

Background: The acetyltransferases ATase1 and ATase2 acetylate ER-transiting and -resident proteins.
Results: ATase1 and ATase2 form homo- and heterodimers, associate with the oligosaccharyltransferase (OST) complex, and modify correctly folded polypetides.
Conclusion: The ATases are novel members of the “broad” ER translocation machinery.
Significance: The ATases act in concert with (and perhaps sequentially to) the OST to “mark” correctly folded glycoproteins.
Keywords: Acetyl Coenzyme A (acetyl-CoA), Acetyltransferase, Endoplasmic Reticulum (ER), Oligosaccharyltransferase, Post-translational Modification (PTM)
Abstract
The endoplasmic reticulum (ER) has two membrane-bound acetyltransferases responsible for the endoluminal Nϵ-lysine acetylation of ER-transiting and -resident proteins. Mutations that impair the ER-based acetylation machinery are associated with developmental defects and a familial form of spastic paraplegia. Deficient ER acetylation in the mouse leads to defects of the immune and nervous system. Here, we report that both ATase1 and ATase2 form homo- and heterodimers and associate with members of the oligosaccharyltransferase (OST) complex. In contrast to the OST, the ATases only modify correctly folded polypetides. Collectively, our studies suggest that one of the functions of the ATases is to work in concert with the OST and “select” correctly folded from unfolded/misfolded transiting polypeptides.
Introduction
Membrane and secreted proteins that are synthesized in the endoplasmic reticulum (ER)3 often undergo transient or permanent modification either during or after translation. These modifications can influence the folding as well as the activity of the proteins; in the case of transiting proteins, they can also affect correct trafficking along the secretory pathway. Our group has recently reported that mammalian cells are able to acetylate Nϵ-lysine residues of nascent proteins in the lumen of the ER (reviewed in Ref. 1). This post-translational event, which was initially discovered on a type I membrane protein (2), affects both ER-resident and -transiting proteins (3, 4).
The Nϵ-lysine acetylation in the lumen of the ER requires a membrane transporter, which translocates acetyl-CoA from the cytosol into the ER lumen (5), and specific ER-based acetyltransferases, which transfer the acetyl group from the donor (acetyl-CoA) to the acceptor (the lysine residue) (Ref. 6; reviewed in Ref. 1). The transferases, ATase1 and ATase2, are single-pass type II membrane proteins; the C-terminal catalytic domain faces the lumen of the organelle, where the reaction of lysine acetylation occurs (6). Deficient ER acetylation in the mouse leads to defects of the immune and nervous system, as well as increased propensity to cancer (7). In humans, mutations that affect the input of acetyl-CoA into the ER lumen are associated with developmental defects (8), as well as a familial form of spastic paraplegia (9).
Lysine acetylation is known to affect biochemical activity as well as stability and conformational assembly of a protein (reviewed in Refs. 1, 10, and 11). Although these general functions might be shared by the different acetylation machineries (i.e. in the cytosol, nucleus, mitochondria, and ER), it is likely that individual machineries also play specific functions related to the organelle where the modification occurs. An example is the role that the mitochondria acetylation machinery plays in cell metabolism (12). So far, the role of lysine acetylation in the lumen of the ER has only been dissected for two type I membrane proteins, BACE1 and CD133 (2, 13). BACE1 is involved in the pathogenesis of Alzheimer disease (14, 15), whereas CD133 appears to regulate the “stemness potential” of cancer cells (16–18). Both are active targets for therapeutics. Although there are subtle differences, in both cases deficient acetylation of the nascent protein resulted in deficient trafficking, retention in the early secretory pathway, and rapid degradation (2, 13).
Here, we report that both ER-based acetyltransferases, ATase1 and ATase2, form homo- and heterodimers and associate with the oligosaccharyltransferase (OST) complex. However, in contrast to the OST, the ATases only modify correctly folded polypeptides. Collectively, our studies suggest that one of the functions of the ATases is to work in concert with the OST and “select” correctly folded from unfolded/misfolded transiting polypeptides.
EXPERIMENTAL PROCEDURES
Cell Lines
Cells overexpressing ATase1, ATase2, or both were described before (6, 19).
Plasmid Constructs
Constructs and deletion mutants were generated as described before (4, 6, 20). The following cloning primers were used: for bacterial studies (ATases): MBP-ATase158–227-F, ACTTGCGGATCCCTGGTTCCGCGTGGATCTGGCTCCTGGATTCTGGCC; MBP-ATase158–227-R, GGCTGCGAATTCTCATAGACGCCCTGCCTGAGCAGAAGGGAGGTG; MBP-ATase158–192-F, ACTTGCGGATCCCTGGTTCCGCGTGGATCTGGCTCCTGGATTCTGGCC; MBP-ATase158–192-R, GGCTGCGAATTCTCAGCCCAAGCTCTGGTAGAGGCC; MBP-ATase158–199-F, ACTTGCGGATCCCTGGTTCCGCGTGGATCTGGCTCCTGGATTCTGGCC; MBP-ATase158–199-R, GGCTGCGAATTCTCAGGACTGGCCCGTCTTCTTGAAGCCCAAG; MBP-ATase258–227-F, ACTTGCGGATCCCTGCTGGTTCCGCGTGGATCTGGATCCTGGCTTCTA; MBP-ATase258–227-R, GGCTGCGAATTCTCACAGACTCCCTACC; for mammalian studies (ATases): ATase11–192-myc-F, GCCGAATTCATGGCTCCTTATCACATCC; ATase11–192-myc-R, GAACTCGAGGCCCAAGCTCTGGTAGAGGC; ATase11–207-myc-F, GCCGAATTCATGGCTCCTTATCACATCC; ATase11–207-myc-R, GAACTCGAGCAGCCTGGCCCACAC; ATase1-Halo, insert fragment was obtained by cleaving MBP-ATase1 (pet-18b vector); ATase2-Halo-F, GACGAATTCATGGCTCCTTGTCAC; ATase2-Halo-R, CGGCTCGAGCAGACTCCCTACCTTAG; for BACE1 deletion mutant studies: mt1-F, GGAGCCTTTCTTTGACTCTCTCATGACCATAGCCTATG; mt1-R, CATAGGCTATGGTCATGAGAGAGTCAAAGAAAGGCTCC; mt2-F, CAAGAGCATTGTGGACAGTCTCATGACCATAGCCTATG; mt2-R, CATAGGCTATGGTCATGAGACTGTCCACAATGCTCTTG; mt3-F, GCAGCCTCCTCCACGCTCATGACCATAGCC; mt3-R, CATAGGCTATGGTCATGAGACTGTCCACAATGCTCTTG; and for CD133 deletion mutant studies, 5′-TGTGCGGGAACTCCTTTTCAGCAAATCACCAGG-3′.
Expression in mammalian cells was conducted as described (4, 6, 20). The following vectors were used throughout this study: pMAL-c5E (New England Biolabs) for bacteria, pcDNATM3.1/myc-His (Invitrogen), and pHTC HaloTag® CMV-neo (Promega) for mammalian cells. The CD133-myc expression clone for mammalian cells was obtained from Origene (RC221611; variant 1).
MBP-ATases Expression in Escherichia coli
RosettaTM(DE3)pLysS cells (Novagen) containing MBP-ATases plasmids were grown at 37 °C until A600 of 1.2–1.4; they were then induced with 200 μm isopropyl β-d-thiogalactopyranoside at 22 °C for 16 h. Bacteria were collected by centrifugation and resuspended with AT buffer (20 mm HEPES, pH 8.0, 150 mm NaCl, 1 mm EDTA, 1 mm DTT) containing 2% DDM, followed by 10 pulses of sonication (10 s each time). Cell lysates were spun at 15,000 rpm for 20 min; the supernatant was then collected and incubated with amylose resin (E8021; New England Biolabs) for 2 h at 4 °C. The resin was washed three times with 10 volumes of AT buffer containing 0.02% DDM and then eluted with 20 mm maltose in AT buffer.
Western Blot Analysis
Western blotting was performed on a 4–12% Bis-Tris SDS-PAGE system (NuPAGE, Invitrogen) as described (2, 4, 6). The following primary antibodies were used in this study: anti-myc (sc-40, Santa Cruz Biotechnology; C3950, Sigma-Aldrich), anti-Halo tag (G921A, Promega), anti-ATases (AP4957c, Abgent), anti-OST48 (sc-74408, Santa Cruz Biotechnology), anti-RPN I (sc-25559, Santa Cruz Biotechnology), anti-RPN II (sc-166421, Santa Cruz Biotechnology), anti-OSTC (AP53125, Acris), anti-STT3A/ITM1 (sc-134685, Santa Cruz Biotechnology), anti-STT3B/SIMP (sc-100148, Santa Cruz Biotechnology), anti-DAD1 (ab23836, Abcam), anti-KRTCAP2 (ab108010, Abcam), and anti-BiP (C50B12, Cell Signaling). Goat anti-rabbit Alexa Fluor 680-conjugated or anti-mouse Alexa Fluor 800-conjugated secondary antibodies were used in infrared imaging at a 1:10,000 dilutions. HRP-conjugated anti-mouse or anti-rabbit secondary antibodies were used at 1:8,000 dilutions in chemiluminescent detection. Images were obtained with LICOR Odyssey Infrared Imaging System (LI-COR Biosciences) or ImageQuant LAS4000 (GE Healthcare).
AcetylCoA:Lysine Acetyltransferase Activity
Acetyltransferase activity was assayed as described before (6, 19). Concentrations of acetyl-CoA and affinity-purified ATases are shown in the appropriate figures. Lineweaver-Burk plots were generated using the GraphPad Prism software.
Analytical Ultracentrifugation
Affinity-purified MBP-ATase58–227
MBP-ATase158–227 was purified as described above and then separated on a 10-ml 8–30% glycerol gradient for 48 h at 4 °C as described (7, 21, 22). Fractions of 0.35 ml were collected from the top to the bottom. Carbonic anhydrase, bovine serum albumin, and alcohol dehydrogenase (Sigma) were used as sedimentation controls.
Microsomal OST
CHO cells stably expressing ATase1-myc and ATase2-myc were collected by centrifugation and resuspended in homogenization buffer containing 1 mm triethanolamine, 10 mm acetic acid, 250 mm sucrose, 1 mm EDTA, pH7.4, and 1 mm dithiothreitol. Cells were homogenized with 20 strokes on ice in a Dounce Teflon homogenizer, followed by 20 passes in a syringe with 25-gauge 5/8 needles. The homogenate was spun at 1,500 × g for 5 min at 4 °C to isolate post nuclear supernatant. The supernatant was then centrifuged at 100,000 × g, 25 p.s.i. for 15 min in a Beckman Coulter AirfugeTM air-driven ultracentrifuge. Microsomal membrane proteins were extracted in GTIP buffer (100 mm Tris·HCl, pH 7.6, 20 mm EDTA, 1.5 m NaCl, 1% Triton X-100, 0.25% Nonidet P-40). Extracted proteins were then separated on sucrose or glycerol gradients by using a SW41 Ti rotor in a Beckman Coulter OptimaTM L-90K ultracentrifuge. Sucrose solutions were made in buffer containing 50 mm Tris·HCl (pH 7.5), 1 mm EDTA, and 0.25% Nonidet P-40. The gradient layers were (top to bottom): 0.5-ml sample, 1 ml of 5, 10, 20, 50, and 60% sucrose solutions. The gradient was centrifuged at 132,000 × g for 16.5 h at 4 °C with slow acceleration profile and non-break deceleration profile. Fractions of 250 μl were collected. Glycerol solutions were made in buffer containing 1 mm Tris·HCl (pH 7.0), 1 mm MgCl2, 1 mm dithiothreitol, 1% glycerol, and 0.5% Triton X-100. The gradients layers were (top to bottom): 0.5-ml sample and 1 ml of 8, 10.2, 12.4, 14.6, 30, 35, 40, 50, 60, and 70% glycerol solutions. The gradient was centrifuged at 260,000 × g for 48 h at 4 °C. Fractions of 350 μl were collected.
Statistical Analysis
Results are always expressed as means ± S.D. of the indicated number of determinations. Data analysis was performed using GraphPad InStat 3.06 statistical software (GraphPad Software Inc.). Differences were declared statistically significant if p ≤ 0.05.
RESULTS
ATase 1 and ATase2 Form Homo- and Heterodimers through the C-terminal Tail
To study the functional assembly state in the ER membrane and further understand mechanisms of action of the ATases, we generated in silico structures of the catalytic domain of ATase1 and ATase2 by homology modeling with the Modeler 9.1 software, using the coordinates of three acetyltransferases as individual templates (from Bacillus subtilis, Protein Data Bank entry code 1YVK; Salmonella enteritidis, Protein Data Bank entry code 1S3Z; and Bacillus anthracis, Protein Data Bank entry code 3PP9). Specifically, we first searched the Protein Data Bank for proteins of known structure bearing the highest sequence identity/similarity with the amino acid sequence of ATase1 and ATase2. We then selected, as templates for homology modeling, the acetyltransferases with higher sequence identity/similarity with the ATases that were crystallized in the presence of CoA (1YVK and 1S3Z) or acetyl-CoA (3PP9). All three selected templates present a homo-oligomeric biological assembly in the crystal, 1YVK as tetramer (dimer of dimers) and 1S3Z and 3PP9 as dimers. The resulting homology models of ATase1 (Fig. 1, A and B) and ATase2 (Fig. 1, C and D) reflect the oligomerization states of the templates. Importantly, visual inspection of the homology models suggested that residues 199–205 (KKTGQSFFHVWA) form the core of the dimer interface regardless of the template used (Fig. 1, B and D).
FIGURE 1.

Structure modeling of ATase1 and ATase2. Homology modeling of the ATases suggests that they are dimers and that the dimerization involves the 194KKTGQSFFHVWA205 sequence at the C-terminal end of the protein. A and B, ATase1. C and D, ATase2. B and D show close-ups of the dimerization domain. Solvent-accessible surface area analysis is shown in Table 1.
To confirm the above models experimentally, we co-expressed myc- and halo-tagged versions of the ATases in the same cells and determined whether the immunoprecipitation of the myc-tagged version was also able to pull down the halo-tagged version. This was indeed the case for both ATase1 (Fig. 2A) and ATase2 (Fig. 2B), suggesting that each acetyltransferase is able to oligomerize (presumably as homodimers) in vivo. Co-immunoprecipitation (co-IP) was also observed when we co-expressed ATase1 and ATase2 (Fig. 2C), suggesting that the two transferases can form hetero-oligomers (presumably heterodimers) in vivo. To confirm these results and assess whether the oligomerization causes the formation of dimers and/or other high molecular mass species, we also assessed the migration profile of ATase1 and ATase2 on gel electrophoresis under native conditions. Surprisingly, both acetyltransferases migrated with an apparent molecular mass of ∼300–350-kDa (Fig. 2, D and E). From this, we concluded that either the ATases form high molecular mass oligomers or they assemble with other proteins into a high molecular mass complex.
FIGURE 2.

ATase1 and ATase2 exist as homo- and heterodimers and are part of a high molecular mass complex. A–C, immunoblots showing co-IP of myc- and halo-tagged versions of ATase1 and ATase2 when co-expressed in the same cells. A, ATase1 can form homodimers. B, ATase2 can form homodimers. C, ATase1 and ATase2 can form heterodimers. A schematic view of each experiment is shown in the lower panels. D and E, affinity-purified ATase1 (D) and ATase2 (E) migrate with the molecular mass of 300–350 kDa under native conditions. IP, immunoprecipitation; Wb, Western blot.
To differentiate between the above two possibilities, we generated a fusion protein where a truncated version of ATase1, starting at Gly58 and lacking the transmembrane domain, was placed at the C-terminal end of the maltose-binding protein (MBP) (Fig. 3A). MBP was chosen because it allows high mass/one-step purification for biochemical studies, and it is known to exist as a monomer (see also later). The truncated version of ATase1 contained the entire catalytic domain, as well the C-terminal KKTGQSFFHVWA domain required for oligomerization. The fusion protein (MBP-ATase158–227) was expressed in E. coli and then purified using amylose affinity chromatography and fast protein size exclusion liquid chromatography. As expected, the fused protein maintained its biochemical signature in vitro (Fig. 3, B and C), indicating that the protein was correctly folded.
FIGURE 3.

The dimerization of the ATases occurs through the C terminus. A, schematic view of the MBP-ATase1 fusion protein (referred to as MBP-ATase158–227). The membrane domain of ATase1 is shown in yellow. B and C, MBP-ATase158–227 retains acetyl-CoA:lysine acetyltransferase activity. The transfer activity of the fusion protein at 25 μm concentration of acetyl-CoA ranged between 80 and 300 pmol/min/ng of enzyme. D, MBP-ATase158–227 migrates as a dimer under native conditions. E, MBP-ATase158–227 migrates as a dimer on sedimentation gradients. E, molecular standards (Mol. St.) were run in parallel; their sedimentation profile is shown as bars on top. CA, carbonic anhydrase (29 kDa); BSA, bovine serum albumin (66 kDa); AlchD, alcohol dehydrogenase (150 kDa). F, schematic view of the C-terminal deletions used to generate MBP-ATase158–192 and MBP-ATase158–199 fusion proteins. The domain predicted to be involved in the dimerization is shown in red. MBP is shown in blue. G, MBP-ATase158–192 migrates as a monomer under native conditions. H, acetyltransferase activity of the different MBP-ATase1 fusion proteins tested here. MBP-ATase158–192 displays reduced activity. The values are the averages (n = 5) ± S.D. **, p < 0.005.
Next, we assessed the migration profile of MBP-ATase158–227 on gel electrophoresis under both denaturing and native conditions. MBP-ATase158–227 migrated with the expected molecular mass of ∼60 kDa under denaturing and ∼120 kDa under native conditions supporting a functional assembly state of a homodimer (Fig. 3D). A similar outcome was observed when we analyzed the migration profile of MBP-ATase258–227 (Fig. 4). Because under the same conditions MBP alone migrates as a monomer (data not shown), we concluded that ATase1 and ATase2 were responsible for the dimerization process. To support this conclusion, we analyzed the migration profile of MBP-ATase158–227 and MBP alone on analytical ultracentrifugation. A similar system was used in the past to assess the assembly state of other proteins (7, 21). Under the same gradient conditions, MBP-ATase158–227 migrated as a homodimer, whereas MBP migrated as a monomer (Fig. 3E), thus confirming that ATase1 was responsible for the homodimerization process.
FIGURE 4.
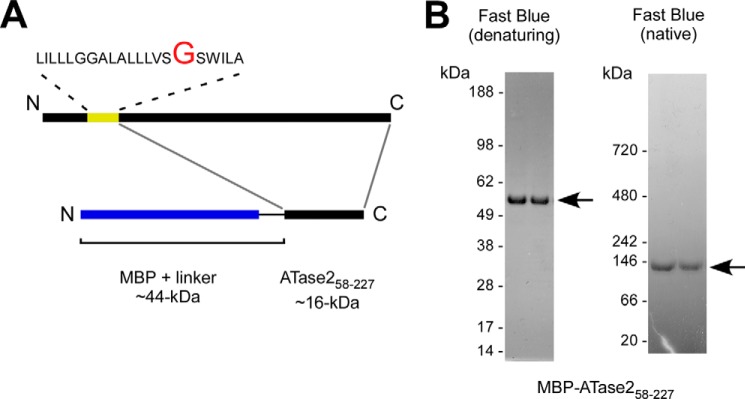
The dimerization of ATase2 occurs through the C terminus. A, schematic view of the MBP-ATase258–227 fusion protein. The membrane domain of ATase2 is shown in yellow. B, MBP-ATase258–227 migrates as a dimer under native conditions.
As stressed above, the KKTGQSFFHVWA sequence at the C-terminal end of the protein is predicted to form the dimerization interface in all homology models, regardless of the template used (Fig. 1, B and D). To confirm this, we generated a truncated version of the MBP-ATase158–227 protein missing the C-terminal tail that included the homodimerization domain (Fig. 3F; referred to as MBP-ATase158–192). This new version of the protein migrated with the predicted molecular mass of ∼49 kDa under both denaturing and native conditions, indicating that the C-terminal tail of ATase1 was responsible for the homodimerization (Fig. 3G). Homology modeling also predicted the stretch 194KKTGQS199 to constitute the core of the dimer interface in all models. Therefore, any truncation that left these initial six amino acids in place would not affect the dimerization process. To confirm this prediction, we generated a second C-terminal deletion mutant that retained amino acids 194–199 (Fig. 3F; referred to as MBP-ATase158–199). As predicted, MBP-ATase158–199 migrated as a homodimer under native conditions (Fig. 3G). We then calculated the solvent-accessible surface area using the Getarea server (Sealy Center for Structural Biology, University of Texas Medical Branch, Galveston, TX). The solvent-accessible surface area analysis confirmed that the solvent-accessible surface area of residues 194–205 diminishes as a result of dimerization (Table 1), particularly for residues 194–201, in agreement with the biochemical analysis. Finally, biochemical assessment showed that MBP-ATase158–199 retained catalytic activity, whereas MBP-ATase158–192 lost most of its activity (Fig. 3H).
TABLE 1.
SASA changes for ATase1
The table shows the percentages of solvent-accessible surface area (SASA) of residues forming the dimerization interface in ATase1 homology models based on templates 3PPA, 1YVK, and 1S3Z. m, monomer; d, dimer; t, tetramer.
| Amino acid | 3PPA_m | 3PPA_d | 1YVK_m | 1YVK_t | 1S3Z_m | 1S3Z_d |
|---|---|---|---|---|---|---|
| Lys194 | 81.8 | 46.4 | 73.8 | 33.8 | 98.7 | 74.4 |
| Lys195 | 5.8 | 8.7 | 0.2 | 2.9 | 71.8 | 64.1 |
| Thr196 | 63.1 | 0 | 72.2 | 5.9 | 41.9 | 8.6 |
| Gly197 | 18.3 | 3.6 | 56.4 | 2.7 | 34 | 44.1 |
| Gln198 | 24.9 | 12.7 | 82.6 | 4 | 66.6 | 18 |
| Ser199 | 51.5 | 8.5 | 30.8 | 1.2 | 52.8 | 14.7 |
| Phe200 | 50.9 | 21.6 | 67.2 | 12 | 86.6 | 17.8 |
| Phe201 | 94.7 | 17.3 | 76.3 | 6 | 72.1 | 6.5 |
| His202 | 68.7 | 58.1 | 66.7 | 68.3 | 88.7 | 0 |
| Val203 | 0.7 | 26.5 | 18.1 | 3.9 | 98.9 | 6.7 |
| Trp204 | 81.3 | 45.4 | 38.3 | 33.6 | 87 | 41.9 |
| Ala205 | 95.6 | 13.8 | 43.6 | 46.4 | 92.9 | 68.2 |
In conclusion, when taken together, the above results indicate that ATase1 and ATase2 exist either as homo- or heterodimers. The dimerization status is maintained by a short segment of the C-terminal tail (amino acids 194–205), as predicted by homology modeling. Finally, they also indicate that the dimerization is essential for their biochemical activity.
ATase1 and ATase2 Associate with Members of the OST in a High Molecular Mass Complex
Next, we decided to investigate the nature of the ∼300–350-kDa complex that was observed when the assembly state of the ATases was studied in mammalian cells (Fig. 2, D and E). Specifically, the complex was isolated and analyzed by mass spectrometry (MS). In addition to the ATases, the analysis identified five different proteins: OST48 (oligosaccharyltransferase 48-kDa subunit), RPN1 (ribophorin 1), RPN2 (ribophorin 2), STT3A (oligosaccharyltransferase subunit 3A), and DAD1 (defender against cell death 1) (Table 2). All identified proteins are integral components of the large oligosaccharyltransferase (OST) complex, a multisubunit ER membrane complex responsible for the N-glycosylation of nascent polypeptides entering the ER through the Sec61/translocon (23, 24). To confirm the association, we performed co-IP experiments in CHO cells overexpressing a myc-tagged version of full-length ATase1 (referred to as ATase11–227; see Fig. 5A for schematics). Immunoprecipitation of ATase1 successfully pulled down OST48, RPN1, RPN2, and STT3A but not DAD1 (Fig. 5B, lane 1). The fact that DAD1 was identified by MS but not by co-IP might be due to the low levels of the endogenous protein, which impeded visualization following co-IP and immunoblotting. As control, we performed the same experiments in cells transfected with an empty plasmid (mock transfection; Fig. 5B, lane 3). We also analyzed whether other OST-associated proteins that were not identified in the 300–350-kDa ATase complex by MS could be pulled down with ATase1. Specifically, we targeted STT3B, a close variant of STT3A, KCP2/KRTCAP2 (keratinocyte-associated protein 2), and OSTC (oligosaccharyltransferase subunit C). Finally, we also targeted BiP, a chaperone that acts in synergy with the translocation of the nascent polypeptide through the translocon. Neither STT3B, nor KCP2, nor OSTC, nor BiP was found to co-IP with ATase1 (Fig. 5B), providing additional validity to our findings. Of particular interest is the fact that STT3B was not identified by either MS or co-IP analysis (discussed later). The molecular mass of the individual members of the 300–350-kDa ATase complex is shown in Table 2; the predicted molecular mass of the complex (Table 2) is in line with the migration profile determined under native conditions (Fig. 2, D and E). In agreement with the above results, immunoprecipitation of endogenous OST48 was also able to pull down ATase1 and ATase2 (Fig. 5C).
TABLE 2.
The OST-ATase1/ATase2 complex
FIGURE 5.

ATase1 and ATase2 associate with OST members in a high-molecular-mass complex. A, schematic view of the C-terminal deletions used for the co-IP experiments described in B, G, and H. TM, transmembrane domain; myc, myc tag. B, immunoblots showing co-IP of OST48, RPN1, RPN2, and STT3A with transgenic ATase1 (referred to as ATase11–227-myc). Lanes 1 and 2 are from cells stable transfected with ATase1. Lane 1, immunoprecipitated product; lane 2, INPUT (prior to IP); lane 3, immunoprecipitation from mock-transfected cells (with empty plasmid). Proteins that were identified by MS analysis in the ATases high molecular mass complex are indicated. C, immunoblots showing co-IP of ATase1 and ATase2 with OST48. D and E, co-migration of OST48 and the ATases on two different sedimentation gradients. Microsomes from CHO cells overexpressing human ATase1 and ATase2 were used as starting material. F, co-migration of OST48 with endogenous ATases on a 5–70% glycerol sedimentation gradient. Microsomes from canine pancreas were used as starting material. G, immunoblots showing co-IP of OST48, RPN2, and STT3A with transgenic ATase11–192-myc. Lanes 1 and 2 are from cells stable transfected with ATase11–192. Lane 1, immunoprecipitated product; lane 2, INPUT (prior to IP); lane 3, immunoprecipitation from mock-transfected cells (with empty plasmid). H, immunoblots showing that the C-terminal tail (amino acids 208–227) of ATase1 is required for the ATase-RPN1 interaction. Co-IP of OST48 served as control. Lanes 1 and 2 are from cells stable transfected with ATase11–207. Lane 1, immunoprecipitated product; lane 2, INPUT (prior to IP); lane 3, immunoprecipitation from mock-transfected cells (with empty plasmid). IP, immunoprecipitation.
To further confirm the above results, we isolated the OST from cell microsomes and then assessed whether transgenic ATase1 and ATase2 co-migrated with OST48 on sucrose and glycerol density gradients. A similar approach has been used to confirm functional association of several members of the OST complex (25–27). Co-migration was determined under two different conditions (Fig. 5, D and E). We always observed co-migration of both ATases with OST48, which was used as a “migration marker” of the OST. Interestingly, the co-migration with OST48 was slightly different when we used a 5–60% sucrose gradient, suggesting that ATase1 and ATase2 might associate with a specific subset of the OST complex (Fig. 5, compare D with E). Finally, we assessed the migration profile of endogenous ATase1 and ATase2 by analyzing canine pancreas microsomes. Both acetyltransferases displayed co-migration with OST48 (Fig. 5F), thus confirming the results obtained with cells expressing transgenic ATases. As already determined in other cellular systems, endogenous ATase1 is more easily detectable than ATase2, which appears to display a more selective expression profile (19).
Finally, we assessed whether the ability of the ATases to dimerize was required for the association with OST members. For this purpose, we transfected ATase11–192, a truncated version of ATase1 lacking amino acids 193–227 (Fig. 5A), into CHO cells and then assessed its co-IP profile. The immunoprecipitation of ATase11–192 was able to pull down OST48, RPN2, and STT3A, confirming functional association with members of the OST subcomplex (Fig. 5G). Again, we were unable to co-immunoprecipitate DAD1 (Fig. 5G). Surprisingly, we were not able to co-IP RPN1 (Fig. 5G), suggesting that the C-terminal domain favors the interaction between the ATases and RPN1 but not the other members of the complex. This result provides specificity to the ATase-OST association described above and suggests that different ATase structural motifs might be required to maintain the functional association with the OST complex members. To further map the segment of the protein responsible for interacting with RPN1, we generated a new deletion mutant version of ATase1 that retained the oligomerization domain but lacked the last 20 amino acids of the C terminus (referred to as ATase11–207). The results show that ATase11–207 was able to interact with OST48 (used here as IP control) but not RPN1 (Fig. 5H). Therefore, we conclude that amino acids 208–227 are necessary for the ATase-RPN1 interaction.
When taken together, the above results suggest that the ATases associate with the OST complex. This conclusion is supported by four different lines of evidence: first, members of the OST complex were identified by MS in a 300–350-kDa complex that contained the ATases; second, the same OST members could also be co-immunoprecipitated with ATase1; third, the ATases co-migrated with OST48 on sedimentation gradients when the OST complex was functionally isolated from cell microsomes; and finally, the site of interaction between ATase1 and one member of the above complex was mapped to a specific domain of the acetyltransferase. Of interest is the fact that the association of the ATases with the OST complex members requires different domains. Specifically, the C-terminal end (amino acids 208–227) is required for the interaction with RPN1 but not OST48, RPN2, or STT3A.
ATase1 and ATase2 Only Modify Correctly Folded Polypeptides
We recently analyzed the ER-based acetylome and reported that OST48, RPN2, and STT3A are acetylated (4). Therefore, it might be possible that they associate with the ATases while being modified. However, in the above mentioned study (4), we identified a total of 143 proteins that undergo acetylation in the ER lumen, whereas only five (OST48, RPN1, RPN2, STT3A, and DAD1) were found to associate with ATase1 and ATase2 in a high molecular mass complex. In addition, although acetylated (4), STT3B does not associate with the ATases (Fig. 5B). Therefore, it is difficult to conceive that the association of the ATases with OST members in the ∼300–350-kDa complex is only based on a simple enzyme-substrate type of interaction. It is likely that OST and the ATases share common substrates and interact while modifying nascent polypeptides in the ER lumen. The OST recognizes the NX(T/S) consensus sequence within nascent polypeptides and transfers a preformed GlcNAc2Man9Glc3 oligosaccharide from the dolichol pyrophosphate-linked oligosaccharide, which acts as donor, to the asparagine residue of the above consensus sequence, which acts as acceptor of the tetradecasaccharide (23, 24). Importantly, the modification occurs while the protein is being translated (hence, co-translational), and the consensus sequence is ∼30–40 Å away from the ER membrane (28). However, Nϵ-lysine acetylation has no consensus motif, and therefore, the specificity of the reaction is likely to be provided by the structural signature of the protein being acetylated. To define this process in detail, we studied both N-glycosylation and Nϵ-lysine acetylation of BACE1, a known and already characterized substrate of the ER acetylation machinery (2, 6). Specifically, we designed different deletion mutant versions of BACE1 lacking (i) large segments of the protein, (ii) key cysteine residues required for disulfide bonds, and (iii) different asparagine residues required for N-glycosylation (see Fig. 6A for schematics). The overall objective was to cause significant changes to the folding and structure of BACE1 so that we could compare structural requirements for both N-glycosylation and Nϵ-lysine acetylation.
FIGURE 6.

Nϵ-Lysine acetylation of BACE1 is post-translational and recognizes structural features of the folded protein. A, schematic view of the different deletion mutants of BACE1. The signal peptide (SP) and pro-domain (PD) at the N terminus, the single transmembrane domain (TM), and the myc tag at the C terminus are shown. The region of the protein containing the TM and the short cytosolic tail of BACE1 were left intact so as not to disrupt membrane insertion. All deletion mutants retained both SP and PD so as not to disturb correct translation and topology of the protein. *, Asn residues that are normally glycosylated; c, Cys residues required for disulfide bonds; bars, Lys residues that are normally acetylated; fl, full-length protein; mt1–mt3, deletion mutants 1–3. B, immunoblotting showing expression (left panel) and lysine acetylation (right panel) of full-length BACE1 and the deletion mutants 1–3. Mature (m) and immature (im) forms of full-length BACE1 are indicated. Ac.K., acetylated lysine. C, immunoblotting showing migration shift of all mutants following PNGase F digestion. Wb, Western blot.
Fig. 6B shows that only full-length BACE1 (presumably correctly folded) could be acetylated. Importantly, although all deletion mutants retained at least one site for acetylation (Fig. 6A), they were not acetylated (Fig. 6B, right panel). As expected (2), only the immature, ER-based form of BACE1 was found to be acetylated (Fig. 6B, right panel). Interestingly, only full-length BACE1 appeared to be able to complete all post-translational modifications, as shown by the presence of the mature version of the protein, which is known to arise from the Golgi apparatus (Fig. 6B, left panel). In contrast to Nϵ-lysine acetylation, all deletion mutants were found to be glycosylated, as shown by the shift in migration following digestion with peptide N-glycosidase F (PNGase F) (Fig. 6C). PNGase F cleaves between the asparagine residue and the very first N-acetyl-glucosamine, thus removing the entire oligosaccharide chain from the polypeptide. When taken together, the above results indicate that, in contrast to N-glycosylation, Nϵ-lysine acetylation of a polypeptide is post-translational and can only recognize correctly folded polypeptides.
To assess whether the above conclusions could be extended to other ATase substrates, we decided to target CD133. Specifically, we removed a stretch of 159 amino acids of the N terminus of the protein that included the first two transmembrane domains (Fig. 7, A and B). The removal of the two transmembrane domains was expected to affect the structure of the N-terminal domain containing the three lysine residues that are normally acetylated (Fig. 7B). Importantly, the new deletion mutant version of CD133 retained the signal peptide and all sites of N-glycosylation to preserve topology and glycosylation of the protein. Fig. 7C shows that only full-length CD133 could be acetylated. However, although not acetylated, the deletion mutant version of CD133 was still able to undergo N-glycosylation, as shown by the shift in migration following digestion with PNGase F (Fig. 7D). In conclusion, the studies performed with CD133 reproduced the same outcomes of those performed with BACE1 and suggest that our conclusions can be generalized to other nascent polypeptides that undergo Nϵ-lysine acetylation in the ER lumen.
FIGURE 7.
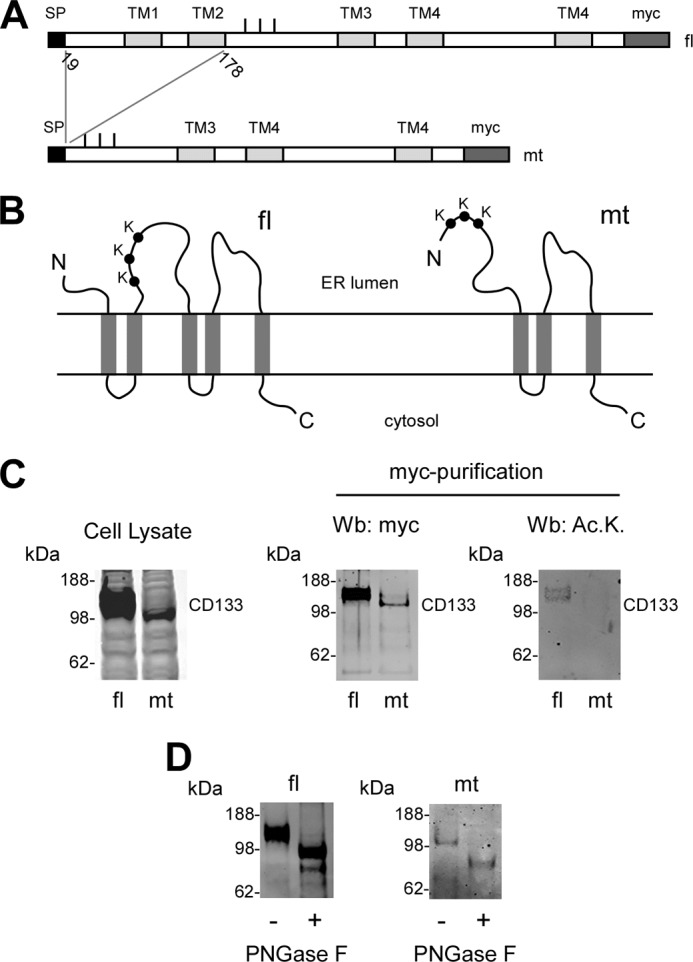
Nϵ-Lysine acetylation of CD133 is post-translational and recognizes structural features of the folded protein. A, schematic view of full-length and deleted version of CD133. The signal peptide (SP) at the N terminus, the transmembrane domains (TM), and the myc tag at the C terminus are shown. Bars indicate the three Lys residues that are normally acetylated. fl, full-length protein; mt, deletion mutant. The deletion mutant retained the signal peptide so as not to disturb correct translation and topology of the protein. All predicted N-glycosylation sites (Asn220, Asn274, Asn395, Asn414, Asn548, Asn580, Asn729, and Asn730) were retained. B, schematic view of the topology of CD133. The truncation eliminates the first two transmembrane domains causing significant changes in the structural organization of the mutant protein. C, immunoblotting showing expression (left and middle panels) and lysine acetylation (right panel) of full-length CD133 and the deletion mutant. The expression of CD133 is shown in a total cell lysate (left panel) and after affinity purification (middle panel) with immobilized anti-myc antibodies. The slightly different migration profile of mutant CD133 is due to the deletion. Ac.K., acetylated lysine. D, immunoblotting showing migration shift of full-length and mutant CD133 following PNGase F digestion. Wb, Western blot.
DISCUSSION
The acetylation of transiting proteins in the lumen of the ER was initially described in 2007 (2). Since then, many proteins that undergo the same process have been identified, either through proteomics or direct analysis (4, 13, 29). Here, we report that the ER-based acetyltransferases, ATase1 and ATase2, assemble to form homo- and heterodimers and associate with four (perhaps five) members of the OST complex. The OST glycosylates nascent, still unfolded polypetides while they enter the ER lumen through the translocon (23, 24). In contrast, the ATases only acetylate correctly folded polypeptides that have already received the GlcNAc2Man9Glc3 oligosaccharide. Therefore, in contrast to N-glycosylation, which is a co-translational event, Nϵ-lysine acetylation is post-translational and requires structural features to maintain specificity.
One of the many functions of the ER is to ensure that nascent membrane and secreted polypeptides fold correctly. Although the essential information for folding is present in the primary amino acid sequence, co-translational events (such as N-glycosylation and disulfide bonds) help ensure the fidelity of the process (1, 30). Incorrectly folded polypeptides must be “sorted” and disposed of. To preserve the function of membrane and secreted proteins, the sorting process must occur with high efficiency, thus ensuring that only correctly folded polypeptides are allowed to leave the organelle and traffic along the secretory pathway. For this purpose, transient modifications have been designed to recognize unfolded/misfolded polypeptides. One example is UDP-glucose:glycoprotein glucosyltransferase and the chaperone calnexin system. UDP-glucose:glycoprotein glucosyltransferase attaches one glucose residue to improperly folded polypeptides; the sugar, in turn, serves as a marker for calnexin, which retains the modified polypeptide in the ER (23, 30, 31). If the polypeptide can eventually fold, the glucose residue is removed, and the polypeptide is free to move along the secretory pathway.
Previous studies with BACE1 revealed that the transient Nϵ-lysine acetylation was able to regulate the ability of the ER-based polypeptide to leave the organelle. Indeed, acetylated intermediates of BACE1 could move through the secretory pathway, whereas nonacetylated intermediates could not (2). Similar conclusions were recently reached with CD133 (13). Therefore, in light of the above findings, we can interpret the association of the ATases with the OST complex as follows: the OST initially recognizes the NX(T/S) consensus motif on nascent glycoproteins and proceeds to transfer the GlcNAc2Man9Glc3 oligosaccharide to the Asn residue (Fig. 8). Following correct folding, the glycoprotein is recognized by the ATases, which proceed to attach one or more acetyl groups to the protein (Fig. 8A). This will “mark” the glycoprotein for correct trafficking along the secretory pathway. Incorrectly folded glycoproteins will not be recognized by the ATases (Fig. 8B) and therefore will not be allowed to proceed through the secretory pathway (2, 13). Interestingly enough, as with the UDP-glucose:glycoprotein glucosyltransferase/calnexin system, Nϵ-lysine acetylation is also a transient event. In fact, the acetyl group is removed once the protein reaches the Golgi apparatus by a Golgi-based deacetylase (2), probably amfion/C6orf89, a recently identified deacetylase that resides in the cis-Golgi (32). Although Nϵ-lysine acetylation shares some similarities with the UDP-glucose:glycoprotein glucosyltransferase/calnexin system, it differs in the fact that it “marks” correctly folded and not unfolded/misfolded polypeptides. The biological relevance of the ER-based acetylation machinery is shown by the fact that deficient ER acetylation in the mouse leads to defects of the immune and nervous system, as well as increased propensity to cancer (7). Finally, in humans, mutations that affect the input of acetyl-CoA into the ER lumen are associated with developmental defects (8), as well as a familial form of spastic paraplegia (9).
FIGURE 8.

Proposed model for our conclusions. Newly synthesized membrane proteins translocate across the translocon channel to enter the ER lumen. As soon as the NX(T/S) consensus motif for glycosylation is 30–40 Å (∼12–15 amino acids) away from the ER membrane, the OST will transfer the preformed GlcNAc2Man9Glc3 oligosaccharide to the asparagine residue of the consensus sequence. Following correct folding, the glycoprotein is recognized by the ATases, which proceed to attach one or more acetyl groups to the protein (A). This will “mark” the glycoprotein for correct trafficking along the secretory pathway. Incorrectly folded glycoproteins will not be recognized by the ATases and therefore will be “retained” for disposal (B). Therefore, Nϵ-lysine acetylation appears to act as a “selection marker” for correctly folded polypeptides. The model is based on results reported here and published elsewhere (2, 13). The model is not to scale and is only a “working interpretation” of the results.
The OST is a multisubunit complex that is structurally positioned in close proximity to the translocon (33). Purification of the OST complex has yielded different subcomplexes with different subunits and different molecular mass (reviewed in Ref. 23). However, it is generally accepted that in eukaryotic cells, the OST assembles as three main subcomplexes where individual protein members interact with proteins of the same subcomplex, as well as with proteins of different subcomplexes (23, 24). Based on studies performed in yeast, OST48, RPN1, RPN2, STT3, and DAD1 are considered essential subunits. The sedimentation profile of the ATases shown here might suggest that they preferentially interact with a certain subcomplex. It is also highly possible that the interaction and association of the ATases with the OST is dynamic, sequential, and (perhaps) dictated by other intrinsic events. Indeed, it is already known that the interaction of the OST with the signal peptidase complex during translation is sequential and dependent on the successful cleavage of the signal peptide (34). Of particular interest is the fact that the ATases were not found to associate with STT3B. Direct assessment in both yeast and mammalian systems has shown that STT3 proteins provide the catalytic activity to the OST complex; studies in mammalian systems have also revealed that different OST complexes, with either STT3A or STT3B, exist in the ER membrane (24). STT3A- and STT3B-containing OST complexes differ in substrate specificity (35) and temporal order in the processing of nascent polypeptides (36). Therefore, it is possible either that the ATases only act upon STT3A-OST substrates or that they only act sequentially to STT3A-OST activity. Obviously, the two possibilities are not mutually exclusive.
In conclusion, our results suggest that the ATases are novel members of the “broad” ER translocation machinery, where they act in concert with (and perhaps sequentially to) the OST to “mark” correctly folded glycoproteins. Additional studies are required to determine molecular aspects of the interaction, as well as define whether or not the acetyl-CoA:lysine acetyltransferase activity of the ATases is limited to glycosylated proteins.
Acknowledgment
We thank Dr. Carlos Hirschberg for critical reading of an early version of this manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants AG033514 (to L. P.) and NS34727 (to C. C.). This work was also supported by Veterans Affairs Merit Award BX001638 (to L. P.) and funds from the Thome Memorial Foundation (to L. P.).
- ER
- endoplasmic reticulum
- ATase1
- acetyltransferase 1
- ATase2
- acetyltransferase 2
- MBP
- maltose-binding protein
- OST
- oligosaccharyltransferase
- PNGase F
- peptide N-glycosidase F
- co-IP
- co-immunoprecipitation.
REFERENCES
- 1. Pehar M., Puglielli L. (2013) Lysine acetylation in the lumen of the ER: a novel and essential function under the control of the UPR. Biochim. Biophys. Acta 1833, 686–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Costantini C., Ko M. H., Jonas M. C., Puglielli L. (2007) A reversible form of lysine acetylation in the ER and Golgi lumen controls the molecular stabilization of BACE1. Biochem. J. 407, 383–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 4. Pehar M., Lehnus M., Karst A., Puglielli L. (2012) Proteomic assessment shows that many endoplasmic reticulum (ER)-resident proteins are targeted by Nϵ-lysine acetylation in the lumen of the organelle and predicts broad biological impact. J. Biol. Chem. 287, 22436–22440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jonas M. C., Pehar M., Puglielli L. (2010) AT-1 is the ER membrane acetyl-CoA transporter and is essential for cell viability. J. Cell Sci. 123, 3378–3388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ko M. H., Puglielli L. (2009) Two endoplasmic reticulum (ER)/ER Golgi intermediate compartment-based lysine acetyltransferases post-translationally regulate BACE1 levels. J. Biol. Chem. 284, 2482–2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Peng Y., Li M., Clarkson B. D., Pehar M., Lao P. J., Hillmer A. T., Barnhart T. E., Christian B. T., Mitchell H. A., Bendlin B. B., Sandor M., Puglielli L. (2014) Deficient import of acetyl-CoA into the ER lumen causes neurodegeneration and propensity to infections, inflammation, and cancer. J. Neurosci. 34, 6772–6789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huppke P., Brendel C., Kalscheuer V., Korenke G. C., Marquardt I., Freisinger P., Christodoulou J., Hillebrand M., Pitelet G., Wilson C., Gruber-Sedlmayr U., Ullmann R., Haas S., Elpeleg O., Nürnberg G., Nürnberg P., Dad S., Møller L. B., Kaler S. G., Gärtner J. (2012) Mutations in SLC33A1 cause a lethal autosomal-recessive disorder with congenital cataracts, hearing loss, and low serum copper and ceruloplasmin. Am. J. Hum. Genet. 90, 61–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin P., Li J., Liu Q., Mao F., Li J., Qiu R., Hu H., Song Y., Yang Y., Gao G., Yan C., Yang W., Shao C., Gong Y. (2008) A missense mutation in SLC33A1, which encodes the acetyl-CoA transporter, causes autosomal-dominant spastic paraplegia (SPG42). Am. J. Hum. Genet. 83, 752–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kouzarides T. (2000) Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 19, 1176–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang X. J., Grégoire S. (2007) Metabolism, cytoskeleton and cellular signalling in the grip of protein Nϵ- and O-acetylation. EMBO Rep. 8, 556–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhao S., Xu W., Jiang W., Yu W., Lin Y., Zhang T., Yao J., Zhou L., Zeng Y., Li H., Li Y., Shi J., An W., Hancock S. M., He F., Qin L., Chin J., Yang P., Chen X., Lei Q., Xiong Y., Guan K. L. (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mak A. B., Pehar M., Nixon A. M., Williams R. A., Uetrecht A. C., Puglielli L., Moffat J. (2014) Post-translational regulation of CD133 by ATase1/ATase2-mediated lysine acetylation. J. Mol. Biol. 426, 2175–2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang H., Li R., Shen Y. (2013) β-Secretase: its biology as a therapeutic target in diseases. Trends Pharmacol. Sci. 34, 215–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yan R., Vassar R. (2014) Targeting the β secretase BACE1 for Alzheimer's disease therapy. Lancet Neurol. 13, 319–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rappa G., Fodstad O., Lorico A. (2008) The stem cell-associated antigen CD133 (Prominin-1) is a molecular therapeutic target for metastatic melanoma. Stem Cells 26, 3008–3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ferrandina G., Petrillo M., Bonanno G., Scambia G. (2009) Targeting CD133 antigen in cancer. Expert Opin. Ther. Targets 13, 823–837 [DOI] [PubMed] [Google Scholar]
- 18. Takenobu H., Shimozato O., Nakamura T., Ochiai H., Yamaguchi Y., Ohira M., Nakagawara A., Kamijo T. (2011) CD133 suppresses neuroblastoma cell differentiation via signal pathway modification. Oncogene 30, 97–105 [DOI] [PubMed] [Google Scholar]
- 19. Ding Y., Ko M. H., Pehar M., Kotch F., Peters N. R., Luo Y., Salamat S. M., Puglielli L. (2012) Biochemical inhibition of the acetyltransferases ATase1 and ATase2 reduces β-secretase (BACE1) levels and Aβ generation. J. Biol. Chem. 287, 8424–8433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pehar M., Jonas M. C., Hare T. M., Puglielli L. (2012) SLC33A1/AT-1 protein regulates the induction of autophagy downstream of IRE1/XBP1 pathway. J. Biol. Chem. 287, 29921–29930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Puglielli L., Mandon E. C., Hirschberg C. B. (1999) Identification, purification, and characterization of the rat liver golgi membrane ATP transporter. J. Biol. Chem. 274, 12665–12669 [DOI] [PubMed] [Google Scholar]
- 22. Puglielli L., Mandon E. C., Rancour D. M., Menon A. K., Hirschberg C. B. (1999) Identification and purification of the rat liver Golgi membrane UDP-N-acetylgalactosamine transporter. J. Biol. Chem. 274, 4474–4479 [DOI] [PubMed] [Google Scholar]
- 23. Dempski R. E., Jr., Imperiali B. (2002) Oligosaccharyl transferase: gatekeeper to the secretory pathway. Curr. Opin. Chem. Biol. 6, 844–850 [DOI] [PubMed] [Google Scholar]
- 24. Kelleher D. J., Gilmore R. (2006) An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology 16, 47R–62R [DOI] [PubMed] [Google Scholar]
- 25. Kelleher D. J., Kreibich G., Gilmore R. (1992) Oligosaccharyltransferase activity is associated with a protein complex composed of ribophorins I and II and a 48 kd protein. Cell 69, 55–65 [DOI] [PubMed] [Google Scholar]
- 26. Kumar V., Heinemann F. S., Ozols J. (1994) Purification and characterization of avian oligosaccharyltransferase: complete amino acid sequence of the 50-kDa subunit. J. Biol. Chem. 269, 13451–13457 [PubMed] [Google Scholar]
- 27. Kelleher D. J., Gilmore R. (1997) DAD1, the defender against apoptotic cell death, is a subunit of the mammalian oligosaccharyltransferase. Proc. Natl. Acad. Sci. U.S.A. 94, 4994–4999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nilsson I. M., von Heijne G. (1993) Determination of the distance between the oligosaccharyltransferase active site and the endoplasmic reticulum membrane. J. Biol. Chem. 268, 5798–5801 [PubMed] [Google Scholar]
- 29. Jonas M. C., Costantini C., Puglielli L. (2008) PCSK9 is required for the disposal of non-acetylated intermediates of the nascent membrane protein BACE1. EMBO Rep. 9, 916–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Trombetta E. S., Parodi A. J. (2003) Quality control and protein folding in the secretory pathway. Annu. Rev. Cell Dev. Biol. 19, 649–676 [DOI] [PubMed] [Google Scholar]
- 31. Kleizen B., Braakman I. (2004) Protein folding and quality control in the endoplasmic reticulum. Curr. Opin. Cell Biol. 16, 343–349 [DOI] [PubMed] [Google Scholar]
- 32. Lalioti V. S., Vergarajauregui S., Villasante A., Pulido D., Sandoval I. V. (2013) C6orf89 encodes three distinct HDAC enhancers that function in the nucleolus, the Golgi and the midbody. J. Cell. Physiol. 228, 1907–1921 [DOI] [PubMed] [Google Scholar]
- 33. Pfeffer S., Dudek J., Gogala M., Schorr S., Linxweiler J., Lang S., Becker T., Beckmann R., Zimmermann R., Förster F. (2014) Structure of the mammalian oligosaccharyl-transferase complex in the native ER protein translocon. Nat. Commun. 5, 3072. [DOI] [PubMed] [Google Scholar]
- 34. Chen X., VanValkenburgh C., Liang H., Fang H., Green N. (2001) Signal peptidase and oligosaccharyltransferase interact in a sequential and dependent manner within the endoplasmic reticulum. J. Biol. Chem. 276, 2411–2416 [DOI] [PubMed] [Google Scholar]
- 35. Kelleher D. J., Karaoglu D., Mandon E. C., Gilmore R. (2003) Oligosaccharyltransferase isoforms that contain different catalytic STT3 subunits have distinct enzymatic properties. Mol. Cell 12, 101–111 [DOI] [PubMed] [Google Scholar]
- 36. Ruiz-Canada C., Kelleher D. J., Gilmore R. (2009) Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell 136, 272–283 [DOI] [PMC free article] [PubMed] [Google Scholar]