

Background: An IGF2/IGF1R autocrine loop regulates beta cell glucose competence, proliferation, and apoptosis, and GLP-1 increases IGF1R expression.
Results: Glutamine enhances IGF2 biosynthesis, secretion, and IGF2-dependent Akt phosphorylation.
Conclusion: Glutamine controls the activity of the IGF2/IGF1R autocrine loop through IGF2 up-regulation.
Significance: The IGF2/IGF1R autocrine loop integrates changes in feeding and metabolic state to adapt beta cell mass and function.
Keywords: Beta Cell (B-cell), Glucose, Glutamine, Insulin, Insulin Secretion, Insulin-like Growth Factor (IGF)
Abstract
IGF2 is an autocrine ligand for the beta cell IGF1R receptor and GLP-1 increases the activity of this autocrine loop by enhancing IGF1R expression, a mechanism that mediates the trophic effects of GLP-1 on beta cell mass and function. Here, we investigated the regulation of IGF2 biosynthesis and secretion. We showed that glutamine rapidly and strongly induced IGF2 mRNA translation using reporter constructs transduced in MIN6 cells and primary islet cells. This was followed by rapid secretion of IGF2 via the regulated pathway, as revealed by the presence of mature IGF2 in insulin granule fractions and by inhibition of secretion by nimodipine and diazoxide. When maximally stimulated by glutamine, the amount of secreted IGF2 rapidly exceeded its initial intracellular pool and tolbutamide, and high K+ increased IGF2 secretion only marginally. This indicates that the intracellular pool of IGF2 is small and that sustained secretion requires de novo synthesis. The stimulatory effect of glutamine necessitates its metabolism but not mTOR activation. Finally, exposure of insulinomas or beta cells to glutamine induced Akt phosphorylation, an effect that was dependent on IGF2 secretion, and reduced cytokine-induced apoptosis. Thus, glutamine controls the activity of the beta cell IGF2/IGF1R autocrine loop by increasing the biosynthesis and secretion of IGF2. This autocrine loop can thus integrate changes in feeding and metabolic state to adapt beta cell mass and function.
Introduction
Preserving glucose homeostasis over a lifetime requires that beta cells adapt their number and secretion capacity in response to different diets as well as to variations in insulin sensitivity of peripheral tissues that occur in obesity, pregnancy, and aging (1). Defects of this beta cell compensatory response trigger the onset of diabetic hyperglycemia. Therefore, identifying molecular mechanisms controlling adult beta cell proliferation and glucose competence, i.e. the insulin secretion response to an increase in glucose concentration, should provide novel targets for the treatment of type 2 diabetes (2). Several pathways that control this beta cell plasticity have been described over the recent years. For instance, studies of mice with inactivation of genes involved in the insulin and IGF1 signaling pathways have revealed that the insulin receptor and insulin receptor substrate 2 are required for the compensatory increase in beta cell mass in insulin resistance conditions (3–6). Glucose metabolism also participates in the control of beta cell mass and function (7–10). This signaling pathway depends on glucose metabolism, and it is controlled by glucokinase, beta cell secretion activity (11), as well as glucose and Ca2+-induced calcineurin/NFAT signaling leading to an increase of insulin receptor substrate 2 expression (12–14).
The gluco-incretin hormones GLP-1 and glucose-dependent insulinotropic polypeptide, secreted by intestinal L- and K-cells, respectively, also control beta cell mass and function. These hormones bind to specific Gs protein-coupled receptors present at the beta cell surface, and most of their actions depend on the initial production of cAMP (15, 16) and signaling through β-arrestin (17–19). The proliferation effect of gluco-incretin hormones has been attributed to signaling through cAMP-regulated element binding protein-dependent activation of IRS-2 (20, 21) as well as to indirect activation by betacellulin of the EGF receptor (22). More recently, we showed that GLP-1 induces the proliferation of beta cells, increases their glucose competence, and protects them against apoptosis through the induction of IGF1 receptor expression and activation of the IGF1R/Akt signaling pathway. We further showed that activation of IGF1R3 intracellular signaling was dependent on the autocrine secretion of IGF2 (23, 24). These trophic actions of GLP-1 were indeed abolished by suppressing the expression of the IGF1R or of IGF2. Thus, an IGF2/IGF1R autocrine loop controls beta cell mass and function, and its activity is increased by GLP-1 through the induction of IGF1R expression.
Here, we investigated whether the expression and secretion of IGF2 can also be modulated to increase the activity of this autocrine loop. We show that glutamine increased IGF2 biosynthesis and secretion through the regulated pathway, a mechanism augmented by the presence of glucose. Moreover, we show that glutamine induces Akt phosphorylation, an effect strictly dependent on IGF2 secretion. Thus, the activity of the IGF2/IGF1R autocrine loop is also controlled through a glutamine-dependent increase in IGF2 biosynthesis and secretion.
MATERIALS AND METHODS
Reagents
l-glutamine, 100× amino acids mix (Invitrogen, catalog no. 11130-036; composed of 29 mm Arg, 5 mm Cys, 10 mm His, 20 mm Ile, 20 mm Leu, 20 mm Lys, 5 mm Met, 10 mm Phe, 20 mm Thr; 2.5 mm Trp, 10 mm Tyr, and 20 mm Val), diazoxide, nimodipine, cycloheximide, actinomycin, tolbutamide, 6-diazo-5-oxo-l-norleucine (DON), and rapamycin were purchased from Sigma. Radioimmunoassay kits for insulin were from Millipore, and mouse IGF2 enzyme-linked immunosorbent assays (ELISA) were purchased from R&D Systems.
Antibodies and shRNA
Antibodies were purchased from Sigma (actin, A2066); Abcam (Cambridge, UK; IGF2, ab9574; synaptophysin, ab52636); Cell Signaling (Danvers, MA; phospho-Akt (Ser-473), 4051), Biolabs (Allschwil, Switzerland; Akt, 9272). Knockdown of igf2 was performed by adenoviral transduction of igf2-specific or control shRNA, as described previously (23, 24).
MIN6 Cell Culture
MIN6 cells (25) were grown in Dulbecco's modified Eagle's medium containing 15% heat-inactivated FCS, 2 mm glutamine, and 50 μm β-mercaptoethanol and used between passages 20–30. For secretion experiments, 2 × 105 MIN6 cells were seeded in 12-well tissue culture plates, and for Western blot experiments, 2 × 106 cells were seeded in 60-mm tissue culture dishes and used 2 days later.
Cell Lysis and Western Blot Analysis
MIN6 cells in culture dishes were placed on ice, washed twice with phosphate buffer saline (PBS), and lysed in protein lysis buffer (50 mm Tris (pH 7.5) containing 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 10 mm β-glycerophosphate, 1 mm Na3VO4, 50 mm PMSF, 10 μg/ml aprotonin, leupeptin, and pepstatin). The lysates were centrifuged for 15 min at 13,000 rpm at 4 °C, and the supernatant was used for Western blot analysis (26). For detection of IGF2, the antibodies were diluted 1:2000, for actin, the antibodies were diluted 1:4000, and for synaptophysin, the antibodies were diluted 1:1000 in Tris-buffered saline with 0.1% Tween 20 and 5% skim milk and revealed using horseradish peroxidase-conjugated donkey anti-rabbit IgG or horseradish peroxidase-conjugated sheep anti-mouse IgG as secondary antibodies Amersham Biosciences. The band intensities were determined using a Bio-Rad densitometer (Strasbourg, France).
Secretion Tests
MIN6 cells in tissue culture dishes were washed with PBS and preincubated for 2 h at 37 °C in Krebs-Ringer bicarbonate HEPES buffer (120 mm NaCl, 4 mm KH2PO4, 20 mm HEPES, 1 mm MgCl2, 1 mm CaCl2, 5 mm NaHCO3, and 0.5% BSA, pH7.4) supplemented with 2 mm glucose. The medium was then replaced with fresh KRBH-BSA solution containing 2 or 20 mm glucose, in the presence or absence of 2 mm l-glutamine, or 1× amino acids mix (Invitrogen, 11130-036), along with various reagents (cycloheximide (10 μg/ml), actinomycin (1 μg/ml), diazoxide (200 μm), nimodipine (1 μm), tolbutamide (200 μm), KCl (30 mm), rapamycin (100 nm)) for the indicated periods of time. Secreted and cellular insulin and IGF2 levels were assessed by radioimmunoassay and ELISA, respectively.
Subcellular Fractionation
Twelve 10-cm tissue culture dishes were seeded with 2.5 × 106 MIN6 cells and cultured for 3 days. The cells were then detached using a rubber policeman, pelleted by centrifugation at 1000 rpm, and resuspended in 2 ml of a solution consisting of 0.25 m sucrose, 1 mm EDTA, 10 mm Tris-HCL, pH 7.5, and 1 mm PMSF. Cells were homogenized by 100 strokes of a Dounce homogenizer, the homogenate was centrifuged at 1500 rpm, the pellet was discarded, and the supernatant was loaded on top of a continuous 20 to 50% sucrose gradient containing 5 mm Hepes. After centrifugation at 4 °C for 18 h at 30,000 rpm in a SW40Ti rotor Beckman (Brea, CA) 12 1 ml-fractions were collected, and the protein and sucrose concentrations were measured. The insulin content of each fraction was measured by radioimmunoassay. A volume of 800 μl of each fraction was then ultracentrifuged at 4 °C for 15 min at 55,000 rpm in a T55LA rotor (Beckman). The pellets were resuspended in protein lysis buffer, and IGF2 and synaptophysin were detected by Western blot analysis as described (27).
Quantitative PCR Analysis
Total RNA was extracted using RNeasy Plus Micro Kit from Qiagen, and cDNAs were synthesized using SuperScript II RNase H reverse transcriptase (Invitrogen), and 50 pmol of random hexamers (Applied Biosystems, Basel, Switzerland). Real time quantitative PCR was performed using a 7500 Fast Light Cycler technology Roche Applied Science (Basel, Switzerland). Amplification was performed in a 20-μl reaction mixture including 1× QuantiTectTM SYBR Green PCR Master Mix (Qiagen) and 10 pmol of each primer. Primers used for mouse IGF2 were as follows: 5′-GTCGATGTTGGTGCTTCTCA-3′ (forward) and 5′-AAGCAGCACTCTTCCACGAT-3′ (reverse). For the IGF2 leader sequences, L1, 5′-ACTTCAGCAGCTCCCACTTC-3′ (forward); L2, 5′-CGGCTTCCAGGTA-CCAATG-3′ (forward); L3, 5′-ACCTTCCAGCCTTTTCCTGT-3′ (forward). The same reverse primer for all of the leader sequences was used: 5′-TGAAGGCCTGCTGAAGTAGAA-3′ (reverse). Expression was normalized to TBP (5′-ATCCCAAGCGATTTGCTGC-3′ (forward) and 5′-ACTCTTGGCTCCTGTGCACA-3′ (reverse)). All primers were obtained from Microsynth (Balgach, Switzerland).
Translation Reporter Vectors and Cell Transfection
The igf2 leader sequences L1 (380 bp), L2 (1099 bp), and L3 (115 bp) (transcript ref. ENSMUST00000105936, ENSMUST00000121128, ENSMUST00000000033) were amplified by PCR using MIN6 cDNA as template with primers extended with a XhoI site and minimal promoter sequence at the 5′ end and a NcoI site at the 3′ end of the amplified sequences. After digestion with XhoI and NcoI, the leader sequences were cloned upstream of a destabilized form of luciferase in the pGL4.24 reporter vector (Promega, Madison, WI). The plasmids were transfected in MIN6 cells in 24-well plates using Lipofectamine 2000 (Invitrogen). pRL-TK vector, which contains a Renilla luciferase cDNA (Promega), was co-transfected to serve as a control of transfection efficiency.
Recombinant Lentiviruses
The three igf2 leader sequences were cloned into pLentI PGK V5-LUC puromycin vector from Abgene (Pittsburgh, PA). For subcloning, the minimal promoter from the pGL4.24 vectors containing the igf2 leader sequences was removed by HindIII and XhoI digestion and replaced with a BamHI site. The luciferase cassette in pLentI-LUC vector was excised by BamHI and XbaI digestion and replaced by the BamHI and XbaI digested leader (L1, L2, or L3)-Luc2P cassette from the pGL4.24-leader constructs to generate leader (L1, L2, or L3)-pLentI-Luc2P vectors. Lentiviruses were prepared by transient transfection of HEK-293T cells (22). Virus titers were determined by p24 ELISA from PerkinElmer Life Sciences (Schwerzenbach, Switzerland). Mouse islets from C57BL6 mice were dissociated by trypsinization (trypsin 5 mg/ml at 37 °C for 4 min) and incubated in RPMI medium containing 10% FCS, 2 mm glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin for 24 h and then transduced with lentiviral particles at multiplicity of infection of 20. Co-transduction with pLenti-III luciferase (Renilla reniformis) from Abmgood (Richmond, BC, Canada) was used to assess transduction efficiency.
Luciferase Assays
Luciferase activity was measured using the Dual-Luciferase Assay System (Promega, Madison, WI) as per the manufacturer's instructions. Briefly, after transfection or transduction, cells were incubated at 37 °C in a CO2 incubator for 48 h. The cells were then washed with PBS and preincubated for 2 h in KRBH-BSA buffer supplemented with 5 mm glucose. The medium was then replaced with fresh KRBH-BSA solution containing 5 mm glucose or 20 mm glucose in presence or absence of 2 mm l-glutamine for 3 h. Cells were then lysed in the buffer provided by the manufacturer and mixed on a shaker for 15 min at room temperature. The firefly and Renilla luciferase activities of the cell lysates were measured on a Glomax-96 microplate luminometer (Promega, Madison, WI, USA).
Apoptosis
Apoptosis assays were performed as described (24). Briefly, 20 islets were plated on tissue culture dishes coated with an extracellular matrix derived from bovine corneal endothelial cells (Novamed; Jerusalem, Israel) and kept in tissue culture for 7 days to form monolayers. Medium from islets monolayers was replaced with fresh RPMI medium with or without 2 mm glutamine for 24 h, and a cytokine mixture (25 ng/ml TNF-α, 10 ng/ml IL-1β, and 10 ng/ml IFN-γ) was added 16 h before the end of the incubation. At the end of the treatment, apoptosis was assessed by TUNEL (In situ cell death detection kit, catalog no. 11 684 795 910, Roche Applied Science). For each condition, >2500 cells were counted in randomly selected fields.
Statistical Analysis
All experiments were performed at least three times. Results are expressed as means ± S.D. Comparisons were performed using unpaired Student's t test or one-way or two-way analysis of variance for the different groups followed by Tukey's or Bonferroni test, respectively. Single, double, and triple asterisks indicate statistically significant differences (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
RESULTS
Glutamine Stimulates IGF2 Secretion
We measured IGF2 secretion from MIN6 cells exposed to 2 or 20 mm glucose and in the presence of a mixture of amino acids or of 2 mm glutamine. Secretion was measured at 1, 3, and 6 h and expressed as absolute quantities (Fig. 1A) or as percent of intracellular content (Fig. 1B). Secretion was very low in the presence of 2 mm glucose and increased over time in the presence of 20 mm glucose. The addition of glutamine markedly increased IGF2 secretion an effect that was amplified by the presence of 20 mm glucose. In contrast, addition of an amino acid mixture without glutamine had no stimulatory effect. When expressed as percent of intracellular content (Fig. 1B, and see Figs. 3, 4, and 7) secreted IGF2 rapidly represented >10-fold the initial intracellular content (intracellular IGF2 levels at time t = 0), indicating that IGF2 biosynthesis must be activated to sustain this secretion rate.
FIGURE 1.
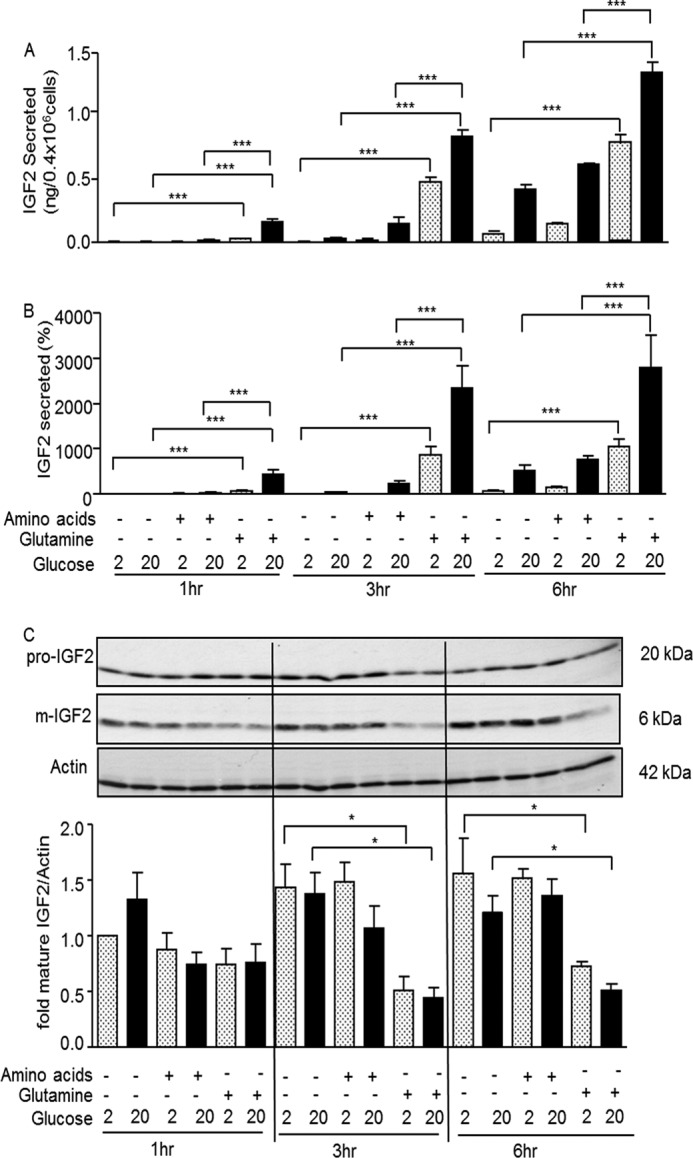
Glutamine stimulates IGF2 secretion. MIN6 cells were exposed for the indicated periods of time to 2 or 20 mm glucose in the presence of an amino acid mixture without glutamine or with 2 mm glutamine, as indicated. A, amounts (ng) of IGF2 secreted in each condition. B, secreted IGF2 expressed as percent of initial intracellular content. C, top: Western blot analysis of intracellular pro-IGF2, mature IGF2, and actin in the different experimental conditions. Bottom: quantification of the Western blot data. The data are the means ± S.D. from three independent experiments.*, p < 0.05; ***, p < 0.001.
FIGURE 3.

Glutamine-induced IGF2 secretion is controlled at the translational level. MIN6 cells were treated as indicated in the presence of 2 or 20 mm glucose, 2 mm glutamine, and in the presence of cycloheximide (CHX) or actinomycin A. Intracellular and secreted IGF2 were then determined. A, the secretion of IGF2 stimulated by glutamine is suppressed in the presence of cycloheximide. B, the presence of cylcoheximide suppresses the intracellular levels of pro-IGF2 and mature IGF2 in glutamine-stimulated cells. C, actinomycin A did not significantly reduce glutamine-stimulated IGF2 secretion. D, the Igf2 mRNA levels were not reduced by actinomycin A in the experimental conditions used for these experiments. The data are the means ± S.D. from three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
FIGURE 4.

Glutamine-induced IGF2 secretion requires glutamine metabolism but not mTOR activation. MIN6 cells were incubated for 3 h in the presence of the indicated concentrations of glucose and glutamine, and in presence of the glutaminase inhibitor DON or of rapamycin. A, glutamine-induced IGF2 secretion is dose-dependently inhibited by DON. B, rapamycin did not impact glutamine-induced IGF2 secretion. C, Western blot detection of phospho-4E-BP1 in the presence of glucose (mm), glutamine (2 mm), and rapamycin (100 nm). The data are the means ± S.D. from three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001. DMSO, dimethyl sulfoxide.
FIGURE 7.

IGF2 secretion is blocked by the insulin secretion inhibitors diazoxide and nimodipine. A, glutamine-induced IGF2 secretion by MIN6 cells in the indicated conditions was blocked by the addition of diazoxide or nimodipine. B, insulin secretion by the same cells used in A was similarly blocked by diazoxide or nimodipine. C, Western blot analysis of the intracellular content of pro-IGF2 and mature IGF2. The insulin secretion inhibitors blocked IGF2 secretion and lead to its intracellular accumulation. The data are the means ± S.D. from three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
The intracellular content of pro-IGF2 and mature IGF2 was measured by Western blot analysis in the same conditions as described above (Fig. 1C). The level of pro-IGF2 did not change in the different experimental conditions, whereas mature IGF2 levels were significantly decreased when secretion was stimulated by glutamine. This suggests that glutamine and glucose also increase the rate of IGF2 secretion in addition to stimulating its biosynthesis.
As a control, we measured insulin secretion in the same experimental conditions. Fig. 2, A and B, shows that secretion of insulin was markedly increased by glucose in a time-dependent manner and was also significantly increased in the presence of glutamine and amino acids. However, in contrast to the secretion of IGF2, the amount of secreted insulin did not exceed its initial intracellular content, in line with the known large intracellular insulin store of MIN6 cells.
FIGURE 2.
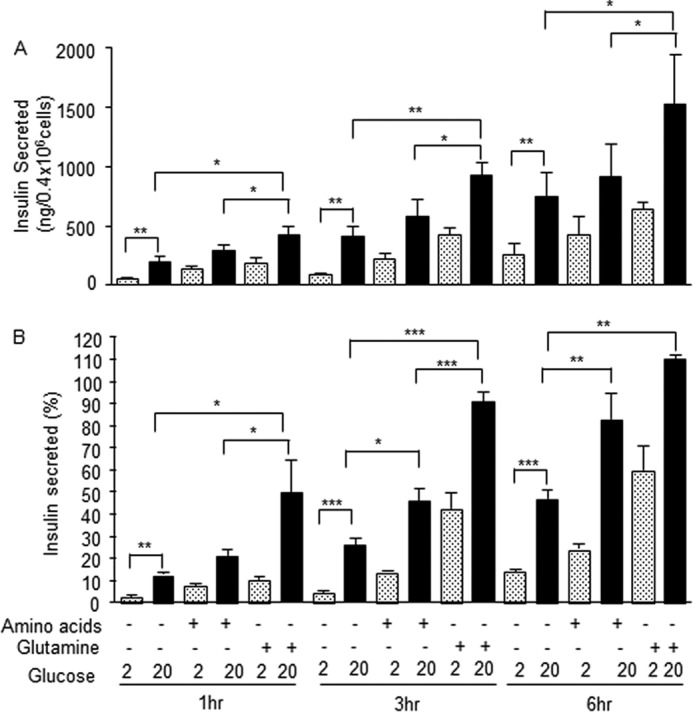
Glutamine induction of insulin secretion. MIN6 cells were treated as in Fig. 1, and insulin levels were determined by radioimmunoassay. A, amounts (ng) of insulin in each condition. B, secreted insulin expressed as percent of intracellular content. The data are the means ± S.D. from three independent experiments.*, p < 0.05; **, p < 0.01; ***, p < 0.001.
Translational Control of IGF2 Biosynthesis in MIN6 and Primary Islet Cells
To confirm that IGF2 secretion was dependent on ongoing protein synthesis, we measured secreted IGF2 from cells exposed to 2 and 20 mm glucose and in the presence of glutamine and cycloheximide. Fig. 3A shows that the time-dependent increase in IGF2 secretion stimulated by glucose and glutamine was suppressed in the presence of cycloheximide. Western blot analysis of intracellular pro- and mature IGF2 showed that both forms of IGF2 were markedly reduced in the presence of cycloheximide (Fig. 3B). Inhibition of transcription by actinomycin A did not affect the secretion of IGF2 stimulated by glutamine (Fig. 3C), and the IGF2 mRNA levels remained stable under the different stimulatory conditions (Fig. 3D).
To determine whether glutamine metabolism was required for induction of IGF2 secretion, we measured secreted IGF2 in the presence or absence of the glutaminase inhibitor DON. Fig. 4A shows a dose-dependent inhibition of IGF2 secretion by DON. Furthermore, in similar incubation conditions the mTOR inhibitors rapamycin (Fig. 4B) and pp224 (data not shown) did not inhibit IGF2 secretion. In the presence of rapamycin, the total amounts of IGF2 secreted over 3 h were 1.54 ± 0.19, 1.62 ± 0.06, 1.58 ± 0.2 ng (means ± S.D.) for control, dimethyl sulfoxide, and rapamycin-treated cells, respectively. The efficacy of rapamycin treatment was ascertained by measuring the inhibition of glutamine-induced 4E-BP1 phosphorylation (Fig. 4C).
Transcription of the IGF2 gene can be initiated at three distinct promoters, generating mRNAs with distinct leader sequences (Fig. 5A). Using quantitative RT-PCR analysis, we identified igf2 mRNA species present in MIN6 cells and primary beta cells (igf2 is not detected in non-beta cells (28)). Fig. 5, B and C, shows that MIN6 cells expressed the three mRNA species with the L2 mRNA being the most abundant one. In primary islets, there is a marked predominance of the L2 mRNA.
FIGURE 5.
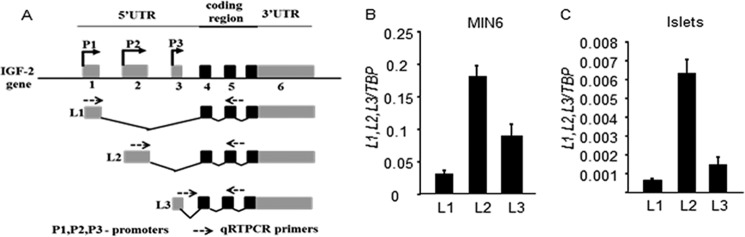
Differential usage of igf2 leader sequences in MIN6 cells and mouse islets. A, schematic drawing of the mouse igf2 gene; gray, noncoding exons; black, coding exons. The three promoters (P1–3) controlling expression of the leader sequences L1–3 are indicated as well as the primers used for quantitative RT-PCR analysis. Shown is a quantitative analysis of the expression of each leader-containing igf2 mRNA in MIN6 cells (B) and in primary mouse islets (C). The data are the means ± S.D. from three independent experiments.
To determine which leader sequence is required for glucose and glutamine translational control in MIN6 cells and primary islets cells, we generated reporter constructs containing the L1, L2, and L3 sequences subcloned between the minimal promoter and the destabilized firefly luciferase gene present in the pGL4.24 vector (Fig. 6A). These plasmids were co-transfected in MIN6 cells with a plasmid encoding Renilla luciferase to allow normalization of firefly luciferase activity. The data of Fig. 6B show that luciferase activity expressed from the three reporter constructs were not different in the presence of 5 or 20 mm glucose. However, addition of glutamine in the presence of either glucose concentration increased luciferase activity from each reporter construct. We furthermore showed that the effect of glutamine was suppressed by the glutaminase inhibitor DON but was insensitive to mTOR inhibition by rapamycin (Fig. 6C). To determine whether the same leader sequences were also subject to translational control by glutamine in primary islets, the reporter constructs (Fig. 6A) were subcloned in a lentiviral vector. Recombinant lentiviruses were then used to co-transduce primary islets with a lentivirus encoding Renilla luciferase. Fig. 6D shows that glutamine robustly induced luciferase activity from the L2 leader-containing reporter construct, followed by L3, and with no significant effect on the L1-containing reporter construct.
FIGURE 6.

Glutamine stimulates translation of igf2 mRNA mostly from the L2 leader sequence. A, schematic drawing of the translational reporter constructs containing the leader sequences subcloned upstream of the destabilized luciferase of the pGL4.24 vector. B, normalized luciferase activities were determined in lysates of transfected in MIN6 cells incubated for 3 h in the indicated incubation conditions. C, normalized luciferase activities were measured in cells exposed to glucose (Gluc) and glutamine as indicated and in the presence of DON or rapamycin. D, the Igf2 leader sequences were subcloned in a luciferase reporter lentivector, and the recombinant lentiviruses were used to co-infect dispersed mouse islets cells with a Renilla luciferase encoding lentivirus. Relative luciferase activity was measured after incubation for 3 h in the indicated conditions. The activities were expressed relative to the normalized luciferase measured in 5 mm glucose. The data are the means ± S.D. from three independent experiments. *, p < 0.05; **p < 0.01; ***, p < 0.001; RLU, relative light units; w.r.t., fold induction over basal activity.
Collectively, the above data show that IGF2 biosynthesis is regulated at the translational level by glutamine in MIN6 cells and in primary islet cells. The glutamine effect is blocked by glutaminase inhibition but is independent of mTOR activity. Finally, in islet cells, the L2 mRNA is the predominant species.
IGF2 Is Secreted through the Regulated Secretory Pathway
To determine whether IGF2 secretion occurs through the regulated pathway, we first assessed the effect of the secretion inhibitors diazoxide and nimodipine. The secretion of IGF2 induced by glucose plus glutamine was almost completely blocked by either inhibitor at all of the time points studied (Fig. 7A); a similar inhibition of insulin secretion was observed in all conditions (Fig. 7B). Western blot analysis showed that intracellular pro-IGF2 levels were identical in all experimental conditions (Fig. 7C). Intracellular levels of mature IGF2 decreased in the maximal stimulatory conditions but were increased in the presence of the secretion inhibitors.
Next, we performed subcellular fractionation of MIN6 cells on sucrose density gradients in conditions that separate insulin granules from synaptic-like vesicles (29). Fig. 8A shows the sucrose and protein concentrations in each collected fractions. Fig. 8, B and C, show the distribution of insulin, mature IGF2, pro-IGF2, and synaptophysin in the gradient fractions. The insulin peak was clearly separated from the low density synaptic-like vesicles, and the data showed association of mature IGF2 with the insulin-containing fractions.
FIGURE 8.
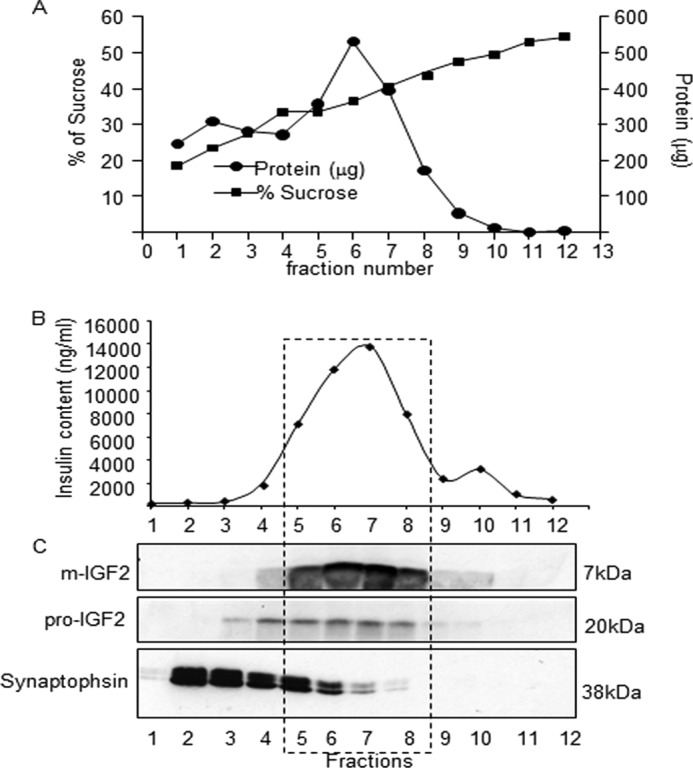
IGF2 co-localizes with the insulin secretory granules. A, subcellular fractionation of MIN6 cells homogenates by ultracentrifugation on a 20–50% sucrose density gradient. Each gradient fraction was analyzed for protein and sucrose concentration. B, insulin content of each fraction. C, Western blot immunodetection of mature IGF2, pro-IGF2, and synaptophysin in each gradient fraction. The mature form of IGF2 co-localizes with the insulin granule fractions.
Finally, we compared the effect of tolbutamide and high external K+ concentrations on the time-dependent induction of IGF2 and insulin secretion in the presence of 3 mm glucose. Fig. 9, A and B, shows that tolbutamide and 30 mm KCl induce a time-dependent increase in insulin but not in IGF2 secretion, in agreement with the presence of a very low intracellular pool of IGF2.
FIGURE 9.
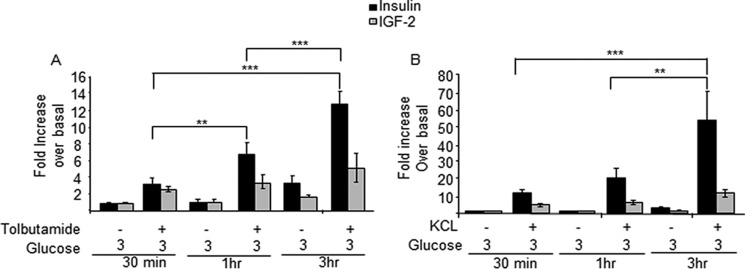
Sulfonylureas and high K+ induce insulin but not IGF2 secretion. MIN6 cells were incubated for the indicated periods of time with 3 mm glucose and in the presence of tolbutamide (A) or 30 mm KCl (B). Secreted IGF2 and insulin were then determined. Secretion is calculated as percent of intracellular content and reported in the figure as fold increase over basal (3 mm). Tolbutamide and KCl increase insulin but not IGF2 secretion, in agreement with the high intracellular store of insulin and the requirement of stimulated biosynthesis, and low intracellular IGF2 pools. The data are the means ± S.D. from three independent experiments. **, p < 0.01; ***, p < 0.001.
Glutamine Induces IGF2-dependent Activation of Akt Signaling in MIN6 Cells and Primary Islets
To determine whether glutamine-induced IGF2 expression can cooperate with GLP-1 to stimulate IGF1R intracellular signaling, we measured Akt phosphorylation in MIN6 cells transduced with an adenovirus expressing a control or an igf2-specific shRNA and treated with glutamine for 3 h. Fig. 10A shows that glutamine increases Akt phosphorylation, an effect that is markedly reduced by knocking down IGF2 expression. Fig. 10, B and C, shows a similar experiment performed with primary mouse islets. To obtain a significant induction of Akt phosphorylation by glutamine, mouse islets had to be first incubated in the presence of exendin-4 for 18 h, a condition that increases IGF-1R expression (24). A marked induction of Akt phosphorylation was then observed following glutamine treatment for 3 h. When the same experiment was performed with islets from mice with beta cell-specific inactivation of igf2,4 the induction of Akt phosphorylation by glutamine was completely suppressed. Finally, we tested whether glutamine could protect islet cells against cytokine-induced apoptosis. As shown in Fig. 10D, incubation of islet cells in the presence of high glutamine concentration significantly reduced the percent of TUNEL-positive beta cells, an effect not seen in islets lacking igf2 expression.
FIGURE 10.

Glutamine-induced IGF2 secretion leads to enhanced Akt signaling and protection against apoptosis. A, MIN6 cells were transduced with igf2-specific or unrelated (luc) shRNAs expressing adenoviruses. 48 h later, the cells were preincubated for 2 h in 2 mm glucose then for 3 h in 2 mm glucose with or without 2 mm glutamine. Western blot analysis of Pi-Akt, total Akt, pro-IGF2, and actin is shown on the left panel. Right panel: quantification of Pi-Akt/Akt ratio. B, islets from control (Ctrl) and beta cell-specific Igf2KO mice (βIgf2KO) were pretreated with exendin-4 for 18 h to increase IGF1R expression and then incubated as the MIN6 cells were described in A). Left: Western blot analysis of Pi-Akt, Akt, and actin. Right: quantification of the results. C, quantitative PCR analysis of Igf2 mRNA expression in islets from control and βIgf2KO mice. D, apoptosis measured in islet cells from control (Ctrl) and beta cell-specific Igf2KO mice (βIgf2KO) exposed, as indicated, to cytokines and glutamine for 16 h before TUNEL analysis. The data are the means ± S.D. from three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
DISCUSSION
Here, we show that igf2 mRNA translation is induced by glutamine and that secretion of mature IGF2 is rapidly and maximally stimulated when both glutamine and glucose concentrations are high. Secretion of IGF2 occurs through the regulated secretory pathway but, in contrast to insulin, the intracellular pool of IGF2 is not sufficient to ensure sustained secretion and requires continuous biosynthesis of the hormone. Furthermore, glutamine-induced IGF2 secretion leads to increased Akt signaling. Thus, nutrient-regulated induction of IGF2 secretion by beta cells contributes to the activation of the IGF2/IGF1R autocrine loop that regulates beta cell mass and function.
The effect of glutamine on IGF2 expression is mostly at the translational level and in MIN6 cells this induction is observed with each of the three leader sequence-containing reporter constructs. In mouse islets, the L2-containing igf2 mRNA, which is the predominant transcript, is also the most induced by glutamine. The mechanism by which glutamine stimulates IGF2 biosynthesis is not yet understood. However, glutamine metabolism is required because inhibition of glutaminase, which catalyzes the first step in glutamine utilization, prevents the glutamine effect. Recent studies have shown that glutamine can activate mTOR (30) and that, in HEK293 cells, mTOR induces IGF2 translation by phosphorylating IMP2 (also called IGF2BP2), a mRNA binding protein that binds to the L2 leader sequence of igf2 mRNA to enhance its translation (31); IGF2BP2 is also a diabetes susceptibility gene identified in genome-wide association studies (32, 33). This mechanism is, however, unlikely to operate in beta cells because the mTOR inhibitors rapamycin and pp242 failed to reduce glutamine-induced IGF2 biosynthesis and secretion. An alternative possibility is that glutamine metabolism and the production of Kreb's cycle intermediates may increase IGF2 biosynthesis by a mechanism analogous to that used by glucose to stimulate insulin biosynthesis, which depends on mitochondrial metabolites to induce the binding of a regulatory protein to the 5′-untranslated region of the insulin mRNA (34–36). However, whether a similar metabolic signaling pathway is involved in the glutamine effect needs to be further investigated.
Two striking features of IGF2 secretion are, first, that it depends on its continuous biosynthesis and, second, that it is secreted through the regulated pathway. Indeed, when maximally stimulated by glutamine, secreted IGF2 accumulates in the extracellular medium at levels that rapidly exceeds by several folds its intracellular content. In addition, inhibition of translation by cycloheximide suppresses IGF2 secretion at all time points tested. Thus, the intracellular pool of IGF2 is insufficient to sustain significant secretion in the absence of neosynthesis. The fact that secretion occurs through the regulated pathway is indicated by the co-localization of mature IGF2 with insulin granules as determined by subcellular fractionation experiments. This is in agreement with previous reports that found, by immunoelectron microscopy (37, 38) and biochemical methods (39), that IGF2 is localized in insulin granules. This is also consistent with our observation that the insulin secretion inhibitors nimodipine and diazoxide block IGF2 secretion leading to its accumulation in the cells. The fact that tolbutamide or high extracellular K+, which induce marked secretion of insulin, do not significantly increase IGF2 secretion also confirms that the intracellular store of IGF2 is very small.
The above observations show that the biosynthesis and release of IGF2 are acutely modulated and that the latter proceeds through the regulated secretory pathway. This ensures correct processing of pro-IGF2 into mature IGF2 by the proconvertases present in insulin granules. Studies of the dynamics of insulin granule exocytosis have shown that the last granules to be synthesized are the first to be secreted (40, 41). Our data on the kinetics of IGF2 biosynthesis and secretion suggest that this mechanism is of particular interest for the secretion of an autocrine regulator of beta cell mass and function, which should be rapidly responsive to changes in the metabolic environment. This mechanism would be well suited to constantly adapt the insulin secretion capacity of the endocrine pancreas to feeding and insulin resistance conditions.
In this context, it is interesting to note that recent publications (42–44), analyzing three different human cohorts, have found that glutamine plasma levels are inversely correlated with insulin resistance and risks of type 2 diabetes. Furthermore, experimental studies showed that glutamine administration in mice result in increased glucose tolerance (42). Thus, in addition to the well known effect of glutamine on glucose-stimulated insulin secretion (45–47) and the recent demonstration that glutamine can induce GLP-1 secretion (48, 49), our data show that glutamine may also have a beneficial impact on beta cells via the activation of the IGF2/IGF1R autocrine loop. This is supported by the fact that both in MIN6 cells and primary islets, glutamine supplementation induced Akt phosphorylation through an IGF2-dependent mechanism and reduces cytokine-induced apoptosis.
Another observation of potential clinical relevance is that, whereas tolbutamide induces robust insulin secretion, no such effect is observed on IGF2 secretion. The therapeutic effect of sulfonylureas is known to subside over time due to beta cell exhaustion (50). The lack of induction of IGF2 secretion by sulfonylureas may contribute to the beta cell demise when this treatment is used, and this negative effect may be further enhanced by the reduced plasma glutamine levels in people with insulin resistance, as noted above (42).
The studies presented here have been obtained mostly with MIN6 cells because the level of expression of IGF2 in mouse islets is very low and below the detection limit of the ELISA. Thus, it was not possible to perform a detailed analysis of the kinetics of IGF2 secretion in mouse islets. However, we previously demonstrated that IGF2 is produced by primary islet beta cells (24) and that in mouse islets IGF2 enhances glucose-stimulated insulin secretion and is required for the trophic effects of GLP-1 on apoptosis and proliferation (23, 24). Here, we further demonstrate that in primary islet cells, glutamine increases the level of igf2 mRNA reporter constructs. In addition, using islets from mice with a beta cell-specific inactivation of igf2, we showed that glutamine-induced Akt phosphorylation and protection against cytokine-induced apoptosis depend on igf2 expression. There is thus strong evidence for a similar regulation of IGF2 biosynthesis and secretion as well as for the autocrine effect of this hormone in MIN6 cells and primary beta cells.
Collectively, our data show that biosynthesis of IGF2, an autocrine regulator of beta cell mass and function, is controlled by glutamine. This is rapidly followed by secretion through the regulated secretory pathway. Furthermore, glutamine activates Akt phosphorylation and protects against cytokine-induced apoptosis, responses that depend on IGF2 secretion. Thus, the activity of the IGF2/IGF1R autocrine loop can be up-regulated both by GLP-1, which increases IGF-1R expression, and by glutamine, which stimulates the secretion of IGF2. This autocrine loop may thus play an important role in the adaptation of beta cell mass and function in response to food intake and changes in metabolic state of the internal milieu.
Acknowledgments
The expert technical help of Wanda Dolci and Guy Niederhauser is gratefully acknowledged. We thank Dr. Romano Regazzi for careful reading of the manuscript.
This work was supported by Innovative Medicine Initiative Joint Undertaking Grant 155005 (IMIDIA), resources of which are composed of financial contributions from the European Union's Sevenths Framework Programme (FP7/2007-2013) and EFPIA companies in kind contribution; Swiss National Science Foundation (3100A0B-128657), and NCCR Frontiers in Genetics; European Union Seventh Framework Program Integrated Project BetaBat; and European Research Council Advanced Grant INSIGHT.
H. Modi and B. Thorens, unpublished data.
- IGF1R
- IGF1 receptor
- DON
- 6-diazo-5-oxo-l-norleucine
- GLP1
- glucagon-like peptide 1.
REFERENCES
- 1. Prentki M., Nolan C. J. (2006) Islet beta cell failure in type 2 diabetes. J. Clin. Invest. 116, 1802–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thorens B. (2013) The required beta cell research for improving treatment of type 2 diabetes. J. Int. Med. 274, 203–214 [DOI] [PubMed] [Google Scholar]
- 3. Okada T., Liew C. W., Hu J., Hinault C., Michael M. D., Krtzfeldt J., Yin C., Holzenberger M., Stoffel M., Kulkarni R. N. (2007) Insulin receptors in beta cells are critical for islet compensatory growth response to insulin resistance. Proc. Natl. Acad. Sci. U.S.A. 104, 8977–8982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ueki K., Okada T., Hu J., Liew C. W., Assmann A., Dahlgren G. M., Peters J. L., Shackman J. G., Zhang M., Artner I., Satin L. S., Stein R., Holzenberger M., Kennedy R. T., Kahn C. R., Kulkarni R. N. (2006) Total insulin and IGF-I resistance in pancreatic beta cells causes overt diabetes. Nat. Genet. 38, 583–588 [DOI] [PubMed] [Google Scholar]
- 5. Kulkarni R. N. (2005) New insights into the roles of insulin/IGF-I in the development and maintenance of beta cell mass. Rev. Endocr. Metab. Disord. 6, 199–210 [DOI] [PubMed] [Google Scholar]
- 6. Withers D. J., Gutierrez J. S., Towery H., Burks D. J., Ren J. M., Previs S., Zhang Y., Bernal D., Pons S., Shulman G. I., Bonner-Weir S., White M. F. (1998) Disruption of IRS-2 causes type 2 diabetes in mice. Nature 391, 900–904 [DOI] [PubMed] [Google Scholar]
- 7. Alonso L. C., Yokoe T., Zhang P., Scott D. K., Kim S. K., O'Donnell C. P., Garcia-Ocaña A. (2007) Glucose infusion in mice: a new model to induce beta-cell replication. Diabetes 56, 1792–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Susini S., Roche E., Prentki M., Schlegel W. (1998) Glucose and glucoincretin peptides synergize to induce c-fos, c-jun, junB, zif-268, and nur-77 gene expression in pancreatic beta(INS-1) cells. FASEB J. 12, 1173–1182 [PubMed] [Google Scholar]
- 9. Swenne I. (1982) The role of glucose in the in vitro regulation of cell cycle kinetics and proliferation of fetal pancreatic B-cells. Diabetes 31, 754–760 [DOI] [PubMed] [Google Scholar]
- 10. Scharfmann R., Basmaciogullari A., Czernichow P. (1990) Effect of growth hormone and glucose on rat islet cells replication using 5-bromo-2-deoxyuridine incorporation. Diabetes Res. 15, 137–141 [PubMed] [Google Scholar]
- 11. Porat S., Weinberg-Corem N., Tornovsky-Babaey S., Schyr-Ben-Haroush R., Hija A., Stolovich-Rain M., Dadon D., Granot Z., Ben-Hur V., White P., Girard C. A., Karni R., Kaestner K. H., Ashcroft F. M., Magnuson M. A., Saada A., Grimsby J., Glaser B., Dor Y. (2011) Control of pancreatic beta cell regeneration by glucose metabolism. Cell Metab. 13, 440–449 [DOI] [PubMed] [Google Scholar]
- 12. Heit J. J., Apelqvist A. A., Gu X., Winslow M. M., Neilson J. R., Crabtree G. R., Kim S. K. (2006) Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature 443, 345–349 [DOI] [PubMed] [Google Scholar]
- 13. Lingohr M. K., Briaud I., Dickson L. M., McCuaig J. F., Alárcon C., Wicksteed B. L., Rhodes C. J. (2006) Specific regulation of IRS-2 expression by glucose in rat primary pancreatic islet beta-cells. J. Biol. Chem. 281, 15884–15892 [DOI] [PubMed] [Google Scholar]
- 14. Demozay D., Tsunekawa S., Briaud I., Shah R., Rhodes C. J. (2011) Specific glucose-induced control of insulin receptor substrate-2 expression is mediated via Ca2+-dependent calcineurin/NFAT signaling in primary pancreatic islet beta-cells. Diabetes 60, 2892–2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thorens B. (1992) Expression cloning of the pancreatic beta cell receptor for the gluco-incretin hormone glucagon-like peptide I. Proc. Natl. Acad. Sci. U.S.A. 89, 8641–8645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Usdin T. B., Mezey E., Button D. C., Brownstein M. J., Bonner T. I. (1993) Gastric inhibitory polypeptide receptor, a member of the secretin-vasoactive intestinal peptide receptor family, is widely distributed in peripheral organs and the brain. Endocrinology 133, 2861–2870 [DOI] [PubMed] [Google Scholar]
- 17. Talbot J., Joly E., Prentki M., Buteau J. (2012) beta-Arrestin1-mediated recruitment of c-Src underlies the proliferative action of glucagon-like peptide-1 in pancreatic beta INS832/13 cells. Mol. Cell Endocrinol. 364, 65–70 [DOI] [PubMed] [Google Scholar]
- 18. Dalle S., Ravier M. A., Bertrand G. (2011) Emerging roles for beta-arrestin-1 in the control of the pancreatic beta-cell function and mass: new therapeutic strategies and consequences for drug screening. Cell Signal 23, 522–528 [DOI] [PubMed] [Google Scholar]
- 19. Jorgensen R., Martini L., Schwartz T. W., Elling C. E. (2005) Characterization of glucagon-like peptide-1 receptor beta-arrestin 2 interaction: a high-affinity receptor phenotype. Mol. Endocrinol. 19, 812–823 [DOI] [PubMed] [Google Scholar]
- 20. Park S., Dong X., Fisher T. L., Dunn S., Omer A. K., Weir G., White M. F. (2006) Exendin-4 uses Irs2 signaling to mediate pancreatic beta cell growth and function. J. Biol. Chem. 281, 1159–1168 [DOI] [PubMed] [Google Scholar]
- 21. Jhala U. S., Canettieri G., Screaton R. A., Kulkarni R. N., Krajewski S., Reed J., Walker J., Lin X., White M., Montminy M. (2003) cAMP promotes pancreatic beta-cells survival via CREB-mediated induction of IRS2. Genes Dev. 17, 1575–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Buteau J., Foisy S., Joly E., Prentki M. (2003) Glucagon-like peptide 1 induces pancreatic beta-cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes 52, 124–132 [DOI] [PubMed] [Google Scholar]
- 23. Cornu M., Modi H., Kawamori D., Kulkarni R. N., Joffraud M., Thorens B. (2010) Glucagon-like peptide-1 increases beta-cell glucose competence and proliferation by translational induction of insulin-like growth factor-1 receptor expression. J. Biol. Chem. 285, 10538–10545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cornu M., Yang J. Y., Jaccard E., Poussin C., Widmann C., Thorens B. (2009) Glucagon-like peptide-1 protects beta-cells against apoptosis by increasing the activity of an IGF-2/IGF-1 receptor autocrine loop. Diabetes 58, 1816–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Miyazaki J., Araki K., Yamato E., Ikegami H., Asano T., Shibasaki Y., Oka Y., Yamamura K. (1990) Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology 127, 126–132 [DOI] [PubMed] [Google Scholar]
- 26. Klinger S., Poussin C., Debril M. B., Dolci W., Halban P. A., Thorens B. (2008) Increasing GLP-1-induced beta-cell proliferation by silencing the negative regulators of signaling cAMP response element modulator-α and DUSP14. Diabetes 57, 584–593 [DOI] [PubMed] [Google Scholar]
- 27. Uldry M., Steiner P., Zurich M. G., Béguin P., Hirling H., Dolci W., Thorens B. (2004) Regulated exocytosis of an H+/myo-inositol symporter at synapses and growth cones. EMBO J. 23, 531–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cornu M., Thorens B. (2009) GLP-1 protects beta-cells against apoptosis by enhancing the activity of an IGF-2/IGF1-receptor autocrine loop. Islets 1, 280–282 [DOI] [PubMed] [Google Scholar]
- 29. Thorens B., Gérard N., Dériaz N. (1993) GLUT2 surface expression and intracellular transport via the constitutive pathway in pancreatic beta cells and insulinoma: evidence for a block in trans-Golgi network exit by brefeldin A. J. Cell Biol. 123, 1687–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nicklin P., Bergman P., Zhang B., Triantafellow E., Wang H., Nyfeler B., Yang H., Hild M., Kung C., Wilson C., Myer V. E., MacKeigan J. P., Porter J. A., Wang Y. K., Cantley L. C., Finan P. M., Murphy L. O. (2009) Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136, 521–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dai N., Rapley J., Angel M., Yanik M. F., Blower M. D., Avruch J. (2011) mTOR phosphorylates IMP2 to promote IGF2 mRNA translation by internal ribosomal entry. Genes Dev. 25, 1159–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Christiansen J., Kolte A., Hansen T., Nielsen F. (2009) IGF2 mRNA-binding protein 2 - biological function and putative role in type 2 diabetes. J. Mol. Endocrinol. [DOI] [PubMed] [Google Scholar]
- 33. Groenewoud M. J., Dekker J. M., Fritsche A., Reiling E., Nijpels G., Heine R. J., Maassen J. A., Machicao F., Schäfer S. A., Häring H. U., 't Hart L. M., van Haeften T. W. (2008) Variants of CDKAL1 and IGF2BP2 affect first-phase insulin secretion during hyperglycaemic clamps. Diabetologia 51, 1659–1663 [DOI] [PubMed] [Google Scholar]
- 34. Wicksteed B., Uchizono Y., Alarcon C., McCuaig J. F., Shalev A., Rhodes C. J. (2007) A cis-element in the 5′ untranslated region of the preproinsulin mRNA (ppIGE) is required for glucose regulation of proinsulin translation. Cell Metab. 5, 221–227 [DOI] [PubMed] [Google Scholar]
- 35. Alarcon C., Wicksteed B., Prentki M., Corkey B. E., Rhodes C. J. (2002) Succinate is a preferential metabolic stimulus-coupling signal for glucose-induced proinsulin biosynthesis translation. Diabetes 51, 2496–2504 [DOI] [PubMed] [Google Scholar]
- 36. Wicksteed B., Herbert T. P., Alarcon C., Lingohr M. K., Moss L. G., Rhodes C. J. (2001) Cooperativity between the preproinsulin mRNA untranslated regions is necessary for glucose-stimulated translation. J. Biol. Chem. 276, 22553–22558 [DOI] [PubMed] [Google Scholar]
- 37. Portela-Gomes G. M., Höög A. (2000) Insulin-like growth factor II in human fetal pancreas and its co-localization with the major islet hormones: comparison with adult pancreas. J. Endocrinol. 165, 245–251 [DOI] [PubMed] [Google Scholar]
- 38. Reinecke M., Broger I., Brun R., Zapf J., Maake C. (1995) Immunohistochemical localization of insulin-like growth factor I and II in the endocrine pancreas of birds, reptiles, and amphibia. Gen. Comp. Endocrinol. 100, 385–396 [DOI] [PubMed] [Google Scholar]
- 39. Buchanan C. M., Phillips A. R., Cooper G. J. (2001) Preptin derived from proinsulin-like growth factor II (proIGF-II) is secreted from pancreatic islet beta-cells and enhances insulin secretion. Biochem. J. 360, 431–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Halban P. A. (1982) Differential rates of release of newly synthesized and of stored insulin from pancreatic islets. Endocrinology 110, 1183–1188 [DOI] [PubMed] [Google Scholar]
- 41. Seino S., Shibasaki T., Minami K. (2011) Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J. Clin. Invest. 121, 2118–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cheng S., Rhee E. P., Larson M. G., Lewis G. D., McCabe E. L., Shen D., Palma M. J., Roberts L. D., Dejam A., Souza A. L., Deik A. A., Magnusson M., Fox C. S., O'Donnell C. J., Vasan R. S., Melander O., Clish C. B., Gerszten R. E., Wang T. J. (2012) Metabolite profiling identifies pathways associated with metabolic risk in humans. Circulation 125, 2222–2231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sookoian S., Pirola C. J. (2012) Alanine and aspartate aminotransferase and glutamine-cycling pathway: their roles in pathogenesis of metabolic syndrome. World J. Gastroenterol. 18, 3775–3781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stancáková A., Civelek M., Saleem N. K., Soininen P., Kangas A. J., Cederberg H., Paananen J., Pihlajamäki J., Bonnycastle L. L., Morken M. A., Boehnke M., Pajukanta P., Lusis A. J., Collins F. S., Kuusisto J., Ala-Korpela M., Laakso M. (2012) Hyperglycemia and a common variant of GCKR are associated with the levels of eight amino acids in 9,369 Finnish men. Diabetes 61, 1895–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vetterli L., Carobbio S., Pournourmohammadi S., Martin-Del-Rio R., Skytt D. M., Waagepetersen H. S., Tamarit-Rodriguez J., Maechler P. (2012) Delineation of glutamate pathways and secretory responses in pancreatic islets with beta-cell-specific abrogation of the glutamate dehydrogenase. Mol. Biol. Cell 23, 3851–3862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Odegaard M. L., Joseph J. W., Jensen M. V., Lu D., Ilkayeva O., Ronnebaum S. M., Becker T. C., Newgard C. B. (2010) The mitochondrial 2-oxoglutarate carrier is part of a metabolic pathway that mediates glucose- and glutamine-stimulated insulin secretion. J. Biol. Chem. 285, 16530–16537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Henquin J. C., Dufrane D., Nenquin M. (2006) Nutrient control of insulin secretion in isolated normal human islets. Diabetes 55, 3470–3477 [DOI] [PubMed] [Google Scholar]
- 48. Samocha-Bonet D., Wong O., Synnott E. L., Piyaratna N., Douglas A., Gribble F. M., Holst J. J., Chisholm D. J., Greenfield J. R. (2011) Glutamine reduces postprandial glycemia and augments the glucagon-like peptide-1 response in type 2 diabetes patients. J. Nutr. 141, 1233–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tolhurst G., Zheng Y., Parker H. E., Habib A. M., Reimann F., Gribble F. M. (2011) Glutamine triggers and potentiates glucagon-like peptide-1 secretion by raising cytosolic Ca2+ and cAMP. Endocrinology 152, 405–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Turner R. C., Cull C. A., Frighi V., Holman R. R. (1999) Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus: progressive requirement for multiple therapies (UKPDS 49). UK Prospective Diabetes Study (UKPDS) Group. JAMA 281, 2005–2012 [DOI] [PubMed] [Google Scholar]