

Background: Activation of ribonucleotide reductase Ib depends on the flavodoxin-like maturase NrdI.
Results: The redox properties of Bacillus anthracis NrdI allow isolation of the semiquinone state, NrdIsq, which can catalyze formation of the manganese-tyrosyl radical cofactor.
Conclusion: The maturation capacity of NrdIsq provides evidence that Mn-NrdF is activated via a superoxide radical.
Significance: Novel antibiotics may be designed to selectively target the maturation mechanism.
Keywords: Bacillus, Free Radicals, Manganese, Ribonucleotide Reductase, Superoxide Ion, NrdF, NrdI, Cyclic Voltammetry, Maturase, Semiquinone
Abstract
Ribonucleotide reductases (RNRs) catalyze the conversion of ribonucleotides to deoxyribonucleotides, and represent the only de novo pathway to provide DNA building blocks. Three different classes of RNR are known, denoted I-III. Class I RNRs are heteromeric proteins built up by α and β subunits and are further divided into different subclasses, partly based on the metal content of the β-subunit. In subclass Ib RNR the β-subunit is denoted NrdF, and harbors a manganese-tyrosyl radical cofactor. The generation of this cofactor is dependent on a flavodoxin-like maturase denoted NrdI, responsible for the formation of an active oxygen species suggested to be either a superoxide or a hydroperoxide. Herein we report on the magnetic properties of the manganese-tyrosyl radical cofactor of Bacillus anthracis NrdF and the redox properties of B. anthracis NrdI. The tyrosyl radical in NrdF is stabilized through its interaction with a ferromagnetically coupled manganese dimer. Moreover, we show through a combination of redox titration and protein electrochemistry that in contrast to hitherto characterized NrdIs, the B. anthracis NrdI is stable in its semiquinone form (NrdIsq) with a difference in electrochemical potential of ∼110 mV between the hydroquinone and semiquinone state. The under anaerobic conditions stable NrdIsq is fully capable of generating the oxidized, tyrosyl radical-containing form of Mn-NrdF when exposed to oxygen. This latter observation strongly supports that a superoxide radical is involved in the maturation mechanism, and contradicts the participation of a peroxide species. Additionally, EPR spectra on whole cells revealed that a significant fraction of NrdI resides in its semiquinone form in vivo, underscoring that NrdIsq is catalytically relevant.
Introduction
Ribonucleotide reductases (RNRs)2 catalyze the conversion of ribonucleotides to deoxyribonucleotides. This represents the only de novo pathway to provide DNA building blocks, and consequently RNR is essential in all living organisms (1, 2). Three different RNR classes (I-III) are known, and although they differ in cofactor requirement, oxygen dependences, and quaternary structures, their catalytic core structures demonstrate that they all have a common origin.
Class I RNRs are heteromeric proteins, and their two components are generally referred to as the α- and the β-subunits respectively. The reduction of ribose, via replacement of the 2′-hydroxyl group of the ribose with a hydrogen resulting in a deoxyribose moiety, occurs in the α-subunit, and proceeds via a radical mechanism. The β-subunit is responsible for harboring a strongly oxidizing species in the form of a radical or a high valent metal complex, essential for initiating the reaction in the α-subunit via long-range electron abstraction. Class I RNRs have a set of subclasses with comparable structures and reaction pathways, but with remarkably different β-subunit metallo-cofactors. The subclassification is partially based on these differences. In most class I RNRs, e.g. as found in all eukaryotes including mammals, many bacteria, and a few archaea, the radical is housed on a tyrosine side chain in the form of a tyrosyl radical, which in turn is stabilized via its interaction with a dinuclear FeIIIFeIII complex (this form is sometimes called subclass Ia). In contrast, subclass Ic RNRs, primarily found in chlamydia and halobacteria, do not harbor an amino acid based radical. Instead the necessary electron acceptor is a MnIVFeIII complex (3).
In subclass Ib RNRs, primarily found in some actinobacteria, firmicutes, and proteobacteria, the α- and β-subunits are denoted NrdE and NrdF, respectively. The make-up of the metallo-cofactor in subclass Ib long remained controversial. In vivo studies indicated a dependence on manganese, while initial in vitro studies with heterologously expressed subunits supported the presence of an iron cofactor, and early attempts to reconstitute NrdF with manganese were unsuccessful (4–6). Recent work reconciled these observations by showing that in vitro activation of manganese loaded NrdF (Mn-NrdF) is not a spontaneous process, but is dependent on an additional maturase denoted NrdI (7). This confirmed earlier studies in Streptococcus pyogenes, which demonstrated the essentiality of NrdI in vivo (8). Recently manganese-containing forms of NrdF have been homologously expressed and isolated from Corynebacterium ammoniagenes (9), Escherichia coli (10), and Bacillus subtilis (11). The manganese cofactor from C. ammoniagenes was studied in depth by EPR spectroscopy leading to a MnIII2-Y· oxidation state assignment (9). Additionally, in the case of Bacillus anthracis class Ib RNR only the manganese form responds to physiologically relevant reductants in in vitro enzymatic assays (12, 13). A similar observation was recently reported for Streptococcus sanguinis Ib RNR, where a 4-fold increase in specific activity is observed with Mn-NrdF as compared with Fe-NrdF when endogenous reductants are employed (14). It appears that active NrdF does indeed harbor a manganese cofactor under most, if not all, conditions in vivo. Hence, there are to date three well-established metallo-cofactors utilized to store the necessary electron hole in the β-subunit in class I RNRs. Conversely, the post-translational processes responsible for forming the active β-subunits are less explored. For example the NrdI protein which is found in all organisms encoding a class Ib RNR, was only recently shown to be crucial for in vitro activation of NrdF (7). NrdI is a flavodoxin-like protein shown to be responsible for the generation of an active dioxygen species via reduction, but the mechanism of the activation is still not settled.
Previous studies have only evaluated the maturation capacity of the fully reduced hydroquinone state of the maturase, as B. subtilis and E. coli NrdI are unable to stabilize the one-electron reduced semiquinone state. The predicted isoelectric point (pI) of 5.4 for B. anthracis NrdI is significantly more acidic than those for B. subtilis and E. coli, of 8.0 and 9.4, respectively. The acidic pI value of B. anthracis NrdI is close to what is generally observed for flavodoxins (pI ≈ 4.5), implying a greater propensity for one-electron chemistry and an increased stability of the semiquinone state of B. anthracis NrdI (15). Thus, the B. anthracis system provides a powerful tool in studying the mechanism of maturation. Moreover, B. anthracis is a severe mammalian pathogen, which is dependent on subclass Ib RNR for aerobic growth. As discussed above we have previously shown that only the manganese reconstituted form of B. anthracis NrdF shows a substantial enzymatic activity with the physiologically relevant reductants TrxA and NrdH in enzymatic assays (12, 13). Additionally, it has been demonstrated that inhibiting B. anthracis RNR using N-methylhydroxylamine efficiently knocks out growth of the organism, making it an attractive target for novel antibiotics (16).
Herein we report the EPR spectroscopic properties of the oxidized manganese cofactor in B. anthracis NrdF, and show that the catalytically relevant state features a tyrosyl radical in magnetic communication with a ferromagnetically coupled manganese dimer. Our results are in line with what has previously been reported by Cox et al. for NrdF from C. ammoniagenes (9), albeit noticeable differences imply subtle variations in the geometry of the active site. Furthermore, we have investigated the redox properties of B. anthracis NrdI, using a combination of redox titrations and protein electrochemistry. In contrast to other NrdI enzymes studied thus far, the two redox potentials of the FMN cofactor in B. anthracis NrdI differ by ∼110 mV and it was possible to stabilize stoichiometric amounts of the semiquinone state, NrdIsq. Likewise, we observed substantial amounts of NrdIsq in vivo. We have utilized these advantageous NrdI properties to probe the maturation mechanism of Mn-NrdF. We show that the NrdIsq form is capable of generating active NrdF, strongly supporting the notion that the oxidized manganese-tyrosyl radical cofactor is formed via a superoxide intermediate.
EXPERIMENTAL PROCEDURES
General
Chemical reagents were purchased from Sigma Aldrich and used as received unless otherwise stated. Protein purity was assessed by gel electrophoresis by loading samples on PhastGelTM; Gradient 10–15 precast gels (GE Healthcare) with Precision Plus ProteinTM standards (Bio-Rad). Migration was achieved on a PhastSystem Separation and Control apparatus (Pharmacia). Protein concentrations were determined with the Bio-Rad Protein Assay, using bovine serum albumin as a standard. In vivo quantification of NrdI in cell paste was performed by densitometry measurements from SDS page gels run on whole cell lysis material, using purified B. anthracis NrdI as internal standard. Images were recorded on a Molecular Imager® ChemiDocTM XRS+ system and calculations performed using the ImagelabTM software from Bio-Rad. UV-visible spectra were recorded on a PerkinElmer Life Sciences Lambda 35 spectrophotometer.
Expression and Purification of B. anthracis NrdI and NrdF
B. anthracis NrdI was obtained following literature protocols (12, 15). The FMN content in purified NrdI was determined via acid treatment of the protein to release the FMN cofactor, in accordance with the method of Mayhew and Massey (17). NrdI expressed and purified as outlined above was found to contain one FMN per protein, with ϵ447 = 10 750 (±500) m−1 cm−1 for the oxidized form. Following titration of NrdI with sodium dithionate under anaerobic conditions the extinction coefficient of the semiquinone state (ϵ580 = 4,500 (±400) m−1 cm−1) was determined by UV/visible spectroscopy with concomitant spin-quantification of the radical content using EPR (as described below).
The apo-form of B. anthracis NrdF was purified as described previously (16) with the following modification: the intermediate purification product obtained following phenyl-Sepharose chromatography was concentrated and the solution was treated with 2.5 mm dithionite and 5 mm EDTA under strictly anaerobic conditions at 4 °C for 2–3 h. Following this treatment the resulting reaction mixture was purified by size exclusion chromatography on a Superose column, eluting with 50 mm Tris-HCl buffer, pH 7.6, with 100 mm NaCl. Finally, apo-NrdF was loaded onto a Q-Sepharose column and the column was washed with three column volumes of Tris-HCl buffer before apo-NrdF was eluted using a linear gradient from 0 to 500 mm KCl. Thereby apo-NrdF was isolated in >95% purity as determined by SDS-page gel.
Electrochemistry
Cyclic square wave voltammetry (SWV) and cyclic voltammetry (CV) were recorded under strictly anaerobic conditions in a Jacomex glovebox using a potentiostatic three-electrode assembly and a computer-controlled BioLogic potentiostat PS 200. The working electrodes were a 3-mm diameter disc of gold (BioLogic) for SWV experiments and 3-mm diameter graphite disc (Goodfellow) in the case of CV experiments, the reference electrode was a Ag/AgCl electrode (RE1b from BioLogic) while the counter-electrode was a platinum electrode (BioLogic). The setup used is based on a system described previously (18). In our case, a 10–15 μl droplet of NrdI (180 μm) in 50 mm Tris-HCl buffer at pH 7.6 was deposited on the working electrode positioned upside. The other two electrodes were immersed directly into the droplet. The gold working electrode was polished with different sizes of alumina (1, 0.3, 0.05 micrometer, respectively) prior to each experiment. The graphite electrode was polished on sandpaper. Potentials versus the normal hydrogen electrode (NHE) were obtained by adding 0.210 V. In cyclic SWV, the ground potential may be constructed by the superposition of a rectangular pulse potential of constant amplitude and of Δt duration, at the beginning of each step (duration 2Δt) of a staircase potential. The height of the pulse is denoted by ΔE and the height of the stair potential ΔEs. The current is measured at the end of the pulse (Ifor) and the end of the stair potential (Irev) on which the pulse is superimposed. ΔI or Inet = Ifor − Irev is plotted versus E + ΔE/2 or E, where E is the potential of the stair.
Titration of NrdI
NrdI (20 μm) in 50 mm Tris-HCl buffer was reduced by titration with sodium dithionite under strictly anaerobic conditions. The solution was allowed to equilibrate for 15–20 min after each addition and the extent of reduction was monitored by UV-visible spectroscopy in the 300–750 nm range.
Metal Reconstitution of apo-NrdF, Preparation of MnIII2-Y·
EPR samples with ∼1.75 molar equivalents of Mn2+ and 0.5 molar equivalents of NrdI per NrdF were produced in micro vials on an argon line. Apo-NrdF was added to an anaerobic Tris-HCl buffer (50 mm, pH 7.5) with Mn2+ and left to equilibrate for 30 to 60 min. Then the hydroquinone form of NrdI (NrdIhq) was added and left for ∼5 min where after ∼500 μl O2 was injected into the reaction mixture to form the MnIII2-Y· center. Samples with final NrdF concentrations of 90–100 μm were then subjected to a brief centrifugation before transfer to EPR tubes and frozen with liquid nitrogen ∼30 min after addition of oxygen. In one reconstituted sample the tyrosyl radical containing NrdF was separated from NrdI by anionic exchange chromatography (12).
Additionally, to compare the activation capacity of NrdIhq and NrdIsq, solutions of apo-NrdF (200 μm) in Tris-HCl buffer (50 mm, pH 7.0) were incubated with NrdI (100 μm) in the presence of Mn2+ (300 μm) under anaerobic conditions in a Forma Scientific glove box with less than 1 ppm O2. The FMN cofactor present in NrdI was reduced to either its semiquinone or hydroquinone form through titration with dithionite, as described above. Once the desired oxidation state of the flavin cofactor had been obtained the reaction mixture was flushed with O2 for 2 min and stirred open to air for an additional 8 min. The amount of tyrosyl radical formed was quantified by the dropline method (12). Furthermore the effect of catalase or superoxide dismutase on the reconstitution reaction was evaluated by injecting 150 μl of an anaerobic solution containing NrdIhq (120 μm), NrdF (120 μm), and MnCl2 (180 μm) into 20 μl of air saturated Tris-HCl buffer containing either 1275 units/ml of catalase, or 1275 units/ml of superoxide dismutase (final concentration 150 units/ml). The amount of tyrosyl radical formed was compared with that obtained by injecting the anaerobic solution into pure buffer.
EPR Spectroscopy
EPR spectra at 5 to 50K were recorded either on a Bruker ELEXSYS 530 X-band spectrometer (Bruker Biospin), equipped with an Oxford Instruments ESR 900 cryostat, or on an ELEXSYS E500 spectrometer (Bruker Biospin) equipped with a SuperX bridge and a SHQ4122 cavity and an Oxford Instruments ESR900 cryostat. For measurements at 77 K a cold finger Dewar was used. For temperatures above 77 K the temperature was controlled by flowing nitrogen gas of known temperature past the sample in a Dewar inserted in the cavity. Quantifications were made by comparing the double integral of the samples with that of a Cu2+/EDTA (1 mm and 10 mm, respectively) standard at non-saturating conditions.
Evaluation of the magnetic properties of the NrdIsq radical and the MnIII2-Y· center at different temperatures was performed by monitoring the power at half saturation (P1/2) according to the equation I = 1/(1+P/P1/2)b/2 (19). I denotes the normalized intensity of the signal (double integral or as in this study the amplitude of the central peak), P is the applied power and b is the inhomogeneity factor. The factor b is 1 for completely inhomogeneous broadening and 3 for completely homogeneous broadening. We observe that the b factor (that informs of the type of broadening that occurs) for the evaluated saturation curves of MnIII2-Y·-NrdF always is below one. This most likely reflects that relaxation processes from the ground state and excited states in the ferromagnetically coupled MnIII2 spin state ladder influences the relaxation process of the tyrosyl radical. P1/2 was evaluated using b = 0.5 since this gave the best fits of the saturation curves. Values of b below one has been reported for the Mn-NrdF proteins from B. cereus (b = 0.6) and E. coli (b = 0.91) (7, 20) but to our knowledge there is no theoretical evaluation of the underlying processes resulting in b-values below one.
P1/2 reflects the relaxation process according to the equation P1/2 = α/T1T2 where T1 is the spin-lattice relaxation rate and reflects the interaction between the paramagnetic center and the surrounding ligands. T2 is a spin-spin relaxation rate, which in some cases does not change with temperature. If this is the case the measured P1/2 is inversely proportional to T1. The proportionality factor α depends on the particular settings of the EPR system used and the geometry and nature of the sample (21). In this study we have measured saturation curves on two different EPR spectrometers and different samples; hence we can mirror the temperature dependences of T1T2 from plots of P1/2 as a function of temperature.
RESULTS
Isolation and Characterization of B. anthracis NrdI
When FMN was included in the growth medium (12, 15), the purified B. anthracis NrdI protein was found to contain stoichiometric amounts of its FMN cofactor, thus circumventing the need for in vitro reconstitution. Importantly this observation also allowed the study of the FMN-NrdI complex in vivo (see below).
Reducing the flavin cofactor by titration with dithionite at pH 7.0 allowed the isolation of two oxygen sensitive states. Addition of one equivalent of dithionite generated the one-electron reduced semiquinone state (NrdIsq). The formation of this species is readily observable from the broad rise in absorbance between 500 and 650 nm and the appearance of a new peak at 350 nm, with a concomitant decrease in absorbance at 375 nm and around 450 nm (Fig. 1, top). Further reduction generates the two-electron reduced hydroquinone state (NrdIhq), inferred from the disappearance of semiquinone features and the increase in absorbance below 320 nm (Fig. 1, bottom).
FIGURE 1.
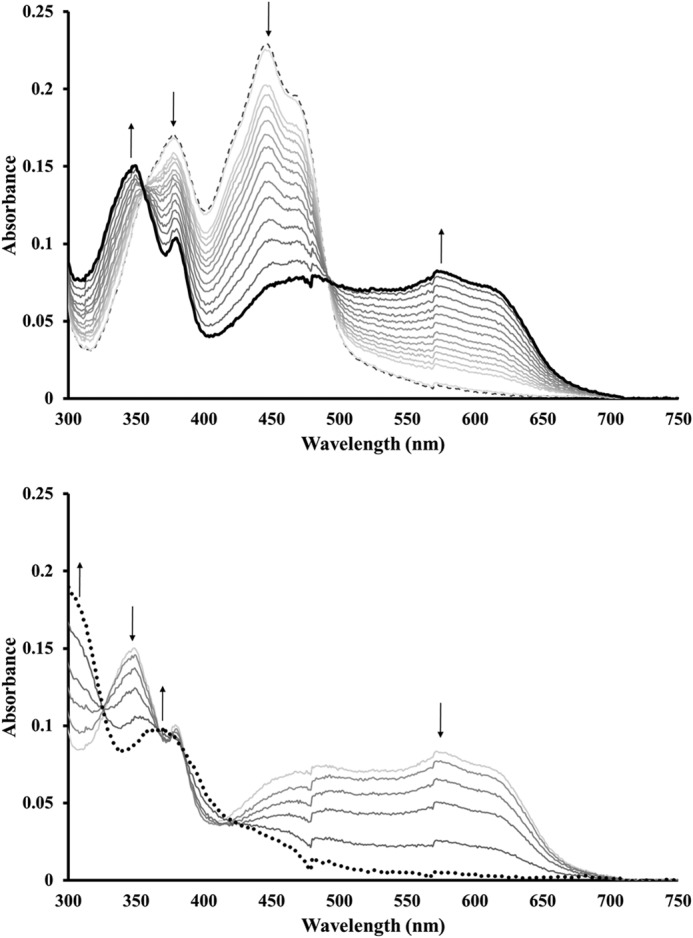
B. anthracis NrdI reduced through titration with dithionite. Top: formation of the semiquinone state (NrdIsq, thick black line) from NrdIq (dashed line); Bottom: continued titration resulting in the formation of the fully reduced hydroquinone state (NrdIhq, dotted black line). The concentration of NrdI was 20 μm in 50 mm Tris-HCl buffer, pH 7.0.
The maximum obtainable yield of the semiquinone form was found to be pH-dependent in the studied pH range (7.0–8.8), with the yield decreasing with higher pH. Close to stoichiometric amounts of NrdIsq (95 ± 5%) were obtained at pH 7.0 based on ϵ580 = 4500 m−1 cm−1. Furthermore this form of the protein was stable with regards to disproportionation, as no changes were observed in the UV-visible spectrum during one hour at 17 °C. More basic pH lowered the yield of the semiquinone state and only ∼75–80 and 40% of the total NrdI pool could be observed in the NrdIsq state at pH 7.6 and 8.8, respectively. The addition of the NrdF protein with or without MnII under anaerobic conditions did not influence the observed ratios of the NrdI oxidation states.
Based on the intense absorbance in the 500–650 nm region observed for NrdIsq (Fig. 1, top), we conclude that in vitro reduction of NrdIq (the oxidized, quinone, form of NrdI) generated the protonated, neutral state of the semiquinone. This assignment is further supported by the observed singlet EPR signal with a g-value of 2.00 (see below).
The redox properties of NrdI were further studied by electrochemical methods. When NrdI was analyzed by cyclic voltammetry (CV) on a graphite electrode without a promoter present, the protein displayed two broad quasi-reversible processes. One with a midpoint potential of ≈ −260 mV (versus NHE; peak cathodic current, ip,c = −280 mV; peak anodic current, ip,a ≈ −235 mV; Δ ip,a − ip,c ≈ 45 mV), and a second with a midpoint potential of ≈ −370 mV (versus NHE; peak cathodic current, ip,c = −400 mV; peak anodic current, ip,a ≈ −340 mV; Δ ip,a − ip,c ≈ 60 mV) (Fig. 2A). The potential of the first reduction process is similar to what has been reported for E. coli NrdI (22) and the second is in the expected range for flavodoxin proteins (23), thus we attribute the two redox couples to FMN bound to the protein. However, the cyclic voltammograms can potentially be complicated by free FMN, liberated from the protein (18). We therefore also show the distinctly different CV trace recorded for free FMN (Fig. 2A, scan c), which features a reduction potential of ≈ −240 mV.
FIGURE 2.
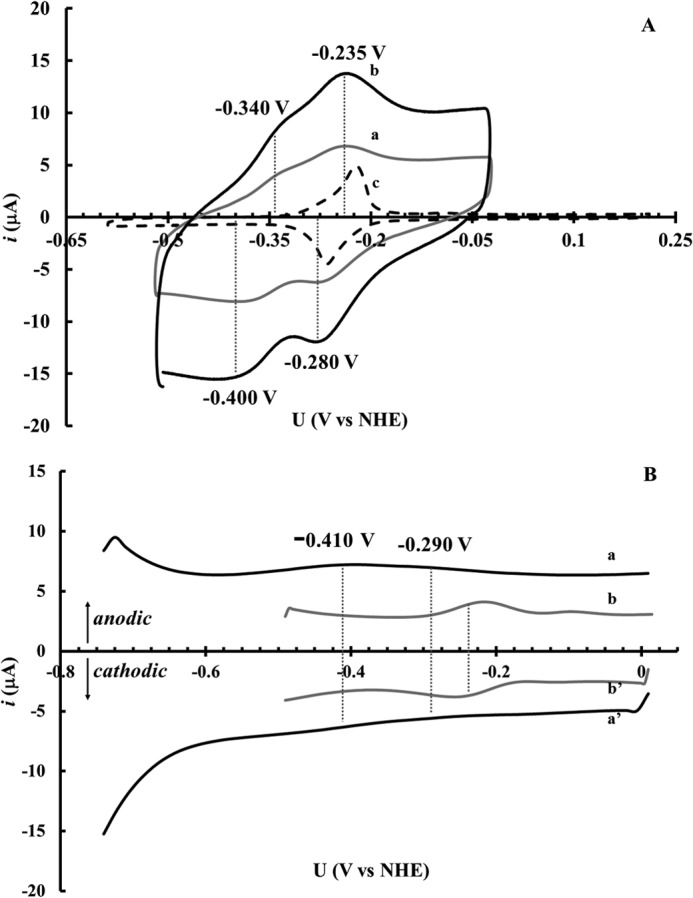
A, cyclic voltammetry traces of B. anthracis NrdI and of free FMN. Recorded under anaerobic conditions using a graphite electrode in 50 mm Tris-HCl, pH 7.6. [NrdI] = 180 μm, scan rate 60 mV s−1 (a); [NrdI] = 180 μm, scan rate 100 mV s−1 (b); [FMN] = 150 μm, scan rate 10 mV s−1 (c). B, cyclic square wave voltammetry traces of B. anthracis NrdI (solid black line) and of free FMN (solid gray line). Recorded under anaerobic conditions using a gold electrode in 50 mm Tris-HCl, pH 7.6. a and a′ anodic and cathodic scans, respectively, of NrdI (180 μm); b and b′ anodic and cathodic scans respectively of free FMN (150 μm). Square wave voltammograms were obtained using a square wave frequency of 10 Hz, pulse height = 25 mV, pulse width = 50 ms, and step height = 15 mV.
To further substantiate the electrochemical behavior of NrdI, we changed the electrode material and performed cyclic square wave voltammetry (SWV) using a gold electrode. The SWV potential sweep may be performed in the negative direction (cathodic sweep) or the positive direction (anodic sweep). It is known that for a reversible electrochemical reaction, the net current peak potential corresponds to the redox potential (24–27). Thus for a reversible system the anodic and cathodic SWV voltammograms lie on either side of the potential axis and are mirror images. Both the initial anodic as well as the cathodic SWV scans display two broad waves with midpoint potentials of ≈ −410 mV and ≈ −290 mV. The cyclic SWV scans for NrdI are shown in Fig. 2B (scans a and a′), together with that of free FMN recorded under identical conditions (scans b and b′). The reduction potentials obtained from SWV are close to what is observed in the CV experiments, and the differences are tentatively attributed to variances in interactions with the two electrode materials. Importantly, under these conditions the potentials are clearly separated from that of free FMN. In summary, the combined data from SWV and CV reveal two redox processes for B. anthracis NrdI separated by about 110 mV, attributable to the NrdIq/NrdIsq (Eq/sq = −270 ± 30 mV versus NHE) and NrdIsq/NrdIhq (Esq/hq = −385 ± 30 mV versus NHE) couples.
In Vivo Characterization of B. anthracis NrdI
The strong overproduction of B. anthracis NrdI in E. coli cells prompted us to analyze intact cells before and after IPTG-induction using EPR. Samples taken before induction showed no trace of NrdIsq, and only a minor background signal was visible in the X-band EPR spectrum (Fig. 3, trace a). Conversely, after induction the cell paste showed an intense EPR signal characteristic of NrdIsq (Fig. 3, trace b) with a g-value of 2.00 and an overall line shape identical to the EPR spectrum observed following in vitro generation of the NrdI semiquinone (Fig. 3, trace c). The peak to trough width is 2.1 mT as expected for neutral FMN semiquinone protein radicals, which display peak to trough widths ≥1.9 mT (28). The microwave saturation behavior for NrdI in cells and NrdIsq obtained in vitro via dithionite reduction is within error limit identical, i.e. P1/2 210 and 207 μW, respectively, at 77K. The radical concentration in the cell paste was quantified to 175 μm, while the total concentration of NrdI in the lysed cell paste was estimated to 600 μm. Thus NrdIsq represents a significant fraction (≈30%) of the total NrdI pool in vivo.
FIGURE 3.
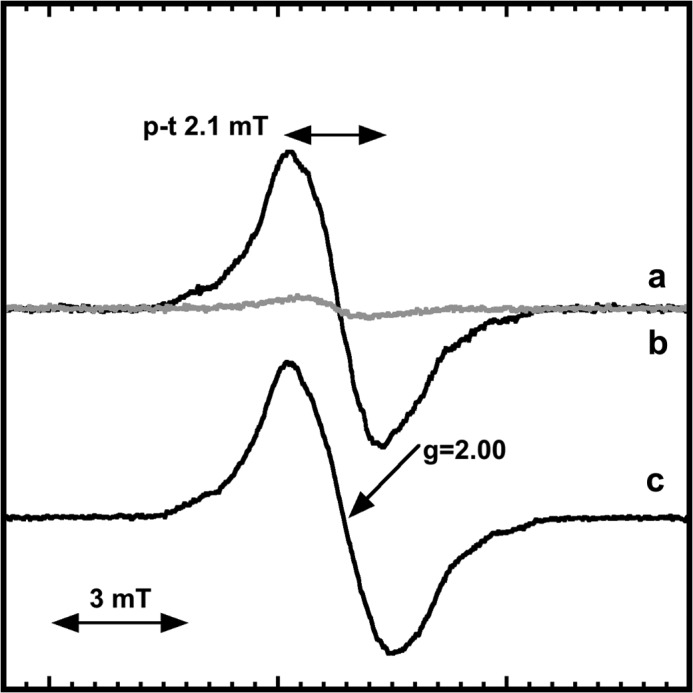
X-band EPR spectra of B. anthracis NrdIsqin vivo and in vitro. In vivo EPR recorded on a frozen cell pellet obtained prior to IPTG addition (a); and following 4 h overexpression of NrdI in E. coli (b); NrdIsq generated in vitro through dithionite reduction (c). The spectra were recorded at 98K under non-saturating conditions. In a and b recording conditions were identical, (c) has been scaled to the same signal amplitude as for NrdIsq in b for comparison.
Preparation and Characterization of Mn-reconstituted B. anthracis NrdF
The apo-NrdF protein was purified via a modified literature procedure (16), by adding an incubation with 5 mm EDTA under reducing conditions for 2–3 h after which time adventitiously bound metal ions were removed by size-exclusion chromatography. Finally, apo-NrdF was isolated in >95% purity by anionic exchange chromatography. The reconstitution reaction was performed by mixing NrdF with Mn2+ and pre-formed NrdIhq under anaerobic conditions. The oxygen-free reaction mixture was incubated for 5 min before O2 gas was injected into the reaction mixture to generate the oxidized form of Mn-NrdF.
The EPR spectrum of the tyrosyl radical in Mn-reconstituted B. anthracis NrdF, recorded at 77K is a singlet with peak to trough width of 3.05 mT (Fig. 4A). In addition, poorly resolved hyperfine couplings (0.6–0.7 mT) from the 3,5-ring protons of the tyrosine can be observed. The shape of the singlet is independent of the presence the NrdI. The signal remains unsaturated at the full available microwave power indicating a strong interaction with a fast relaxing metal center. The EPR spectrum is very similar but not identical to those described for E. coli, B. subtilis, B. cereus, S. sanguinis, and C. ammoniagenes MnIII2-Y· (7, 9, 11, 14, 20).
FIGURE 4.
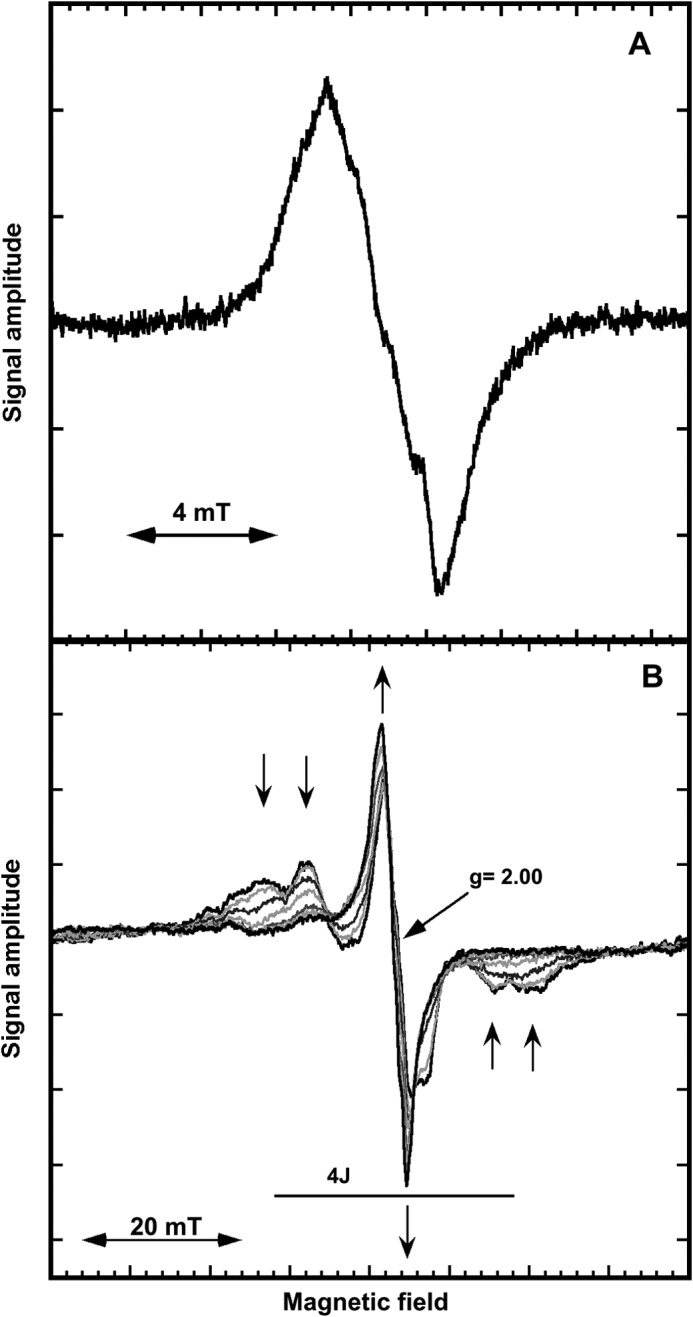
X-band EPR spectra of B. anthracis MnIII2-Y·-NrdF at 77K (A) and in the temperature interval T = 6.7 to 40 K (T = 6.7, 10, 14, 20, 25, 35, and 40 K) (B). Arrows in Fig. 4B indicate the temperature dependence at the five lines of the “split tyrosyl radical” with increasing temperature. Recording conditions: modulation amplitude, 0.5 mT; 4 scans; microwave power was 10 milliwatt in A and 20 milliwatt in B.
The EPR spectrum is strongly temperature dependent as shown in Fig. 4B for the temperature interval 7 to 40 K. The singlet observed in Fig. 4A is shown to increase with increasing temperature and its peak to trough width decreases from ∼3.5 to 3 mT at the applied microwave power. At temperatures ≤10K four additional lines flanking the central peak are clearly resolved. Both the exchange narrowing of the central part of the B. anthracis Mn-NrdF spectrum and accompanying decrease of the flanking lines in spectra with increasing temperature are similar to that observed for C. ammoniagenes NrdF (9). Cox et al. denoted this a “split-tyrosyl radical” and presented a model for the magnetic interaction between the radical and the dimanganese site (9). The five lines we observe at 10K are consistent with coupling of the tyrosyl radical to a S = 2 ground state of the MnIII2 manifold ladder. The observed splitting gives a rough estimate of 6.8 mT for J and 29.8 mT for 4J (Fig. 4B), slightly narrower than we estimate for C. ammoniagenes NrdF from Fig. 9A in Ref. 9. The observed increase in resolution of the hyperfine splitting with decreasing temperature is consistent with a ferromagnetic coupling between the two manganese ions. These drastic changes in the spectra of the B. anthracis Mn-NrdF radical are in contrast to the temperature behavior of the tyrosyl radical in B. anthracis Fe-NrdF. The latter does not display any change in line shape in the temperature region 10 to 136 K (data not shown) indicative of a rather weak interaction with the assumed antiferromagnetically coupled diiron site.
The relaxation properties of the split-tyrosyl radical were evaluated from microwave saturation studies in the temperature range 6.7 to 77 K. The relaxation characteristic P1/2 was calculated both in the absence and the presence of NrdI with no significant difference. As mentioned above the radical does not saturate at all at 77K and even at 40K the available microwave power was not enough to determine a P1/2 value, again manifesting a strong interaction with the dimanganese site. Fig. 5 shows a comparison of the relaxation properties of the tyrosyl radical in Mn-reconstituted B. anthracis NrdF to that of the Fe-reconstituted form (16), as well as to the more thoroughly studied tyrosyl radical in E. coli FeIII2-NrdB with an antiferromagnetically coupled diiron site (21, 29). In the case of a ferromagnetic metal site, the lowest lying states are non-zero and can enhance the relaxation of the tyrosyl radical, and non-direct processes such as a Raman mechanism become dominant already at low temperatures. For B. anthracis Mn-NrdF we see no direct process as expected for a S = 2 ferromagnetic ground state. In the presence of an antiferromagnetically coupled metal site with ground state S = 0 a direct relaxation process will operate until a substantial amount of excited states in the coupled metal center is populated and can enhance the relaxation of the tyrosyl radical. Hence at low temperatures only a direct process will be operational at an antiferromagnetic site and we will expect different temperature dependences for the direct and non-direct processes. For B. anthracis Fe-NrdF the direct relaxation mechanism persists to at least 50K and the possible Raman process dominates above 80K as judged from extrapolation (see dashed lines in Fig. 5). In E. coli NrdB a break between the direct and non-direct process occurs in the 25K region (Fig. 5), displaying species-specific differences of rather similar diiron-radical sites. The temperature dependence of the evaluated P1/2 is consistent with a Raman mechanism for the spin lattice relaxation proportional to a temperature dependence (Tn) of the radical, where n can vary between 3 and 9. The fitted lines for the temperature dependences for possible Raman relaxation mechanism regimes are T4.9, T5.5, and T5.3 for Mn-NrdF and Fe-NrdF from B. anthracis and Fe-NrdB from E. coli, respectively. It is striking that the temperatures at which the tyrosyl radical in the different proteins start to experience the neighboring metal site varies widely (Fig. 5), but once the possible Raman process occurs its temperature dependence for the three species is rather similar. We can however not exclude that this similarity is coincidental.
FIGURE 5.
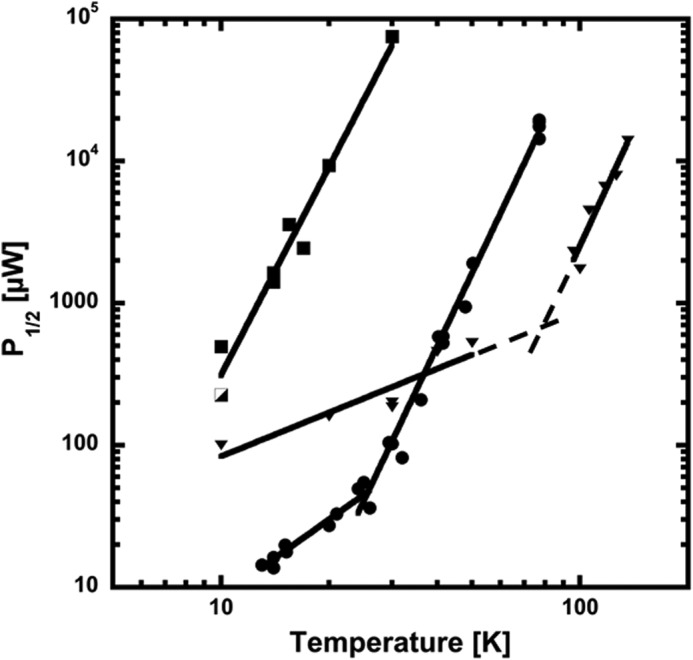
Power of half saturation (P1/2) for the Mn and Fe reconstituted forms of B. anthracis NrdF and for E. coli NrdB. B. anthracis MnIII2-Y·-NrdF (squares); purified MnIII2-Y·-NrdF (half-filled square); FeIII2-Y·-NrdF (triangles); E. coli FeIII2-Y·-NrdB (filled circles). Dashed lines are extrapolation from the fitted curves for the different relaxation regimes for FeIII2-Y·-NrdF.
The observed differences in magnetic and electronic properties between B. anthracis Mn-NrdF and other Mn-NrdF species (7, 9, 11, 14, 20) may reflect differences in the micro-environment for the described tyrosyl radicals. Such differences can be attributed to minor differences in the properties of the metal sites in the different proteins (see for instance ref 9 showing differences in structure for Mn and Fe loaded NrdF in C. ammoniagenes) and/or the geometry of the tyrosine carrying the radical. The latter can reflect the angle of the β-protons versus the plane of the tyrosyl ring as well as differences in the hydrogen bonding networks.
Studies on the Maturation Mechanism of the MnIII2-Y· Cofactor of B. anthracis NrdF
The importance of the maturase protein NrdI in vivo and during in vitro activation of Mn-NrdF has been reported (7, 8). However, which redox forms of NrdI that are capable of generating the MnIII2-Y· cofactor and the nature of the oxidizing oxygen molecule is still not settled. The possibility to stabilize the B. anthracis NrdI protein in either its one-electron or two-electron reduced state allowed us to address these issues.
The maturation capacity of NrdIsq, i.e. its capacity to generate MnIII2-Y·-NrdF, was evaluated by generating NrdIsq in the presence of two molar equivalents of NrdF monomer and three molar equivalents of MnII with regards to NrdI. When this reaction mixture was exposed to air over the course of 10 min, formation of a new species was observable from a broad rise in absorbance in the UV-visible spectrum around 400 nm concomitant with re-oxidation of NrdIsq (Fig. 6, black line). The observed difference spectrum before and after O2 incubation contains the same spectral features previously attributed to formation of MnIII2-Y· (12), with broad increases in the <550 nm region and a peak at 409 nm assigned to formation of Y· (Fig. 6, inset). X-band EPR spectroscopy was further employed to confirm that the changes observed in the UV-visible spectrum were attributable to the formation of MnIII2-Y·-NrdF (data not shown). The formation of the manganese-tyrosyl radical moiety under these conditions strongly supports the notion of a superoxide intermediate. We consider formation of hydrogen peroxide starting from the NrdIsq state highly unlikely, as the reduction of dioxygen to peroxide is a two-electron process.
FIGURE 6.
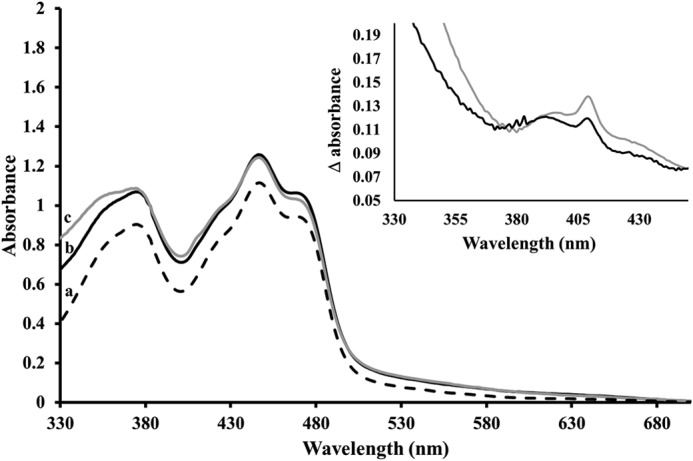
UV-visible absorption spectra of Mn reconstituted NrdF and NrdIq. Starting spectrum containing NrdIq (100 μm), NrdF (200 μm), and MnII (300 μm) in Tris-HCl, pH 7.0 (dashed line, a); Formation of MnIII2-Y·-NrdF (≈6 μm) from NrdIsq (black solid line, b); Formation of MnIII2-Y·-NrdF (≈10 μm) from NrdIhq (gray solid line, c). Inset: absorption difference spectra (obtained by subtraction of the starting spectrum from the respective end spectra) highlighting changes attributable to the formation of MnIII2-Y·. Oxidation product formed from NrdIsq (b-a) (black line); oxidation product formed from NrdIhq (c-a) (gray line).
With this obvious maturation capacity of NrdIsq established, the same experiment was performed starting from the NrdIhq state under otherwise identical reaction conditions. In this case an initial fast reoxidation of NrdIhq generating the NrdIsq state was followed by a substantially slower oxidation of NrdIsq. As expected, the final spectrum upon complete re-oxidation of NrdIhq displayed changes in the same region as that with NrdIsq, albeit the increase in absorbance at 409 nm was larger with NrdIhq (Fig. 6). The inset of figure 6 shows the difference spectra obtained by subtracting the starting spectra from the end spectra recorded following oxidation of Mn loaded NrdF using NrdIsq (black line) or NrdIhq (gray line), respectively.
The amount of MnIII2-Y·-NrdF generated was quantified using the dropline method from the peak at 409 nm observed in the difference spectra (12). Based on this method, the yield of tyrosyl radical was 6 (±1) μm using NrdIsq as maturase and 10 (±1) μm using NrdIhq. Thus generating the enzymatically active Mn-cofactor from NrdIhq increased the yield by ∼65% as compared with NrdIsq, in agreement with a two-step reoxidation process of NrdIhq, generating up to two equivalents of superoxide.
The kinetics of the latter reaction was further studied in a separate experiment, where a deoxygenated solution containing MnII2-NrdF and NrdIhq was injected with a small aliquot of O2-saturated buffer and the reaction was monitored by UV-visible spectroscopy. The spectra recorded from 10 s to 15 min after initiating the oxidation are shown in Fig. 7. Already at the first time point, 10 s, after exposing the reduced reaction mixture to oxygen a significant amount of NrdIsq (≈70% of the total NrdI content) was clearly visible in the spectrum (Fig. 7, solid black line). Subtracting the NrdIsq and NrdIq spectral features allowed quantification of the tyrosyl radical content, and the yield was found to be ∼5.3 (±0.6)% with regards to NrdF (Fig. 7, inset, 10 s). Extended reaction times resulted in disappearance of the NrdIsq features from the spectrum (Fig. 7, solid gray lines), with concomitant increase in tyrosyl radical concentration (Fig. 7, inset). Thus, the biphasic behavior observed during NrdIhq oxidation is also clearly reflected in the formation of the tyrosyl radical. The former observation clearly shows that a substantial fraction (≥70%) of NrdIhq is oxidized in a two-step fashion proceeding via an NrdIsq intermediate. Moreover, the fact that the formation of the tyrosyl radical is also biphasic strongly supports a superoxide intermediate during the maturation also in the case of NrdIhq, with each one-electron-oxidation step generating one equivalent of superoxide.
FIGURE 7.
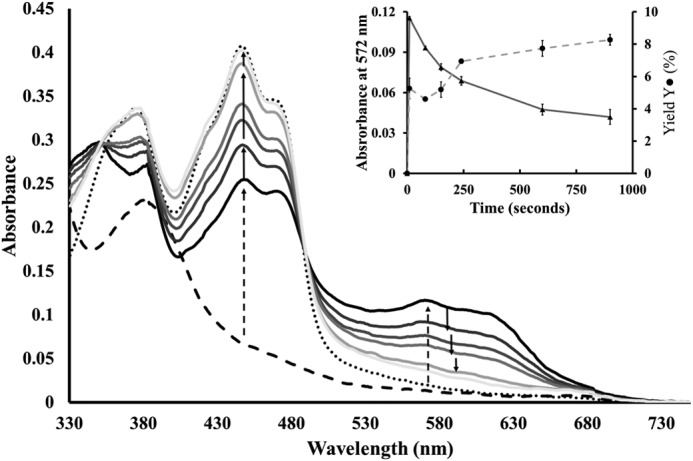
UV-visible spectra following the reoxidation of NrdIhq in the presence of MnII2-NrdF. Starting spectrum containing NrdIq (38 μm), NrdF (65 μm), and MnII (100 μm) in Tris-HCl, pH 7.0 (dotted black line); NrdI reduced to NrdIhq (dashed black line); Spectra recorded during the re-oxidation of NrdIhq, showing the appearance and disappearance of NrdIsq (solid lines). Direction of change indicated by arrows, initial changes observed 10 s after exposure to oxygen (dashed arrows); subsequent changes during 15 min (solid arrows). Spectra recorded after 10, 80, 150, 240, 600, and 900 s. Inset: absorbance at 572 nm followed over time as a measure of NrdIsq concentration (triangles, left Y-axis); Yield of MnIII2-Y·-NrdF with regards to NrdF concentration (circles, right Y-axis), time points as above.
DISCUSSION
The magnetic and electronic properties of the MnIII2-Y· cofactor in B. anthracis have been studied by X-band EPR spectroscopy. The observed spectra showed a strong temperature dependence, and the power saturation behavior was not influenced by the maturase NrdI. Both signal shape and power saturation studies are in line with the coupling of the tyrosyl radical signal to a ferromagnetically coupled manganese dimer. The observed properties of the split tyrosyl radical are similar, but not identical, to what has been reported for other forms of Mn-NrdF (7, 9, 11, 14, 20).
Interestingly, in contrast to what is observed for the Fe forms of NrdB and NrdF, oxidative activation of Mn-NrdF by O2 does not occur spontaneously but instead requires the flavodoxin-like maturase NrdI (7). The general consensus is that NrdI in line with other flavin containing proteins initiates the reaction by reducing O2 into a more reactive form. This would generate either superoxide (O2⨪) or peroxide (HOO−(H)), via a one or two electron reduction, respectively. The activated oxygen species is then delivered to the active site of NrdF via a hydrophilic channel. X-ray crystallographic studies of the Mn-NrdF-NrdI complex from E. coli have shown the presence of electron density assigned to a peroxide species in the channel (30). Moreover, in the case of E. coli NrdI, the doubly reduced hydroquinone form (NrdIhq) has in fact a lower reduction potential than the singly reduced semiquinone form, NrdIsq, −255 versus −264 mV (22). Consequently, it was argued that the protein is designed to perform two-electron reductions, i.e. catalyze the formation of HOO−(H) rather than O2⨪ (7). Combined, these observations originally led to the hypothesis that the MnIII2-Y· cofactor is generated by two consecutive peroxide oxidations, potentially proceeding via a di-(μ-O)-MnIV2 intermediate species. Conversely, a recent study of the kinetics of the cluster assembly in B. subtilis NrdF, using stopped flow optical absorption and rapid freeze quench EPR spectroscopies to analyze the flavin and manganese cofactors, supported a mechanism in which NrdIhq reduces O2 to O2⨪ with concomitant formation of NrdIsq (31). It was suggested that the superoxide in turn reacts with MnII2-NrdF to form the MnIII2-Y· cofactor in a three-electron redox reaction.
If superoxide is the oxidizing species, the semiquinone state of NrdI would be sufficient for generating the required reduced dioxygen intermediate. Nevertheless, the capacity of NrdIsq with regards to NrdF maturation has so far not been established, to a large extent attributable to difficulties in preparing this state in vitro (7, 11). We have shown herein that the redox chemistry of B. anthracis NrdI is markedly different to what has been reported for E. coli NrdI (22), as expected from their theoretical pI values (15). While E. coli NrdI has a predicted pI value of 9.4 the corresponding value for B. anthracis is 5.4. The latter is close to that of B. cereus NrdI (5.0), which has been found capable of stabilizing the semiquinone state in crystallo (32). Indeed, for B. anthracis NrdI it is possible to obtain stoichiometric amounts of the neutral semiquinone form, NrdIsq (Fig. 1). This observation is further supported by our electrochemical experiments, which showed two distinct quasi-reversible redox processes at −270 and −385 mV versus NHE, attributable to the NrdIq/NrdIsq and NrdIsq/NrdIhq couples, respectively. Both these observations underline how NrdI from B. anthracis is tuned to be an efficient one-electron donor.
The advantageous redox properties of B. anthracis NrdI allowed us to examine the maturation capacity of NrdIsq, and by extension also the activation mechanism. As discussed above earlier studies have suggested either a peroxide (7, 9, 30), or a superoxide intermediate (31). We have shown herein that both the semiquinone and hydroquinone generate the active radical center. The maturation capacity of NrdIsq strongly support the superoxide intermediate, as the formation of a two-electron reduced peroxide species would require the joint action of two NrdIsq. Moreover, we observe an approximate 65% increase in yield of MnIII2-Y· from NrdIhq when compared with an equimolar amount of NrdIsq, suggesting a two-step reoxidation process from the NrdIhq state generating up to two equivalents of superoxide. The latter hypothesis is further supported by the clearly biphasic behavior we observed during the maturation employing NrdIhq, with regards to both formation of NrdIq and the MnIII2-Y· cofactor. Interestingly, our 65% increase in yield obtained from B. anthracis NrdIhq agrees well with an earlier study by Cotruvo et al., where the reaction between B. subtilis NrdIhq and O2 was reported to generate 30% H2O2 and 70% O2⨪ based on spectroscopic analysis of the flavin and manganese cofactors (31).
The reaction pathway we suggest for NrdI catalyzed activation of Mn-NrdF is summarized in Scheme 1. A single superoxide would be sufficient to perform the three-electron oxidation of the initial MnII2-Y state to form the active MnIII2-Y· cofactor. This reactive oxygen species can be generated by the one-electron reduced semiquinone form of NrdI, via reduction of dioxygen and concomitant reoxidation to NrdIq. The two-electron reduced hydroquinone state can instead react with dioxygen via two different pathways: in a one-electron reaction generating a superoxide and NrdIsq (Scheme 1, bottom, solid arrow), or in a two-electron reaction forming an unproductive peroxide and NrdIq (Scheme 1, bottom, dashed arrow). Assuming a 2:1 ratio between the superoxide and peroxide pathway for the reoxidation of NrdIhq, this scheme is consistent with our observations as well as those reported by Cotruvo et al. (31). It should also be noted that we observe no effect on the reconstitution when the reaction is performed in the presence of either catalase or superoxide dismutase. This strongly supports the hypothesis that the active oxygen species is delivered to the metal site via a continuous shielded protein channel, as suggested from x-ray crystallography and earlier enzymatic assay studies (7, 30).
SCHEME 1.
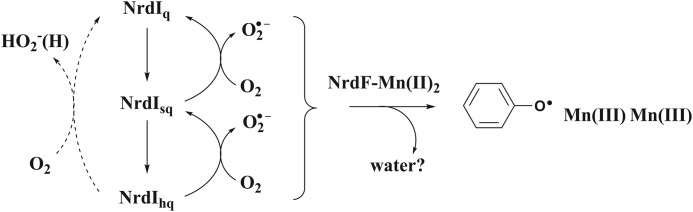
Suggested reaction pathway for NrdI-catalyzed maturation of Mn-NrdF, where Mn-NrdF is activated by a superoxide radical formed from NrdIsq. Alternatively NrdIhq can generate either two equivalents of the active O2⨪ species (right), or regenerate NrdIq through formation of one equivalent of the unproductive H2O2 (left, dashed line). Note that the scheme is not balanced with regard to protons.
Based on our findings there is a priori no need for a hydroquinone intermediate from an electron-economy perspective. Consequently, there should be a strong evolutionary pressure toward evolving a system utilizing NrdIsq rather than NrdIhq, to prevent an excess production of potentially deleterious superoxide. We have measured the oxidation state of NrdI in whole cells by EPR spectroscopy, and showed that a significant fraction (≈30%) of the protein existed as NrdIsq, the EPR active one-electron reduced form. Thus it is likely that in the case of B. anthracis, NrdIsq is the active reductant in vivo. Still, it should be noted that different redox properties have been reported for NrdIs of different origin. Thus, depending on the organism, NrdIhq might be relevant in vivo in certain cases. Future studies will address these key issues, as blocking the maturation reaction provides an intriguing target for novel highly selective antibiotics by knocking out RNR activity without targeting RNR itself.
Acknowledgments
We thank Sten Sjöberg for help with overexpression of B. anthracis Mn-NrdF in E. coli, and Fikret Mamedov and Stenbjörn Styring for access to their EPR equipment.
This study was supported by grants from the Swedish Research Council, the Swedish Cancer Foundation, Carl Trygger Foundation, Magn. Bergvall Foundation, and the Wenner-Gren Foundations.
- RNR
- ribonucleotide reductase
- apo-NrdF
- NrdF without bound metal ions
- Fe-NrdB
- the iron-reconstituted form of NrdB
- Fe-NrdF
- the iron-reconstituted form of NrdF
- Mn-NrdF
- the manganese-reconstituted form of NrdF
- NrdIhq
- NrdI with the hydroquinone form of FMN
- NrdIq
- NrdI with the oxidized (quinone) form of FMN
- NrdIsq
- NrdI with the semiquinone form of FMN.
REFERENCES
- 1. Nordlund P., Reichard P. (2006) Ribonucleotide Reductases. Annu. Rev. Biochem. 75, 681–706 [DOI] [PubMed] [Google Scholar]
- 2. Hofer A., Crona M., Logan D. T., Sjöberg B. M. (2012) DNA building blocks: keeping control of manufacture. Crit. Rev. Biochem. Mol. Biol. 47, 50–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cotruvo J. A., Stubbe J. (2011) Class I Ribonucleotide Reductases: Metallocofactor Assembly and Repair In Vitro and In Vivo. Annu. Rev. Biochem. 80, 733–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schimpff-Weiland G., Follmann H. (1981) A new manganese-activated ribonucleotide reductase found in gram-positive bacteria. Biochem. Biophys. Res. Commun. 102, 1276–1282 [DOI] [PubMed] [Google Scholar]
- 5. Willing A., Follmann H., Auling G. (1988) Nucleotide and thioredoxin specificity of the manganese ribonucleotide reductase from Brevibucterium ammoniagenes. Eur. J. Biochem. 175, 167–173 [DOI] [PubMed] [Google Scholar]
- 6. Huque Y., Fieschi F., Torrents E., Gibert I., Eliasson R., Reichard P., Sahlin M., Sjöberg B.-M. (2000) The Active Form of the R2F Protein of Class Ib Ribonucleotide Reductase from Corynebacterium ammoniagenes Is a Diferric Protein. J. Biol. Chem. 275, 25365–25371 [DOI] [PubMed] [Google Scholar]
- 7. Cotruvo J. A., Jr., Stubbe J. (2010) An Active Dimanganese(III)-Tyrosyl Radical Cofactor in Escherichia coli Class Ib Ribonucleotide Reductase. Biochemistry 49, 1297–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Roca I., Torrents E., Sahlin M., Gibert I., Sjöberg B.-M. (2008) NrdI Essentiality for Class Ib Ribonucleotide Reduction in Streptococcus pyogenes. J. Bacteriol. 190, 4849–4858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cox N., Ogata H., Stolle P., Reijerse E., Auling G., Lubitz W. (2010) A Tyrosyl-Dimanganese Coupled Spin System is the Native Metalloradical Cofactor of the R2F Subunit of the Ribonucleotide Reductase of Corynebacterium ammoniagenes. J. Am. Chem. Soc. 132, 11197–11213 [DOI] [PubMed] [Google Scholar]
- 10. Cotruvo J. A., Stubbe J. (2011) Escherichia coli Class Ib Ribonucleotide Reductase Contains a Dimanganese(III)-Tyrosyl Radical Cofactor in Vivo. Biochemistry 50, 1672–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Y., Stubbe J. (2011) Bacillus subtilis Class Ib Ribonucleotide Reductase Is a Dimanganese(III)-Tyrosyl Radical Enzyme. Biochemistry 50, 5615–5623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Crona M., Torrents E., Røhr A. K., Hofer A., Furrer E., Tomter A. B., Andersson K. K., Sahlin M., Sjöberg B.-M. (2011) NrdH-Redoxin Protein Mediates High Enzyme Activity in Manganese-reconstituted Ribonucleotide Reductase from Bacillus anthracis. J. Biol. Chem. 286, 33053–33060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gustafsson T. N., Sahlin M., Lu J., Sjöberg B.-M., Holmgren A. (2012) Bacillus anthracis Thioredoxin Systems, Characterization and Role as Electron Donors for Ribonucleotide Reductase. J. Biol. Chem. 287, 39686–39697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Makhlynets O., Boal A. K., Rhodes D. V., Kitten T., Rosenzweig A. C., Stubbe J. (2014) Streptococcus sanguinis Class Ib Ribonucleotide Reductase: High activity with both iron and manganese cofactors and structural insights. J. Biol. Chem. 289, 6259–6272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Johansson R., Torrents E., Lundin D., Sprenger J., Sahlin M., Sjöberg B.-M., Logan D. T. (2010) High-resolution crystal structures of the flavoprotein NrdI in oxidized and reduced states – an unusual flavodoxin. FEBS J. 277, 4265–4277 [DOI] [PubMed] [Google Scholar]
- 16. Torrents E., Sahlin M., Biglino D., Gräslund A., Sjöberg B.-M. (2005) Efficient growth inhibition of Bacillus anthracis by knocking out the ribonucleotide reductase tyrosyl radical. Proc. Natl. Acad. Sci. U.S.A. 102, 17946–17951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mayhew S. G., Massey V. (1969) Purification and Characterization of Flavodoxin from Peptostreptococcus elsdenii. J. Biol. Chem. 244, 794–802 [PubMed] [Google Scholar]
- 18. Heering H. A., Hagen W. R. (1996) Complex electrochemistry of flavodoxin at carbon-based electrodes: results from a combination of direct electron transfer, flavin-mediated electron transfer and comproportionation. J. Electroanal. Chem. 404, 249–260 [Google Scholar]
- 19. Sahlin M., Gräslund A., Ehrenberg A. (1986) Determination of relaxation times for a free radical from microwave saturation studies. J. Magn. Reson. 67, 135–137 [Google Scholar]
- 20. Tomter A. B., Zoppellaro G., Bell C. B., 3rd, Barra A.-L., Andersen N. H., Solomon E. I., Andersson K. K. (2012) Spectroscopic Studies of the Iron and Manganese Reconstituted Tyrosyl Radical in Bacillus Cereus Ribonucleotide Reductase R2 Protein. PLoS One 7, e33436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sahlin M., Petersson L., Gräslund A., Ehrenberg A., Sjöberg B. M., Thelander L. (1987) Magnetic interaction between the tyrosyl free radical and the antiferromagnetically coupled iron center in ribonucleotide reductase. Biochemistry 26, 5541–5548 [DOI] [PubMed] [Google Scholar]
- 22. Cotruvo J. A., Jr., Stubbe J. (2008) NrdI, a flavodoxin involved in maintenance of the diferric-tyrosyl radical cofactor in Escherichia coli class Ib ribonucleotide reductase. Proc. Natl. Acad. Sci. U. S. A. 105, 14383–14388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hoover D. M., Drennan C. L., Metzger A. L., Osborne C., Weber C. H., Pattridge K. A., Ludwig M. L. (1999) Comparisons of wild-type and mutant flavodoxins from Anacystis nidulans. Structural determinants of the redox potentials. J. Mol. Biol. 294, 725–743 [DOI] [PubMed] [Google Scholar]
- 24. Helfrick J. C., Jr., Bottomley L. A. (2009) Cyclic Square Wave Voltammetry of Single and Consecutive Reversible Electron Transfer Reactions. Anal. Chem. 81, 9041–9047 [DOI] [PubMed] [Google Scholar]
- 25. Komorsky-Lovric S., Lovric M. (2014) Square-wave Voltammetry of Two-step Electrode Reaction. Int. J. Electrochem. Sci. 9, 435–444 [Google Scholar]
- 26. Armstrong F. A., Hill H. A. O., Oliver B. N., Walton N. J. (1984) Direct electrochemistry of redox proteins at pyrolytic graphite electrodes. J. Am. Chem. Soc. 106, 921–923 [Google Scholar]
- 27. Jeuken L. J. C., McEvoy J. P., Armstrong F. A. (2002) Insights into Gated Electron-Transfer Kinetics at the Electrode-Protein Interface: A Square Wave Voltammetry Study of the Blue Copper Protein Azurin. J. Phys. Chem. 106, 2304–2313 [Google Scholar]
- 28. Palmer G., Müller F., Massey V. (1971) Electron paramagnetic resonance studies on flavoprotein radicals in Flavins and Flavoproteins (Kamin H., ed), pp. 123–140, University Park Press/Butterworths, Baltimore [Google Scholar]
- 29. Petersson L., Gräslund A., Ehrenberg A., Sjöberg B. M., Reichard P. (1980) The iron center in ribonucleotide reductase from Escherichia coli. J. Biol. Chem. 255, 6706–6712 [PubMed] [Google Scholar]
- 30. Boal A. K., Cotruvo J. A., Jr., Stubbe J., Rosenzweig A. C. (2010) Structural Basis for Activation of Class Ib Ribonucleotide Reductase. Science 329, 1526–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cotruvo J. A., Jr., Stich T. A., Britt R. D., Stubbe J. (2013) Mechanism of Assembly of the Dimanganese-Tyrosyl Radical Cofactor of Class Ib Ribonucleotide Reductase: Enzymatic Generation of Superoxide Is Required for Tyrosine Oxidation via a Mn(III)Mn(IV) Intermediate. J. Am. Chem. Soc. 135, 4027–4039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Røhr Å. K., Hersleth H.-P., Andersson K. K. (2010) Tracking Flavin Conformations in Protein Crystal Structures with Raman Spectroscopy and QM/MM Calculations. Angewandte Chemie International Edition 49, 2324–2327 [DOI] [PubMed] [Google Scholar]