenylalanine hydroxylase by simulated redox manipulation. PLoS One 7, e53005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Basappa, Murugan S., Sugahara K. N., Lee C. M., ten Dam G. B., van Kuppevelt T. H., Miyasaka M., Yamada S., Sugahara K. (2009) Involvement of chondroitin sulfate E in the liver tumor focal formation of murine osteosarcoma cells. Glycobiology 19, 735–742 [DOI] [PubMed] [Google Scholar]
- 37. Pettipher E. R., Labasi J. M., Salter E. D., Stam E. J., Cheng J. B., Griffiths R. J. (1996) Regulation of tumour necrosis factor production by adrenal hormones in vivo: insights into the antiinflammatory activity of rolipram. Br. J. Pharmacol. 117, 1530–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cooper H. S., Murthy S. N., Shah R. S., Sedergran D. J. (1993) Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab. Invest. 69, 238–249 [PubMed] [Google Scholar]
- 39. Gonzalez-Rey E., Chorny A., Delgado M. (2006) Therapeutic action of ghrelin in a mouse model of colitis. Gastroenterology 130, 1707–1720 [DOI] [PubMed] [Google Scholar]
- 40. Liu J., Feng Z., Xu J., Wang Y., Zhang P. (2007) Rare biscoumarins and a chlorogenic acid derivative from Erycibe obtusifolia. Phytochemistry 68, 1775–1780 [DOI] [PubMed] [Google Scholar]
- 41. Hagiwara H., Fujimoto N., Suzuki T., Ando M. (2000) Synthesis of methylenebis(4-hydroxy-2-pyrone) or methylenebis(4-hydroxycoumarin) derivatives by organic solid state reaction. Heterocycles 53, 549–552 [Google Scholar]
- 42. Mehrabi H., Abusaidi H. (2010) Synthesis of biscoumarin and 3,4-dihydropyrano[c]chromene derivatives catalysed by sodium dodecyl sulfate (SDS) in neat water. J. Iran. Chem. Soc. 7, 890–894 [Google Scholar]
- 43. Cravotto G., Nano G. M., Palmisano G., Tagliapietra S. (2003) The reactivity of 4-hydroxycoumarin under heterogeneous high-intensity sonochemical conditions. Synthesis 2003, 1286–1291 [Google Scholar]
- 44. Mohd Fauzi F., Koutsoukas A., Lowe R., Joshi K., Fan T.-P., Glen R. C., Bender A. (2013) Chemogenomics approaches to rationalizing the mode-of-action of traditional Chinese and ayurvedic medicines. J. Chem. Inf. Model. 53, 661–673 [DOI] [PubMed] [Google Scholar]
- 45. Koutsoukas A., Simms B., Kirchmair J., Bond P. J., Whitmore A. V., Zimmer S., Young M. P., Jenkins J. L., Glick M., Glen R. C., Bender A. (2011) From in silico target prediction to multi-target drug design: Current databases, methods and applications. J. Proteomics 74, 2554–2574 [DOI] [PubMed] [Google Scholar]
- 46. Koutsoukas A., Lowe R., Kalantarmotamedi Y., Mussa H. Y., Klaffke W., Mitchell J. B. O., Glen R. C., Bender A. (2013) In silico target predictions: defining a benchmarking data set and comparison of performance of the multiclass naïve Bayes and Parzen-Rosenblatt window. J. Chem. Inf. Model. 53, 1957–1966 [DOI] [PubMed] [Google Scholar]
- 47. Rath P. C., Aggarwal B. B. (1999) TNF-induced signaling in apoptosis. J. Clin. Immunol. 19, 350–364 [DOI] [PubMed] [Google Scholar]
- 48. van Horssen R., Ten Hagen T. L., Eggermont A. M. (2006) TNF-α in cancer treatment: molecular insights, antitumor effects, and clinical utility. Oncologist 11, 397–408 [DOI] [PubMed] [Google Scholar]
- 49. Takahashi S., Hakuta M., Aiba K., Ito Y., Horikoshi N., Miura M., Hatake K., Ogata E. (2003) Elevation of circulating plasma cytokines in cancer patients with high plasma parathyroid hormone-related protein levels. Endocr.-Relat. Cancer 10, 403–407 [DOI] [PubMed] [Google Scholar]
- 50. Askling J., van Vollenhoven R. F., Granath F., Raaschou P., Fored C. M., Baecklund E., Dackhammar C., Feltelius N., Cöster L., Geborek P., Jacobsson L. T., Lindblad S., Rantapää-Dahlqvist S., Saxne T., Klareskog L. (2009) Cancer risk in patients with rheumatoid arthritis treated with anti-tumor necrosis factor α therapies: does the risk change with the time since start of treatment? Arthritis Rheum. 60, 3180–3189 [DOI] [PubMed] [Google Scholar]
- 51. Kim H. Y., Park E. J., Joe E.-H., Jou I. (2003) Curcumin suppresses Janus kinase-STAT inflammatory signaling through activation of Src homology 2 domain-containing tyrosine phosphatase 2 in brain microglia. J. Immunol. 171, 6072–6079 [DOI] [PubMed] [Google Scholar]
- 52. Olivier S., Robe P., Bours V. (2006) Can NF-κB be a target for novel and efficient anti-cancer agents? Biochem. Pharmacol. 72, 1054–1068 [DOI] [PubMed] [Google Scholar]
- 53. Keffer J., Probert L., Cazlaris H., Georgopoulos S., Kaslaris E., Kioussis D., Kollias G. (1991) Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 10, 4025–4031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Iqbal M., Verpoorte R., Korthout H. A. J., Mustafa N. (2013) Phytochemicals as a potential source for TNF-α inhibitors. Phytochem. Rev. 12, 65–93 [Google Scholar]
- 55. Singh S., Aggarwal B. B. (1995) Activation of transcription factor NF-κB is suppressed by curcumin (diferuloylmethane). J. Biol. Chem. 270, 24995–25000 [DOI] [PubMed] [Google Scholar]
- 56. Chen Y. R., Tan T. H. (1998) Inhibition of the c-Jun N-terminal kinase (JNK) signaling pathway by curcumin. Oncogene 17, 173–178 [DOI] [PubMed] [Google Scholar]
- 57. Suh H.-W., Kang S., Kwon K.-S. (2007) Curcumin attenuates glutamate-induced HT22 cell death by suppressing MAP kinase signaling. Mol. Cell. Biochem. 298, 187–194 [DOI] [PubMed] [Google Scholar]
- 58. Truelove S. C., Witts L. (1955) Cortisone in ulcerative colitis. Br. Med. J. 2, 1041–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Feuerstein J. D., Cheifetz A. S. (2014) Ulcerative colitis: epidemiology, diagnosis, and management. Mayo Clin. Proc. 10.1016/j.mayocp.2014.07.002 [DOI] [PubMed] [Google Scholar]