

Background: Lipoxygenases (LOX) catalyze the oxygenation of polyunsaturated fatty acids but generate distinct products from a common substrate.
Results: We report the first structure of a LOX-substrate complex.
Conclusion: The structure provides a context for understanding product specificity in enzymes that metabolize arachidonic acid.
Significance: With roles in the production of potent lipid mediators, LOX are targets for drug design.
Keywords: Arachidonic Acid (AA) (ARA), Eicosanoid Biosynthesis, Lipid Signaling, Lipoxygenase Pathway, Protein Structure, X-ray Crystallography
Abstract
Lipoxygenases (LOX) play critical roles in mammalian biology in the generation of potent lipid mediators of the inflammatory response; consequently, they are targets for the development of isoform-specific inhibitors. The regio- and stereo-specificity of the oxygenation of polyunsaturated fatty acids by the enzymes is understood in terms of the chemistry, but structural observation of the enzyme-substrate interactions is lacking. Although several LOX crystal structures are available, heretofore the rapid oxygenation of bound substrate has precluded capture of the enzyme-substrate complex, leaving a gap between chemical and structural insights. In this report, we describe the 2.0 Å resolution structure of 8R-LOX in complex with arachidonic acid obtained under anaerobic conditions. Subtle rearrangements, primarily in the side chains of three amino acids, allow binding of arachidonic acid in a catalytically competent conformation. Accompanying experimental work supports a model in which both substrate tethering and cavity depth contribute to positioning the appropriate carbon at the catalytic machinery.
Introduction
Lipoxygenases (LOX)2 catalyze the highly regio- and stereo-specific reaction of polyunsaturated fatty acids with molecular oxygen, typically forming a single, chiral fatty acid hydroperoxide (1–3). The diversity is such that individual LOX enzymes are known that can account for oxygenation on almost all the available positions on the common polyunsaturated fatty acid substrates (3–5). The different members of the LOX superfamily, whether prokaryotic, plant, fungal, invertebrate, or vertebrate/mammalian, all retain the same conserved amino acids at the catalytic center and share a common structural framework (6–12). Taken together with an in-depth understanding of the basic chemistry involved in LOX catalysis (4, 13–15), this similarity has led to models in which much of the diversity in product formation is accounted for by proposed differences in substrate binding (Fig. 1) and by the direction of oxygen attack on the activated fatty acid. Two variables in fatty acid binding can influence the product outcome. 1) Polyunsaturated fatty acids may have one or more pentadienes available for attack (for example, arachidonic acid (AA) has three chemically equivalent pentadienes), and a primary determinant of regiospecificity is which pentadiene in the double bond system aligns with the LOX catalytic iron (16). 2) The substrate can enter the active site either carboxyl (“head”) or hydrocarbon (“tail”) first (2, 17). This determines which side of the selected pentadiene is accessible to the catalytic iron. In addition to the mode of fatty acid binding, the direction of oxygen attack across the face of the pentadiene determines the R or S chirality of the products (18). As an example, 8R and 12S products have the hydroperoxide moiety on the same side of the carbon chain and the 8S/12R-hydroperoxides are on the opposite side. Taken together, the fatty acid binding issues and direction of the reaction with O2 can “on paper” explain all the observed LOX-catalyzed specific oxygenations of AA.
FIGURE 1.
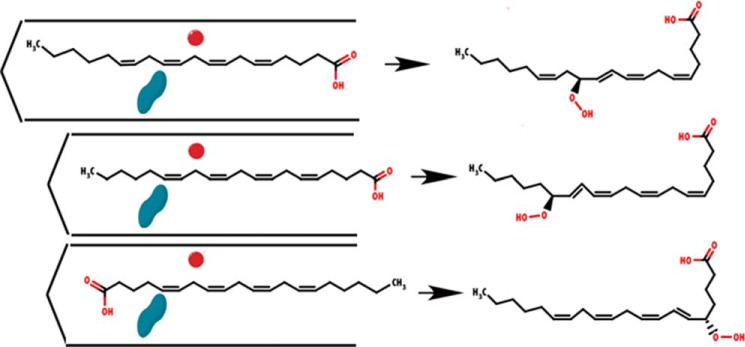
Schematic of the current model for product specificity in lipoxygenases. Cavity depth and substrate orientation combine to confer regio- and stereo- specificity. The substrate (AA shown here) must align between the catalytic iron (red sphere) and O2 channel (blue peanut). Differences in cavity depth and head-to-tail orientation determine the hydroperoxyeicosatetraenoic acid generated.
Although clues to LOX-substrate recognition have emerged from a wealth of mechanistic chemistry, enzyme mutagenesis, inhibitor structure-activity relationships, and crystal structures of inhibitor complexes, what is missing is a melding of protein structural information with a detailed picture of substrate binding. The fundamental deterrent has been the difficulty in obtaining the crystal structure of an enzyme-substrate complex. Acting against the attempted crystallization of the enzyme-substrate complex are (i) LOX enzymes require no cofactor for the dioxygenase reaction and are therefore primed for instant activity, (ii) the ready oxidation of the resting state ferrous iron to the active ferric state by traces of peroxide, and (iii) the O2 co-substrate is ubiquitous under standard conditions of crystallography. Therefore, a combination of these factors has thwarted attempts to halt the one-turnover transformation that will metabolize a substrate molecule in the LOX active site. Crystals of an 8R-LOX from Plexaura homomalla, which generates 8R-hydroperoxyeicosatetraenoic acid from AA, proved to be to sufficiently robust to survive the manipulations required to introduce the hydrophobic substrate at concentrations of AA and carrier that do not compromise diffraction quality, but are adequate for complex formation.
Combined with the previously reported structures of the mammalian lipoxygenases with inhibitors, the model provides a robust framework for the design of isoform-specific inhibitors. Furthermore, mutagenesis and activity assays support the proposed model.
MATERIALS AND METHODS
Mutagenesis
Mutants were constructed in pseudo-wild-type 8R-LOX (1) by whole plasmid polymerase chain reaction using Pfu UltraII Hot Start polymerase (Agilent Technologies). Where possible, silent mutations were included in the primers for rapid identification of mutant plasmids. Plasmid DNA was purified using the Wizard system (Promega).
Protein Expression and Purification
Pseudo-wild-type 8R-LOX (19) and variants were expressed and purified essentially as described in the literature with the following modifications (11). BL21(DE3) cells were transformed with the appropriate pet3a construct. Single colonies were picked to grow overnight starter cultures in Luria Broth, 50 μg/ml ampicillin. The starter culture was then diluted 100× into 2× Yeast Tryptone and grown at 37 °C to an A600 of 0.6, at which point the temperature was changed to 22 °C, and expression was induced with 0.25 mm isopropyl 1-thio-β-d-galactopyranoside. The cells were harvested after 22 h, frozen in liquid nitrogen, and stored at −80 °C. The frozen cells were resuspended in B-PER bacterial protein extraction reagent (Thermo Scientific) supplemented with DnaseI, PMSF, leupeptin, and pepstatin, according to the manufacturer's instructions, and sonicated at 5-s intervals and 50% power for 5 min. The cell lysate was centrifuged at 38,000 × g for 30 min at 4 °C. The supernatant was cleared with a 0.45-μm Millipore PES syringe filter, and applied on a Co-nitrilotriacetic acid column using AKTA FPLC. The 8R-LOX was eluted using a gradient of 20 mm to 200 mm imidazole in 20 mm Tris (pH 8.0) and 500 mm NaCl. The fractions containing the prominent peak of the absorbance chromatogram were pooled and dialyzed against 20 mm Tris (pH 8.0) overnight with three buffer exchanges. The dialyzed protein was then purified further using a Mono-Q column and AKTA FPLC and a salt gradient of 0 to 500 mm NaCl. Most of 8R-LOX eluted at ∼250 mm NaCl. The purities of fractions were evaluated by SDS-PAGE and Coomassie staining; the fractions containing purest 8R-LOX were collected and concentrated to 5–15 mg/ml using Amicon Ultra centrifugal units.
Kinetic Assays
The kinetic measurements were performed on an Applied Photophysics Stopped Flow instrument SX.18MV, monitoring the increase in absorbance at 237 nm over time in the following buffer: 50 mm Tris (pH 7.4), 150 mm NaCl, 0.5 mm EDTA. The concentration of enzyme was 15 (native), 20 (R182A), or 200 nm (A589M and A620H). For each enzyme at least 10 AA (Cayman Chemical) concentrations, varying from 1–50 μm, were monitored. Initial rates were measured by fitting the average of four to five measurements of the initial linear region of absorbance change to a linear equation. Most of the variants were analyzed by using the Michaelis-Menten equation to determine Vmax and Km values; however, the R182A mutant was analyzed by using the reciprocal plot and substrate inhibition steady state kinetics using the equation shown below.
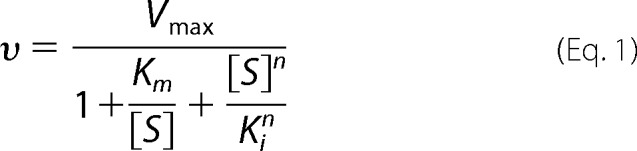 |
Equation 1 has been used before to study other systems that involve cooperative substrate inhibition, and it was the model that gave the best fit to the experimental data (20–22). An extinction coefficient of 23 absorbance units/cm/mm (23) was used for product.
Product Analyses
Incubations were carried out in 1 ml of 50 mm Tris, pH 8.0, containing 500 mm NaCl, 2 mm CaCl2, using 50 μm arachidonic acid (15 μg) added in 5 μl of ethanol. (10-ml incubations were carried out with the slowly reacting A589M and A620H mutants.) The samples were incubated at room temperature for 2–10 min, with the time depending on the rate of reaction observed by UV-235 nm. The incubations were extracted using a Waters 1-cc Oasis HLB cartridge and eluted with 1 ml of methanol. The extracts were treated with triphenylphosphine in methanol at room temperature for 30 min to reduce hydroperoxyeicosatetraenoic acid to the corresponding hydroxtetraenoic acid (HETE). The samples were analyzed (and the individual HETEs collected) by SP-HPLC using a Beckman Ultrasphere 5μ silica column (250 × 4.6 mm) with a solvent system of hexane/isopropanol/glacial acetic acid (100:2:0.1, by volume) running at 1 ml/min, and the eluant was monitored with an Agilent 1100 diode array detector. The individual HETEs were then methylated with diazomethane, and the stereochemistry of the 8-, 11-, 12-, and 15-HETE products was determined using a Chiralpak AD column (250 × 4.6 mm) with a solvent of hexane/methanol (100:2, v/v) running at 1 ml/min (24).
Crystallization
Crystals of 8R-LOX were prepared in an anaerobic chamber maintained below 20 ppm O2. Crystals were grown by vapor diffusion with a well solution of 8% PEG-8000, 5% glycerol, 0.2 m CaCl2, 0.1 m imidazole acetate, pH 8.0. Crystals were then soaked overnight (∼17 h) in a solution of 25% glycerol, 10% PEG-8000, 0.02 m CaCl2, 0.1 m imidazole acetate, pH 8.0, 1% dimethyl sulfoxide, and 1 mg/ml AA. The next morning, crystals were cooled in liquid nitrogen that was brought into the anaerobic chamber. During the cooling process, O2 levels rose to 55 ppm (due to O2 dissolved in the liquid nitrogen).
Structure Determination
Diffraction data from the soaked 8R-LOX crystals were collected with an ADSC Quantum 315 at the NE-CAT 24ID-E beamline located at the Advanced Photon Source. Two data sets were collected from a single crystal. The data were indexed, integrated, and scaled with XDS (25), merged with XSCALE, and analyzed with POINTLESS (26) and AIMLESS (27).
The structure of 8R-LOX (3FG1) described previously was used as the starting model for refinement. Iterative model building and refinement were run in PHENIX (28) and COOT (29). Four monomers are located in the asymmetric unit. Residues for the N-terminal His tag were modeled only where sufficient electron density warranted (one N-terminal His for chains A, B, and D and three N-terminal His for chain C). In addition, residues 307–315 were not modeled in any of the chains, along with residue 316 in chain A, residues 305 and 306 in chain B, and residue 306 in chain D, due to lack of electron density in those regions. Residue 600 was not modeled in chain B, also due to a lack of electron density. Arachidonate was modeled into electron density present in the active site of chain C. An omit map, calculated after three cycles of coordinate refinement of the model without AA, shows clear density for the substrate (Fig. 2C). Electron density in the active sites of the other monomers was suggestive of AA, but insufficient to model the ligand structure in these sites with confidence. The final Rfactor/Rfree was 16.21/21.45%. Data processing and refinement statistics are provided in Table 1. Although the AA B-factor (39 Å2) is higher than the average protein B-factor (29 Å2), it is within the range of those of the neighboring amino acid side chains (21 to 46 Å2). The coordinate and structure factors have been deposited to the Protein Data Bank and assigned the identifier 4QWT.
FIGURE 2.

8R-LOX with AA. A, ribbon drawings of 8R-LOX with (blue) and without (gray) AA in the active site. Fe2+ is shown as a red sphere. The helical insertion described in all LOX structures is shown in violet. B, detail of the outward shift of the arched helix observed in the presence of AA. C, an omit map contoured +2/−2σ (green/magenta) reveals clear electron density for AA. D, the steady state rate dependence on substrate concentration of 8R-LOX and the mutant R182A. 8R-LOX data were fit to the Michaelis-Menten equation, whereas the R182A data were fit to Equation 1. Equation 1 describes cooperative substrate inhibition for the fit, n = 2.4 ± 0.3.
TABLE 1.
Data collections and refinement statistics
Statistics for the highest resolution shell are shown in parentheses.
| Data processing | |
| PDB code | 4QWT |
| Wavelength (Å) | 0.98 |
| Resolution range (Å) | 50–2.0 (2.11–2.0) |
| Space group | P21 |
| Unit cell | |
| a, b, c (Å) | 104.0, 170.6, 104.7 |
| B (°) | 95.4 |
| Total reflections | 914,097 (129,971) |
| Unique reflections | 242,473 (34,734) |
| Multiplicity | 3.8 (3.7) |
| Completeness (%) | 99.5 (96.1) |
| Mean I/σ(I) | 12.9 (1.9) |
| Wilson B-factor | 27.01 |
| Rmergea | 0.102 (0.813) |
| Rmeasb | 0.119 (0.951) |
| Rp.i.mc | 0.061 (0.490) |
| CC1/2 (44) | 0.996 (0.615) |
| Refinement statistics | |
| Rwork (%) | 16.21 |
| Rfree (%) | 26.36 |
| No. of non-H atoms | |
| Protein | 21,834 |
| Ligands | 242 |
| Water | 1795 |
| Protein residues | 2725 |
| RMS (bonds) (Å) | 0.007 |
| RMS (angles) (°) | 1.03 |
| Ramachandran | |
| Favored (%) | 97 |
| Outliers (%) | 0.33 |
| Average B-factor | 29.30 |
| Protein | 28.80 |
| Ligands | 38.10 |
| Arachidonate | 38.96 |
| Solvent | 34.40 |
a .
b .
c .
RESULTS
Invariant Amino Acids Position the Pentadiene for Attack
The crystal structure 8R-LOX was determined to 2.0 Å resolution. Lipoxygenases are composed of an amino-terminal β-barrel domain and a largely α-helical catalytic domain, which is ∼80% of the polypeptide (6) and harbors the catalytic iron. Those enzymes that metabolize AA are roughly 650 amino acids in length. Although in general the placement of elements of secondary structure in lipoxygenases is conserved, helix α2, which helps define the active site, has been observed in two strikingly distinct orientations: it can be a long single α-helix that runs the length of the catalytic domain or a broken helix in which some side chains are inserted into the common active site to conceal it from bulk solvent. Helix α2 is one long helix in 8R-LOX and substrate can enter without invoking a major conformational change.
The catalytic iron in 8R-LOX is positioned by three invariant His (384, 389, 570) side chains and the terminal main chain carboxylate of an invariant Ile (693). A large U-shaped cavity, in which Fe2+ sits at the base of the U, is observed in the structure of 8R-LOX in the absence of substrate (30). An arched helix, kinked by a distinct two-amino acid insertion in one helical turn (first described in detail by Minor et al. (31)), covers this site and contributes one of several invariant leucines that line the proposed substrate binding pocket.
In the structure of 8R-LOX with AA bound (Fig. 2, A and B) this “arched” helix (420–442) is displaced ever-so-slightly outwards (Cα-Cα distance = 1.3 Å for Ile-437) and the side chains of Leu-431 and Ile-437, which frame the insertion, are now ordered (Fig. 2, A and B). These amino acids make van der Waals contacts to secure the AA, Leu-431 with the central carbon of the pentadiene centered at C10. Ile-437 is positioned at the top of the “U” between the carboxyl head (C1) and C17 at the tail. These amino acids side chains fill the U from the side opposite of the catalytic iron, where just across from Leu-431 sits invariant Leu-627. Leu-627 and Leu-431 are equidistant from the substrate, and the two side chains appear to clamp the C10 pentadiene in place. The base of the U is positioned by invariant Leu-385 on one side, and the iron and His-384 and His-389 on the other. Leu-385 and the catalytic iron cradle the base of the U (Fig. 2C).
Pocket depth, which allows the tail to slide in so that C10 is attacked, appears to be conferred by Ala-620, which with Ala-589 makes for a very a deep cavity. Leu-381 and Val-428 define the wall of the cavity at C15, and the polar and charged amino acids Gln-380 and Asp-424 (highly conserved) flank the cluster of histidines that hold the iron in place. In the absence of AA, Tyr-181 and Arg-182 of helix α2 participate in an interhelical charge cluster with Glu-430 of the arched helix. In the AA-bound structure, the AA carboxylate expands this charge network with only the slight shift in the arched helix described above and the reorientation of the side chain of Glu-430 to allow Arg-182 to interact with the substrate carboxylate as well as Glu-430.
Apparent in the crystal structure is a tubular opening that leads to the U-shaped site. It sits on the face of the AA opposite the Fe and is thus positioned to allow molecular oxygen to access the pentadiene. This tubular cavity abuts the shallower side of the U, where C8 of AA sits. Proximal to the substrate it is defined by Gly-427. Access to the pentadiene from the iron side is blocked not only by the residues that position the iron but also by Ile-427, which is invariant.
Thus, the substrate-binding site is formed largely by invariant amino acids, and these amino acids are positioned around the pentadiene that is adjacent to the catalytic iron. The depth of the cavity is set by residues 620 and 589, and the cavity entrance is defined by amino acids from α2.
Mutations to the Binding Site Affect Enzyme Activity and Product Specificity
Site-directed mutants were prepared to test the structural interpretation. Arg-182, which appears to charge pair with the substrate carboxylate, was mutated to Ala. Ala-589 and Ala-620, positioned deep in the cavity to allow the AA tail slide into the enzyme active site to position C10 at the catalytic site, were mutated to methionine and histidine, respectively, to introduce bulk and charge at the putative hydrocarbon end of the active site. Whereas the mutation of R182A had only a modest impact on product specificity (Table 2), the kinetic properties of the enzyme were dramatically different from that of the wild type enzyme, as this mutant displayed striking substrate inhibition (Fig. 2D). In contrast, mutations deep in the cavity lead to 11R-hydroperoxyeicosatetraenoic acid as the primary product, a product consistent with a shallower site. To generate the 11R-product, C13, rather than C10, must be at the catalytic center.
TABLE 2.
The impact of 8R-LOX mutations on the enzyme product profile and kinetic parameters
ND indicates not determined. Major products are underlined, and tail-first entry stereo-chemistry is indicated with boldface type.
| Enzyme/Product | % of products | % R:S | Km | kcat | kcat/Km | Fold change | Ki |
|---|---|---|---|---|---|---|---|
| μm | s−1 | μm | |||||
| 8R-LOX | |||||||
| 8-HETE | 97.9 | 99:1 | 30 ± 7 | 210 ± 23 | 6.9 ± 1.7 | 1 | |
| 12-HETE | 2.1 | ND | |||||
| R182A | |||||||
| 8-HETE | 88.8 | 98:2 | 67 ± 11 | 193 ± 8.7 | 2.9 ± 0.5 | 0.42 | 13 ± 2 |
| 12-HETE | 4.6 | ND | |||||
| 11-HETE | 2.9 | ND | |||||
| 15-HETE | 3.7 | ND | |||||
| A589M | |||||||
| 8-HETE | 21 | 92:8 | 59 ± 19 | 32 ± 6 | 0.54 ± 0.23 | 0.079 | |
| 12-HETE | 5 | 31:69 | |||||
| 11-HETE | 64 | 94:6 | |||||
| 15-HETE | 10 | 9:91 | |||||
| A620H | |||||||
| 8-HETE | 20 | 81:18 | 58 ± 22 | 3.2 ± 0.8 | 0.055 ± 0.029 | 0.008 | |
| 12-HETE | 12 | 46:54 | |||||
| 11-HETE | 62 | 92:8 | |||||
| 15-HETE | 6 | 32:68 | |||||
The steady state kinetic parameters for wild type 8R-LOX and the single amino acid substitution variants are shown in Table 2. The kcat for A620H is <2% of that for 8R-LOX, whereas the kcat for A589M is reduced to ∼15%.
The R182A mutant displays conspicuous substrate inhibition for the reaction with AA. The rate versus substrate concentration plots of R182A and the wild type are shown in Fig. 2D, along with the curve fit for Michaelis-Menten and substrate inhibition kinetics. Prior to fitting the data to the substrate inhibition model, the Km of R182A was first determined by the double reciprocal plot of 1/νo versus 1/[S] as described (32) and fitting a linear line to the data at intermediate substrate concentrations. The Km determined from the double reciprocal plot was then used as a constant in fitting the substrate inhibition curve using Equation 1.
DISCUSSION
Cavity Depth as a Determinant of Product Formation
The LOX reaction proceeds with the abstraction of hydrogen from the sp3 carbon of a pentadiene via a water molecule that fills the metal coordination sphere. The resulting free radical is delocalized along the pentadiene, and oxygenation occurs at the carbon ±2 from the site of attack. Hydrogen abstraction and oxygenation occur on opposite faces of the substrate, and the structural basis for this relationship is clearly elucidated in the structure of 8R-LOX with AA. The substrate fills a U-shaped cavity, and at the base of the U, the catalytic iron is positioned by the side chains of conserved amino acids. Of the 16 amino acids that define this cavity and make contacts with the AA six are invariant: two His (384, 389) that also bind the iron, three Leu (385, 431, 627), and an Ile (627). All of these amino acids are positioned to envelope the pentadiene centered at C10 for attack. AA enters 8R-LOX tail first and slides deep into the active site, which is exceptionally deep when compared with that described for other LOX structures due to an Ala at position 620 as compared with Met, His, Val, or Gln in other LOX structures (Table 3). Consistent with tail first entry, the deepest part of the binding site is formed by hydrophobic amino acids (Fig. 3A). Moreover, the mutation of A589M, deep in the cavity, gives 11R-HETE, indicating attack at C13, rather than C10, as expected for a shallower cavity with tail-first entry of AA (Fig. 1 and Table 2). The introduction of a histidine at position 620 not only shortens the deepest part of the U-shaped cavity, but adds a positive charge, making it challenging for the substrate to bind in a tail-first orientation. As a result, the Km increases, and the turnover rate plummets. Similar to A589M, the major product is 11R-HETE along with minor amounts of predominantly 12S- and 15S-HETES, all consistent with a shallower tail-first entry into the active site (Table 2).
TABLE 3.
The amino acids that define the AA binding site in 8R-LOX and their counterparts in other LOX structures
Overall sequence identity with P. homomalla 8R-LOX is indicated in parentheses. The two far right columns include the highly homologous 15-LOX-1 and 12-LOX pair. Residues in bold are invariant.
| 8R-LOX | 11R-LOX (47%) | 5-LOX (38%) | 15-LOX-2 (33%) | 15-LOX-1 (30%) | 12-LOX (30%) |
|---|---|---|---|---|---|
| 4QWTa | 3FG1a | 3O8Ya | 4NREa | 2P0Ma | 3RDEa |
| Tyr-178 | Phe-185 | Phe-177 | Phe-184 | Phe-175 | Phe-175 |
| Arg-182 | Gly-188 | Tyr-181 | Ala-188 | Leu-179 | Leu-179 |
| Gln-380 | Gln-369 | Gln-363 | Glu-369 | Gln-357 | Glu-357 |
| His-384 | 373 | 367 | 373 | 361 | 361 |
| Leu-385 | 374 | 368 | 374 | 362 | 362 |
| His-389 | 378 | 372 | 378 | 366 | 366 |
| Ile-423 | 412 | 406 | 412 | 400 | 400 |
| Gly-427 | Gly-416 | Ala-410 | Ala-416 | Ala-404 | Ala-404 |
| Leu-431 | 420 | 414 | 420 | 408 | 408 |
| Ile-437 | Ala-426 | Leu-420 | Val-426 | Ile-414 | Ile-414 |
| Val-438 | Ala-427 | Phe-421 | Val-427 | Phe-415 | Phe-415 |
| Leu-442 | Leu-431 | Asn-425 | Thr-431 | Met-419 | Leu-419 |
| Ala-589 | Thr-575 | Pro-569 | Pro-572 | Cys-560 | Cys-560 |
| Ala-620 | Met-606 | His-600 | Val-603 | Gln-590 | Gln-590 |
| Thr-623 | Val-609 | Ala-603 | Ala-606 | Ile-593 | Ile-593 |
| Ile-626 | Ala-612 | Ala-606 | Leu-609 | Gln-596 | Gln-596 |
| Leu-627 | 613 | 607 | 610 | 597 | 597 |
a Protein Data Bank codes.
FIGURE 3.
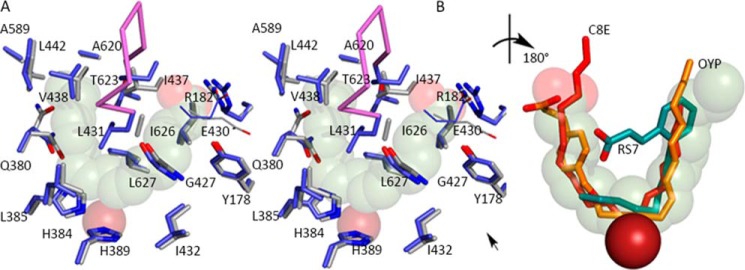
The AA-binding site. A, the substrate adopts a horseshoe shape in the U-shaped channel (stereo). The side chains of highly conserved amino acids line the base of the active site, along with Gly-427. Glu-430, part of an inter-helical charge cluster that includes the substrate carboxylate, is shown in line rendering. The Fe2+ (transparent dark red sphere) is positioned behind the substrate. B, detail of the superposition of inhibitors (rotated ∼180° with respect to A) observed in 15-LOX-2 (red) 12-LOX (gold, C; red, O) and 15-LOX-1 (teal, C; red, O). The 15-LOX-2 and 12-LOX inhibitors conform to the AA placement, whereas the 15-LOX-1 inhibitor overlaps partially. The Fe2+, solid dark red sphere, is in front of the substrate.
The Importance of Substrate Tethering
Whereas cavity depth determines how deep the AA slides into the active site, our data suggest that van der Waals contacts alone (the steric complementarity of the AA and the U-shaped active site) are not sufficient to adequately fix the geometry of the pentadiene at the catalytic iron in 8R-LOX. The long hydrocarbon tail of the substrate makes extensive contacts with the amino acid side chains lining the site, but helix α2 must provide a polar/charged amino acids to interact with the AA carboxyl group, perhaps to prevent AA from penetrating deeper into the cavity, which extends beyond C20 of the AA. Although substitution of Arg-182 with Ala has only a modest effect on the Km for AA or the product generated, the mutant displays dramatic substrate inhibition. Substrate inhibition in LOX has been reported previously (for example, Refs. 33 and 34), but it was unexpected that a single substitution of R182A would have such a large impact on enzyme activity. However, substrate inhibition by a compound with few hydrogen bond donors/acceptors for precise positioning can be explained by a non-productive binding mode that makes many of the same substrate-protein contacts observed in the productive binding mode. The AA, untethered by the polar/charge contact with the carboxylate, is not positioned precisely at the catalytic center.
Substrate inhibition has been studied in other enzymes (20, 35–37), and its analysis can be complex (32, 38, 39). We observed that the R182A data are best explained by cooperative substrate inhibition (Equation 1). In this case, the cooperativity observed may be due to an untethered hydrophobic substrate that can assume multiple non-productive conformations in the hydrophobic cavity and be retained there without having to reenter the aqueous milieu before positioning itself into an alternate conformation. Thus, we suggest that Arg-182 securely positions the substrate in a catalytically competent orientation at the active site machinery. The modest loss of product specificity (88 versus 98% 8-HETE), is consistent with this interpretation.
Can Cavity Depth Be Predicted?
The obvious question, given the structural similarity observed in the LOX family, is whether this 8R-active site is consistent with the other enzyme structures that have been reported. There are now structures for six AA-metabolizing LOX. In addition to the 8R-LOX, an 11R-, a 5S-, two 15S- (15-LOX-1 and 15-LOX-2), and 12S- lipoxygenases have been described (7, 9, 12, 40, 41). With the exception of the 15-LOX-1/12-LOX pair, which share 79% sequence identity, pairwise identity for these enzymes is at most 47%. It is the 8R-/11R pair that has 47% sequence identity, and in terms of cavity depth, the most significant difference is presence of a Met in the latter enzyme, instead of Ala-620 (Table 3). The Met protrudes directly in U-shaped cavity and limits its depth so that C13 is positioned for attack in the 11R enzyme. For the 15-LOX-2 enzyme, which also must position C13 at the catalytic machinery, the back end of the cavity is shortened by Val-610 and a Leu at 607.
However, this simple relationship between amino acid side chain volume deepest in the U and cavity depth is not straightforward as overall sequence identity decreases. For instance, both porcine 12-LOX and 8R-LOX (∼30% identity) are tail first enzymes that position C10 at the catalytic center; thus, they should have similar cavity depths. Yet 12-LOX has a Gln at the deepest part of the cavity (Ala-620 in 8R-LOX). Unlike the Met side chain in 11R-, the Gln lies perpendicular to the tail end of the cavity, resulting in cavity depth comparable with that in 8R-LOX.
Further complicating comparisons among these structures is the fact that porcine 12-LOX and rabbit 15-LOX-1 share ∼80% identity and have only one conservative difference in the amino acids that line the U-shaped site (Table 3, Met/Leu-419). Yet in the rabbit enzyme, the AA can only slide deep enough to position C13, rather than C10 at the catalytic iron. One significant conformational difference in this highly homologous pair is a conspicuous constriction conferred by Phe-415 (Val-438 in 8R-LOX) and Leu-597 in 15-LOX-1. Whereas the amino acids are present in the 12-LOX, and Leu-597 is invariant, placement of the latter in 15-LOX-1 is anomalous with respect to the other LOX structures in Table 3.
Substrate- and Inhibitor-induced Conformational Changes
8R-LOX is the only LOX for which structures are available in the absence and presence of substrate; only modest conformational changes are necessary to accommodate the AA. Yet this is clearly not the case for all family members, as in 5-LOX and 11R-LOX the cavities are “corked,” and the active site is concealed from bulk solvent. Inhibited structures have been reported for rabbit 15-LOX-1 (2P0MB), human 15-LOX-2 (Protein Data Bank code 4NRE), and porcine 12-LOX (Protein Data Bank code 3RDE). The 8R-pocket agrees well with those defined for the inhibited 15-LOX-2 and 12-LOX structures. However, 15-LOX-1 shows a dramatic reorientation of α2 in the presence of the inhibitor RS7. Although the cavity must be reconfigured for the substrate to enter to position the C13 pentadiene because of the constriction described above, whether it requires displacement of α2 is not clear. The RS7 conformation may be unique to the inhibitor, which is constrained in a conformation inconsistent with the U-shaped site (Fig. 3B).
An O2 Access Pathway
Also common to the LOX structures is the long tubular opening that emanates from the cavity at the base of the U, from the side opposite the catalytic iron (31). In the 8R-enzyme, this channel extends to the surface and reveals a likely path for O2 to access the activated pentadiene. In each of the reported structures, a similar “channel” is observed. Work by Knapp et al. (42) demonstrated the importance of the counterpart of 8R-LOX Leu-431, positioned proximal to the intersection of the channel and the catalytic site, in controlling access to the activated pentadiene for soybean LOX-1.
Subsequently, Coffa and Brash (18) reported a Gly/Ala switch in LOX that directs the stereochemistry of the product. 8R- and 11R-enzymes have a Gly at this residue, whereas the 12S-, 15S-, and 5S-enzymes all have an alanine. One way to think about this “switch” is that it directs O2 to the carbon that is either +2 or −2 from the central carbon of the pentadiene. The 8R-, 11R-, 15S-, and 12S-LOX are all tail-first enzymes, and for the R enzymes, the oxygen channel is tilted “above” the iron to allow access to the more shallow C-2 carbon. For the S enzymes, the channel is nudged toward the deeper C +2 carbon (Fig. 4). Note that Gly-427 (and its counterparts) defines the top of the putative O2 channel where it abuts the U-shaped cavity. The presence of Ala shields this position and molds the channel deeper into the cavity, so that O2 no longer has access to C-2. Placement of the shielding invariant Leu (8R-LOX Leu-431), one turn of the helix from the Gly/Ala switch, is coupled to the switch.
FIGURE 4.
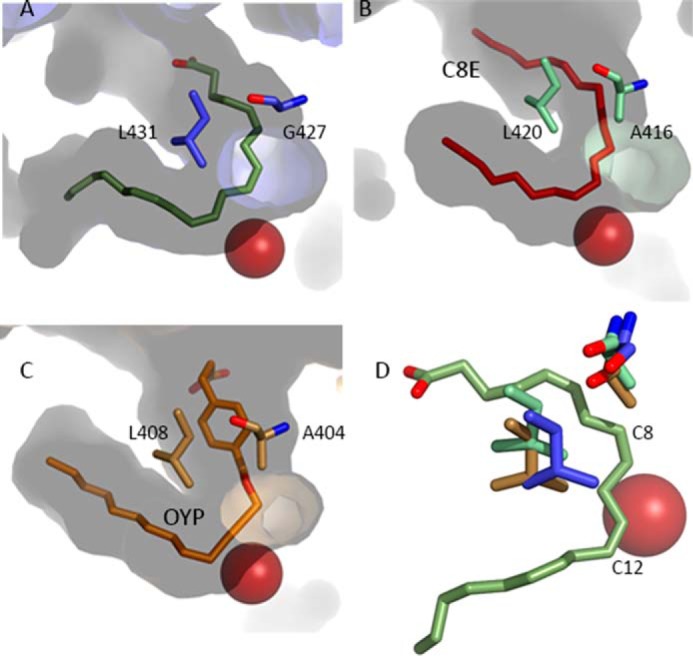
The antarafacial relationship between Fe2+ and a putative O2 access channel. A, in 8R-LOX the direction of the channel is set by Gly-427 and the orientation of the shielding amino acid Leu-431. B and C, similar channels in 15-LOX-2 and 12-LOX. Note the presence of an Ala pushes the tubular opening deeper into the body of the enzyme. D, detail of the relationship between the Gly/Ala switch and the Leu-431 shielding residues. Note the absence of the side chain in the 8R-enzyme (blue) and positioning of Leu-431 leave C8 (C-2) of the AA unprotected. Blue, 8R-LOX; green, 15-LOX-2; beige, 12-LOX. In contrast, in the 15-LOX-2 and 12-LOX enzymes, the same carbon would be shielded by the Ala and the invariant Leu no longer shields C12 (C+2) at the opposite end of the pentadiene.
Concluding Remarks
Over the years, individual LOX structures have led to models for substrate recognition by this enzyme family. The “boot-shaped” model was derived from the inhibited 15-LOX structure and suggested that Ile-418, Phe-353, and Ile-593 define cavity depth and that Arg-403 tethered the substrate carboxylate (43). Of these four amino acids, two are included in Table 3. Although the boot-shaped model nicely explained the supporting mutagenesis data, in retrospect, it was incomplete due to what was then interpreted as disordering of helix α2 (10, 40). Additionally, it was based on an inhibitor that lacks the structural flexibility of AA and so might include conformational changes not required for the natural substrate to enter. Subsequently, a U-shaped model was described (30), which included ∼60% of the amino acids in Table 3; this model was flawed as well, as it set the direction of the tail of AA more in the direction of the O2 channel. Eek et al. (7), tested both these models with the 11R structure, and the data are consistent with our 8R-LOX-AA structure. However, 11R-LOX is corked in the absence of substrate so the complete U shape could not be described in that enzyme either. The structure of 8R-LOX with substrate builds on these models to reveal a robust structural context for mechanistic studies that have provided tremendous insight into how similar enzymes can generate distinct products from a common substrate. It is clear that subtle structural differences among family members are not readily inferred with sequence information alone, and the design of isoform-specific inhibitors that might lead to the development of novel therapeutics remains a challenge.
Acknowledgments
This work includes research conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines, which are supported by a grant from the Nation Institute of General Medical Sciences (P41 GM103403) from the National Institutes of Health. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U. S. Department of Energy Office of Science by Argonne National Laboratory, was supported by the U. S. Department of Energy under Contract DE-AC02-06CH11357.
This work was supported in part by National Institutes of Health Grants HL R01 107887 (to M. E. N.) and GM 15431 (to A. R. B.) and in its early stages by National Science Foundation Grant MCB-0818387.
The atomic coordinates and structure factors (code 4QWT) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- LOX
- lipoxygenase(s)
- AA
- arachidonic acid
- HETE
- hydroxyeicosatetraenoic acid.
REFERENCES
- 1. Yamamoto S. (1992) Mammalian lipoxygenases: molecular structures and functions. Biochim. Biophys. Acta 1128, 117–131 [DOI] [PubMed] [Google Scholar]
- 2. Brash A. R. (1999) Lipoxygenases: occurrence, functions, catalysis, and acquisition of substrate. J. Biol. Chem. 274, 23679–23682 [DOI] [PubMed] [Google Scholar]
- 3. Ivanov I., Heydeck D., Hofheinz K., Roffeis J., O'Donnell V. B., Kuhn H., Walther M. (2010) Molecular enzymology of lipoxygenases. Arch. Biochem. Biophys. 503, 161–174 [DOI] [PubMed] [Google Scholar]
- 4. Schneider C., Pratt D. A., Porter N. A., Brash A. R. (2007) Control of oxygenation in lipoxygenase and cyclooxygenase catalysis. Chem. Biol. 14, 473–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Andreou A., Feussner I. (2009) Lipoxygenases: structure and reaction mechanism. Phytochemistry 70, 1504–1510 [DOI] [PubMed] [Google Scholar]
- 6. Boyington J. C., Gaffney B. J., Amzel L. M. (1993) The three-dimensional structure of an arachidonic acid 15-lipoxygenase. Science 260, 1482–1486 [DOI] [PubMed] [Google Scholar]
- 7. Eek P., Järving R., Järving I., Gilbert N. C., Newcomer M. E., Samel N. (2012) Structure of a calcium-dependent 11R-lipoxygenase suggests a mechanism for Ca2+ regulation. J. Biol. Chem. 287, 22377–22386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Garreta A., Val-Moraes S. P., García-Fernández Q., Busquets M., Juan C., Oliver A., Ortiz A., Gaffney B. J., Fita I., Manresa À., Carpena X. (2013) Structure and interaction with phospholipids of a prokaryotic lipoxygenase from Pseudomonas aeruginosa. FASEB J. 27, 4811–4821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gilbert N. C., Bartlett S. G., Waight M. T., Neau D. B., Boeglin W. E., Brash A. R., Newcomer M. E. (2011) The structure of human 5-lipoxygenase. Science 331, 217–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gillmor S. A., Villaseñor A., Fletterick R., Sigal E., Browner M. F. (1997) The structure of mammalian 15-lipoxygenase reveals similarity to the lipases and the determinants of substrate specificity. Nat. Struct. Biol. 4, 1003–1009; Correction (1998) Nat. Struct. Biol. 5, 242 [DOI] [PubMed] [Google Scholar]
- 11. Oldham M. L., Brash A. R., Newcomer M. E. (2005) Insights from the x-ray crystal structure of coral 8R-lipoxygenase: calcium activation via a C2-like domain and a structural basis of product chirality. J. Biol. Chem. 280, 39545–39552 [DOI] [PubMed] [Google Scholar]
- 12. Xu S., Mueser T. C., Marnett L. J., Funk M. O., Jr. (2012) Crystal structure of 12-lipoxygenase catalytic-domain-inhibitor complex identifies a substrate-binding channel for catalysis. Structure 20, 1490–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Klinman J. P. (2007) How do enzymes activate oxygen without inactivating themselves? Acc. Chem. Res. 40, 325–333 [DOI] [PubMed] [Google Scholar]
- 14. Hamberg M., Samuelsson B. (1967) On the specificity of the oxygenation of unsaturated fatty acids catalyzed by soybean lipoxidase. J. Biol. Chem. 242, 5329–5335 [PubMed] [Google Scholar]
- 15. Brash A. R., Schneider C., Hamberg M. (2012) Applications of stereospecifically-labeled Fatty acids in oxygenase and desaturase biochemistry. Lipids 47, 101–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kühn H., Sprecher H., Brash A. R. (1990) On singular or dual positional specificity of lipoxygenases. The number of chiral products varies with alignment of methylene groups at the active site of the enzyme. J. Biol. Chem. 265, 16300–16305 [PubMed] [Google Scholar]
- 17. Egmond M. R., Vliegenthart J. F., Boldingh J. (1972) Stereospecificity of the hydrogen abstraction at carbon atom n-8 in the oxygenation of linoleic acid by lipoxygenases from corn germs and soya beans. Biochem. Biophys. Res. Commun. 48, 1055–1060 [DOI] [PubMed] [Google Scholar]
- 18. Coffa G., Brash A. R. (2004) A single active site residue directs oxygenation stereospecificity in lipoxygenases: stereocontrol is linked to the position of oxygenation. Proc. Natl. Acad. Sci. U.S.A. 101, 15579–15584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Neau D. B., Gilbert N. C., Bartlett S. G., Dassey A., Newcomer M. E. (2007) Improving protein crystal quality by selective removal of a Ca2+-dependent membrane-insertion loop. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 63, 972–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dewal M. B., Firestine S. M. (2013) Site-directed mutagenesis of catalytic residues in N(5)-carboxyaminoimidazole ribonucleotide synthetase. Biochemistry 52, 6559–6567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. LiCata V. J., Allewell N. M. (1997) Is substrate inhibition a consequence of allostery in aspartate transcarbamylase? Biophys. Chem. 64, 225–234 [DOI] [PubMed] [Google Scholar]
- 22. Willemoës M., Larsen S. (2003) Substrate inhibition of Lactococcus lactis cytidine 5′-triphosphate synthase by ammonium chloride is enhanced by salt-dependent tetramer dissociation. Arch. Biochem. Biophys. 413, 17–22 [DOI] [PubMed] [Google Scholar]
- 23. Boutaud O., Brash A. R. (1999) Purification and catalytic activities of the two domains of the allene oxide synthase-lipoxygenase fusion protein of the coral Plexaura homomalla. J. Biol. Chem. 274, 33764–33770 [DOI] [PubMed] [Google Scholar]
- 24. Schneider C., Boeglin W. E., Brash A. R. (2000) Enantiomeric separation of hydroxy eicosanoids by chiral column chromatography: effect of the alcohol modifier. Anal. Biochem. 287, 186–189 [DOI] [PubMed] [Google Scholar]
- 25. Kabsch W. (2010) Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66, 133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 27. Evans P. R., Murshudov G. N. (2013) How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 30. Neau D. B., Gilbert N. C., Bartlett S. G., Boeglin W., Brash A. R., Newcomer M. E. (2009) The 1.85 A structure of an 8R-lipoxygenase suggests a general model for lipoxygenase product specificity. Biochemistry 48, 7906–7915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Minor W., Steczko J., Stec B., Otwinowski Z., Bolin J. T., Walter R., Axelrod B. (1996) Crystal structure of soybean lipoxygenase L-1 at 1.4 A resolution. Biochemistry 35, 10687–10701 [DOI] [PubMed] [Google Scholar]
- 32. Lineweaver H., Burk D. (1934) The determination of enzyme dissociation constants. J. Am. Chem. Soc. 56, 658–666 [Google Scholar]
- 33. Peng S., van der Donk W. A. (2003) An unusual isotope effect on substrate inhibition in the oxidation of arachidonic acid by lipoxygenase. J. Am. Chem. Soc. 125, 8988–8989 [DOI] [PubMed] [Google Scholar]
- 34. Berry H., Debat H., Larreta-Garde V. (1997) Excess substrate inhibition of soybean lipoxygenase-1 is mainly oxygen-dependent. FEBS Lett. 408, 324–326 [DOI] [PubMed] [Google Scholar]
- 35. Chen C., Joo J. C., Brown G., Stolnikova E., Halavaty A. S., Savchenko A., Anderson W. F., Yakunin A. F. (2014) Structure-based mutational studies of substrate inhibition of betaine aldehyde dehydrogenase BetB from Staphylococcus aureus. Appl. Environ. Microbiol. 80, 3992–4002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Szegletes T., Mallender W. D., Thomas P. J., Rosenberry T. L. (1999) Substrate binding to the peripheral site of acetylcholinesterase initiates enzymatic catalysis: substrate inhibition arises as a secondary effect. Biochemistry 38, 122–133 [DOI] [PubMed] [Google Scholar]
- 37. Ziegler J., Brandt W., Geissler R., Facchini P. J. (2009) Removal of substrate inhibition and increase in maximal velocity in the short chain dehydrogenase/reductase salutaridine reductase involved in morphine biosynthesis. J. Biol. Chem. 284, 26758–26767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kühl P. W. (1994) Excess-substrate inhibition in enzymology and high-dose inhibition in pharmacology: a reinterpretation [corrected]. Biochem. J. 298, 171–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marangoni A. G. (2003). in Enzyme Kinetics: A Modern Approach, Wiley-Interscience; pp 184–190 [Google Scholar]
- 40. Choi J., Chon J. K., Kim S., Shin W. (2008) Conformational flexibility in mammalian 15S-lipoxygenase: Reinterpretation of the crystallographic data. Proteins 70, 1023–1032 [DOI] [PubMed] [Google Scholar]
- 41. Kobe M. J., Neau D. B., Mitchell C. E., Bartlett S. G., Newcomer M. E. (2014) The structure of human 15-lipoxygenase-2 with a substrate mimic. J. Biol. Chem. 289, 8562–8569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Knapp M. J., Seebeck F. P., Klinman J. P. (2001) Steric control of oxygenation regiochemistry in soybean lipoxygenase-1. J. Am. Chem. Soc. 123, 2931–2932 [DOI] [PubMed] [Google Scholar]
- 43. Borngräber S., Browner M., Gillmor S., Gerth C., Anton M., Fletterick R., Kühn H. (1999) Shape and specificity in mammalian 15-lipoxygenase active site. The functional interplay of sequence determinants for the reaction specificity. J. Biol. Chem. 274, 37345–37350 [DOI] [PubMed] [Google Scholar]
- 44. Karplus P. A., Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]