

Background: Inhibitors of cellular sialic acid expression offer substantial therapeutic promise for diseases associated with oversialylation.
Results: 2-Acetylamino-2-deoxy-3-O-methyl-d-mannose reduces the sialic acid concentration in cells and inhibits the UDP-GlcNAc-2-epimerase/ManNAc kinase.
Conclusion: Inhibition of the key enzyme of sialic acid biosynthesis by a ManNAc analog decreases cellular sialic acid expression.
Significance: ManNAc analogs represent a new class of sialic acid inhibitors.
Keywords: Enzyme Inhibitor, Flow Cytometry, Glycosylation Inhibitor, High-performance Liquid Chromatography (HPLC), Sialic Acid, Surface Plasmon Resonance (SPR)
Abstract
Due to its position at the outermost of glycans, sialic acid is involved in a myriad of physiological and pathophysiological cell functions such as host-pathogen interactions, immune regulation, and tumor evasion. Inhibitors of cell surface sialylation could be a useful tool in cancer, immune, antibiotic, or antiviral therapy. In this work, four different C-3 modified N-acetylmannosamine analogs were tested as potential inhibitors of cell surface sialylation. Peracetylated 2-acetylamino-2-deoxy-3-O-methyl-d-mannose decreases cell surface sialylation in Jurkat cells in a dose-dependent manner up to 80%, quantified by flow cytometry and enzyme-linked lectin assays. High-performance liquid chromatography experiments revealed that not only the concentration of membrane bound but also of cytosolic sialic acid is reduced in treated cells. We have strong evidence that the observed reduction of sialic acid expression in cells is caused by the inhibition of the bifunctional enzyme UDP-GlcNAc-2-epimerase/ManNAc kinase. 2-Acetylamino-2-deoxy-3-O-methyl-d-mannose inhibits the human ManNAc kinase domain of the UDP-GlcNAc-2-epimerase/ManNAc kinase. Binding kinetics of the inhibitor and human N-acetylmannosamine kinase were evaluated using surface plasmon resonance. Specificity studies with human N-acetylglucosamine kinase and hexokinase IV indicated a high specificity of 2-acetylamino-2-deoxy-3-O-methyl-d-mannose for MNK. This substance represents a novel class of inhibitors of sialic acid expression in cells, targeting the key enzyme of sialic acid de novo biosynthesis.
Introduction
Sialic acid is an essential constituent of the glycocalyx. Due to its chemical characteristics, sialic acid interacts with the environment of the cell. Its terminal position in glycoconjugates enables this amino sugar to disguise or create recognition sites for receptors or ligands and thus modulates biological cell functions (1). Sialic acid is a ligand for the selectin and the siglec family of adhesion molecules, mediating immune regulation and leukocyte rolling (2–5). Various pathogenic microorganisms, similar to the influenza virus with hemagglutinin, use sialylated glycans to recognize and bind host cells (6). Most tumor cells demonstrate hypersialylation and an alteration of their sialic acid pattern, allegedly to protect themselves from immune surveillance (7, 8). There is evidence that oversialylated tumor cells are more resistant to anti-cancer drugs (9).
The bifunctional enzyme UDP-GlcNAc-2-epimerase/ManNAc4 kinase is the major determinant of cell surface sialylation (10). It catalyzes the first two steps in the de novo biosynthesis of sialic acid, namely the epimerization of UDP-GlcNAc to ManNAc and the phosphorylation of ManNAc to ManNAc-6-phosphate. Former studies succeeded in synthesizing inhibitors of the UDP-GlcNAc-2-epimerase/ManNAc kinase (11–14). However, these inhibitors were membrane-impermeable and could not be used in cell-based assays. Consequently, we focused on developing membrane-permeable analogs of N-acetylmannosamine that inhibit sialic acid expression in intact cells.
Non-natural ManNAc analogs are well established precursors of modified sialic acid on the cell surface. They, for example carrying a keto or an azido group, can be utilized to visualize sialic acid in cells (15–17). These analogs are either modified at the C-2 or the C-4 position of ManNAc (18, 19).
In our study, we introduced C-3-modified ManNAc analogs and tested their effect on cell surface sialylation. C-3 derivatives of ManNAc are not likely to be metabolized by cells into the respected sialic acids (20). Inhibition studies for the ManNAc kinase domain of the UDP-GlcNAc-2-epimerase/ManNAc kinase were performed.
EXPERIMENTAL PROCEDURES
Preparation of ManNAc Analogs
Chemical synthesis of C-3-modified N-acetyl-d-mannosamines (Fig. 1) started from azido mannopyranosides 1 and 2, respectively (21, 22). Attachment of the alkoxy residue at C-3 was achieved by Williamson ether synthesis to give the alkylated derivatives 3 and 4a–c in good to excellent yields. Reduction of the azido function with H2-Pd/C or through Staudinger reaction and subsequent acylation under standard conditions yielded the N-acetylated derivatives. In the case of methyl glycoside 3 cleavage of the 4,6-O-benzylidene group by treatment with acetic acid at elevated temperature provided ManNAc analog 5. In addition benzyl glycosides 6a–c were also synthesized and then subjected to acidic hydrolysis to afford derivatives 7a–c. Cleavage of the benzyl group through palladium-catalyzed hydrogenolysis furnished the free N-acetyl-3-O-alkylmannosamine derivatives 8a–c.
FIGURE 1.

Synthesis of C-3-modified mannosamine analogs. 2-Acetylamino-2-deoxy-3-O-methyl-d-mannose (8a), methyl 2-acetylamino-2-deoxy-3-O-methyl-α-d-mannopyranoside (5), 2-acetylamino-2-deoxy-3-O-ethyl-d-mannose (8b), and 2-acetylamino-2-deoxy-3-O-propyl-d-mannose (8c) were prepared in three to four steps. For cell experiments, the given ManNAc analogs have been peracetylated (9, 10a, 10b, and 10c, respectively) to increase their permeability for the plasma membrane of the cells: (a) NaH, alkyl halide (MeI, EtI, and allyl bromide), 0 °C → room temperature, dimethylformamide; (b) 1) H2 (10% v/v), Pd/C, MeOH (for reduction of 4c; PPh3, THF/H2O); 2) AcCl, NEt3, DMAP, DCM; (c) AcOH (80%), 80 °C; (d) H2 (11 bar), Pd/C, MeOH; and (e) Ac2O, pyridine.
To increase their membrane permeability, peracetylated ManNAc analogs have been synthesized for cell experiments. In the cytoplasm, the O-acetyl groups of these molecules are removed by cytoplasmic esterases releasing the active monosaccharides (23–25). Peracetylation of 5 and 8a–c with acetic anhydride in pyridine afforded compounds 9 and 10a–c.
Details are included in the supplemental data. Identity and purity of all intermediates and final compounds were verified by spectroscopic and spectrometric methods (data not shown).
Determination of Cell Surface Sialylation
Cell Preparation
Approximately 20,000 Jurkat cells (ATCC) in 100 μl of RPMI 1640 culture medium (PAN Biotech) with 10% FBS (PAN Biotech) and 2 mm l-glutamine, containing peracetylated ManNAc analogs in varying concentrations, were cultured for 72 h.
After incubation, cells were washed with PBS containing 0.5% bovine serum albumin (PBS + 0.5% BSA) and labeled with various lectins. Labeling was carried out for 1 h at 4 °C and completed by three washing steps with PBS + 0.5% BSA. Obtained data were normalized to untreated cells, and cells were treated for 1 h with 0.2 units/ml sialidase from Clostridium perfringens (Sigma Aldrich). All experiments were performed in triplicate.
In Cell Enzyme-linked Lectin Assay
Alkaline phosphatase-conjugated Arachis hypogaea agglutinin (2 μg/ml, EY Laboratories) and alkaline phosphatase-conjugated Sambucus nigra agglutinin (3 μg/ml, EY Laboratories) were used in an in cell enzyme-linked lectin assay. 200 μl of p-nitrophenyl phosphate solution (Sigma-Aldrich) was added to start the reaction. Alkaline phosphatase activity was recorded at a wavelength of 405 nm.
Flow Cytometry
Flow cytometry experiments were conducted using FITC-conjugated Polyporus squamosus lectin (0.1 μg/ml, a gift from Prof. H.-J. Gabius, Technische Universität München).
Cytotoxicity Assays
The cell proliferation assay AlamarBlue© (AbD Serotec) was used to determine the cytotoxicity of given peracetylated ManNAc analogs. Samples of 100-μl cell suspension in RPMI 1640 + 10% FBS and 2 mm l-glutamine (20,000 Jurkat cells per well), containing different concentrations of peracetylated ManNAc analogs, were prepared. Cells were cultured for 72 h. Then, 10 μl of AlamarBlue© solution was added. After 4 h of incubation, samples were analyzed at wavelengths of 570 and 620 nm. Experiments were performed in triplicate and normalized to untreated control cells.
Quantification of Cytosolic and Membrane-associated Sialic Acid
Concentrations of free, CMP-conjugated and membrane-bound sialic acid were measured by DMB HPLC (26). Jurkat cells, cultured 72 h in 5 ml of RPMI 1640 (10% FBS, 2 mm l-glutamine) containing 500 μm 10a, were harvested and homogenized by sonication in ice-cold lysis buffer (150 mm NaCl, 10 mm Tris, 5 mm EDTA, 1 mm PMSF, 40 μm leupeptin, 1.5 μm aprotinin, pH 8.0). Cytosolic fractions were separated by centrifugation at 21,000 × g and 4 °C for 2 h. Subsequently, a chloroform-methanol precipitation was performed to isolate the glycan moiety. The aqueous phase was filtered via a 3-kDa size exclusion filter to remove residual macromolecules. Half of the samples were subjected to sodium borohydride reduction (200 mm sodium borohydride in 200 mm sodium borate buffer, pH 8.0) for 12 h to reduce non-CMP-conjugated sialic acid.
All membrane and the cytosolic fractions were hydrolyzed with 1 m trifluoroacetic acid for 4 h at 80 °C. Hydrolyzed samples were dissolved in 2 m acetic acid. Subsequently, samples were labeled with 50 μl of ice-cold DMB solution (6.9 mm DMB, 0.67 mm β-mercaptoethanol, 0.19% sodium bisulfide).
Labeled samples were analyzed on a Dionex Ultimate 3000 HPLC System (Thermo Scientific) using a Gemini-NX C18 column (110 Å, 3 μm particle size, 4.6 mm × 150 mm, Phenomenex). Probes were separated at 1 ml/min flow rate with methanol/acetonitrile/water (6:8:86) as eluent. The detector was configured with 373 nm for excitation and 448 nm for emission.
Cloning and Protein Purification
Cloning and protein purification of the MNK were performed according to an established method (27). His-tagged MNK was expressed in BL21-CodonPlus (DE3)-RIL Escherichia coli (Stragene), followed by nickel-nitrilotriacetic acid affinity chromatography and gel filtration with a SuperdexTM HighLoad 16/600 column (GE Healthcare) (28). Human N-acetyl-glucosamine kinase was purified as described previously (29). GST-tagged N-acetyl-glucosamine kinase was expressed in E. coli followed by glutathione affinity chromatography, gel filtration, and GST tag cleavage by thrombin. Human hexokinase IV was purchased from Sigma-Aldrich.
Spectrophotometric Assays (Enzyme Activity, Kinetics, and Inhibition)
Enzyme activity, kinetics, and inhibition were assessed via a coupled optical assay using purified MNK. 120 μl of reaction mixture contained the following buffers and reagents: 66.5 μl of buffer A (65 mm MgCl2, 10 mm Tris-HCl, pH 8.1), 10.0 μl of ATP (varying concentrations), 7.5 μl of NADH (15 mm), 5.0 μl of phosphoenolpyruvate (100 mm), 2 units of pyruvate-kinase, 2 units of lactate dehydrogenase, 10 μl of ManNAc of varying concentrations, and 10 μl of ManNAc analogs of varying concentrations. The reaction was started by adding 80 μl of MNK (1.25 μm) in buffer B (10 mm Tris-HCl, 150 mm NaCl, pH 8.0).
All spectrophotometric experiments were performed at 37 °C, and the results obtained were normalized to blanks, consisting of 120 μl of reaction mixture and 80 μl of buffer B without MNK. For hexokinase IV and N-acetyl-glucosamine kinase, the activity assay was adapted using glucose or N-acetyl-glucosamine as educts, respectively.
Dynamic Light Scattering Analysis
Dynamic light scattering experiments have been performed using a Laser Spector Scatter 201 (RiNA). Cuvettes were prepared with 30 μl of purified MNK (10 μm) in buffer B containing ManNAc or 8a at concentrations of 5 mm. All measurements were performed at 21 °C. Results were compared with negative controls containing untreated MNK only and positive controls containing MNK incubated for 10 min at 45 °C to induce protein aggregation.
Surface Plasmon Resonance
Experiments were conducted using a Biacore T100 biosensor instrument (GE Healthcare) and a nitrilotriacetic acid sensor chip (GE Healthcare). The following buffers were used: (a) running buffer (10 mm HEPES, 150 mm NaCl, 0.05 mm EDTA, 0.005% Tween, pH 7.4) and (b) protein regeneration solution (10 mm taurodeoxycholic acid, 100 mm Tris-HCl, pH 9.0).
His-tagged MNK was covalently immobilized to a level of ∼12,000 resonance units (1 resonance unit = 1 pg/mm2). The immobilization was carried out according to an established procedure with slight modifications, using a Biacore amine coupling kit (GE Healthcare) (30). Prior to the amine coupling procedure, the nitrilotriacetic acid sensor chip was activated with 0.5 mm NiCl2. After covalent attachment of the protein to the surface of the sensor chip, the coating procedure was completed by injecting 350 mm EDTA in running buffer. A flow cell without immobilized protein was used as reference.
All binding experiments were performed at 25 °C and a flow rate of 20 μl/min. Analytes of different concentrations were injected for 180 s followed by a 1000-s dissociation. In case of ManNAc and its analogs, regeneration was carried out by two injections of protein regeneration solution (10 μl/min, 60 s). With nucleosides 10 mm ManNAc in running buffer and 100 mm EDTA were injected (each 10 μl/min, 60 s) to assure regeneration. Each cycle was followed by a stabilization period of 1000 s. Binding data were evaluated with BIAevaluation (version 4.1, GE Healthcare), using 1:1 Langmuir binding model after reference cell and buffer injection subtraction.
RESULTS
Inhibition of Sialic Acid on the Cell Surface
10a decreases cell surface sialylation in a dose-dependent manner. Treatment of Jurkat cells for 72 h with 10a led to a substantial reduction of sialic acid expression on the cell surface. The 6′-sialyl-LacNAc entity was reduced by nearly 80%, determined by Polyporus squamosus lectin binding (Figs. 2a and 3) and by ∼70%, detected using Sambucus nigra agglutinin binding (Fig. 2b). The exposure of non-sialylated T antigen (Galβ1–3GalNAcα-Ser/Thr) was increased under treatment with 10a, verified by Arachis hypogaea agglutinin binding, which equals a reduction of the sialylated epitope of ∼80% (Fig. 2c). An IC50 for 10a of 176 μm in medium was determined. Other tested ManNAc derivatives (9, 10b–c) did not influence the sialic acid expression on the cell surface, even at final concentrations of 500 μm (Fig. 2).
FIGURE 2.
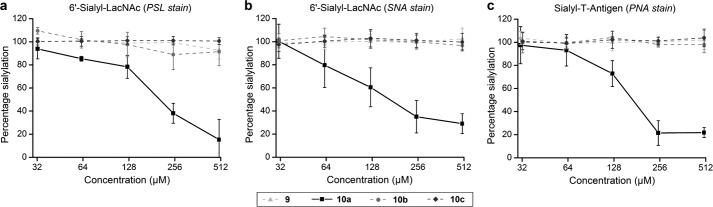
Peracetylated 2-acetylamino-2-deoxy-3-O-methyl-d-mannose (10a) is an inhibitor of cell surface sialylation. Jurkat cells were treated with varying concentrations of peracetylated ManNAc analogs for 72 h. Sialylated epitopes were analyzed with glycan specific lectins via flow cytometry (a) and in cell enzyme-linked lectin assay (b and c). Obtained data were normalized to untreated cells (100%) and cells treated for 1 h with 0.2 units/ml sialidase (0%). Data shown represent the mean values and S.D. of three independent experiments (n = 3). Only 10a shows inhibition of cell surface sialylation. The peracetylated forms of methyl 2-acetylamino-2-deoxy-3-O-methyl-α-d-mannopyranoside (9), 2-acetylamino-2-deoxy-3-O-ethyl-d-mannose (10b), and 2-acetylamino-2-deoxy-3-O-propyl-d-mannose (10c) had no effects on cell surface sialylation. PSL, P. squamosus lectin; SNA, S. nigra agglutinin; PNA, A. hypogaea agglutinin.
FIGURE 3.
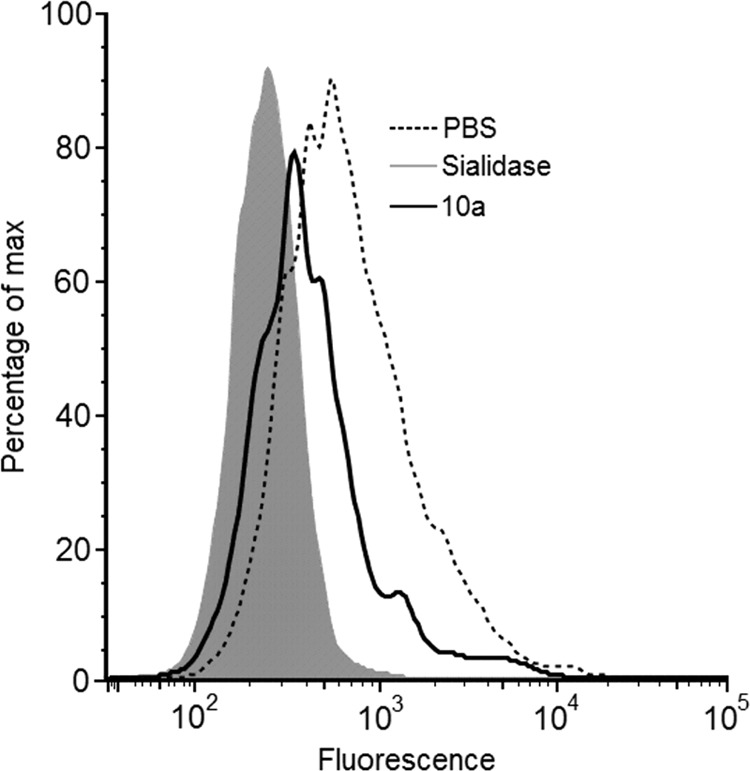
Flow cytometry analysis of Jurkat cells with FITC-conjugated P. squamosus lectin. Jurkat cells were treated for 72 h with peracetylated 2-acetylamino-2-deoxy-3-O-methyl-d-mannose (10a) in a concentration of 125 μm. To evaluate their 6′-sialyl-LacNAc content, cells were labeled with Polyporus squamosus lectin (PSL). Results obtained were compared with untreated cells (PBS, dashed line) and cells treated for 1 h with 0.2 units/ml sialidase (gray shading).
Quantification of Sialic Acid in Cell Lysates
Cell lysates of Jurkat cells treated for 72 h with 500 μm 10a showed decreased concentrations of free, CMP-conjugated and membrane-bound N-acetyl-neuraminic acid (Neu5Ac, Fig. 4). Comparing obtained HPLC chromatograms of treated cells and control cells, no additional peaks appeared, showing that 10a was not metabolized into a modified sialic acid (Fig. 5).
FIGURE 4.
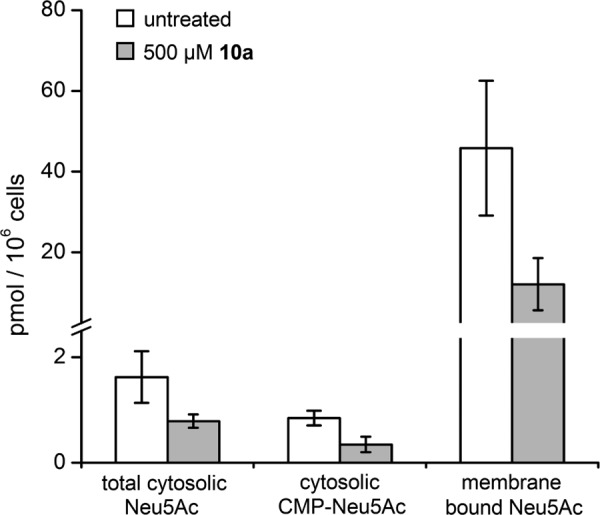
Analysis of cytosolic and membrane bound sialic acid in Jurkat cell lysates. Jurkat cells were treated for 72 h with 500 μm peracetylated 2-acetylamino-2-deoxy-3-O-methyl-d-mannose (10a). Concentrations of Neu5Ac were measured by DMB-HPLC. CMP-activated Neu5Ac was determined after sodium borohydride reduction of the cytosolic fraction. Data shown represent the mean values and S.D. of three independent experiments (n = 3). 10a inhibits the expression of cytosolic and membrane-bound Neu5Ac.
FIGURE 5.

Representative chromatograms of DMB-labeled sialic acid residues from Jurkat cell lysates. Jurkat cells were treated for 72 h with 500 μm peracetylated 2-acetylamino-2-deoxy-3-O-methyl-d-mannose (10a). Contents of CMP-activated Neu5Ac were determined after sodium borohydride reduction of the cytosolic fraction. Chromatograms of treated cells (orange) and untreated cells (blue) are plotted against a standard of 14 ng of Neu5Ac. Comparing treated and untreated cells no additional fluorescence peak appeared, indicating that 10a is not metabolized into a modified sialic acid.
Cytotoxicity of Peracetylated ManNAc analogs
The effect of tested peracetylated ManNAc analogs on cell viability was negligible under given conditions. The proliferation rate of Jurkat cells after 72 h of treatment with the peracetylated sugars at final concentrations of 1 mm was not significantly compromised. It is known that other peracetylated ManNAc derivatives such as peracetylated N-propionylmannosamine show comparable low cytotoxicity (31).
Inhibition of MNK Enzyme Activity
In spectrophotometric assays with purified MNK, the Km values of the enzyme have been measured to be 54 μm for ManNAc (specific activity, 0.77 units/mg) and 782 μm for ATP (specific activity, 0.70 units/mg), respectively.
8a inhibits purified MNK in a dose-dependent manner with an IC50 value of 1.29 mm (Fig. 6a). Other ManNAc derivatives tested (5, 8b–c) revealed no inhibition of enzyme activity, even at maximum concentrations of 10 mm. Dynamic light scattering experiments of purified MNK with 8a demonstrated that the enzyme inhibition of this substance is not caused by unspecific protein aggregation (Fig. 7).
FIGURE 6.
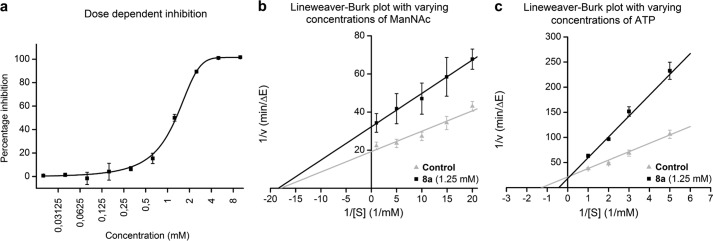
2-Acetylamino-2-deoxy-3-O-methyl-d-mannose (8a) inhibits purified MNK. A coupled optical assay using enzymatic consumption of NADH was performed to show the effect of 8a on MNK activity. In a, purified MNK was incubated with ManNAc (125 μm), ATP (2 mm), and different concentrations of 8a. Obtained activity data were normalized to controls without inhibitor (0%). In b and c, MNK was tested with 8a (1.25 mm) and varying concentrations of ManNAc (b) or ATP (c), respectively. Measured data were compared with controls without inhibitor (gray triangles). Data shown represent the mean values and S.D. obtained in three independent experiments (n = 3) for a and b and five independent experiments (n = 5) in c. Linear (b and c) and sigmoidal dose response (a) curve approximations were performed to determine Km, Ki, and IC50 values.
FIGURE 7.

Density light-scattering experiments. Cuvettes were prepared with purified MNK (10 μm) and ManNAc or 2-acetylamino-2-deoxy-3-O-methyl-d-mannose (8a) at concentrations of 5 mm. All measurements have been performed at 21 °C. As a positive control, MNK was preincubated for 10 min at 45 °C to induce protein aggregation. The radius of the highest peak for each graph is shown. Data depicted represent the mean values obtained in three independent experiments (n = 3). 8a does not cause any protein aggregation under given experimental conditions.
To evaluate the selectivity of 8a, its inhibition was assessed for human N-acetyl-glucosamine kinase and hexokinase IV (29). For these two related sugar kinases, inhibition of enzyme activity could be observed only at significantly higher concentrations (IC50 > 10 mm, data not shown).
Linear curve approximation, using the Lineweaver-Burk plot for MNK activity, shows that 8a is non-competitive for ManNAc, but competitive for ATP (Fig. 6, b and c). The Ki value of 8a for the interaction with ATP is 649 μm. Spectrophotometric assays revealed that 5 and 8a–c were not metabolized by the following enzymes: MNK, N-acetyl-glucosamine kinase, and hexokinase IV.
Binding Affinity to MNK
The binding affinity between 8a and MNK was determined using surface plasmon resonance. The immobilization of MNK to a level of 12,000 resonance units resulted in expected maximal response of ∼120 resonance units after injection of 8a (Fig. 8a and Table 1). Assuming a 1:1 interaction, a dissociation constant (KD) of 755 μm was calculated for the interaction between MNK and 8a. Under the given experimental conditions, no binding of ManNAc to MNK could be detected. ATP and ADP bound MNK with KD values of 429 and 306 μm, respectively (Fig. 8b and Table 1). The slightly lower KD value of ADP is due to its higher off rate (kd). UDP and 5, which were used as controls, did not bind to MNK.
FIGURE 8.
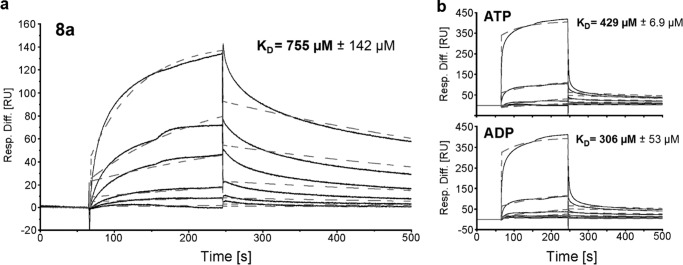
2-Acetylamino-2-deoxy-3-O-methyl-d-mannose (8a), ATP, and ADP binding to MNK. In a, a representative sensorgram of 8a binding to MNK is depicted. The analyte was injected in a 3-fold dilution series starting from 5 mm. Representative sensorgrams of ATP and ADP binding to MNK are shown in b. The analytes were injected in a 2-fold dilution series starting from 6.25 mm. Obtained data (solid lines) was fitted using a 1:1 Langmuir binding model (dashed lines) and was calculated. Depicted KD values represent means and S.D. of three independent experiments (n = 3). Resp. Diff., response difference; RU, response units.
TABLE 1.
Comparison of rate constants for 2-acetylamino-2-deoxy-3-O-methyl-m-mannose (8a) binding to covalently immobilized, His-tagged MNK
SPR-binding sensorgrams were fitted using a 1:1 Langmuir binding model. Data was obtained in three independent experiments (n = 3) at a temperature of 25 °C. For ManNAc and methyl 2-acetylamino-2-deoxy-3-O-methyl-α-d-mannopyranoside (5), no binding could have been observed under given experimental conditions (NA, data not available). ka, on rate; kd, off rate; KD, dissociation constant.
| ManNAc | 8a | 5 | ATP | ADP | UDP | |
|---|---|---|---|---|---|---|
| ka (m−1 s−1) | NA | 2.92 | NA | 2.48 | 2.76 | NA |
| kd (s−1) | NA | 2.25 × 10−3 | NA | 1.06 × 10−3 | 8.40 × 10−3 | NA |
| KD (μm) | NA | 755 | NA | 429 | 306 | NA |
| Χ2 | NA | 7.65 | NA | 19.20 | 18.87 | NA |
DISCUSSION
Cell surface sialic acid expression is involved in key processes between cells and their environment. Thus, having simple tools to decrease cell surface sialylation will greatly benefit research in finding roles of this sugar in biological systems and diseases. Although traditional methods are available to decrease sialic acid expression on the cell surface, e.g. sialyltransferase inhibition or UDP-GlcNAc-2-epimerase/ManNAc kinase-deficient cells (10, 32–34), a systemic inhibitor for the de novo biosynthesis of sialic acid is still not available.
Within this study, we presented a small molecule inhibitor of sialic acid expression, namely 2-acetylamino-2-deoxy-3-O-methyl-d-mannose (8a). Applying its peracetylated form (10a), cell surface sialic acid concentration in Jurkat cells was decreased in a dose-dependent manner (Fig. 2). At final concentrations of 500 μm in culture medium, the sialic acid concentration was reduced significantly to ∼20%. Total inhibition of cell surface sialylation could not be observed, most likely due to uptake and reutilization of sialic acid from the serum supplement (35). In addition, a slow turnover of sialylated glycoproteins could also contribute to this finding (36). The IC50 of the peracetylated analog (10a) is 176 μm. With a molecular weight of 235 g/mol, 8a has the potential for further chemical modification to improve binding affinity.
Performing DMB high-performance liquid chromatography on Jurkat cells treated with 10a revealed that this ManNAc analog causes a substantial reduction of Neu5Ac both on the cell surface and in the cytosol. Cytosolic concentrations of free and CMP-conjugated Neu5Ac were decreased. 10a was not metabolized into a modified sialic acid in cells (Fig. 5). These results indicate that 10a inhibits sialic acid expression at the beginning of the de novo biosynthesis.
Other tested C-3-modified ManNAc analogs (9, 10b–c) did not alter cell surface sialylation in treated cells. The low cytotoxicity of 10a underlines the fact that inhibition of sialic acid biosynthesis does not cause cell viability defects (10, 32).
We suggest that the reduction of sialic acid expression achieved by 10a is a consequence of MNK inhibition. The enzyme activity of MNK was inhibited by 8a. The IC50 for the interaction is 1.29 mm. Dynamic light scattering experiments verified that the inhibition of 8a is not due to protein aggregation. Related substances 5, 8b, and 8c, which in their peracetylated forms (9, 10b–c) did not alter cell surface sialylation, also showed no inhibitory effects on the MNK.
The mode of inhibition for 8a is non-competitive for ManNAc, but competitive for ATP (Ki, 649 μm). Apparently, this substance has its binding pocket in the ATP-binding site of MNK.
Surface plasmon resonance confirms binding of 8a to MNK with a KD of 755 μm. While covalently immobilized to the nitrilotriacetic acid sensor chip, MNK does not bind ManNAc. This may be caused by conformational changes at the ManNAc binding site due to the immobilization procedure, or because of the technical set-up of the experiment. The selective binding of ADP, ATP and 8a but not ManNAc also indicates that the inhibitor has the same binding site as ADP/ATP. The fact that 5 as well as UDP did not bind to MNK shows that the ATP-binding pocket of the enzyme was still intact.
The selectivity of 8a for MNK was affirmed by evaluating its inhibitory capacity on other pyranose kinase enzymes (N-acetyl-glucosamine kinase, hexokinase IV). 8a shows significantly weaker inhibitory effects on these enzymes (IC50 > 10 mm). MNK, N-acetyl-glucosamine kinase, and hexokinase IV did not metabolize any of the tested ManNAc analogs (5 and 8a–c).
Based on our results, we conclude that 10a decreases sialic acid expression in cells by inhibiting cytosolic MNK. Being the key enzyme of sialic acid biosynthesis, its inhibition ultimately leads to the observed reduction of cell surface sialylation (10). The proposed model of sialic acid reduction in cells caused by 10a is depicted in Fig. 9.
FIGURE 9.

Proposed mechanism of inhibition for peracetylated 2-acetylamino-2-deoxy-3-O-methyl-d-mannose (10a). 10a is taken up by cells and converted to the corresponding non-peracetylated sugar (8a) by unspecific esterases. 8a inhibits the kinase domain of the bifunctional UDP-GlcNAc-2-epimerase/ManNAc kinase (GNE/MNK). Inhibition of the MNK lowers the de novo biosynthesis of sialic acid. The depicted glycan (3′-sialyllactose) is representative for sialylated glycans in general.
Peracetylated 2-acetylamino-2-deoxy-3-O-methyl-d-mannose (10a) is the first membrane permeable inhibitor of the UDP-GlcNAc-2-epimerase/ManNAc kinase described. With an inhibitor of the de novo biosynthesis of sialic acid, access to biological effects of global cell surface sialic acid reduction is facilitated. It paves the way for deeper understanding on the role of sialic acid in health and disease.
Supplementary Material
Acknowledgments
We thank Prof. Hans-Joachim Gabius (Technische Universität München) for providing FITC-conjugated Polyporus squamosus lectin. We also thank the Freie Universität Berlin for supporting our research.
This work was supported by the Wilhelm-Sander Stiftung, by the Sonnenfeld Stiftung, and the Roland und Elfriede Schauer Stiftung.
This work is dedicated to Professor Konrad Sandhoff on the occasion of his 75th birthday.

This article contains supplemental Methods and data.
- ManNAc
- N-acetylmannosamine
- DMB
- 1,2-diamino-4,5-methylenedioxybenzene
- GlcNAc
- N-acetyl-glucosamine
- GNE
- human N-acetyl-glucosamine-2-epimerase
- MNK
- human N-acetylmannosamine kinase
- Neu5Ac
- N-acetyl-neuraminic acid.
REFERENCES
- 1. Schauer R. (2009) Sialic acids as regulators of molecular and cellular interactions. Curr. Opin. Struct. Biol. 19, 507–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Varki A. (2007) Glycan-based interactions involving vertebrate sialic-acid-recognizing proteins. Nature 446, 1023–1029 [DOI] [PubMed] [Google Scholar]
- 3. Lehmann F., Tiralongo E., Tiralongo J. (2006) Sialic acid-specific lectins: occurrence, specificity and function. Cell Mol. Life Sci. 63, 1331–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Crocker P. R., Paulson J. C., Varki A. (2007) Siglecs and their roles in the immune system. Nat. Rev. Immunol. 7, 255–266 [DOI] [PubMed] [Google Scholar]
- 5. Hakomori S. (1989) Aberrant glycosylation in tumors and tumor-associated carbohydrate antigens. Adv. Cancer Res. 52, 257–331 [DOI] [PubMed] [Google Scholar]
- 6. Russell C. J., Webster R. G. (2005) The genesis of a pandemic influenza virus. Cell 123, 368–371 [DOI] [PubMed] [Google Scholar]
- 7. Grewal P. K. (2010) The Ashwell-Morell receptor. Methods Enzymol. 479, 223–241 [DOI] [PubMed] [Google Scholar]
- 8. Chen S., Fukuda M. (2006) Cell type-specific roles of carbohydrates in tumor metastasis. Methods Enzymol. 416, 371–380 [DOI] [PubMed] [Google Scholar]
- 9. Chen L., Liang J. F. (2012) Metabolic monosaccharides altered cell responses to anticancer drugs. Eur. J. Pharm. Biopharm. 81, 339–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Keppler O. T., Hinderlich S., Langner J., Schwartz-Albiez R., Reutter W., Pawlita M. (1999) UDP-GlcNAc 2-epimerase: a regulator of cell surface sialylation. Science 284, 1372–1376 [DOI] [PubMed] [Google Scholar]
- 11. Al-Rawi S., Hinderlich S., Reutter W., Giannis A. (2004) Synthesis and biochemical properties of reversible inhibitors of UDP-N-acetylglucosamine 2-epimerase. Angew. Chem. Int. Ed. Engl. 43, 4366–4370 [DOI] [PubMed] [Google Scholar]
- 12. Stolz F., Reiner M., Blume A., Reutter W., Schmidt R. R. (2004) Novel UDP-glycal derivatives as transition state analogue inhibitors of UDP-GlcNAc 2-epimerase. J. Org. Chem. 69, 665–679 [DOI] [PubMed] [Google Scholar]
- 13. Zhu X., Stolz F., Schmidt R. R. (2004) Synthesis of thioglycoside-based UDP-sugar analogues. J. Org. Chem. 69, 7367–7370 [DOI] [PubMed] [Google Scholar]
- 14. Zeitler R., Giannis A., Danneschewski S., Henk E., Henk T., Bauer C., Reutter W., Sandhoff K. (1992) Inhibition of N-acetylglucosamine kinase and N-acetylmannosamine kinase by 3-O-methyl-N-acetyl-d-glucosamine in vitro. Eur. J. Biochem. 204, 1165–1168 [DOI] [PubMed] [Google Scholar]
- 15. Saxon E., Bertozzi C. R. (2000) Cell surface engineering by a modified Staudinger reaction. Science 287, 2007–2010 [DOI] [PubMed] [Google Scholar]
- 16. Mahal L. K., Yarema K. J., Bertozzi C. R. (1997) Engineering chemical reactivity on cell surfaces through oligosaccharide biosynthesis. Science 276, 1125–1128 [DOI] [PubMed] [Google Scholar]
- 17. Niederwieser A., Späte A. K., Nguyen L. D., Jüngst C., Reutter W., Wittmann V. (2013) Two-color glycan labeling of live cells by a combination of DielsAlder and click chemistry. Angew. Chem. Int. Ed. Engl. 52, 4265–4268 [DOI] [PubMed] [Google Scholar]
- 18. Möller H., Böhrsch V., Bentrop J., Bender J., Hinderlich S., Hackenberger C. P. (2012) Glycan-specific metabolic oligosaccharide engineering of C7-substituted sialic acids. Angew. Chem. Int. Ed. Engl. 51, 5986–5990 [DOI] [PubMed] [Google Scholar]
- 19. Du J., Meledeo M. A., Wang Z., Khanna H. S., Paruchuri V. D., Yarema K. J. (2009) Metabolic glycoengineering: sialic acid and beyond. Glycobiology 19, 1382–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tanner M. E. (2005) The enzymes of sialic acid biosynthesis. Bioorganic Chem. 33, 216–228 [DOI] [PubMed] [Google Scholar]
- 21. Paulsen H., Helpap B., Lorentzen J. P. (1987) Bausteine von Oligosacchariden, LXXXIII. Synthese der Repeating Unit der Teichuronsäure von Micrococcus luteus. Liebigs Ann. 10.1002/jlac.198719870349 [DOI] [Google Scholar]
- 22. Hartlieb S., Günzel A., Gerardy-Schahn R., Münster-Kühnel A. K., Kirschning A., Dräger G. (2008) Chemoenzymatic synthesis of CMP-N-acetyl-7-fluoro-7-deoxy-neuraminic acid. Carbohydr. Res. 343, 2075–2082 [DOI] [PubMed] [Google Scholar]
- 23. Schwartz E. L., Hadfield A. F., Brown A. E., Sartorelli A. C. (1983) Modification of sialic acid metabolism of murine erythroleukemia cells by analogs of N-acetylmannosamine. Biochim. Biophys. Acta 762, 489–497 [DOI] [PubMed] [Google Scholar]
- 24. Sarkar A. K., Fritz T. A., Taylor W. H., Esko J. D. (1995) Disaccharide uptake and priming in animal cells: inhibition of sialyl Lewis X by acetylated Gal β1←4GlcNAc β-O-naphthalenemethanol. Proc. Natl. Acad. Sci. U.S.A. 92, 3323–3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jones M. B., Teng H., Rhee J. K., Lahar N., Baskaran G., Yarema K. J. (2004) Characterization of the cellular uptake and metabolic conversion of acetylated N-acetylmannosamine (ManNAc) analogues to sialic acids. Biotechnol. Bioeng. 85, 394–405 [DOI] [PubMed] [Google Scholar]
- 26. Galuska S. P., Geyer H., Weinhold B., Kontou M., Röhrich R. C., Bernard U., Gerardy-Schahn R., Reutter W., Münster-Kühnel A., Geyer R. (2010) Quantification of nucleotide-activated sialic acids by a combination of reduction and fluorescent labeling. Anal. Chem. 82, 4591–4598 [DOI] [PubMed] [Google Scholar]
- 27. Martinez J., Nguyen L. D., Hinderlich S., Zimmer R., Tauberger E., Reutter W., Saenger W., Fan H., Moniot S. (2012) Crystal structures of N-acetylmannosamine kinase provide insights into enzyme activity and inhibition. J. Biol. Chem. 287, 13656–13665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Reinke S. O., Lehmer G., Hinderlich S., Reutter W. (2009) Regulation and pathophysiological implications of UDP-GlcNAc 2-epimerase/ManNAc kinase (GNE) as the key enzyme of sialic acid biosynthesis. Biol. Chem. 390, 591–599 [DOI] [PubMed] [Google Scholar]
- 29. Weihofen W. A., Berger M., Chen H., Saenger W., Hinderlich S. (2006) Structures of human N-acetylglucosamine kinase in two complexes with N-acetylglucosamine and with ADP/glucose: insights into substrate specificity and regulation. J. Mol. Biol. 364, 388–399 [DOI] [PubMed] [Google Scholar]
- 30. de Mol N. J., Fischer M. J. (2010) Surface Plasmon Resonance: Methods and Protocols, pp. 91–100, Springer Verlag GmbH, Heidelberg, Germany [Google Scholar]
- 31. Kim E. J., Sampathkumar S. G., Jones M. B., Rhee J. K., Baskaran G., Goon S., Yarema K. J. (2004) Characterization of the metabolic flux and apoptotic effects of O-hydroxyl- and N-acyl-modified N-acetylmannosamine analogs in Jurkat cells. J. Biol. Chem. 279, 18342–18352 [DOI] [PubMed] [Google Scholar]
- 32. Rillahan C. D., Antonopoulos A., Lefort C. T., Sonon R., Azadi P., Ley K., Dell A., Haslam S. M., Paulson J. C. (2012) Global metabolic inhibitors of sialyl- and fucosyltransferases remodel the glycome. Nat. Chem. Biol. 8, 661–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rillahan C. D., Brown S. J., Register A. C., Rosen H., Paulson J. C. (2011) High-throughput screening for inhibitors of sialyl- and fucosyltransferases. Angew. Chem. Int. Ed. Engl. 50, 12534–12537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Preidl J. J., Gnanapragassam V. S., Lisurek M., Saupe J., Horstkorte R., Rademann J. (2014) Fluorescent mimetics of CMP-Neu5Ac are highly potent, cell-permeable polarization probes of eukaryotic and bacterial sialyltransferases and inhibit cellular sialylation. Angew. Chem. Int. Ed. Engl. 53, 5700–5705 [DOI] [PubMed] [Google Scholar]
- 35. Oetke C., Hinderlich S., Brossmer R., Reutter W., Pawlita M., Keppler O. T. (2001) Evidence for efficient uptake and incorporation of sialic acid by eukaryotic cells. Eur. J. Biochem. 268, 4553–4561 [DOI] [PubMed] [Google Scholar]
- 36. Mendla K., Baumkötter J., Rosenau C., Ulrich-Bott B., Cantz M. (1988) Defective lysosomal release of glycoprotein-derived sialic acid in fibroblasts from patients with sialic acid storage disease. Biochem. J. 250, 261–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.