

Background: Phosphoglucomutase 1 deficiency is a newly described inherited disorder in humans.
Results: In vitro studies of 13 missense variants associated with disease are presented.
Conclusion: Compromised catalysis and differential effects on recombinant protein folding/stability are found.
Significance: Biochemical defects are consistent with previous in vivo studies and may have clinical relevance.
Keywords: Enzyme Mutation, Genetic Disease, Kinetics, Metabolic Disease, N-linked Glycosylation, Protein Misfolding
Abstract
Recent studies have identified phosphoglucomutase 1 (PGM1) deficiency as an inherited metabolic disorder in humans. Affected patients show multiple disease phenotypes, including dilated cardiomyopathy, exercise intolerance, and hepatopathy, reflecting the central role of the enzyme in glucose metabolism. We present here the first in vitro biochemical characterization of 13 missense mutations involved in PGM1 deficiency. The biochemical phenotypes of the PGM1 mutants cluster into two groups: those with compromised catalysis and those with possible folding defects. Relative to the recombinant wild-type enzyme, certain missense mutants show greatly decreased expression of soluble protein and/or increased aggregation. In contrast, other missense variants are well behaved in solution, but show dramatic reductions in enzyme activity, with kcat/Km often <1.5% of wild-type. Modest changes in protein conformation and flexibility are also apparent in some of the catalytically impaired variants. In the case of the G291R mutant, severely compromised activity is linked to the inability of a key active site serine to be phosphorylated, a prerequisite for catalysis. Our results complement previous in vivo studies, which suggest that both protein misfolding and catalytic impairment may play a role in PGM1 deficiency.
Introduction
Human phosphoglucomutase 1 (PGM1)2 (EC 5.4.2.2) plays a central role in glucose homeostasis, mediating the switch between glycolysis and gluconeogenesis through the reversible conversion of glucose 1-phosphate and glucose 6-phosphate. Expressed in tissues throughout the body, PGM1 is also required for protein N-glycosylation (1, 2) as glucose 1-phosphate is a precursor for the formation of nucleotide sugars used in glycan biosynthesis. Recent studies have identified PGM1 deficiency as a hereditary genetic disorder, with characteristics of both a glycogen storage disease (GSDXIV, MIM 612934) and a congenital disorder of glycosylation of types I and II (1, 3, 4). Affected individuals show multiple clinical phenotypes (3), including hepatopathy, dilated cardiomyopathy, hypoglycemia, muscle weakness, exercise intolerance, growth retardation, and congenital malformations of the head such as cleft palate. Life threatening complications may also occur, as seen in four patients (ages 8–33 years) who suffered cardiac arrest (3).
PGM1 deficiency is autosomal recessive in inheritance, and associated with various types of mutations, including frame shifts, aberrant splicing, and missense variants (1–5). In the latter category, 13 different mutations were identified (3), affecting 12 residue positions in the protein. Analyses from patient-derived samples show that the mutations associated with PGM1 deficiency produce varying effects on the enzyme (3). Enzyme activity, measured from cell extracts, was reduced for all cases studied. Protein expression levels in fibroblasts, however, varied widely, with some of the mutant proteins expressing similarly to wild-type (WT) PGM1, whereas others had poor or no expression, despite mRNA levels ∼40% or more of controls (3). Although not definitive, this pattern is consistent with early degradation of certain mutant proteins by the proteasome due to misfolding. As has been shown for a growing number of metabolic disorders (6–9), the available in vivo data suggest that inherited PGM1 deficiency fits a “loss-of-function” phenotype that may include protein misfolding, in addition to conventional catalytic defects. Other possibilities, such as problems with protein oligomerization or cellular trafficking are unlikely, because PGM1 is a monomeric protein found in the cytosol of the cell.
Because of its fundamental importance in metabolism, the reaction catalyzed by human PGM1 has been appreciated for decades (10). Its reaction entails two consecutive phosphoryl transfers (Fig. 1A). The first takes place from phosphoserine 117 of the enzyme to substrate (α-d-glucose 1-phosphate or glucose 6-phosphate), to create the bisphosphorylated sugar intermediate, α-d-glucose 1,6-bisphosphate (Glc-1,6-P). The second phosphoryl transfer is from the intermediate back to the enzyme, creating product and regenerating the active state of the enzyme. The protein has 562 amino acids, and although its crystal structure is unknown, a high quality model is provided by the structure of PGM from rabbit (97% identity to human enzyme; Fig. 1B) (11). The large active site cleft of the protein contains key functional regions necessary, including the catalytic phosphoserine, a loop that binds a Mg2+ ion required for activity, as well as residues involved in contacting the sugar hydroxyls and phosphate group of the substrates (12–14).
FIGURE 1.

Overview of PGM1 mechanism and structure. A, schematic of the reversible reaction catalyzed by human PGM1. B, the PGM1 deficiency mutants highlighted on the crystal structure of rabbit PGM (97% sequence identity; Protein Data Bank code 3PMG). The catalytic phosphoserine residue (Ser-117) is shown in green, and the Mg2+ ion as an orange sphere. The 12-residue positions associated with disease are shown either in red (catalytic defects) or blue (apparent folding defects) as described in text. Positions affected by the three common functional polymorphisms of PGM1 (see “Results”) are shown in yellow.
To help shed light on the molecular defects associated with PGM1 deficiency, we conducted comprehensive, in vitro biochemical analyses of WT PGM1, three functional polymorphic variants (15, 16), and the 13 missense mutations associated with disease as reported in Ref. 3. Recombinantly expressed, affinity purified proteins were used to eliminate the complications present in in vivo studies, including heterozygosity, use of cell extracts from varying tissue types, and the presence of homologous enzymes (1, 3, 4, 17). Proteins were characterized for expression, solubility, conformational flexibility, and stability. We establish in this recombinant system that PGM1 missense variants segregate into two categories: those with severe catalytic defects and those that primarily appear to affect folding/solubility relative to the behavior of WT enzyme. The biochemical phenotypes are consistent with the three-dimensional structural context of the missense variants and available data from patient samples. This work provides an essential baseline for future in vivo and clinical studies of PGM1 variants associated with disease.
EXPERIMENTAL PROCEDURES
Materials
Leuconostoc mesenteroides glucose 6-phosphate dehydrogenase, α-d-glucose 1-phosphate, glucosamine 1-phosphate, and Glc-1,6-P were obtained from Sigma. Tritirachium album proteinase K was obtained from Fisher Scientific and 8-anilino-1-naphthalene sulfonate (ANS) from Acros Organics. Contaminants of Glc-1,6-P in the α-d-glucose 1-phosphate were removed by anion exchange column chromatography (18).
Protein Expression, Mutagenesis, and Purification
The gene for human PGM1 (GI:189926) was commercially synthesized (GenScript) and inserted into the pMCSG7 vector with N-terminal His6 affinity tag and tobacco etch virus protease site. PGM1 mutants were constructed using the QuikChange kit (Stratagene) and verified by automated DNA sequencing. For expression of WT PGM1 and mutants, Escherichia coli BL21(DE3) cells were transformed with corresponding plasmids.
For small scale expression tests, E. coli cultures were grown at 37 °C in 20 ml of LB media, supplemented with 0.1 mg/ml of ampicillin, to an A600 of 0.8–1.0. Prior to induction with isopropyl 1-thio-β-d-galactopyranoside (final concentration 0.2 mm), cultures were cooled at 4 °C for at least 30 min. Cells were induced for ∼17 h at 19 °C. At the end of induction, 1.5 ml of each cell culture was centrifuged, and pellets were resuspended in 200 μl of B-PER Protein Extraction Reagent (Thermo Scientific) and processed according to the manufacturer's instructions. Equal volumes of samples obtained from the soluble and insoluble fractions (10 μl each) were mixed with 10 μl of 2× SDS sample buffer, and 9 μl of this mixture loaded on a 10% SDS-PAGE gel for analysis. Gels were scanned using Universal Hood II and ChemiDoc XRS in the Quantity One software suite (Bio-Rad). The white light imaging protocol was employed, and corrected by the subtract background function. Constant area rectangles were used to select bands and densities determined using the volume analysis function (intensity/mm2). Data were plotted as a histogram using Excel 2010 (Microsoft Office).
For protein purification, 0.5–1.0-liter cell cultures were grown as described above, except that following completion of induction, cell pellets were flash frozen in liquid N2 and stored at −80 °C. Frozen cell pellets were resuspended in Buffer A (20 mm sodium phosphate, 0.3 m NaCl, pH 7.8) containing 14.4 mm β-mercaptoethanol, 0.5 mm phenylmethylsulfonyl fluoride (PMSF), 0.5 mm tosyllysine chloromethyl ketone, 2 mm CaCl2, 2 mm MgSO4, and 10 μg/ml of DNase. Cell lysis was performed with a French press, and the soluble fraction containing PGM1 was obtained through centrifugation. Protamine sulfate was added at 5 mg/g of cell pellet over 15 min, stirred for 30 min, and centrifuged. The supernatant was mixed with Ni2+ affinity resin (His-Select, Sigma), which had been previously equilibrated in Buffer A, and incubated for 30 min on a two-way orbital rocker. The mixture was transferred into a gravity-packed column and washed with Buffer A containing 5 mm imidazole, pH 7.8. Protein was eluted using Buffer A supplemented with 250 mm imidazole, pH 7.8. Purified proteins were dialyzed by a slow NaCl gradient into 50 mm MOPS, 1 mm MgCl2, 0.1 mm EDTA, pH 7.4, and then into the final buffer (50 mm MOPS, 1 mm MgCl2, pH 7.4).
Circular Dichroism Spectroscopy and Thermal Denaturation
Protein samples (7 μm) in 10 mm MOPS, pH 7.4, were analyzed at 25 °C in a 0.1-cm quartz cuvette with an Aviv 62DS spectrometer. Background subtraction was performed using buffer dialysate as the reference. Data were collected at 1-nm intervals from 200 to 250 nm and the signal averaged for 30 s. For thermal denaturation, samples were heated from 25 to 90 °C while monitoring ellipticity θ at 222 nm. As thermal denaturation of PGM1 is not reversible, the apparent Tm reports on both thermal stability and the kinetics of unfolding in this system.
Dynamic Light Scattering (DLS)
Protein samples at 1 mg/ml in 50 mm MOPS, pH 7.4, and 1 mm MgCl2 were prepared and centrifuged prior to data collection. Data were collected on a Protein Solutions DynaPro 99 instrument at a wavelength of 8363 Å for at least 200 s (10 s each for 20 acquisitions) at 25 °C. Polydispersity of samples ranged from 0 to 39%. Samples that gave larger than expected Mr estimates (T19A, N38Y, Q41R, and D263G) were additionally filtered through a 0.2-μm filter and data were re-measured. For T19A and D263G, the expected Mr was then obtained, however, aggregation for N38Y and Q41R remained (data on Table 1 for these four samples is post-filtration). In a further effort to remove aggregates, the Q41R sample was applied to a Superdex 200 size-exclusion column in 50 mm MOPS, pH 7.4, 0.1 m NaCl, and 1 mm MgCl2, and the protein in a fraction corresponding to the expected size was collected. However, subsequent analysis of this fraction by DLS (Table 1) showed that aggregation remained.
TABLE 1.
Summary of expression and biochemical data on PGM1 deficiency mutants
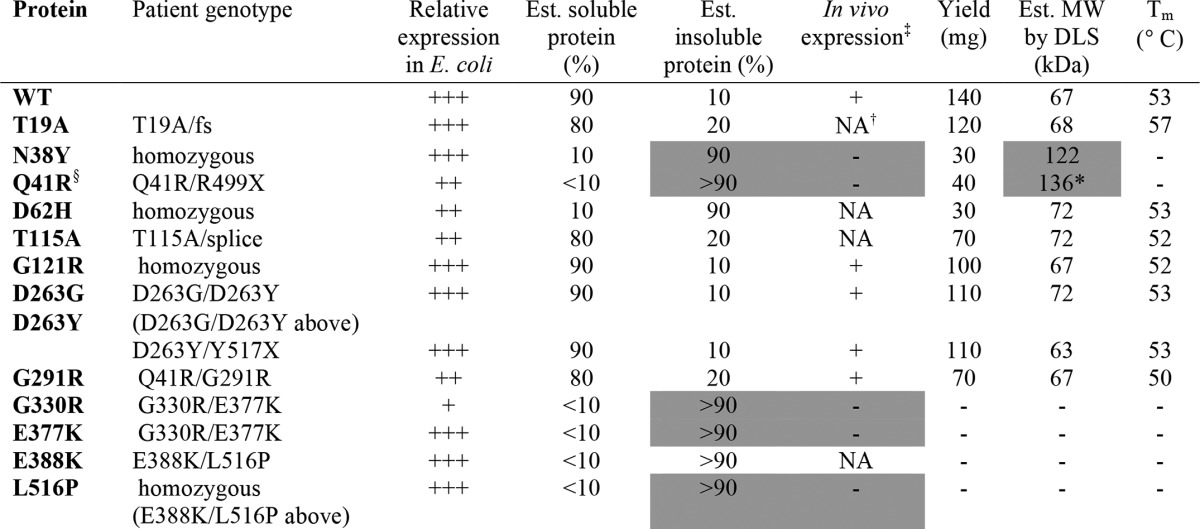
‡ In vivo data on protein expression levels obtained from patient fibroblasts as described in Ref. 4; NA indicates sample not available.
† Description of in vivo protein expression protein for T19A in Ref. 3 is ambiguous, so we list as NA. Alleles with frame shift (fs) or premature stop codons (X) are unlikely to contribute to in vivo protein expression levels; for detailed information on patient genotypes, see Ref. 3.
§ Additional genotype for this mutant's shown under G291R; good in vivo protein expression in sample heterozygous for Q41R/G291R is consistent with reduced expression of Q41R but normal expression of the G291R variant.
* Data shown for this sample is post-purification by size exclusion chromatography. Relative expression in E. coli reflects a combination of soluble and insoluble protein fractions. Gray shading highlights the correlation between poor in vivo expression of certain PGM1 mutants with poor solubility/aggregation of recombinantly expressed proteins. The G291R mutant was also characterized in a patient heterozygous for a fs mutant (1), where it showed reduced but visible protein expression in vivo.
ANS Binding
ANS-binding assays were performed by incubating 10 μm protein with 0.5 mm ANS in 50 mm MOPS, pH 7.4, for 1 h at 25 °C. Data were collected using the BioTek Synergy Mx Microplate Reader. The excitation wavelength was 365 nm, and emission spectra were recorded from 400 to 560 nm. Relative fluorescence intensities of samples were corrected for ANS emission spectra in buffer. To control for possible effects of phosphorylation of catalytic serine on enzyme flexibility (20),3 all protein samples were first treated with Glc-1,6-P as described below (see mass spectrometry). Glc-1,6-P was removed by dialysis in 50 mm MOPS, pH 7.4, 1 mm MgCl2 prior to ANS-binding assays.
Enzyme Activity Assays
Enzymatic activities for WT PGM1, three polymorphic variants, and the missense mutants were quantified by measuring the phosphoglucomutase activity by coupling the formation of glucose 6-phosphate from glucose 1-phosphate to NADH formation via glucose 6-phosphate dehydrogenase. Reactions were conducted at 25 °C in 50 mm MOPS, pH 7.4, with 1 mm dithiothreitol, 1.5 mm MgSO4, and 0.9 mm NAD+. The enzyme concentration ranged from 7.8 to 780 nm, depending on activity. The substrate (α-d-glucose 1-phosphate) concentration was varied from 2.5 to 800 μm, depending on the amount of enzyme used. The activator Glc-1,6-P was present at 1.0 μm, which was sufficient to relieve substrate inhibition in all reactions. Data were fitted to the Michaelis-Menten equation using SigmaPlot version 12.0®. A control assay using WT enzyme was performed in parallel with the characterization of each mutant, to ensure that differences in the kinetic parameters observed for the mutant proteins were not due to changes in experimental conditions. All assays were performed in duplicate.
Assessment of Phosphorylation by Mass Spectrometry
The phosphorylation of the active site phosphoserine (Ser-117) before and after treatment with Glc-1,6-P was analyzed using electrospray ionization mass spectrometry (ESI-MS). Protein samples were used at concentrations between 100 and 240 μm in 50 mm MOPS, pH 7.4, 1 mm MgCl2 and incubated with a 6-fold molar excess of Glc-1,6-P for 16 h at 4 °C. For mass spectrometric analyses, 10-μl protein samples at 1 pmol/μl in 1% formic acid were analyzed by NanoLC-Nanospray QTOF (Agilent 6520) in positive ion mode with a Zorbax C8 trap column. Data were examined using the Qual software provided with the instrument. The mass error between samples is 0.11 Da (2.1 ppm) and quantification error is 2%. Percent phosphorylation was calculated by normalizing the sum of the dephosphorylated and phosphorylated peak heights to 1.0. In all samples, no more than one phosphorylation modification was observed.
Dephosphorylation of WT PGM1
Following a protocol established for a related enzyme (20), glucosamine 1-phosphate was tested as a dephosphorylating agent for the active site serine of human PGM1. The protein sample at 160 μm was incubated with a 10-fold molar excess of glucosamine 1-phosphate for 16 h at 4 °C, and then dialyzed extensively against 50 mm MOPS, pH 7.4, and 1 mm MgCl2. Reduction in phosphorylation from 30 to 13% was achieved, as confirmed by ESI-MS.
Limited Proteolysis
Proteins were first treated with Glc-1,6-P as above to phosphorylate Ser-117; WT PGM1 was also dephosphorylated using glucosamine 1-P (see above section). Protein samples at 1.5 mg/ml were incubated with proteinase K in 50 mm MOPS, pH 7.4, at a 200:1 (w/w) ratio. Digestion was conducted at room temperature, aliquots were removed at various time points, and the reaction terminated by the addition of PMSF (final concentration: 3 mm). The reaction mixtures were subject to SDS-PAGE on 14% polyacrylamide gels.
RESULTS
Recombinant Mutant Proteins Vary Widely in Solubility and Aggregation
The gene for WT human PGM1 was commercially synthesized with codon optimization for bacterial expression and an N-terminal His6 tag for affinity purification (see “Experimental Procedures”). WT PGM1 expresses well, is largely soluble (∼90% of total protein; Fig. 2A), and yields ∼140 mg of purified protein from 1 liter of bacterial culture.
FIGURE 2.

Comparison of the expression levels and solubility of WT PGM1 and deficiency mutants in E. coli. A, SDS-PAGE (bottom) and corresponding histogram (top) of the soluble and insoluble fractions of cell extracts from E. coli cultures of WT and mutant PGM1 proteins. Relative protein levels on the histogram are normalized to a value of 1.0, for the combined amounts of soluble/insoluble protein obtained from WT PGM1. Asterisks highlight samples with corresponding mutations in patient fibroblasts that are poorly expressed in vivo (see Table 1 for more detail). B, overlay of near UV CD spectra for WT PGM1 and six mutants. Spectrum of the D263G mutant (not shown) is identical to that of D263Y. C, overlay of thermal melting curves as monitored at 222 nm by CD. For clarity, only WT and the two mutants with the greatest differences in Tm are shown (T19A and G291R). For others, see Table 1. Midpoint of each curve corresponding to the Tm is indicated by a vertical line.
The PGM1 deficiency mutants also generally express well in this recombinant system. Wide variation, however, is seen in the relative expression levels of soluble versus insoluble protein for the various mutants. Seven (N38Y, Q41R, D62H, G330R, E377K, E388K, and L516P) mutants show significantly reduced expression of soluble protein relative to WT (Fig. 2A) with a concurrent increase in the production of insoluble protein. As seen on Table 1, available in vivo data from patient fibroblasts (3) indicate no or poor expression of the same mutant enzymes that produce largely insoluble/aggregated recombinant protein (e.g. N38Y, Q41R, G330R, E377K, and L516P).
Four of the mutant enzymes (T19A, G121R, D263Y, and D263G) express soluble protein at levels ≥WT, and also yielded purified protein at levels similar to WT (>100 mg from 1 liter of culture). These mutants also tend to show good protein expression in vivo (Table 1) (3). In addition to these four, sufficient soluble protein from five other mutants was also obtained for purification (Table 1), yielding 30–70 mg from 1 liter of culture. Purification was not pursued for mutants without an apparent visible band for soluble protein in the expression gels (G330R, E377K, E388K, and L516P).
WT PGM1 and the nine mutants purified to homogeneity were characterized by DLS to assess their behavior in solution (Table 1). WT PGM1 is monomeric, and DLS gives an estimated molecular mass of 67 kDa. This agrees closely with the calculated molecular mass of the expression construct of 64.1 kDa (WT sequence plus affinity tag). Most of the mutants purified had apparent molecular masses similar to WT enzyme (ranging from 63 to 72 kDa). However, DLS indicated aggregation for the N38Y and Q41R mutants, both with estimated molecular masses >120 kDa. In the case of Q41R, the sample was further purified over a size exclusion column in an unsuccessful attempt to remove aggregation. This mutant, along with N38Y, was not further characterized due to its poor behavior in solution.
Well Folded PGM1 Mutants Show Modest Differences in Conformation/Stability
WT enzyme and the seven well behaved PGM1 mutants from above were further characterized with regard to conformational properties and stability. Near-UV circular dichroism (CD) shows that all proteins exhibit a similar minimum at 222 nm (Fig. 2B). Generally small differences between the mutants and WT enzyme are seen, indicating similarity in secondary structural content. The mutant with the greatest difference in its spectrum from WT is D62H.
Apparent melting temperatures (Tm) for the mutants were also determined via thermal denaturation by CD monitored at 220 nm. Overall, only small changes in Tm from WT were found (Table 1). The G291R mutant had the lowest Tm of 50 °C, a few degrees below that of WT, whereas the T19A mutant had a modestly increased Tm of 57 °C (Fig. 2C). These results suggest that these seven mutants have similar stability to WT enzyme, paralleling their good behavior in solution.
To probe possible effects of the missense mutations on protein conformation, binding of WT and mutant enzymes to the amphipathic dye ANS was monitored by fluorescence (Fig. 3A). Relative to WT enzyme, the G291R, D62H, and T115A mutants show somewhat increased fluorescence due to ANS-binding. This behavior is consistent with increased structural flexibility in these proteins, perhaps due to local structural perturbations caused by the mutations.
FIGURE 3.
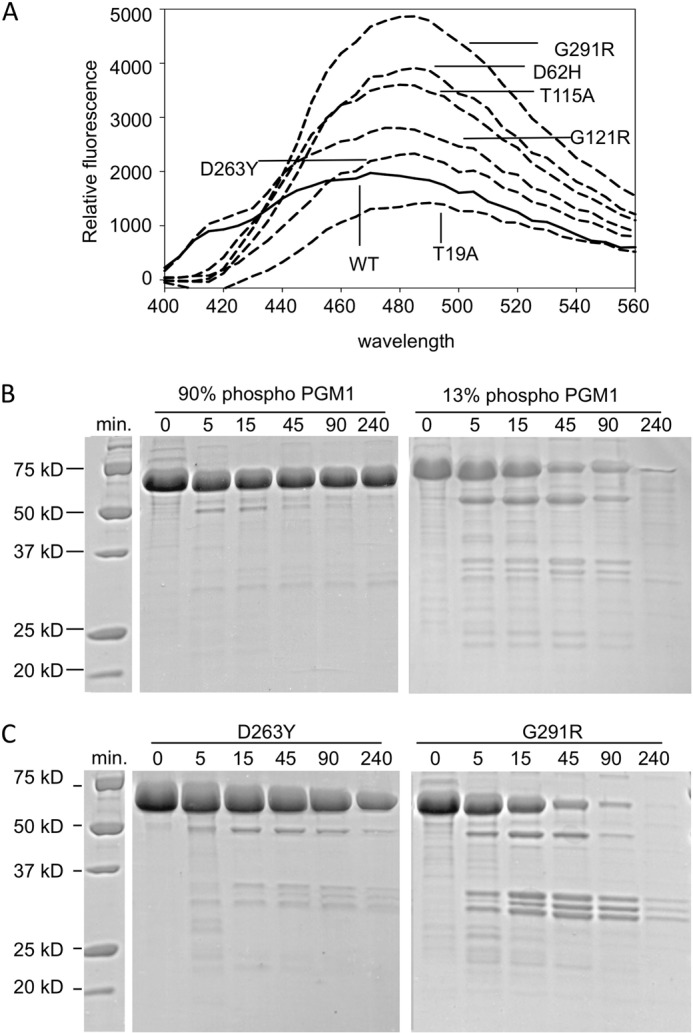
Effects of mutations on PGM1 flexibility and susceptibility to proteolysis. A, binding of ANS to WT PGM1 and the deficiency mutants (λex = 365 nm). SDS-PAGE showing limited proteolysis of (B) phosphorylated (90%) and dephosphorylated (13%) WT PGM1 (left and right panels, respectively), and (C) the D263Y (left) and G291R mutants (right). Length of digestion with proteinase K is given in minutes. Zero incubation time indicates untreated (control) samples. Note the increased susceptibility to proteolysis for both dephosphorylated PGM1 and the G121R mutant, which cannot be phosphorylated (Table 2).
A Subset of PGM1 Deficiency Mutations Have Severely Compromised Enzyme Activity
Steady state kinetic parameters for recombinantly expressed, affinity-purified WT PGM1 were determined and are reported here for the first time (Table 2). Kinetic assays were also performed for the seven PGM1 deficiency mutants that were soluble and not aggregated in solution. Except for T115A, all of the mutant enzymes showed dramatic catalytic impairment, with kcat often <1% that of WT enzyme. Effects on Km were generally modest suggesting that decreased affinity for substrate may be a less significant factor in the reduced activity of the mutant enzymes than impairment of the catalytic steps. Two of the mutants, however, are markedly different from the rest. One of these is G291R, which is so impaired in catalysis that its steady state kinetic parameters could not be determined. The other is T115A, which has only mildly reduced activity (kcat/Km ∼50% of WT).
TABLE 2.
Kinetic parameters and phosphorylation state of WT PGM1 and missense variants
Enzyme concentrations were 7.8 nm for WT and the functional polymorphisms, 15.6 nm for T115A and 780 nm for others. Percent phosphorylation refers to level of phosphorylation of Ser-117, the active site residue responsible for phosphoryl transfer.
| Protein | kcat | Km | % of kcat/Km | kcat of WT | Phosphorylation before/after G16P |
|---|---|---|---|---|---|
| s−1 | μm | μm−1s−1 | % | ||
| WT | 143 ± 2 | 80 ± 4 | 1.8 ± 0.1 | 35/90 | |
| Deficiency-associated mutations | |||||
| T19A | 0.685 ± 0.009 | 130 ± 5 | 0.0053 ± 0.0002 | 0.5 | 40/83 |
| D62H | 0.197 ± 0.002 | 23.3 ± 1.5 | 0.0085 ± 0.0006 | 0.1 | 15/90 |
| T115A | 36.0 ± 0.6 | 41 ± 3 | 0.88 ± 0.07 | 25 | 25/85 |
| G121R | 0.368 ± 0.004 | 56 ± 3 | 0.0066 ± 0.0004 | 0.3 | 20/90 |
| D263G | 3.0 ± 0.1 | 115 ± 10 | 0.026 ± 0.002 | 2.1 | 15/92 |
| D263Y | 1.54 ± 0.04 | 170 ± 14 | 0.0091 ± 0.0008 | 1.1 | 35/90 |
| G291R | BDa | BD | BD | BD | 0/0 |
| Functional polymorphisms | |||||
| K68M | 153 ± 1 | 63.8 ± 1.5 | 2.40 ± 0.06 | 107 | 22/92 |
| R221C | 200 ± 4 | 94 ± 7 | 2.1 ± 0.2 | 140 | 30/92 |
| Y420H | 162 ± 2 | 75 ± 4 | 2.1 ± 0.1 | 113 | 18/70 |
a BD, below detection.
Functional Polymorphic Variants of PGM1 Are Highly Active
To compare the catalytic impairment of the disease-related missense mutants with other known variants of PGM1, three functional polymorphisms were kinetically characterized. At least eight alleles of PGM1 have been characterized in humans, representing various combinations of three single amino acid substitutions of the protein: K68M, R221C, and Y419H (15, 16). Each of these were expressed and purified (see “Experimental Procedures”), resulting in soluble, well behaved proteins of the expected molecular mass according to DLS (62–72 kDa); additional biochemical assays were not conducted. Steady state kinetic characterization (Table 2) reveals that all three of these common enzyme variants have activity essentially equivalent to that of WT PGM1, with highly similar values for both kcat and Km.
Phosphorylation of Catalytic Ser-117
Initiation of catalysis by PGM1 is dependent on phosphoryl transfer from phosphoserine 117 to substrate, either glucose 1-phosphate or glucose 6-phosphate, to create the intermediate Glc-1,6-P (Fig. 1A). Hence phosphorylation of Ser-117 is a pre-requisite for enzyme activity. To determine whether this was a factor in the reduced activities of the PGM1 mutants, proteins were incubated with Glc-1,6-P, a known activator for enzymes in the phosphoglucomutase superfamily (10, 18, 21), and their phosphorylation state as monitored by ESI-MS. (In the kinetic assays described above, Glc-1,6-P is routinely included to ensure phosphorylation and maximal activity.) ESI-MS of all proteins showed the correct Mr based on amino acid sequence, and either a single modification by phosphorylation or none. This result is consistent with phosphorylation of Ser-117 and the known mechanism of the enzyme. (Human PGM1 has been reported to be phosphorylated at a second site, Thr-467, by the eukaryotic kinase Pak1 (22); as expected, no evidence for this was observed in the recombinant enzymes.)
Phosphorylation of Ser-117 was assessed both upon purification (prior to addition of Glc-1,6-P) and after treatment with Glc-1,6-P. Upon purification, WT enzyme was 30% phosphorylated; following treatment with Glc-1,6-P, its phosphorylation level was 90% (Table 2). All of the functional polymorphic variants and most of the mutant enzymes showed similar phosphorylation profiles for Ser-117: 15–40% prior to addition of Glc-1,6-P, and 83–92% following. However, the G291R mutant was notably different, with no detectable phosphorylation either before or after treatment with Glc-1,6-P. This phosphorylation defect reflects the lack of quantifiable in vitro enzyme activity from this mutant protein.
G291R Variant Shows Increased Susceptibility to Proteolysis
To further investigate the consequences of the G291R mutation and its lack of active site phosphorylation, limited proteolysis was used to evaluate potential differences in protein flexibility and/or structural disorder. For comparison, WT PGM1 and the D263Y mutant were also characterized. Proteinase K digestions of the WT PGM1 and the D263Y mutant, both of which had been pre-treated with Glc-1,6-P to fully phosphorylate Ser-117, are shown in Fig. 3, B and C (left panels). Only small differences in the patterns of proteolytic digestion are apparent between these two samples, with D263Y showing a slight increase in the production of cleavage fragments of ∼30 kDa in size over time. In contrast, the G291R mutant shows a significant increase in proteolysis, including many more sites of cleavage and much more rapid degradation (Fig. 3C, right panel), such that after 240 min, no intact enzyme remains. Some of these differences might be a direct result of the radical steric change of the Gly → Arg mutation, which could produce locally unstructured regions not apparent by methods such as DLS or CD. However, the lack of phosphorylation of G291R may also be relevant, as significant increased proteolytic cleavage is also observed for the dephosphorylated version of WT PGM1 (Fig. 3B, right panel). This concurs with previous detailed studies of a related protein in the PGM1 superfamily, which correlated the dephosphorylated state of the enzyme with an increase in structural flexibility (20),3 an observation with potential mechanistic implications. Thus, in addition to a detrimental effect on enzyme activity, the lack of active site serine phosphorylation in the G291R mutant may contribute to overall changes in the flexibility of this protein.
Missense Mutants Affect Highly Conserved Residues in the PGM Family
Earlier reports (1, 3, 4, 13) noted that many of the PGM1 deficiency mutations involve changes to conserved residues of the enzyme. To address this further, we conducted a sequence analysis over the large PGM family, which is found in a wide range of organisms from bacteria to mammals. (Alignments of PGM sequences from more closely related organisms, such as mammals, show extremely high sequence identity, often >90% overall, and so do not provide much insight into residue conservation.) The starting point for this analysis was a set of 409 PGM homologs, compiled in the Protein Information Resource as family PIRSF001493. From this large set, “seed members” (highly diverse sequences within the family) pre-selected by PIR were used to create a multiple sequence alignment.
This diverse alignment (supplemental Fig. S1) clearly highlights key functional regions of PGM, consistent with the conserved catalytic mechanism of the enzyme across evolution. Many of the missense mutants affect residues in/near conserved functional loops (e.g. the phosphoserine and metal-binding loops; see legend), and are strictly or very highly conserved. These include Thr-19, Asp-62, Thr-115, Asp-263, and Glu-377 (Fig. 1B), mutations of which primarily affect catalytic efficiency in these in vitro studies. (Glu-377 is an exception to this, as it produces insoluble protein.) Not surprisingly, other mutants located within the active site, including Gly-121, Gly-291, and Glu-388 are also highly conserved. Glu-388, for example, is found as either a glutamate or aspartate, retaining the negative charge of the side chain. Even for residues outside the active site, including Asn-38, Gly-41, Gly-330, and Leu-516, sequence conservation is generally still quite strong, possibly reflecting key roles of these residues in protein folding, as suggested in Ref. 13. In contrast to the residues with disease-associated mutations, sites of functional polymorphisms in PGM1 are less conserved (supplemental Fig. S1), presumably due to their lack of key structural or functional roles in the enzyme (13).
DISCUSSION
The in vitro biochemical phenotypes of 13 missense mutants associated with inherited PGM1 deficiency are reported here for the first time. Although the behavior of the protein produced in this recombinant system is not directly applicable to the situation in eukaryotic cells, such studies are essential for eliminating the complications of patient-derived data, such as heterozygosity and the presence of cellular homologs with PGM activity (1, 3, 4, 17). Overall, our in vitro data support the view derived from previous in vivo studies (1, 3) that folding defects and/or catalytic impairment may contribute to PGM1 deficiency, depending on the missense variant involved.
The utility of the recombinantly expressed proteins as a tool for understanding PGM1 deficiency is supported by a strong correlation between the missense mutants with poor in vivo protein expression (3) and those that produce insoluble/aggregated protein in E. coli (e.g. N38Y, Q41R, G330R, and E377K; Table 1 and Fig. 1A). Similar correlations between mutants that misfold in vivo and also show problems in recombinant expression have been observed in many other inherited metabolic diseases, including deficiencies in cystathione β-synthase (23), medium-chain acyl-CoA dehydrogenase (24, 25), phosphomannomutase 2 (26), and phenylalanine hydroxylase (27–29).
In this recombinant system, it appears that the 13 PGM1 missense mutants segregate almost equally into those with catalytic defects and those with possible folding problems. A few, however, such as D62H, show changes in both the expression level of soluble protein and enzyme activity. In total, six of the 13 PGM1 deficiency mutants were found to be impaired in the recombinant expression of soluble protein or to exhibit aggregation after purification. This group includes all four of the mutants that lie outside of the active site of the enzyme (N38Y, Q41R, G330R, and L516P), as well as both of the Glu → Lys mutants, found within the active site cleft (Fig. 1B). The other seven missense mutations characterized here were found to affect in vitro catalytic activity to varying degrees. All of these residues are found in/near the active site of PGM1 (Fig. 1B), and, as discussed further below, are associated with key regions important for catalysis or ligand binding.
A structural overview of the missense mutations associated with folding problems in E. coli is shown in Fig. 1B. Using the crystal structure of rabbit PGM, as a model (Protein Data Bank code 3PGM), the four mutants located outside the active site are shown to have varying structural roles (for a detailed review of the three-dimensional context of the missense variants and preliminary genotype-phenotype analyses, see Ref. 13). Asn-38 and Gln-41 are found in helices near the N terminus of the protein, and participate in multiple contacts with neighboring residues. Gly-330 and Leu-516 are buried residues found in β-strands within the folding cores of domains 3 and 4 of the enzyme. These important structural roles are consistent with the observed reduction in expression of soluble protein and/or aggregation observed for these mutant proteins in the recombinant expression system. Patients affected with the N38Y and Q41R mutations (both of which have poor in vivo expression and produce soluble but aggregated protein here) show generally mild disease phenotypes (3, 13), suggesting that in vivo perhaps a small fraction of the mutant enzyme is able to fold correctly and support catalysis. Patients with the G330R and L516P mutations (which include individuals heterozygous for other missense variants of PGM1) show moderate-severe clinical pathology (3, 13).
In contrast to the other residues associated with expression of insoluble protein, Glu-377 and Glu-388 are located directly within the active site cleft of PGM1 and make limited intra-residue interactions (13). Both of these missense variants produce a net change of +2 in the charge of the side chain as a result of the Glu → Lys mutation. Hence, electrostatic disruption may contribute to the impaired folding of E377K and E388K, perhaps due to unfavorable interactions with the many other positively charged residues found within the active site cleft (13). Moreover, in the case of these two mutants, even if small amounts of folded protein were successfully produced in vivo, it seems likely that substrate binding/catalysis would nevertheless, be impaired, due to the location and nature of the mutations. Indeed, patients with the E377K and E388K mutations have generally severe clinical manifestations (3, 13), including dilated cardiomyopathy and cardiac arrest, perhaps reflecting dual defects in folding and catalysis.
With one exception, the PGM1 missense mutants with kinetic defects all exhibit dramatic reductions in activity. Overall, the activities of the mutants determined from the in vitro studies are similar, but generally reduced, relative to those measured from cell extracts of patient samples (0.3–12% of control) (3). We find here that the mutants with impaired catalysis affect residues in/near key loops in the active site cleft (Fig. 1B), including the phosphoserine loop (D62H, T115A, and G121R), the metal binding loop (D263G, D263Y, and G291R), and an intra-domain latch (T19A). The least impaired of all the mutants is T115A, found only two residues away from the key phosphoserine 117. With ∼50% WT activity in vitro, the clinical pathology resulting from this conservative Thr → Ala mutation is unexplained. To date, only one individual with the T115A mutation (who is heterozygous for a splicing defect in PGM1) has been identified. This person lacks several clinical abnormalities common to other patients, such as small stature and congenital malformations of the head, but still suffers from exercise intolerance (3). The other missense mutations located in/near the phosphoserine loop (D62H and G121R) both had kcat/Km <0.5% of WT, presumably due to structural perturbations of this key functional region. The similarly reduced activity of the T19A mutant is consistent with its important structural role in formation of a multidomain interface necessary for correct binding of substrate (13). However, despite the overall similarity of their in vitro activities, clinical phenotypes of patients with these mutations vary, with D62H being relatively mild, T19A moderate, and G121R severe (3, 13).
Several of the mutations with kinetic defects are associated with the metal-binding loop in the active site of PGM1. This loop contains the conserved motif DGDGDR, spanning residues 288–292 (supplemental Fig. S1), where the three aspartates serve to coordinate the Mg2+ ion. The D263G and D263Y mutants are found outside but nearby this loop, whereas G291R affects the second glycine within the motif. Mutations of residue 263 are the most commonly found in PGM1 deficiency to date, affecting 3 of the 19 total patients in Ref. 3. In the crystal structure of rabbit PGM, the side chain of this aspartate interacts with that of Arg-293, the residue at the end of the conserved motif (13). This side chain-side chain interaction would be disrupted by mutation to either glycine or tyrosine, but the basis for its critical importance is not readily apparent from structural analysis. On the other hand, it is easy to envision a significant structural disruption caused by the G291R mutation, which affects a conserved glycine in the midst of the tight turn directly responsible for Mg2+ binding. As described above, G291R is the most catalytically impaired of all the missense mutants, is incompetent for phosphorylation of Ser-117, and also demonstrates increased susceptibility to proteolytic digestion in vitro (Table 1 and Fig. 3). The only patient identified with this mutation to date is heterozygous for Q41R (one of the mutants prone to aggregation in vitro), and has a mild overall phenotype (3).
We also show here for the first time using purified proteins that three common polymorphisms of PGM1 (K68M, R221C, and Y419H) have enzymatic activity essentially equivalent to that of WT PGM1, the most common allele in the human population (15, 16). Although some studies have suggested that certain alleles of PGM1 may be associated with differences in body mass (30, 31), these polymorphisms have no known association with disease. This is consistent with their high enzymatic activity in vitro (Table 2).
The biochemical characterizations of the missense mutants associated with human PGM1 deficiency provide a baseline for correlating patient genotype and clinical phenotype. Previous studies of PGM1 mutants were done using samples derived from patients, which are complicated by a number of factors, as noted under the Introduction. Additional caveats to these analyses include the diverse clinical manifestations of PGM1 deficiency, the observed variation in phenotype even in patients with the same genotype, and the small number of affected individuals known to date (3, 5). Even so, our studies suggest that mild biochemical impairment of several mutants (e.g. N38Y, Q41R in folding; T115A in catalysis) tends to correlate with mild clinical phenotype. In several other cases, catastrophic molecular defects (e.g. E377K, E388K in folding and likely also catalysis) are linked to severe clinical manifestations. These results help clarify those of Ref. 3, where reductions in enzyme activity from tissue-derived samples were apparent, but not clearly correlated with phenotypic severity. In the future, as the number of individuals identified with PGM1-deficiency grows, biochemical characterizations of disease-related mutants should prove invaluable to unraveling genotype-phenotype relationships that may benefit patient prognosis.
Although additional work is needed to establish relevance in vivo, knowledge of the biochemical phenotypes of the missense mutants may prove to be of assistance in targeting appropriate therapies to patients with PGM1 deficiency, which to date are largely unexplored except for dietary intervention (3, 32). As previously suggested for PGM1 (1), therapeutic approaches to increase protein folding/stability may be of benefit. Our data suggest this could be especially pertinent for the four mutants located outside of the active site of PGM1 (N38Y, Q41R, G330R, and L516). Such approaches, including proteasome inhibitors and pharmacological chaperones, are currently being used with success with other metabolic diseases (7, 8, 19). In contrast, the missense variants associated with severe catalytic defects, as established by these in vitro studies, would likely require additional or alternative approaches, such as enzyme replacement therapy, that could compensate for impaired activity.
Supplementary Material
Acknowledgments
We thank Ritcha Mehra-Chaudhary of the University of Missouri, Structural Biology Core for assistance with mutant preparation, and Brian Mooney and Beverly DaGue of the University of Missouri Charles W. Gehrke Proteomics Center for mass spectrometric analyses.
This work was supported National Science Foundation Grant MCB-1409898 (to L. J. B.).

This article contains supplemental Fig. S1.
J. Xu, Y. Lee, L. J. Beamer, and S. R. Van Doren, submitted for publication.
- PGM1
- phosphoglucomutase 1
- Glc-1,6-P
- α-d-glucose 1,6-bisphosphate
- ANS
- 8-anilino-1-naphthalene sulfonate
- ESI-MS
- electrospray ionization mass spectrometry
- DLS
- dynamic light scattering.
REFERENCES
- 1. Pérez B., Medrano C., Ecay M. J., Ruiz-Sala P., Martínez-Pardo M., Ugarte M., Pérez-Cerdá C. (2013) A novel congenital disorder of glycosylation type without central nervous system involvement caused by mutations in the phosphoglucomutase 1 gene. J. Inherit. Metab. Dis. 36, 535–542 [DOI] [PubMed] [Google Scholar]
- 2. Timal S., Hoischen A., Lehle L., Adamowicz M., Huijben K., Sykut-Cegielska J., Paprocka J., Jamroz E., van Spronsen F. J., Körner C., Gilissen C., Rodenburg R. J., Eidhof I., Van den Heuvel L., Thiel C., Wevers R. A., Morava E., Veltman J., Lefeber D. J. (2012) Gene identification in the congenital disorders of glycosylation type I by whole-exome sequencing. Hum. Mol. Genet. 21, 4151–4161 [DOI] [PubMed] [Google Scholar]
- 3. Tegtmeyer L. C., Rust S., van Scherpenzeel M., Ng B. G., Losfeld M.-E., Timal S., Raymond K., He P., Ichikawa M., Veltman J., Huijben K., Shin Y. S., Sharma V., Adamowicz M., Lammens M., Reunert J., Witten A., Schrapers E., Matthijs G., Jaeken J., Rymen D., Stojkovic T., Laforêt P., Petit F., Aumaître O., Czarnowska E., Piraud M., Podskarbi T., Stanley C. A., Matalon R., Burda P., Seyyedi S., Debus V., Socha P., Sykut-Cegielska J., van Spronsen F., de Meirleir L., Vajro P., DeClue T., Ficicioglu C., Wada Y., Wevers R. A., Vanderschaeghe D., Callewaert N., Fingerhut R., van Schaftingen E., Freeze H. H., Morava E., Lefeber D. J., Marquardt T. (2014) Multiple phenotypes in phosphoglucomutase 1 deficiency. N. Engl. J. Med. 370, 533–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stojkovic T., Vissing J., Petit F., Piraud M., Orngreen M. C., Andersen G., Claeys K. G., Wary C., Hogrel J.-Y., Laforêt P. (2009) Muscle glycogenosis due to phosphoglucomutase 1 deficiency. N. Engl. J. Med. 361, 425–427 [DOI] [PubMed] [Google Scholar]
- 5. Ondruskova N., Honzik T., Vondrackova A., Tesarova M., Zeman J., Hansikova H. (2014) Glycogen storage disease-like phenotype with central nervous system involvement in a PGM1-CDG patient. Neuro. Endocrinol. Lett. 35, 137–141 [PubMed] [Google Scholar]
- 6. Gregersen N. (2006) Protein misfolding disorders: pathogenesis and intervention. J. Inherit. Metab. Dis. 29, 456–470 [DOI] [PubMed] [Google Scholar]
- 7. Muntau A. C., Leandro J., Staudigl M., Mayer F., Gersting S. W. (2014) Innovative strategies to treat protein misfolding in inborn errors of metabolism: pharmacological chaperones and proteostasis regulators. J. Inherit. Metab. Dis. 37, 505–523 [DOI] [PubMed] [Google Scholar]
- 8. Jorge-Finnigan A., Brasil S., Underhaug J., Ruíz-Sala P., Merinero B., Banerjee R., Desviat L. R., Ugarte M., Martinez A., Pérez B. (2013) Pharmacological chaperones as a potential therapeutic option in methylmalonic aciduria cblB type. Hum. Mol. Genet. 22, 3680–3689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Parenti G., Pignata C., Vajro P., Salerno M. (2013) New strategies for the treatment of lysosomal storage diseases (review). Int. J. Mol. Med. 31, 11–20 [DOI] [PubMed] [Google Scholar]
- 10. Quick C. B., Fisher R. A., Harris H. (1974) A kinetic study of the isozymes determined by the three human phosphoglucomutase loci PGM1, PGM2 and PGM3. Eur. J. Biochem. 42, 511–517 [DOI] [PubMed] [Google Scholar]
- 11. Liu Y., Ray W. J., Jr., Baranidharan S. (1997) Structure of rabbit muscle phosphoglucomutase refined at 2.4-Å resolution. Acta Crystallogr. D 53, 392–405 [DOI] [PubMed] [Google Scholar]
- 12. Shackelford G. S., Regni C. A., Beamer L. J. (2004) Evolutionary trace analysis of the α-d-phosphohexomutase superfamily. Protein Sci. 13, 2130–2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Beamer L. J. (2014) Mutations in hereditary phosphoglucomutase 1 deficiency map to key regions of enzyme structure and function. J. Inherit. Metab. Dis. 10.1007/s10545-014-9757-9 [DOI] [PubMed] [Google Scholar]
- 14. Whitehouse D. B., Tomkins J., Lovegrove J. U., Hopkinson D. A., McMillan W. O. (1998) A phylogenetic approach to the identification of phosphoglucomutase genes. Mol. Biol. Evol. 15, 456–462 [DOI] [PubMed] [Google Scholar]
- 15. Takahashi N., Neel J. V. (1993) Intragenic recombination at the human phosphoglucomutase 1 locus: predictions fulfilled. Proc. Natl. Acad. Sci. U.S.A. 90, 10725–10729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. March R. E., Putt W., Hollyoake M., Ives J. H., Lovegrove J. U., Hopkinson D. A., Edwards Y. H., Whitehouse D. B. (1993) The classical human phosphoglucomutase (PGM1) isozyme polymorphism is generated by intragenic recombination. Proc. Natl. Acad. Sci. U.S.A. 90, 10730–10733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McAlpine P. J., Hopkinson D. A., Harris H. (1970) The relative activities attributable to the three phosphoglucomutase loci (PGM1, PGM2, PGM3) in human tissues. Ann. Hum. Genet. 34, 169–175 [DOI] [PubMed] [Google Scholar]
- 18. Ray W. J., Jr., Burgner J. W., 2nd, Post C. B. (1990) Characterization of vanadate-based transition-state-analogue complexes of phosphoglucomutase by spectral and NMR techniques. Biochemistry 29, 2770–2778 [DOI] [PubMed] [Google Scholar]
- 19. Boyd R. E., Lee G., Rybczynski P., Benjamin E. R., Khanna R., Wustman B. A., Valenzano K. J. (2013) Pharmacological chaperones as therapeutics for lysosomal storage diseases. J. Med. Chem. 56, 2705–2725 [DOI] [PubMed] [Google Scholar]
- 20. Lee Y., Villar M. T., Artigues A., Beamer L. J. (2014) Promotion of enzyme flexibility by dephosphorylation and coupling to the catalytic mechanism of a phosphohexomutase. J. Biol. Chem. 289, 4674–4682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jolly L., Ferrari P., Blanot D., Van Heijenoort J., Fassy F., Mengin-Lecreulx D. (1999) Reaction mechanism of phosphoglucosamine mutase from Escherichia coli. Eur. J. Biochem. 262, 202–210 [DOI] [PubMed] [Google Scholar]
- 22. Gururaj A., Barnes C. J., Vadlamudi R. K., Kumar R. (2004) Regulation of phosphoglucomutase 1 phosphorylation and activity by a signaling kinase. Oncogene 23, 8118–8127 [DOI] [PubMed] [Google Scholar]
- 23. Majtan T., Liu L., Carpenter J. F., Kraus J. P. (2010) Rescue of cystathionine-synthase (CBS) mutants with chemical chaperones: purification and characterization of eight cbs mutant enzymes. J. Biol. Chem. 285, 15866–15873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jensen T. G., Bross P., Andresen B. S., Lund T. B., Kristensen T. J., Jensen U. B., Winther V., Kølvraa S., Gregersen N., Bolund L. (1995) Comparison between medium-chain acyl-CoA dehydrogenase mutant proteins overexpressed in bacterial and mammalian cells. Hum. Mutat. 6, 226–231 [DOI] [PubMed] [Google Scholar]
- 25. Maier E. M., Gersting S. W., Kemter K. F., Jank J. M., Reindl M., Messing D. D., Truger M. S., Sommerhoff C. P., Muntau A. C. (2009) Protein misfolding is the molecular mechanism underlying MCADD identified in newborn screening. Hum. Mol. Genet. 18, 1612–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vega A. I., Pérez-Cerdá C., Abia D., Gámez A., Briones P., Artuch R., Desviat L. R., Ugarte M., Pérez B. (2011) Expression analysis revealing destabilizing mutations in phosphomannomutase 2 deficiency (PMM2-CDG). J. Inherit. Metab. Dis. 34, 929–939 [DOI] [PubMed] [Google Scholar]
- 27. Leandro J., Simonsen N., Saraste J., Leandro P., Flatmark T. (2011) Phenylketonuria as a protein misfolding disease: the mutation pG46S in phenylalanine hydroxylase promotes self-association and fibril formation. Biochim. Biophys. Acta 1812, 106–120 [DOI] [PubMed] [Google Scholar]
- 28. Waters P. J., Parniak M. A., Akerman B. R., Scriver C. R. (2000) Characterization of phenylketonuria missense substitutions, distant from the phenylalanine hydroxylase active site, illustrates a paradigm for mechanism and potential modulation of phenotype. Mol. Genet. Metab. 69, 101–110 [DOI] [PubMed] [Google Scholar]
- 29. Gámez A., Pérez B., Ugarte M., Desviat L. R. (2000) Expression analysis of phenylketonuria mutations: effect on folding and stability of the phenylalanine hydroxylase protein. J. Biol. Chem. 275, 29737–29742 [DOI] [PubMed] [Google Scholar]
- 30. Gloria-Bottini F., Magrini A., Antonacci E., La Torre M., Di Renzo L., De Lorenzo A., Bergamaschi A., Bottini E. (2007) Phosphoglucomutase genetic polymorphism and body mass. Am. J. Med. Sci. 334, 421–425 [DOI] [PubMed] [Google Scholar]
- 31. Gloria-Bottini F., Lucarini N., La Torre M., Lucarelli P., Bottini E. (2001) Birth weight and parental PGM1 alleles. Am. J. Hum. Biol. 13, 417–420 [DOI] [PubMed] [Google Scholar]
- 32. Morava E. (2014) Galactose supplementation in phosphoglucomutase-1 deficiency: review and outlook for a novel treatable CDG. Mol. Genet. Metab. 112, 275–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.