

Background: Several amyloid proteins form conformationally distinct aggregates.
Results: 23 antibodies raised against fibrillar Aβ42 display 18 unique reactivity profiles.
Conclusion: The immune response to fibrillar amyloid reflects the diversity in amyloid structures.
Significance: The use of a single antibody in immunization therapies of Alzheimer disease may not be effective, as it is unable to target all structural variants of Aβ.
Keywords: Alzheimer Disease, Amyloid, Monoclonal Antibody, Peptide Conformation, Protein Aggregation, Aβ
Abstract
Amyloidogenic proteins generally form intermolecularly hydrogen-bonded β-sheet aggregates, including parallel, in-register β-sheets (recognized by antiserum OC) or antiparallel β-sheets, β-solenoids, β-barrels, and β-cylindrins (recognized by antiserum A11). Although these groups share many common properties, some amyloid sequences have been reported to form polymorphic structural variants or strains. We investigated the humoral immune response to Aβ42 fibrils and produced 23 OC-type monoclonal antibodies recognizing distinct epitopes differentially associated with polymorphic structural variants. These mOC antibodies define at least 18 different immunological profiles represented in aggregates of amyloid-β (Aβ). All of the antibodies strongly prefer amyloid aggregates over monomer, indicating that they recognize conformational epitopes. Most of the antibodies react with N-terminal linear segments of Aβ, although many recognize a discontinuous epitope consisting of an N-terminal domain and a central domain. Several of the antibodies that recognize linear Aβ segments also react with fibrils formed from unrelated amyloid sequences, indicating that reactivity with linear segments of Aβ does not mean the antibody is sequence-specific. The antibodies display strikingly different patterns of immunoreactivity in Alzheimer disease and transgenic mouse brain and identify spatially and temporally unique amyloid deposits. Our results indicate that the immune response to Aβ42 fibrils is diverse and reflects the structural polymorphisms in fibrillar amyloid structures. These polymorphisms may contribute to differences in toxicity and consequent effects on pathological processes. Thus, a single therapeutic monoclonal antibody may not be able to target all of the pathological aggregates necessary to make an impact on the overall disease process.
Introduction
The misfolding of natively folded proteins into amyloid aggregates is associated with a broad range of pathology in humans. One of the most widely studied amyloidogenic peptides is the amyloid-β (Aβ)3 peptide. The aggregation of this peptide is known to be associated with the progression of Alzheimer disease (AD), which is the most common cause of dementia today (1, 2). One of the hallmarks of AD is the presence of extracellular Aβ plaques, which are composed of insoluble fibrillar aggregates of Aβ (3, 4). However, Aβ also forms soluble oligomeric species that are now widely believed to be the primary pathological species in AD (5, 6).
Antibodies against amyloid-forming peptides and aggregates have attracted considerable interest as potential therapeutic agents and for their utility as research tools. Many of the antisera and monoclonal antibodies produced against oligomeric and fibrillar forms of Aβ are directed against conformation-specific epitopes that are specifically associated with the aggregated state and are absent in normal proteins (7–10). We have produced antibodies that are able to distinguish between oligomeric and fibrillar amyloid aggregates. A11 is a rabbit polyclonal antiserum that recognizes prefibrillar oligomers of Aβ, as well as similar prefibrillar oligomers formed by other amyloidogenic peptides (8). A11 does not recognize monomeric peptides or amyloid fibrils. OC is a rabbit polyclonal antiserum that binds to fibrillar Aβ, along with similar structures formed by other amyloid-forming peptides, but it does not bind to monomers or prefibrillar oligomers (10, 11). The structural basis for the mutually exclusive aggregate specificities of A11 and OC is not entirely clear, but the structures that are known to react with A11 have been antiparallel β-sheets, β-barrels, and β-cylindrins (12–14), whereas the structures recognized by OC are known to be parallel, in-register β-sheets, whether they are associated with long fibrils or the smaller oligomers that may represent small segments of a fibril protofilament (15, 16).
Although amyloid structures may be classified into two general groups based on the presence of parallel in-register versus antiparallel β-sheet structures, it is increasingly evident that there is considerable structural diversity or polymorphism within each of these groups that may be analogous to strains in prion diseases. For instance, hydrogen/deuterium exchange studies (17), scanning proline mutagenesis experiments (18), and solid state nuclear magnetic resonance (NMR) data (19) have revealed that Aβ has the ability to form several distinct fibrillar structures with different morphologies depending on the aggregation conditions used in vitro (20). Similarly, other studies have reported the existence of distinct conformations of Aβ oligomers (21), as well as oligomers of other amyloid proteins (22). Of particular significance to this work, we have produced several A11-type monoclonal antibodies that are able to distinguish at least three immunologically unique conformations of Aβ prefibrillar oligomers (16).
The immune response to Aβ oligomers and fibrils is unusual in the sense that the resulting polyclonal sera are highly conformation-dependent, yet display generic sequence-independent immunoreactivity. Both A11 and OC display little or no reactivity with monomeric Aβ or with amyloid precursor protein (APP), and yet they react with oligomers and fibrils formed by a number of unrelated sequences (7, 8, 10). Individual monoclonal antibodies cloned from A11-producing rabbits also display generic sequence-independent immunoreactivity, indicating that the antibodies recognize generic epitopes on amyloid oligomers (16). These results appear to be inconsistent with results from humans vaccinated against Aβ42 fibrils, where the polyclonal antibodies predominantly react with the N-terminal segment of Aβ and preferentially react with Aβ monomer and not the aggregated forms of Aβ (23). Other studies reported that Aβ42-vaccinated humans produced antibodies that only react with aggregated forms of Aβ42 and not Aβ42 monomer or APP, which is consistent with our observations on Aβ42 fibril-vaccinated rabbits (24).
The goal of this study was to characterize the immune response to fibrillar Aβ42 in rabbits to determine the conformation dependence of the individual monoclonal antibodies and whether they uniquely react with any Aβ fibril polymorphisms. To accomplish this, we cloned as many different monoclonal antibodies as we could identify using an unbiased screen. Here, we report that a majority of the 23 unique mOC antibodies react with one or more linear segments of the Aβ peptide. Furthermore, most of the antibodies recognize linear epitopes in the N-terminal region of the Aβ sequence. We also observed that many of the epitopes displayed by the mOC antibodies were discontinuous in nature. In addition, our results show that many of the mOC antibodies that react with a linear Aβ sequence also recognize amyloid fibrils from unrelated sequences such as α-synuclein and islet amyloid polypeptide (IAPP). This suggests that reactivity with a linear Aβ segment may reflect the pervasive ability of short peptide segments to form amyloid-like structures rather than a specificity for the linear sequence per se. After performing Western blot and dot blot assays on 18 different Aβ40 and Aβ42 samples, we observed at least 18 different reactivity profiles among the 23 mOC antibodies. Finally, we find that some of the 23 mOC antibodies show differential reactivity with AD brain tissue versus healthy control brains, indicating that the different structures they recognize are pathologically relevant.
EXPERIMENTAL PROCEDURES
OC Monoclonal Antibody Production
mOC antibodies were made under contract by Epitomics (Burlingame, CA), using a fibrillar Aβ42 preparation as an antigen. To produce these antibodies, we immunized New Zealand White rabbits as described previously (10, 25, 26). Approximately 10,000 pools of hybridoma cells were screened against Aβ42 fibrils, prefibrillar oligomers, or monomeric Aβ, and 120 of these pools having absorbances of at least 3-fold above background in ELISAs were selected for further analysis. Secondary screening consisted of probing dot blots of a medium density array of 130 different amyloid preparations of Aβ42, Aβ40, IAPP, polyglutamine (polyQ) 40, overlapping 15 residue peptide segments of Aβ, and amyloid-forming random peptides. These hybridoma pools were also screened using immunohistochemistry on human AD and age-matched control brain tissues. Pools giving a unique pattern of immunoreactivity on the array or by immunohistochemistry were selected for cloning and further characterization. Antibodies were produced as hybridoma culture supernatants, and the antibody concentration ranged from 3 to 10 μg/ml.
Peptide Synthesis
Side chain protected fluorenylmethyloxycarbonyl (Fmoc) amino acids, Fmoc-PAL-PEG-polystyrene support, and O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate were purchased from Applied Biosystems Inc. (Carlsbad, CA). N,N-Diisopropylethylamine, dithiothreitol (DTT), thioanisole, ethanedithiol, and anisole were purchased from Aldrich. Trifluoroacetic acid (TFA) was purchased from Advanced Chem Tech Inc. (Louisville, KY). All solvents were of HPLC grade, and all chemicals were of Analar grade. Automatic synthesis was performed by the batchwise method on a CS336X (CS Bio, Inc., Menlo Park, CA) peptide synthesizer, employing Fmoc/t-butyl chemistry. Tetramethyluronium tetrafluoroborate/N,N-diisopropylethylamine was used as the coupling reagent for 1 h and 2% piperidine, 2% 1,8-diazabicyclo[5.4.0]undec-7-ene in dimethylformamide was used as the deprotection reagent for 7 min. Cleavage of the peptide from the resin support and the concomitant deprotection of the amino acid side chains were carried out in Reagent R (TFA/thioanisole/ethanediol/anisole at 90:5:3:2) at room temperature for 6 h. This step was followed by removal of the exhausted resin by filtration and precipitation of the peptide product in cold (4 °C) anhydrous ether. The precipitate was allowed to settle overnight at −20 °C and then washed three times with cold water and dried under high vacuum. Preparative reversed phase-HPLC was performed using a Waters system (model 510) with a Vydac C4 (214TP1022) column and a flow rate of 8 ml/min. The crude peptide was loaded after treatment with DTT and eluted using 0.1% TFA/H2O (buffer A) and 0.1% TFA/acetonitrile (buffer B) by gradient (5–95% buffer B). The center cut from the preparative run was frozen in liquid nitrogen immediately after collection and lyophilized under high vacuum.
SPOT Epitope Mapping Assay
The epitope-mapping SPOT assay was carried out as described previously (27). Whatman 50 cellulose support membranes with 40 spots containing an overlapping 10-amino acid-long fragment of the Aβ40 peptide with a C-terminal covalent bond to the membrane were obtained from JPT Peptide Technologies (Berlin, Germany). The membranes were rinsed with methanol for 5 min. The membranes were then washed three times for 10 min in Tris-buffered saline (TBS) (100 mm Tris, 370 mm NaCl, pH 7.5). After the TBS washes, the membranes were blocked for 2 h at room temperature in 10% nonfat dried milk in TBS containing 0.01% Tween 20 (TBS-T). The membranes were then incubated overnight at 4 °C with the appropriate antibody diluted in 5% nonfat dried milk in TBS-T (1:100 for mOC antibodies and 1:10,000 for 6E10 and 4G8 antibodies). The membranes were then washed three times for 5 min with TBS. Following the three washes, the membranes were incubated with the appropriate secondary antibody (anti-rabbit for mOC primary antibodies and anti-mouse for 6E10 and 4G8) diluted 1:10,000 in 5% nonfat dry milk in TBS-T for 2 h at room temperature. After three 5-min washes in TBS, the membranes were incubated in 3,3′,5,5′-tetramethylbenzidene (Promega, Madison, WI) until color development was completed and photographed. 4G8 and 6E10 antibodies were purchased from Covance (Princeton, NJ). Goat anti-mouse secondary antibody was purchased from Jackson ImmunoResearch (West Grove, PA), and goat anti-rabbit secondary antibody was purchased from Invitrogen.
Aβ40 and Aβ42 Aggregation Reactions
Aβ40 and Aβ42 were aggregated under three different conditions over a 10-day time course. Aggregation reactions were carried out in 1.6-ml (conditions A and C) or 2.0-ml (condition B) Eppendorf tubes at room temperature. For aggregation under condition A, 0.3 mg of the lyophilized peptide was resuspended in 33 μl of 100 mm NaOH and incubated for 10 min. This solution was then diluted to a 40 μm final concentration by adding 1.5 ml of 10 mm sodium phosphate buffer, pH 7.4. To aggregate the peptides under condition B, 0.5 mg of the lyophilized peptide was resuspended in 333.33 μl of hexafluoroisopropanol (HFIP) and incubated for 15 min. This solution was then diluted with 1.33 ml of deionized water, and the tube containing the solution was covered with a punctured cap and placed under a hood; this solution was continuously stirred during the aggregation time course using a stir plate. For aggregation under condition C, 0.3 mg of the peptide was resuspended in 33 μl of 100 mm NaOH and incubated for 10 min. This solution was then diluted to 40 μm by the addition of 1.5 ml HEPES/NaCl buffer (10 mm HEPES, 100 mm NaCl, pH 6.0). All of the solutions contained 0.02% sodium azide (NaN3).
α-Synuclein Aggregation Reaction
We aggregated α-synuclein (generous gift from Ralph Langen) over a 6-day time course. To initiate the aggregation reaction, we resuspended 0.3 mg of lyophilized α-synuclein in 200 μl of HFIP in an Eppendorf tube and allowed it to incubate for 15 min at room temperature. We then diluted this solution with 800 μl of deionized water, covered the tube containing the solution with a punctured cap, and placed the tube under a hood; this solution was continuously stirred during the aggregation period using a stir plate. The aggregation solution was treated with 0.02% NaN3.
IAPP Aggregation Reaction
We aggregated IAPP over a 6-day time course. 0.3 mg of IAPP was resuspended in 30 μl of 100 mm NaOH in an Eppendorf tube and allowed to incubate at room temperature for 25 min. This solution was then diluted by the addition of 1 ml of phosphate-buffered saline (PBS) and allowed to aggregate at room temperature for 6 days. The aggregation solution contained 0.02% NaN3.
Dot Blot Assay
1 μl of each sample was pipetted onto a Whatman nitrocellulose membrane (GE Healthcare) at the appropriate time points (time 0 and 3- and 6-day time points for the Aβ preparations, and time 0 and days 1–7 for the α-synuclein and IAPP preparations). After the last sample was deposited, the membranes were allowed to air dry and were subsequently blocked in 10% nonfat dried milk in TBS-T for 1 h at room temperature. The membranes were then incubated with 6E10 and 4G8 (Covance, Princeton, NJ) or with the 23 mOC hybridoma supernatants overnight at 4 °C. The antibodies were diluted to appropriate concentrations (1:10,000 for 4G8 and 6E10 and 1:100 for mOC hybridoma supernatants) in 5% nonfat dried milk in TBS-T. After three 5-min washes in TBS-T, the membranes were incubated with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibody (goat anti-mouse immunoglobulin G (IgG) for 6E10 and 4G8, and goat anti-rabbit IgG for mOC antibodies) for 1 h at room temperature. The secondary antibodies were diluted 1:10,000 in 5% nonfat dried milk in TBS-T. The blots were then washed three times for 5 min in TBS-T, and antibody reactivity was visualized using the enhanced chemiluminescence protocol (GE Healthcare). Images of the membranes were obtained using a Nikon D700 (Nikon Inc., Melville, NY) camera as described previously (28).
Western Blotting
10 μl of each sample from time 0 and the 3- and 10-day time points were mixed with 2× loading buffer (125 mm Tris, pH 6.8, 4% SDS, 16% glycerol, 10% 2-mercaptoethanol, bromphenol blue), and the resulting 20-μl mixtures were loaded onto an 18-well 4–12% precast Triton Gradient eXtended (TGX) gel (Life Science, Hercules, CA). The gel was then run at 250 V, and the resolved proteins were transferred onto a nitrocellulose membrane at 350 mA for 45 min. Nonspecific binding was blocked by incubating the membrane in 10% nonfat dried milk in TBS-T for 1 h at room temperature. The blot was then incubated in the appropriate primary antibody overnight at 4 °C. After three 5-min washes in TBS-T, the membranes were incubated with the appropriate secondary antibody for 1 h at room temperature. Following three 5-min washes in TBS-T, the results were visualized using the ECL protocol, and images of the results were obtained using a Nikon D700 camera as described previously for the dot blot experiments.
Immunostaining
Postmortem paraformaldehyde-fixed brain tissue was obtained from the neuropathological core of the University of California at Irvine Alzheimer's Disease Research Center. Case number 07-03 was selected because it demonstrates robust immunostaining when probed with several of the mOC antibodies. 14-Month-old 3×-Tg-AD brain was also examined to study the immunoreactivity of deposits in transgenic mice. 40-μm-thick sections of fixed tissue from Brodmann's area 9 of the cortex were obtained using a Vibratome Series 1000 vibrating microtome (The Vibratome Co., St. Louis). Sections were stored in PBS containing 0.02% NaN3 at 4 °C. Endogenous peroxidase activity was quenched by incubating the sections in 3% hydrogen peroxide and 3% methanol in TBS for 30 min at room temperature. Nonspecific background staining was blocked with a 1-h incubation in 2% bovine serum albumin (BSA) (Jackson ImmunoResearch) and 0.01% Triton X-100 in TBS at room temperature. Tissues were incubated with 1 mg/liter primary antibodies diluted in blocking solution (23 mOC, OC polyclonal, and 6E10) overnight at 4 °C. The sections were then washed two times for 5 min with 0.01% Triton X-100 in TBS, blocked for 30 min in blocking buffer, and incubated with the proper biotinylated secondary antibody, goat anti-rabbit for OC polyclonal and mOC antibodies and horse anti-mouse for 6E10 (Vector Laboratories, Inc., Burlingame, CA), for 1 h at room temperature. Anti-mouse secondary antibody was diluted 1:200 in blocking buffer containing a 1:75 dilution of normal horse serum (Vector Laboratories, Inc., Burlingame, CA). Similarly, anti-rabbit secondary antibody was diluted 1:200 in blocking buffer containing a 1:75 dilution of normal goat serum (Vector Laboratories). After incubation with the secondary antibodies, the tissue sections were washed two times for 5 min in 0.01% Triton X-100, and an ABC peroxidase kit and 3,3′-diaminobenzidine substrate kit (Vector Laboratories) were used to detect the biotinylated secondary antibodies. Following the 3,3′-diaminobenzidine incubation, the tissue sections were washed five times for 5 min in TBS and allowed to air dry. The sections were then dehydrated using sequential 3-min incubations in 50, 70, and 95% ethanol, followed by a 15-min incubation in 100% ethanol. The sections were then mounted, cover-slipped with DePeX (EMS, Hatfield, PA), and visualized using an Olympus BH-2 light microscope (Olympus America Inc., Center Valley, PA). The omission of either the primary or the secondary antibody was used as the negative control and resulted in no 3,3′-diaminobenzidine staining.
RESULTS
Antibody Production
The humoral immune response to any antigen includes the expansion of a great number of antibody-producing plasma cell clones and the resulting production of many distinct types of immunoglobulin (Ig) molecules by these cells (29, 30). This fact, together with our experience in producing conformation-specific monoclonal antibodies (16), led us to ask whether the immune response to an amyloid antigen can be used to establish the diversity of epitopes associated with fibrillar amyloid aggregates and their exposure on different fibril polymorphisms. To address this question, we immunized New Zealand White rabbits with a fibrillar Aβ42 preparation and produced a pool of ∼10,000 clones. 120 of these clones reacted to a significant degree with preparations of monomeric, fibrillar, or prefibrillar oligomeric Aβ42 in an ELISA for the primary screening step. 24 of these clones were then selected based on unique dot blot reactivity patterns with 130 different amyloid preparations made from Aβ40, Aβ42, polyQ, and IAPP, as well as 15-residue overlapping fragments of Aβ and short amyloid-forming peptides. After sequencing the heavy and light chains, it was found that two clones expressed identical sequences, resulting in 23 unique clones. We then used these OC-type monoclonal antibodies (mOCs) to study the immunological diversity of amyloid structures adopted by Aβ and other amyloid-forming peptides.
Linear Epitope Specificity
To determine whether the 23 mOC antibodies recognize linear segments (epitopes) of Aβ, and what these linear segments may be, we performed an epitope mapping experiment using a SPOT peptide array consisting of a series of overlapping 10-amino acid peptides beginning at −3 and ending at +44 of the Aβ sequence (27) that vary by a single amino acid. The linear epitope for each antibody was defined as the sequence of consecutive amino acids common to the adjacent spots showing positive reactivity with the antibody of interest. Using this definition, we observe that 19 of the 23 mOC antibodies display linear epitopes, although four fail to recognize a linear segment of the Aβ peptide, suggesting that they recognize conformational epitopes not represented in short peptide segments (Fig. 1). Furthermore, 17 of the 19 antibodies recognize a linear Aβ epitope localized to amino acids 3–12 of the N-terminal region of the Aβ peptide similar to the commercially available mouse monoclonal antibody 6E10. This dominance of N-terminal epitopes is consistent with results that have been previously reported for mouse polyclonal antibodies against Aβ (31) and the fact that human polyclonal antibodies from Aβ42 fibril-vaccinated individuals can be adsorbed by a peptide consisting of residues 1–8 of Aβ (23). In addition, six of the antibodies have discontinuous epitopes, consisting of an N-terminal domain and a central domain from residues 17 to 24 (Table 1). mOC78 recognizes three distinct segments of Aβ. Three of the antibodies have epitopes located in the region from residues 17 to 22, like that of 4G8. None of the antibodies cloned react with C-terminal epitopes, although we specifically screened for such antibodies by using Aβ40 and Aβ42 and short peptides ending at residues 40 and 42. Four antibodies (mOC9, mOC29, mOC88, and mOC104) failed to recognize any linear segment in the array. Although the N-terminal and central epitopes overlap substantially, there are numerous differences in the exact location and extent of the linear epitope in both regions for the individual monoclonal antibodies.
FIGURE 1.

SPOT assay. Left, epitope mapping results for the 19 mOC antibodies that reacted with Aβ segments in the SPOT assay, along with 6E10 and 4G8. Right, interpretation of the SPOT assay results. The sequence of the Aβ peptide is shown beginning with the −3 position. The apparent epitope is the sequence contained in common by all positive spots and is shown in red.
TABLE 1.
Characterization of mOC antibody epitopes
An antibody was described as having a linear epitope if it recognized consecutive 10-amino acid-long fragments of the Aβ peptide in the SPOT epitope mapping assay. An epitope was said to be discontinuous if it consisted of two or more linear epitopes separated by one or more intervening amino acids. Antibodies were described as having a generic epitope if they reacted with aggregates of proteins other than Aβ in addition to the Aβ preparations.

mOC Antibodies Display a Range of Immunological Selectivity for Distinct Aβ Aggregates
To further explore differences in immunoreactivity, Aβ40 and Aβ42 were aggregated under three different conditions for a 10-day time course, and the reactivity of each preparation was examined using a dot blot assay. Representative results for eight antibodies are shown in Fig. 2, and the results for all antibodies are shown in Fig. 3. The experiments were performed three times, and the results shown were obtained at least two times. No antibody, even the widely used commercially available 4G8 and 6E10 antibodies, recognizes all samples under all conditions, demonstrating that epitopes are commonly hidden or unavailable under some conditions. mOC87 has the broadest reactivity, comparable with 4G8, although mOC9 and 76 did not react with any of the samples on dot blots. m31 only recognizes Aβ40 aggregation under condition B for 10 days. mOC3 appears to be Aβ42-specific because it does not react with Aβ40 under any condition, but it also fails to recognize Aβ42 under condition B, indicating that its epitope is only displayed by Aβ42 under specific conditions. Several of the antibodies only recognize specific time points, indicating that the exposure of the epitope changes as aggregation ensues. Based on the dot blot results, the mOC antibodies can be divided into 15 groups in the same manner, with each group recognizing a unique subset of the 18 Aβ samples (Table 2).
FIGURE 2.
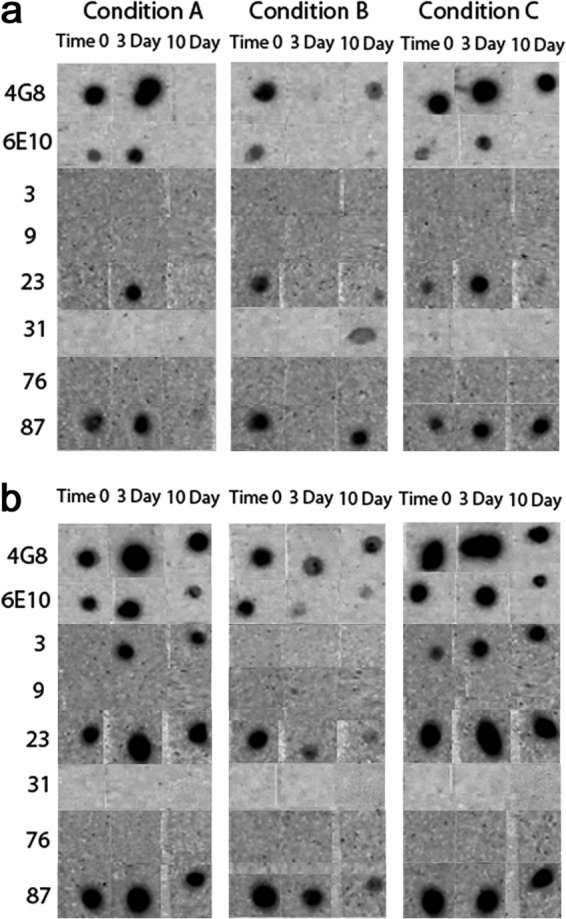
Representative dot blot results. Aβ40 (a) and Aβ42 (b) were aggregated under three different conditions over a 10-day time period. The immunological reactivities of the aggregates from time 0, and the 3- and 10-day time points were then tested with the 23 mOC antibodies, along with 6E10 and 4G8. Results obtained with antibodies 6E10, 4G8, and mOC 3, 9, 23, 31, 76, and 87 are displayed as representative examples. The three aggregation conditions were as follows: Condition A, peptide resuspended in 100 mm NaOH and diluted in phosphate buffer; Condition B, peptide resuspended in HFIP and diluted in water; Condition C, peptide resuspended in 100 mm NaOH and diluted in HEPES/NaCl buffer.
FIGURE 3.

Complete dot blot data. Aβ40 (a) and Aβ42 (b) were aggregated under three different conditions over a 10-day time period. 1-μl aliquots were pipetted onto nitrocellulose membranes at time 0 and at the 3- and 10-day time points. The membranes were then probed with the 23 mOC antibodies, along with 6E10 and 4G8. The three aggregation conditions were as follows: Condition A, peptide resuspended in 100 mm NaOH and diluted in phosphate buffer; Condition B, peptide resuspended in HFIP and diluted in water; Condition C, peptide resuspended in 100 mm NaOH and diluted in HEPES/NaCl buffer.
TABLE 2.
Dot blot immunological profiles of mOC antibodies based on reactivity with different Aβ samples
Check marks indicate reactivity of a sample with the indicated mOC antibody in dot blot assays. Abbreviations used are as follows: 40, Aβ40; 42, Aβ42; A, peptide aggregated using condition A; B, peptide aggregated using condition B; C, peptide aggregated using condition C; t0, time 0; 3d, 3-day time point; 10d, 10-day time point.

Because none of the antibodies react with Aβ dot blots under all conditions and times, we examined whether performing Western blot analysis using SDS-PAGE would expose possible hidden epitopes and reveal selectivity for distinct sizes of aggregates. To answer this question, we carried out Western blots of the same 18 samples used for dot blots. Representative results for eight antibodies are shown in Fig. 4, and the results for all antibodies are shown in Fig. 5, a and b. These experiments revealed that the antibodies that did not react with any of the samples in the dot blot assay recognize one or more of the samples on Western blots following SDS-PAGE. For instance, although antibody mOC9 did not react with any of the 18 Aβ preparations in the dot blot assay, it recognized Aβ40 aggregated under condition B at the 10-day time point, as well as Aβ42 aggregated under condition C at all three time points in the Western blot assay. The Western blot experiments further demonstrated that subjecting the Aβ samples to SDS-PAGE changes the reactivity profiles of the mOC antibodies to these preparations. More subtle examples of epitope exposure upon SDS-gel electrophoresis were also observed. None of the antibodies detectably reacted with Aβ40 aggregated under condition B for 3 days on dot blots, although several recognize the same preparation at time 0 and 10 days. On Western blots, all of the antibodies except 4G8 recognize high molecular weight material at 10 days, although only mOC23 and mOC87 recognize monomeric Aβ40 at all time points.
FIGURE 4.

Representative Western blots results. Aβ40 (a) and Aβ42 (b) were aggregated under three different conditions over a 10-day time period. Aliquots from time 0 and the 3- and 10-day time points were used for Western blotting using the 23 mOC antibodies, along with 4G8 and 6E10. Results obtained using 6E10, 4G8, and antibodies mOC 3, 9, 23, 31, 76, and 87 are displayed here as representative examples. The three aggregation conditions were as follows: Condition (Cond.) A, peptide resuspended in 100 mm NaOH and diluted in phosphate buffer; Condition B, peptide resuspended in HFIP and diluted in water; Condition C, peptide resuspended in 100 mm NaOH and diluted in HEPES/NaCl buffer.
FIGURE 5.
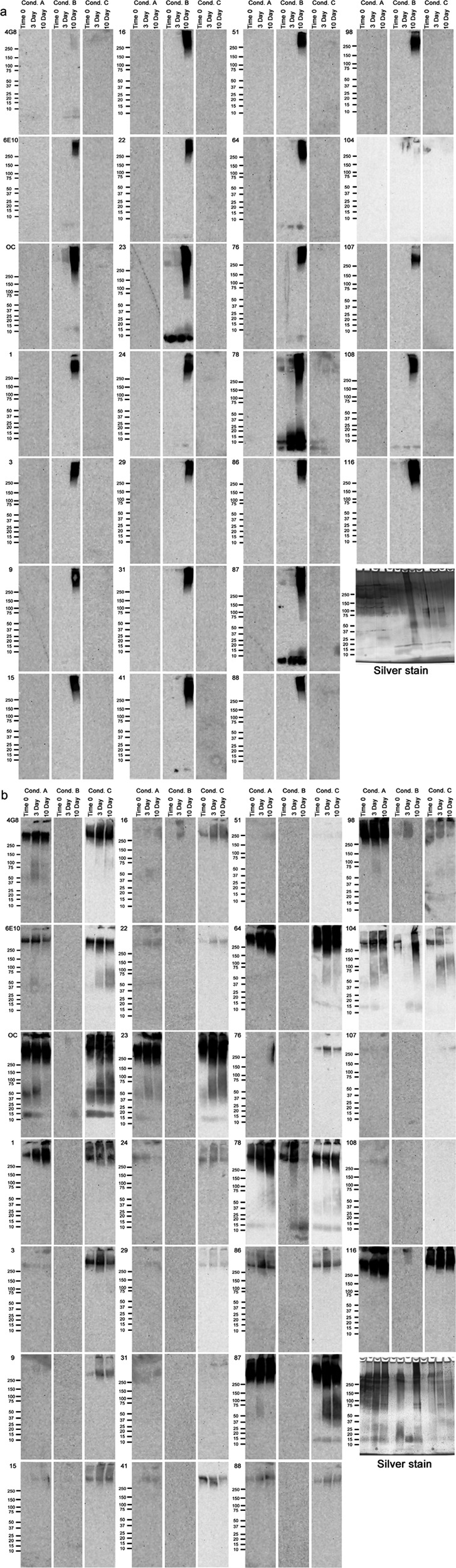
Complete Western blot data. Aβ40 (a) and Aβ42 (b) were aggregated under three different conditions over a 10-day time period. Aliquots from time 0 and the 3- and 7-day time points were used for Western blotting using the 23 mOC antibodies, along with 4G8 and 6E10. The three aggregation conditions were as follows: Condition (Cond.) A, peptide resuspended in 100 mm NaOH and diluted in phosphate buffer; Condition B, peptide resuspended in HFIP and diluted in water; Condition C, peptide resuspended in 100 mm NaOH and diluted in HEPES/NaCl buffer.
Many of the antibodies that recognized samples on dot blots do not recognize the same sample on Western blots. For example, 4G8, 6E10, mOC23, and mOC87 recognize Aβ40 incubated under conditions A and C at one or more time points on dot blots, but none of the antibodies recognize Aβ40 aggregated under these conditions on Western blots at any time point (Figs. 2a and 4a). SDS-PAGE treatment of the Aβ samples alters the reactivity profiles of some of the 23 mOC antibodies and 4G8 and 6E10, indicating that these antibodies recognize diverse epitopes that vary in their exposure depending on the aggregation conditions and the conditions used to detect them. Taken together, the dot blot and Western blot results define 18 reactivity profile groups within the 23 mOC antibodies, with each group having a distinct reactivity profile against the Aβ preparations tested here (Tables 2 and 3).
TABLE 3.
Western blot immunological profiles of mOC antibodies based on reactivity with different Aβ samples
Check marks indicate reactivity of a sample with the indicated mOC antibody in Western blot assays. Abbreviations used are as follows: 40, Aβ40; 42, Aβ42; A, peptide aggregated using condition A; B, peptide aggregated using condition B; C, peptide aggregated using condition C; t0, time 0; 3d, 3-day time point; 10d, 10-day time point.

Silver staining of the gels indicates that roughly equivalent amounts of peptide are present, although the aggregation state varies with incubation time and aggregation condition (Fig. 5, a and b). For instance, Aβ40 aggregated under condition A mostly forms aggregates of 75 kDa and higher, with bands of oligomeric Aβ at 37 and 25 kDa appearing at the 3- and 10-day time points, respectively. In addition, we see distinct bands of high molecular weight aggregates in this preparation at all time points. Similarly, Aβ40 aggregated under condition C forms aggregates of a wide range of molecular weights with more prominent monomer and dimer bands, as well as two oligomeric bands at roughly 65 and 75 kDa. These aggregates are present at all three time points tested, with slight variations in their relative amounts. In contrast, aggregating Aβ40 under condition B leads to the formation of monomeric and dimeric Aβ, and oligomeric Aβ of 75 kDa and higher at time 0- and the 3-day time point, with a dramatic increase in the range of oligomeric Aβ species and the appearance of a prominent oligomer band at roughly 50 kDa at the 10-day time point. Interestingly, although we clearly observed aggregated peptide when Aβ40 is aggregated under conditions A and C, the majority of the antibody reactivity that we observed on Western blots was restricted to Aβ40 aggregated under condition B. When Aβ42 was aggregated under condition A, we observed a broad range of oligomers throughout the reaction time course, with distinct Aβ oligomers of around 15 and 75 kDa, along with distinct high molecular mass bands at all three time points. Similarly, when Aβ42 was aggregated under condition 3, we observed a broad range of oligomers at all three time points, with several distinct oligomer bands and a weak monomer signal with small variations in intensity at the three different time points. In contrast, aggregating Aβ42 under condition C leads to dramatic changes in the composition of the Aβ species detected by silver staining. Specifically, we observed a weak monomer band, an oligomer band around 15 kDa, and distinct high molecular mass bands at all three time points and a broad range of oligomeric aggregates at time 0 and the 10-day time point. This broad oligomer reactivity is largely absent at the 3-day time point, and the 15-kDa band appeared to show much more intense staining. Intriguingly, and in contrast to our observations in the Aβ40 Western blotting experiments, we observed much broader antibody reactivity with Aβ42 aggregated under conditions A and C compared with the preparations formed under condition B. It is also important to note that the intensity with which the individual mOC antibodies react with the aggregated Aβ is not always proportional to the abundance of the specific Aβ species in the overall mixture as seen in silver stains. This observation indicates that the mOC antibodies are indeed conformation-specific and react with distinct structural variants of Aβ within the aggregation mixture. Furthermore, although aggregating Aβ often leads to the formation of several distinct oligomeric bands under different conditions, these bands are generally not read by the mOC antibodies.
Thermal Modulation of Aβ Epitopes
To further investigate the Aβ epitopes, we examined the effect of boiling the membranes prior to Western blotting. We observed that the recognition of the epitopes varied significantly, as demonstrated by three distinct responses to thermal denaturation. The first group of antibodies did not show a significant change in their reactivity pattern after the membrane was boiled and reprobed. An example from this group is antibody 86, which reacts with high molecular weight aggregates formed in the three Aβ42 preparations with no change observed in response to heat denaturation (Fig. 6). This observation indicates that antibody 86 and other members of this group recognize thermo-stable epitopes in these samples. In the second group, most or all of the immunoreactivity was lost after heat denaturation. A representative from this group of antibodies, antibody 87, only maintained its reactivity with the high molecular weight aggregates that it recognizes and lost all of its reactivity to monomeric and oligomeric species of Aβ42 after the membrane was boiled (Fig. 6). These results suggest that the epitope that antibody 87 reacts with is more labile in the low molecular weight aggregates formed by Aβ42 than in the high molecular weight aggregates in these preparations. Finally, in the third group, some of the mOC antibodies only react with the Aβ42 aggregates after they have been subjected to heat denaturation. As an example, antibody 104 reacts with high molecular weight aggregates formed within the three Aβ42 preparations with a pattern similar to the pattern observed with antibody 86; however, this reactivity is only observed after the aggregates have undergone heat denaturation and is absent before the membrane is boiled (Fig. 6). These observations suggest that the epitope recognized by antibody 104 is hidden and is only unmasked and available for binding by the antibody after heat is used as an antigen retrieval mechanism.
FIGURE 6.

mOC antibodies differ in their binding to Aβ fibrils after heat denaturation. Four different fibrillar preparations of Aβ42 were subjected to Western blotting using the 23 mOC antibodies with and without heat denaturation of the membrane prior to the blocking step. Here, we present three representative examples of mOC antibodies with differing responses to the heat denaturation of Aβ42 fibrils.
Several mOC Antibodies Recognize Generic and Sequence-independent Amyloid Epitopes although Others Appear to Be Aβ-specific
Because the OC polyclonal serum from which the monoclonals are derived recognizes amyloid fibrils formed from several different amyloidogenic sequences, we examined whether the individual monoclonal antibodies display this property. To answer this question, we aggregated α-synuclein and IAPP over a 6-day time course and tested the reactivity of the aggregates at time 0, and the 1-, 2-, and 4–6-day time points in a dot blot assay using the 23 mOC antibodies (Fig. 7). None of the antibodies reacted strongly with monomeric α-synuclein or IAPP, but several antibodies displayed strong reactivity with both sequences upon aggregation. We found that six of the 23 mOC antibodies were able to recognize aggregates formed by one or both of these peptides. Specifically, antibodies 3, 22, 78, 87, and 116 recognized both α-synuclein and IAPP aggregates, although antibody 76 recognized the α-synuclein aggregate at the 6-day time point. These experiments indicate that these six antibodies display a generic epitope that is present in a variety of amyloid structures that have little to no sequence identity with Aβ. All of these generic epitope antibodies except 78 map to a single and contiguous sequence of Aβ, indicating that recognition of a linear epitope is not a reliable indicator of sequence specificity. To verify that the binding of the antibodies with a generic amyloid epitope was truly specific to an amyloid conformation, we performed an antibody competition assay wherein the mOC antibodies of interest were incubated with Aβ42 fibrillar aggregates prior to performing Western blots on the reactive α-synuclein and IAPP preparations. As expected, the preincubation of the antibodies with Aβ42 fibrils eliminated reactivity with the α-synuclein and IAPP sample (data not shown). It would be interesting to know if these antibodies also recognize PrPsc, but we did not examine this possibility due to safety concerns.
FIGURE 7.

Dot blot assays of IAPP and α-synuclein aggregates. α-Synculein (left) and IAPP (right) were aggregated over a 6-day time period. The immunoreactivities of samples from time 0 and the 1-, 2-, and 4–6-day time points were tested using the 23 mOC antibodies in a dot blot assay. Antibodies that showed positive reactivity with the α-synuclein and IAPP preparations are shown. IAPP was resuspended in 100 mm NaOH and diluted in PBS, and α-synculein was resuspended in HFIP and diluted in water.
mOC Antibodies Recognize Distinct Types of Amyloid Deposits in Human AD and Transgenic Mouse Brain
We performed immunohistochemistry on human and transgenic mouse brain sections using each of the 23 antibodies to test their abilities to recognize amyloid deposits in brain. Selected images are shown in Fig. 8, and all of the images are shown in Fig. 9, a and b. The mOC antibodies display a broad range of staining morphologies and intensities with little to no staining of WT or control brains that have no amyloid deposits (data not shown). Although most of the antibodies recognize amyloid aggregates in these experiments, 9 of the 23 antibodies failed to significantly stain any of the AD brain samples that stain intensely with other antibodies. Antibodies mOC 9, 15, 29, 41, 51, 88, 104, 107, and 108 failed to stain human brain amyloid deposits even though they react with aggregated Aβ in vitro by Western blotting (Fig. 9a). We tested whether antigen retrieval with formic acid treatment of the sections before immunostaining would uncover a hidden epitope for these antibodies, as SDS-PAGE treatment had done in the in vitro experiments with synthetic Aβ, but in most cases, we were still not able to observe any specific staining with these antibodies after formic acid treatment (data not shown). However, we did observe intracellular staining of human AD brain tissue with antibody 88 after formic acid treatment (data not shown).
FIGURE 8.
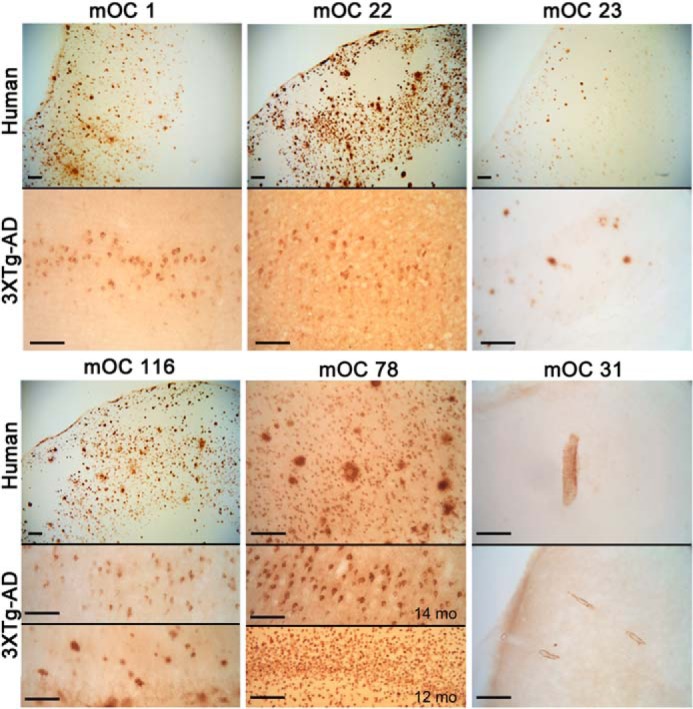
Differential staining of human AD and 3×Tg-AD mouse brain. Sections were stained with the indicated mOC antibodies. Top panels, human AD brain. Bottom panels, 14-month-old 3×Tg-AD mouse brain, except as indicated. Bars, 100 μm.
FIGURE 9.
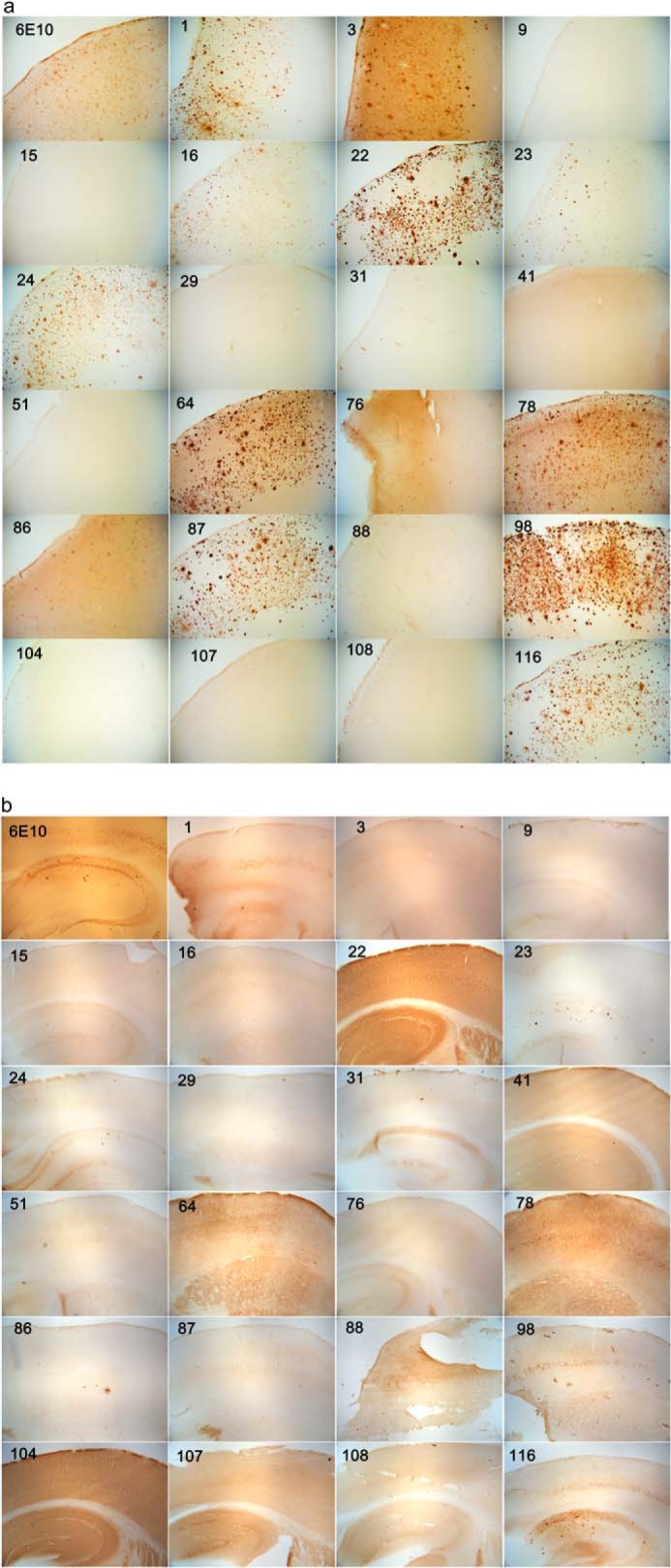
Immunostaining of human AD brain (a) and 3×TgAD mouse brain (b) with mOC antibodies. The 40-μm-thick serial sections from 14-month-old 3×TgAD mice were stained with the 23 mOC antibodies and 6E10. Some of the antibodies reacted with intracellular aggregates; some were specific to extracellular plaques, and some recognized vascular amyloid. For antibodies showing reactivity with more than one type of aggregate, we have included images representative of reactivity with each type of aggregate. Magnification: ×40.
The mOC antibodies identify morphologically and spatially unique types of amyloid deposits in both human AD and 3×Tg-AD mouse brain (Fig. 8). No specific staining was observed in normal control brain or the nontransgenic mouse brain, indicating that the mOC antibodies do not react with APP (data not shown). mOC 1, 22, 23, and 116 all stain parenchymal plaques in the AD frontal cortex, but mOC23 stains a subset of plaques that are morphologically smaller and more spherical than the plaques stained by the other antibodies (Fig. 8). The same antibodies exhibit a different staining pattern in 14-month-old 3×Tg-AD transgenic mice. mOC1 stains a subset of neurons in layer V of the frontal cortex and plaques in the subiculum, but it does not stain plaques in the hippocampus. mOC22 also stains a subset of neurons in layer V, but it additionally stains a subset of pyramidal neurons in CA1. mOC23 stains only a subset of plaques in human and 3×Tg-AD brain but not intraneuronal deposits in the 3×Tg-AD brain that stain with mOC1 and mOC22. mOC116 stains abundant plaques in human brain and plaques and intraneuronal deposits in 3×Tg-AD mice. In contrast, 6E10 exhibits much more extensive intraneuronal labeling (Fig. 9, a and b). The intraneuronal staining by 6E10 is widely interpreted as representing APP and APP C-terminal fragments (32), but the fact that the conformation-dependent aggregation-specific mOC antibodies only stain a subset of these 6E10-positive neurons suggests that the material accumulating in the mOC-positive neurons is misfolded and aggregated. Several of the antibodies that stain parenchymal plaques in human frontal cortex fail to stain plaques in 3×Tg-AD brain but rather stain intraneuronal deposits. These include mOC antibodies 64, 87, and 98 (Fig. 9, a and b). This suggests that the plaques in 3×-TgAD mice are immunologically distinct from human AD plaques.
Two antibodies identify unique amyloid deposits in human and transgenic brain. mOC78 stains intracellular and nuclear deposits in some AD brains (Fig. 8) and at 12 months in 3×Tg-AD mice. The intracellular 78 staining co-localizes with 6E10 staining (data not shown). As reported previously, mOC31 specifically stains vascular amyloid deposits, indicating that these deposits are structurally distinct (Fig. 8) (33). The immunohistochemical data obtained using human AD and 14-month-old 3×Tg-AD mouse brains are summarized in Table 4.
TABLE 4.
Summary of mOC antibody reactivity in human AD frontal cortex and 14-month-old 3×Tg-AD mouse brain sections
| Antibody name | Human AD brain reactivity | 14-Month-old 3×Tg-AD mouse brain reactivity |
|---|---|---|
| 1 | Frontal cortex plaques | Layer V cortical neurons, subiculum plaques |
| 3 | Frontal cortex plaques | NA |
| 9 | NA | NA |
| 15 | NA | NA |
| 16 | Subset of frontal cortex plaques | NA |
| 22 | Frontal cortex plaques | Layer V cortical and CA1 pyramidal neurons |
| 23 | Subset of frontal cortex plaques | Hippocampal plaques |
| 24 | Frontal cortex plaques | Hippocampal plaques |
| 29 | NA | NA |
| 31 | Vascular amyloid | Vascular amyloid |
| 41 | NA | NA |
| 51 | NA | NA |
| 64 | Frontal cortex plaques | NA |
| 76 | Subset of frontal cortex plaques | NA |
| 78 | Intracellular/nuclear, frontal cortex plaques | Layer V cortical neurons |
| 86 | Frontal cortex plaques | Cortical plaques |
| 87 | Frontal cortex plaques | Layer V cortical neurons |
| 88 | NA | NA |
| 98 | Frontal cortex plaques | Layer V cortical neurons |
| 104 | NA | Layer V cortical, CA1, and CA3 neurons |
| 107 | NA | NA |
| 108 | NA | NA |
| 116 | Frontal cortex plaques | Layer V cortical neurons, hippocampal plaques |
DISCUSSION
Recently, there have been several important studies suggesting significant structural polymorphism among fibrillar amyloid aggregates (17, 34–36). Here, we have used the rabbit humoral immune response to a fibrillar Aβ42 amyloid antigen as a tool in studying the structural diversity of β-amyloid fibrils. Based on Western blot and dot blot results with the synthetic Aβ40 and Aβ42 preparations (Figs. 2–5), we were able to classify the mOC antibodies into 18 distinct groups, each with a unique immunological reactivity profile (Tables 2 and 3). This remarkably heterogeneous pool of monoclonal antibodies is a direct reflection of the inherent diversity of amyloid structures present in fibrillar Aβ and is in general agreement with estimates of fibril structural polymorphism by hydrogen/deuterium exchange and solid-state NMR experiments (17, 34).
Our results demonstrate that the mOC antibodies and the commonly used commercially available mouse monoclonal antibodies, 6E10 and 4G8, react with amyloid aggregates in a conformation-dependent fashion. One indicator of the conformation dependence of the antibodies is the fact that they only recognize samples under specific conditions and aggregation times and not other conditions and times. None of the antibodies recognize Aβ aggregated under all conditions and at all times, indicating that epitopes are exposed or hidden differentially in different structures. The fact that thermal denaturation of the Aβ aggregates after SDS-PAGE treatment alters the reactivity profiles of these antibodies is further evidence of their conformational specificity and the fact that hidden epitopes can be revealed by changing the conformation. Antibody mOC23 provides an illustration of the conformation dependence of the mOC antibodies. Although this antibody reacts with a wide range of Aβ40 and Aβ42 aggregates on dot blots, it reacts with a significantly more restricted set of the Aβ samples in Western blots (Figs. 2 and 4 and Tables 2 and 3). 6E10 and 4G8 monoclonal antibodies are generally regarded as “sequence-specific” Aβ antibodies because they react with Aβ monomer and APP and require antigen retrieval for optimal immunoreactivity in some assays (37). However, these antibodies also display a strong preference for Aβ aggregates and react differentially with Aβ preparations used in this study in the absence of heat denaturation (Figs. 2 and 4). In this respect, 6E10 and 4G8 are much like many of the antibodies reported here; they react in both conformation- and sequence-dependent manners with amyloid aggregates. This appears to be a general property of antibodies produced against amyloid aggregates.
Only a few of the mOC antibodies recognize Aβ monomer on Western blots, and those that do stain monomer do not react with monomer under all conditions examined, suggesting that monomeric Aβ can adopt alternative structures that the antibodies distinguish. Although some of the antibodies recognize oligomeric species, none of them is specific for a particular size of oligomer, and all of them stain high molecular weight aggregates at the top of the gel, indicating that the epitope is represented in a broad size range of aggregates. Because of the strong immunoreactivity with high molecular weight aggregates, some of the antibodies appear to be specific for Aβ42, but this is a reflection of the higher aggregation propensity of Aβ42, as the antibodies do not recognize a C-terminal epitope, and they do react with Aβ40 under some aggregation conditions.
A majority (17/23) of mOC antibodies recognize a linear segment in the N-terminal region of Aβ as reported previously for human antibodies generated in response to vaccination in human clinical trials (23). This has been interpreted as indicating that the antibodies are not conformation-dependent or -specific to a particular aggregation state (23). Our results confirm preference for N-terminal epitopes but indicate that virtually all of these antibodies are conformation-dependent as described above. Moreover, the mapping to a linear segment of Aβ is not a reliable indicator of the sequence specificity of the antibodies, as is commonly believed. Several of the mOC antibodies that map to a linear Aβ segment surprisingly react in a generic, conformation-dependent fashion with amyloid fibrils formed from unrelated sequences, like α-synuclein and IAPP (Fig. 7). Thus, their reactivity with short peptides may be more a reflection of the propensity of short peptides to form amyloid-like structures containing generic fibril epitopes than an underlying specificity for the particular sequence.
Immunological subtypes of amyloid also occur in AD and transgenic brain. A conservative estimate is that the mOC antibodies define at least four types of amyloid deposits as follows: two types of parenchymal deposits, intranuclear immunoreactivity and a unique population of vascular amyloid. Antibodies mOC 1, 22, and 116 all stain abundant cortical plaques in humans, although mOC23 and 6E10 stain a much more restricted subset of the plaques. mOC78 stains intraneuronal and intranuclear deposits in a subset of the brain samples, and mOC31 only stains vascular amyloid, indicating that vascular amyloid is structurally distinct. A large number of the antibodies do not display any specific staining of brain tissue, although they all work well on Aβ in vitro. These include mOC 9, 15, 29, 41, 51, 107, and 108. This is consistent with the interpretation that Aβ is able to adopt a larger number of conformations in vitro than occur in vivo. This may be due to the fact that a larger number of different conditions may be probed in vitro and the fact that some of the unique epitopes are detected on kinetically transient species that may not be significantly populated in vivo.
Taken together, our results indicate that all of the antibodies examined only recognize Aβ under specific conditions. Because of this, claims of “pan” Aβ immunoreactivity should be viewed with some suspicion unless independently verified. Otherwise, the immunoreactivity should be optimized for the particular condition or for multiple different antibodies employed to avoid observing only a subpopulation of the amyloid Aβ present.
Several clinical and pre-clinical studies have investigated the use of monoclonal antibodies against Aβ in passive immunization as a therapeutic strategy for the prevention and/or treatment of AD with mixed results. Several different antibodies have been demonstrated to reduce amyloid deposition and improve cognitive function in transgenic animals (38–44). However, clinical trials using humanized versions of some of these antibodies have failed to meet their primary clinical end points in phase III clinical trials. Our results indicate that no single antibody is capable of recognizing all of the different aggregation states of Aβ, suggesting that this may contribute to the lack of clinical effectiveness. To target all of the different immunological subtypes of Aβ, it may be necessary to use several different anti-amyloid monoclonal antibodies. In view of the difficulty and expense of defining an effective mixture of monoclonal antibodies in human clinical trials, it may be more effective to focus efforts on the development of safer active vaccination approaches to stimulate the polyclonal immune response (45, 46).
This work was supported, in whole or in part, by National Institutes of Health Grants AG 033069 and AG00538. This work was also supported by a grant from the Cure Alzheimer's fund.
- Aβ
- amyloid-β
- AD
- Alzheimer disease
- APP
- amyloid precursor protein
- IAPP
- islet amyloid polypeptide
- Fmoc
- fluorenylmethyloxycarbonyl
- HFIP
- hexafluoroisopropanol.
REFERENCES
- 1. Querfurth H. W., LaFerla F. M. (2010) Alzheimer's disease. N. Engl. J. Med. 362, 329–344 [DOI] [PubMed] [Google Scholar]
- 2. Hebert L. E., Scherr P. A., Bienias J. L., Bennett D. A., Evans D. A. (2003) Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch. Neurol. 60, 1119–1122 [DOI] [PubMed] [Google Scholar]
- 3. Katzman R., Saitoh T. (1991) Advances in Alzheimer's disease. FASEB J. 5, 278–286 [PubMed] [Google Scholar]
- 4. Serrano-Pozo A., Frosch M. P., Masliah E., Hyman B. T. (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harbor Perspect. Med. 1, a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hardy J., Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 6. Haass C., Selkoe D. J. (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112 [DOI] [PubMed] [Google Scholar]
- 7. Yang A. J., Knauer M., Burdick D. A., Glabe C. (1995) Intracellular Aβ1–42 aggregates stimulate the accumulation of stable, insoluble amyloidogenic fragments of the amyloid precursor protein in transfected cells. J. Biol. Chem. 270, 14786–14792 [DOI] [PubMed] [Google Scholar]
- 8. Kayed R., Head E., Thompson J. L., McIntire T. M., Milton S. C., Cotman C. W., Glabe C. G. (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 [DOI] [PubMed] [Google Scholar]
- 9. Lambert M. P., Viola K. L., Chromy B. A., Chang L., Morgan T. E., Yu J., Venton D. L., Krafft G. A., Finch C. E., Klein W. L. (2001) Vaccination with soluble Aβ oligomers generates toxicity-neutralizing antibodies. J. Neurochem. 79, 595–605 [DOI] [PubMed] [Google Scholar]
- 10. Kayed R., Head E., Sarsoza F., Saing T., Cotman C. W., Necula M., Margol L., Wu J., Breydo L., Thompson J. L., Rasool S., Gurlo T., Butler P., Glabe C. G. (2007) Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stroud J. C., Liu C., Teng P. K., Eisenberg D. (2012) Toxic fibrillar oligomers of amyloid-β have cross-β structure. Proc. Natl. Acad. Sci. 109, 7717–7722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yoshiike Y., Kayed R., Milton S. C., Takashima A., Glabe C. G. (2007) Pore-forming proteins share structural and functional homology with amyloid oligomers. Neuromolecular Med. 9, 270–275 [DOI] [PubMed] [Google Scholar]
- 13. Cerf E., Sarroukh R., Tamamizu-Kato S., Breydo L., Derclaye S., Dufrêne Y. F., Narayanaswami V., Goormaghtigh E., Ruysschaert J. M., Raussens V. (2009) Antiparallel β-sheet: a signature structure of the oligomeric amyloid β-peptide. Biochem. J. 421, 415–423 [DOI] [PubMed] [Google Scholar]
- 14. Laganowsky A., Liu C., Sawaya M. R., Whitelegge J. P., Park J., Zhao M., Pensalfini A., Soriaga A. B., Landau M., Teng P. K., Cascio D., Glabe C., Eisenberg D. (2012) Atomic view of a toxic amyloid small oligomer. Science 335, 1228–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Head E., Pop V., Vasilevko V., Hill M., Saing T., Sarsoza F., Nistor M., Christie L. A., Milton S., Glabe C., Barrett E., Cribbs D. (2008) A two-year study with fibrillar β-amyloid (Aβ) immunization in aged canines: effects on cognitive function and brain Aβ. J. Neurosci. 28, 3555–3566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kayed R., Canto I., Breydo L., Rasool S., Lukacsovich T., Wu J., Albay R., 3rd, Pensalfini A., Yeung S., Head E., Marsh J. L., Glabe C. (2010) Conformation dependent monoclonal antibodies distinguish different replicating strains or conformers of prefibrillar Aβ oligomers. Mol. Neurodegener. 5, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kheterpal I., Chen M., Cook K. D., Wetzel R. (2006) Structural differences in Aβ amyloid protofibrils and fibrils mapped by hydrogen exchange–mass spectrometry with on-line proteolytic fragmentation. J. Mol. Biol. 361, 785–795 [DOI] [PubMed] [Google Scholar]
- 18. Williams A. D., Portelius E., Kheterpal I., Guo J.-T., Cook K. D., Xu Y., Wetzel R. (2004) Mapping Aβ amyloid fibril secondary structure using scanning proline mutagenesis. J. Mol. Biol. 335, 833–842 [DOI] [PubMed] [Google Scholar]
- 19. Petkova A. T., Yau W.-M., Tycko R. (2006) Experimental constraints on quaternary structure in Alzheimer's β-amyloid fibrils. Biochemistry 45, 498–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goldsbury C. S., Wirtz S., Müller S. A., Sunderji S., Wicki P., Aebi U., Frey P. (2000) Studies on the in vitro assembly of Aβ1–40: implications for the search for a β fibril formation inhibitors. J. Struct. Biol. 130, 217–231 [DOI] [PubMed] [Google Scholar]
- 21. Benilova I., Karran E., De Strooper B. (2012) The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357 [DOI] [PubMed] [Google Scholar]
- 22. Campioni S., Mannini B., Zampagni M., Pensalfini A., Parrini C., Evangelisti E., Relini A., Stefani M., Dobson C. M., Cecchi C., Chiti F. (2010) A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 6, 140–147 [DOI] [PubMed] [Google Scholar]
- 23. Lee M., Bard F., Johnson-Wood K., Lee C., Hu K., Griffith S. G., Black R. S., Schenk D., Seubert P. (2005) Aβ42 immunization in Alzheimer's disease generates Aβ N-terminal antibodies. Ann. Neurol. 58, 430–435 [DOI] [PubMed] [Google Scholar]
- 24. Hock C., Konietzko U., Papassotiropoulos A., Wollmer A., Streffer J., von Rotz R. C., Davey G., Moritz E., Nitsch R. M. (2002) Generation of antibodies specific for β-amyloid by vaccination of patients with Alzheimer disease. Nat. Med. 8, 1270–1275 [DOI] [PubMed] [Google Scholar]
- 25. Nussbaum J. M., Seward M. E., Bloom G. S. (2013) Alzheimer disease: a tale of two prions. Prion 7, 14–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nussbaum J. M., Schilling S., Cynis H., Silva A., Swanson E., Wangsanut T., Tayler K., Wiltgen B., Hatami A., Rönicke R., Reymann K., Hutter-Paier B., Alexandru A., Jagla W., Graubner S., Glabe C. G., Demuth H.-U., Bloom G. S. (2012) Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature 485, 651–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reineke U., Sabat R. (2009) in Epitope Mapping Protocols (Schutkowski M., Reineke U., eds) pp. 145–177, Humana Press Inc., Totowa, NJ [Google Scholar]
- 28. Khoury M. K., Parker I., Aswad D. W. (2010) Acquisition of chemiluminescent signals from immunoblots with a digital single-lens reflex camera. Anal. Biochem. 397, 129–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Murphy K., Travers P., Walport M. (2010) in Janeway's Immunobiology (Lawrence E., ed) 8th Ed., Taylor & Francis, Inc., New York [Google Scholar]
- 30. Fanning L. J., Connor A. M., Wu G. E. (1996) Development of the immunoglobulin repertoire. Clin. Immunol. Immunopathol. 79, 1–14 [DOI] [PubMed] [Google Scholar]
- 31. Spooner E. T., Desai R. V., Mori C., Leverone J. F., Lemere C. A. (2002) The generation and characterization of potentially therapeutic Aβ antibodies in mice: differences according to strain and immunization protocol. Vaccine 21, 290–297 [DOI] [PubMed] [Google Scholar]
- 32. Winton M. J., Lee E. B., Sun E., Wong M. M., Leight S., Zhang B., Trojanowski J. Q., Lee V. M. (2011) Intraneuronal APP, not free Aβ peptides in 3xTg-AD mice: implications for tau versus Aβ-mediated Alzheimer neurodegeneration. J. Neurosci. 31, 7691–7699 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33. McLean D., Cooke M. J., Albay R., 3rd, Glabe C., Shoichet M. S. (2013) Positron emission tomography imaging of fibrillar parenchymal and vascular amyloid-β in TgCRND8 mice. ACS Chem. Neurosci. 4, 613–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Petkova A. T., Leapman R. D., Guo Z., Yau W. M., Mattson M. P., Tycko R. (2005) Self-propagating, molecular-level polymorphism in Alzheimer's β-amyloid fibrils. Science 307, 262–265 [DOI] [PubMed] [Google Scholar]
- 35. Paravastu A. K., Leapman R. D., Yau W.-M., Tycko R. (2008) Molecular structural basis for polymorphism in Alzheimer's β-amyloid fibrils. Proc. Natl. Acad. Sci. 105, 18349–18354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Whittemore N. A., Mishra R., Kheterpal I., Williams A. D., Wetzel R., Serpersu E. H. (2005) Hydrogen-deuterium (H/D) exchange mapping of Aβ 1–40 amyloid fibril secondary structure using nuclear magnetic resonance spectroscopy. Biochemistry 44, 4434–4441 [DOI] [PubMed] [Google Scholar]
- 37. Kim K. S., Miller D. L., Sapienza V. J., Chen C. M. J., Bai C., Grundke-Iqbal I., Currie J. R., Wisniewski H. M. (1988) Production and characterization of monoclonal antibodies reactive to synthetic cerebrovascular amyloid protein. Neurosci. Res. Commun. 2, 121–130 [Google Scholar]
- 38. Bard F., Cannon C., Barbour R., Burke R. L., Games D., Grajeda H., Guido T., Hu K., Huang J., Johnson-Wood K., Khan K., Kholodenko D., Lee M., Lieberburg I., Motter R., Nguyen M., Soriano F., Vasquez N., Weiss K., Welch B., Seubert P., Schenk D., Yednock T. (2000) Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 6, 916–919 [DOI] [PubMed] [Google Scholar]
- 39. Schroeter S., Khan K., Barbour R., Doan M., Chen M., Guido T., Gill D., Basi G., Schenk D., Seubert P., Games D. (2008) Immunotherapy reduces vascular amyloid-β in PDAPP mice. J. Neurosci. 28, 6787–6793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mattson M. P., Chan S. L. (2003) Good and bad amyloid antibodies. Science 301, 1847–1849 [DOI] [PubMed] [Google Scholar]
- 41. Morgan D., Diamond D. M., Gottschall P. E., Ugen K. E., Dickey C., Hardy J., Duff K., Jantzen P., DiCarlo G., Wilcock D., Connor K., Hatcher J., Hope C., Gordon M., Arendash G. W. (2000) Aβ peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature 408, 982–985 [DOI] [PubMed] [Google Scholar]
- 42. Wilcock D. M., Rojiani A., Rosenthal A., Subbarao S., Freeman M. J., Gordon M. N., Morgan D. (2004) Passive immunotherapy against Aβ in aged APP transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J. Neuroinflammation 1, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Neff F., Wei X., Nölker C., Bacher M., Du Y., Dodel R. (2008) Immunotherapy and naturally occurring autoantibodies in neurodegenerative disorders. Autoimmun. Rev. 7, 501–507 [DOI] [PubMed] [Google Scholar]
- 44. Dodart J. C., Bales K. R., Gannon K. S., Greene S. J., DeMattos R. B., Mathis C., DeLong C. A., Wu S., Wu X., Holtzman D. M., Paul S. M. (2002) Immunization reverses memory deficits without reducing brain Aβ burden in Alzheimer's disease model. Nat. Neurosci. 5, 452–457 [DOI] [PubMed] [Google Scholar]
- 45. Rasool S., Albay R., 3rd, Martinez-Coria H., Breydo L., Wu J., Milton S., Misra S., Tran A., Pensalfini A., Laferla F., Kayed R., Glabe C. G. (2012) Vaccination with a non-human random sequence amyloid oligomer mimic results in improved cognitive function and reduced plaque deposition and micro hemorrhage in Tg2576 mice. Mol. Neurodegener. 7, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Goñi F., Prelli F., Ji Y., Scholtzova H., Yang J., Sun Y., Liang F. X., Kascsak R., Kascsak R., Mehta P., Wisniewski T. (2010) Immunomodulation targeting abnormal protein conformation reduces pathology in a mouse model of Alzheimer's disease. PLoS One 5, e13391. [DOI] [PMC free article] [PubMed] [Google Scholar]