

Background: Lipolysis contributes to adipose inflammation.
Results: Lipolysis up-regulates sphingosine kinase 1 (SphK1) in adipocytes. Modulation of SphK1 regulates adipose lipolysis-stimulated interleukin 6 production.
Conclusion: SphK1 plays a pivotal role in adipose inflammation.
Significance: Identification of a role for SphK1 in lipolysis-triggered adipose inflammation. Targeting SphK1 provides a novel intervention for adipose inflammation and associated metabolic syndromes.
Keywords: Adipose Tissue, AP1 Transcription Factor (AP-1), c-Jun N-terminal Kinase (JNK), Inflammation, Interleukin 6 (IL-6), Lipolysis, Beta 3 Adrenergic Receptor, Hormone-sensitive Lipase, Sphingosine Kinase
Abstract
Adipocyte lipolysis can increase the production of inflammatory cytokines such as interleukin-6 (IL-6) that promote insulin resistance. However, the mechanisms that link lipolysis with inflammation remain elusive. Acute activation of β3-adrenergic receptors (ADRB3) triggers lipolysis and up-regulates production of IL-6 in adipocytes, and both of these effects are blocked by pharmacological inhibition of hormone-sensitive lipase. We report that stimulation of ADRB3 induces expression of sphingosine kinase 1 (SphK1) and increases sphingosine 1-phosphate production in adipocytes in a manner that also depends on hormone-sensitive lipase activity. Mechanistically, we found that adipose lipolysis-induced SphK1 up-regulation is mediated by the c-Jun N-terminal kinase (JNK)/activating protein-1 signaling pathway. Inhibition of SphK1 by sphingosine kinase inhibitor 2 diminished the ADRB3-induced IL-6 production both in vitro and in vivo. Induction of IL-6 by ADRB3 activation was suppressed by siRNA knockdown of Sphk1 in cultured adipocytes and was severely attenuated in Sphk1 null mice. Conversely, ectopic expression of SphK1 increased IL-6 expression in adipocytes. Collectively, these data demonstrate that SphK1 is a critical mediator in lipolysis-triggered inflammation in adipocytes.
Introduction
Obesity is a global epidemic that is associated with numerous morbidities such as type 2 diabetes, cardiovascular diseases, hypertension, and certain types of cancers (1–5). The mechanisms that link obesity to disease are poorly understood. However, there is a growing appreciation that for certain individuals, obesity results in low grade chronic inflammation that largely emanates from the adipose tissue (6, 7) and results in systemic insulin resistance (8–10).
Adipose tissue is known to produce signaling molecules that regulate local and systemic proinflammatory responses (11). For example, adipose-derived production of proinflammatory cytokine interleukin-6 (IL-6) drives hepatic insulin resistance (12). However, the signaling pathways involved in adipocyte IL-6 release are not completely known. We previously reported that acute activation of β3-adrenergic receptors (ADRB3)5 triggers expression of proinflammatory genes, including IL-6, MCP-1, PAI-1, among others (13). ADRB3-mediated inflammation mimics inflammation produced by chronic treatments such as high fat feeding and thus offers a tractable model for investigating molecular mechanisms of adipose tissue inflammation. Importantly, this ADRB3-mediated inflammation depends on activation of hormone-sensitive lipase (HSL), suggesting involvement of lipolytic products as proinflammatory mediators.
Sphingosine 1-phosphate (S1P) is a serum-borne bioactive lipid mediator that regulates an array of biological activities in various cell types (14–17). S1P can function either as an extracellular ligand or intracellular mediator (18–20). Increasing evidence indicates that S1P plays important roles in proinflammatory responses. For example, S1P/S1P3 signaling is critical for the late stage proinflammatory responses in dendritic cells (21), whereas S1P/S1P2 signaling in macrophages and endothelial cells promotes vascular inflammation and atherosclerosis (22, 23). In addition, sphingosine kinases (SphKs), which phosphorylate sphingosine to form S1P, have been shown to be involved in many proinflammatory responses such as TNF-α-triggered inflammatory arthritis (24) and endotoxin-induced lung inflammatory injury (25).
In this study, we investigated whether sphingolipid signaling plays an important role in adipose proinflammatory responses that are triggered by lipolytic activation. Our data suggest that adipose lipolysis activates SphK1, which contributes to adipose proinflammatory signaling. Thus, targeting SphK1 may provide a novel means for modulating obesity-induced inflammation.
EXPERIMENTAL PROCEDURES
Reagents
Sphingosine 1-phosphate (BIOMOL) was resuspended in 4% fatty acid-free BSA (Sigma) to make a stock solution of 200 μm (26). Sphingosine kinase inhibitor 2 (SKI-2, Cayman Chemical), a selective inhibitor of SphK (27), was dissolved in dimethylformamide. SP 600125 and JNK inhibitor VIII were purchased from Cayman Chemical. Isoproterenol (specific agonist of β-adrenergic receptor) and CL-316243 (CL) (Sigma) were dissolved in H2O. BAY 59-9435 (BAY, dissolved in 0.5% methylcellulose), a highly selective HSL inhibitor (28–30), was chemically synthesized, as described (30). Novo 13f, another highly selective HSL inhibitor (31), was a generous gift of Dr. Christian Fledelius (Novo Nordisk A/S). Sphk1 antibodies were from Antibody Verify, Inc. (AAS67634C) or Cell Signaling Technology (no. 3297). SphK2 and c-Jun antibodies were from Santa Cruz Biotechnology (sc-22704) and Cell Signaling (no. 9165), respectively. IL-6 antibody was from Novus Biologicals (NB600-1131). AP-1 ChIP assay kit was from SABiosciences. Small interfering RNA (siRNA) oligonucleotides were purchased from Qiagen. Unless otherwise specified, chemicals and reagents were purchased from Sigma-Aldrich.
Cell Culture
3T3-L1 cells and 3T3-L1 cells stably expressing coxsackie and adenovirus receptor (3T3-L1-CAR) were cultured and differentiated as described previously (13, 32). Two days post-differentiation, cells were cultured overnight in serum-free DMEM. Subsequently, cells were treated with 10 μm of isoproterenol or H2O control. Alternatively, cells were pretreated with SKI-2 (5 μm) or BAY (10 μm) for 1 h, followed by stimulating with or without isoproterenol (10 μm) for additional 3 h. Cell pellets were collected, and mRNA or protein levels were measured by qPCR or Western blotting analysis.
Transduction of 3T3-L1-CAR Cells
Differentiated 3T3-L1-CAR cells in 12-well plates were transduced with 200 multiplicity of infection of adenoviral particles carrying SphK1 or β-galactosidase vector (a general gift of Dr. Timothy Hla, Cornell University School of Medicine) for 24 h in serum-free DMEM medium, as described previously (23, 33). Cells were then collected for RNA isolation and qPCR quantification.
Animal Studies
All animal procedures were performed according to the National Institutes of Health and institutional guidelines and were approved by the Wayne State University Animal Use and Care Committee. C57BL/6 (8-week-old male, Harlan Laboratories) and SphK1 null mice (on a C57BL/6 background) (34) were used in this study. To examine the role of ADRB3 signaling in the regulation of SphKs and IL-6 expression, mice (n = 6) were intraperitoneally (i.p.) injected with control H2O or 10 nmol of CL, the highly selective agonist of ADRB3 (28, 29), and tissues were harvested 3 h later as described (13, 29). To examine the role of HSL in the ADRB3-regulated SphKs and IL-6 expression, mice (n = 7–8) were pretreated with the selective HSL inhibitor BAY or methycelluose as described previously (28–30). To examine the effect of SphK inhibition on IL-6 expression, mice (n = 7–8) were pretreated with SphK inhibitor SKI-2 (20 mg/kg body weight) dissolved in 100 μl of dimethylformamide via intraperitoneal injection. Mice injected with dimethylformamide alone were used as a control. One hour later, mice were intraperitoneally injected with 10 nmol of CL or H2O, and tissues were harvested 3 h later. Mice were euthanized, epididymal white adipose tissues (EWAT) pads were collected and processed for real-time PCR analysis and Western blotting analysis.
RT-PCR and Real-time PCR
Total RNA was isolated from cultured cells and mouse EWAT using the RNeasy kit (Qiagen) and was reversely transcribed with an oligo-dT primer (Promega) by M-MLV reverse transcriptase (Promega) for first strand cDNA synthesis. For real-time PCR quantitation, 50 ng of reversely transcribed cDNAs were amplified with the ABI 7500 system (Applied Biosystems) in the presence of TaqMan DNA polymerase. PCR primer pairs used were as follows: mouse IL-6, 5′-AGTGG CTAAG GACCA AGACC-3′ (sense) and 5′-TCTGA CCACA GTGAG GAATG-3′ (antisense); mouse SphK1, 5′-TCTAC CTCCC GCCAT AAAA-3′ (sense) and 5′-CTCCT CCCCA CAACA AAAC-3′ (antisense); mouse SphK2, 5′-CCAAC AAGTG TCTCC TCCAA A-3′ (sense) and 5′-CCTCA GGGAT GTCAA AGTTC A-3′ (antisense); and mouse GAPDH, 5′-CACCT TCGAT GCCGG GGCTG-3′ (sense) and 5′-GGCCA TGAGG TCCAC CACCC-3′ (antisense). The qPCR reaction was performed by using a universal PCR Master Mix (Applied Biosystems) according to manufacturer's instructions. Relative quantification was calculated using the SDS software (Applied Biosystems) based on the following equation: relative quantification = 2−ΔΔCt, where Ct is the threshold cycle to detect fluorescence. Ct values were normalized to the internal GAPDH standard.
Sphingolipid Extraction
Differentiated 3T3-L1 cells were washed with PBS, changed to phenol-red free plain RPMI 1640, and treated with or without isoproterenol for 3 h. Culture media were added with 10 μl (1 μg/ml) of C17-sphingosine internal standard, followed by the addition of equal volume of extraction buffer (isopropanol:ethyl acetate = 15:85, v/v). Mixture was vortexed for 2 min and then centrifuged for 10 min at 4000 rpm. The upper phase was transferred to a new vial, and the aqueous phase was acidified with 100 μl of formic acid. The extraction process was repeated with the aqueous phase by adding another equal volume of extraction buffer, vortexing for 2 min, and centrifuging for 10 min. The upper organic phases from two extractions were combined and speed-vacuum dried. Lipid extracts were reconstituted with 50 μl of solvent A (2 mm NH4CO2H, 0.2% formic acid, in 90% H2O and 10% methanol) and 50 μl of solvent B (95% methanol, 5% H2O, 0.2% formic acid) for LC-MS/MS sphingolipid analysis as we described (23, 35).
LC-MS/MS Quantification
For LC-MS/MS analysis, reverse phase HPLC was performed using BDS HYPERSIL C8 columns (100 × 2.1 mm, 2.4 μm, Thermo Scientific) and gradient elution on Waters Alliance 2695 system (Waters Corp.). The mobile phase consisted of methanol, water, and ammonium formate. Solvent A was 2 mm ammonium formate in methanol with 0.2% formic acid. The column was equilibrated with solvent A for 5 min. Samples were injected using the autosampler maintained at 10 ± 2 °C. The injection volumes were 80 μl for each sample. A complete injection of each sample took 7 min, including column equilibration. The flow rate was 0.3 ml/min. The HPLC eluent was directly introduced to QuattroLC mass spectrometer (Micromass-Waters), equipped with an electrospray ion (ESI) source that was used for electrospray ion-MS/MS. The electrospray ion-MS/MS experiments for the quantitation of sphingolipids were performed as we prescribed (23, 33). Briefly, sphingolipid quantitation was carried out in the positive ion mode with ESI needle voltage, 2.8 kV; source block temperature, 120 °C; desolvation temperature, 350 °C; desolvation gas flow, 540 liters/h; nebulizer gas flow, 80 liters/h; and the collision gas pressure was 3.2 × 10−4 bar. Cone voltage and collision energy for each Multiple Reaction Monitoring transition were optimized. Chromatographic data were analyzed by Quanlynx module of the Masslynx software (Waters Corp.) to integrate the chromatograms for each Multiple Reaction Monitoring transition.
ELISA Measurement of IL-6 Polypeptides
The presence of IL-6 polypeptides in culture media of 3T3-L1 adipocytes was quantitated by ELISA analysis following manufacturer's instructions (BD Biosciences). Briefly, 50 μl of IL-6 polypeptide standard or culture medium was mixed with 50 μl of ELISA diluent. Mixture was loaded into each well of an ELISA plate, which was precoated with anti-mouse IL-6 monoclonal antibody. Reaction was kept at room temperature for 2 h. Subsequently, contents of wells were decanted, and wells were washed with wash buffer for five times. 100 μl of working detector solution was added to each well and kept at room temperature. One hour later, wells were washed for seven times with wash buffer (PBS with 0.05% Tween 20). One hundred μl of 3,3′,5,5′-tetramethylbenzidine One-Step substrate reagent was added to each well and kept in the dark at room temperature. Thirty minutes later, 50 μl of stop solution was added to each well, and absorbance at 450 nm was read within 30 min.
siRNA Transfection
Specific knockdown of SphK1 and SphK2 in 3T3-L1 adipocytes was achieved by employing the siRNA-mediated gene silencing technique, as described previously (36). siRNAs used were mouse si-SphK1 (SI01431283, Qiagen) and si-SphK2 (SI01431283). Non-targeting control siRNA (SI03650318, Qiagen) was used as a negative control. Differentiated 3T3-L1 adipocytes were trypsinized and replated (4 × 105 cells/well) in collagen-coated 12-well plates containing 50 nm of siRNA or non-targeting siRNA. siRNAs were delivered by the HiPerFect transfection reagent (Qiagen). Forty-eight hours later, culture media were changed, and cells were stimulated with 10 μm of isoproterenol for additional 3 h. Cell culture media were collected for ELISA analysis. Cell pellets were collected for RNA isolation, RT-PCR, and real-time PCR as mentioned above.
Immunofluorescence Staining
Cells were cultured in glass-bottomed Petri dish (MatTek, Ashland, MA). After treating with or without isoproterenol (10 μm), cultures were fixed with 4% paraformaldehyde at room temperature for 30 min. Cells were permeabilized with 0.05% Triton X-100 and blocked with 1% bovine serum albumin for 30 min. Subsequently, cells were incubated with primary antibody (1:100) followed by FITC-conjugated secondary antibody (1:500). Fluorescence images were captured by the Leica TCS SP5 confocal system (Leica, Wetzlar, Germany).
Statistical Analysis
Results are shown as mean ± S.D. Differences between various treatments were analyzed by Student's t test. p value < 0.05 was considered significant.
RESULTS
ADRB3/HSL Signaling Induces SphK1 Expression in Adipocytes and White Adipose Tissues
ADRB3 activation stimulates HSL-mediated lipolysis in adipocytes and white adipose tissues (28). We investigated whether sphingolipid signaling plays functional roles in the adipose lipolysis-mediated physiological responses. Initially, we examined whether ADRB3 activation regulates expression of SphKs in adipose tissue. Treatment with CL-316234 (CL), a specific agonist of ADRB3 receptors (28, 29), induced a dramatic increase of SphK1 expression in EWAT (∼120-fold increase) (Fig. 1A). CL did not alter levels of SphK2 mRNA. Western blotting analysis showed that CL administration profoundly increased SphK1 proteins in EWAT but did not change SphK2 protein levels (Fig. 1B). Subsequently, we examined whether the CL-induced SphK1 up-regulation in EWAT is dependent on HSL activity using BAY 59-9435, a highly selective inhibitor of HSL (28–30). As shown in Fig. 1, A and B, pretreatment with BAY completely abrogated the induction of SphK1 expression by CL.
FIGURE 1.
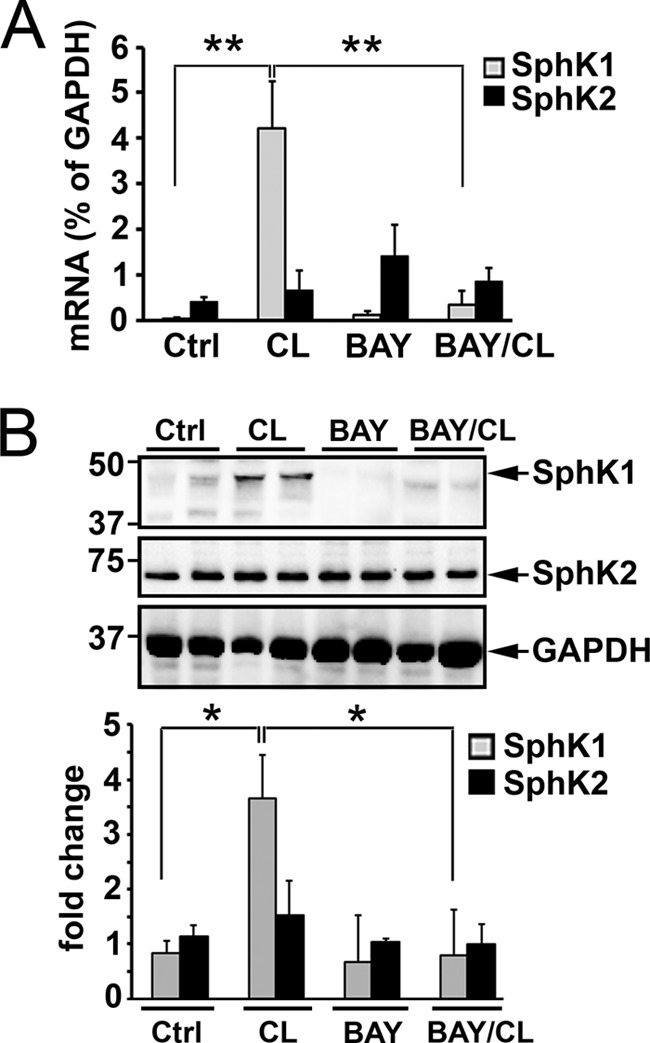
ADRB3/HSL signaling up-regulates SphK1 in white adipose tissues. Mice were injected (i.p.) with vehicle or BAY 59-9435 (BAY) for 1 h. Subsequently, mice were injected (i.p.) with CL-316243 (CL) or control saline. Three hours later, EWAT were collected, and levels of SphK1 and SphK2 were measured by qPCR (A) or Western blotting analysis (B). Note that mRNA and protein levels of SphK1 were dramatically up-regulated after CL-316243 administration. Also note that the CL-316243-increased SphK1 expression was completely abrogated by the pretreatment of BAY 59-9435. Data in A are means ± S.D. of six determinations (**, p < 0.01, t test). Fold change (lower panel of B), levels of SphK1 and SphK2 proteins were quantitated by NIH ImageJ software and normalized to GAPDH loading control (*, p < 0.05, t test, n = 4). Ctrl, control.
Next, cultured mouse 3T3-L1 adipocytes were used as an in vitro system to validate the observation that lipolysis induced SphK1 expression. Differentiated 3T3-L1 adipocytes were treated with or without isoproterenol (10 μm, 3 h), and expression of SphK1 and SphK2 was determined. We observed that isoproterenol treatment induced ∼3-fold increase of SphK1 expression (Fig. 2A). In contrast, isoproterenol had no effect on SphK2 expression (Fig. 2B). Similar to our observation in EWAT, isoproterenol-induced SphK1 expression was blocked by BAY treatment (Fig. 2A). To confirm the specific involvement of HSL in isoproterenol-induced SphK1 up-regulation, 3T3-L1 cells were pretreated with Novo 13f, another selective HSL inhibitor (31). As shown in Fig. 2C, isoproterenol-increased SphK1 levels were completed abrogated by Novo 13f treatment. In addition, Western blot analysis showed that isoproterenol treatment markedly increased SphK1 protein levels, and the isoproterenol-increased SphK1 protein expression was inhibited by the presence of BAY (Fig. 2, D and E). Neither isoproterenol nor BAY treatment altered levels of SphK2 proteins. SphKs are lipid kinases that catalyze the formation of S1P from sphingosine. Concomitant with SphK1 up-regulation, isoproterenol treatment increased S1P production and release into media by 5-fold (Fig. 2F).
FIGURE 2.
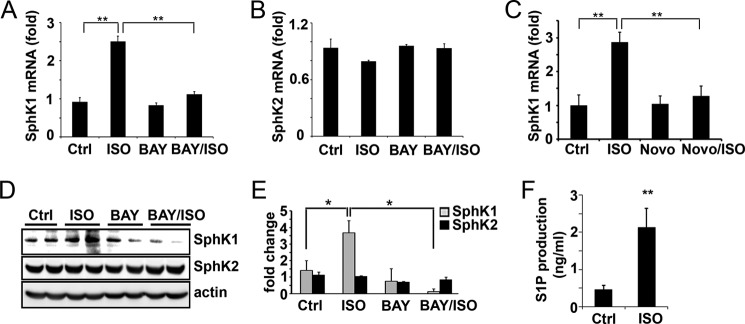
ADRB3/HSL signaling up-regulates SphK1 in cultured adipocytes. 3T3-L1 adipocytes were treated with or without isoproterenol, in the presence and absence of HSL inhibitor BAY. Levels of SphK1 (A) and SphK2 (B) were measured by qPCR or Western blot analysis (D and E). Note that isoproterenol (ISO) treatment significantly up-regulated SphK1 expression, and the isoproterenol-increased SphK1 expression was completely abrogated in the presence of BAY. D, SphK1 and SphK2 protein levels were quantitated by NIH ImageJ software and normalized to actin loading control (*, p < 0.05, n = 4, t test). C, 3T3-L1 adipocytes were pretreated with or without HSL inhibitor Novo 13f (Novo, 10 μm, 0.5 h) followed by stimulation in the presence or absence of isoproterenol. Levels of SphK1 were measured by qPCR. F, quantitation of S1P in adipocyte culture media by LC-MS/MS method. Note that S1P production was significantly increased in isoproterenol-treated cells. Data in A–C are means ± S.D. of triplicate determinations. Panels A-F were repeated at least twice with similar results. **, p < 0.01, t test. Ctrl, control.
ADRB3/HSL Up-regulates SphK1 via the JNK/AP-1 Signaling Pathway
We reported previously that ADRB3/HSL signaling activates stress kinases, including p38 and c-Jun N-terminal kinase (JNK) in adipocytes (13). Also, promoter analysis found several candidate binding sites of activator protein-1 (AP-1, a heterodimeric complex composed of proteins including c-Jun and c-Fos) transcriptional factor and c-Jun in the SphK1 promoter region (see the SABiosciences website, EpiTect ChIP qPCR Primers search). Thus, we examined whether ADRB3/HSL signaling mediated SphK1 up-regulation is controlled by the JNK/c-Jun pathway. ChIP analysis with the c-Jun antibody showed that isoproterenol increased c-Jun binding to the AP-1 site in the SphK1 promoter (Fig. 3A). Moreover, pharmacological inhibition of JNK activity with SP600125 greatly suppressed induction of SphK1 expression by isoproterenol (Fig. 3B). In contrast, pharmacological inhibition of p38 activity with SB203580 had no effect on the induction of SphK1 expression by isoproterenol. Moreover, treatment with another JNK inhibitor, JNK inhibitor VIII (38, 39), completely diminished the isoproterenol-induced SphK1 up-regulation (Fig. 3C). These data suggest that ADRB3/HSL signaling activates the JNK/AP-1 pathway, ultimately leading to transcriptional up-regulation of SphK1.
FIGURE 3.
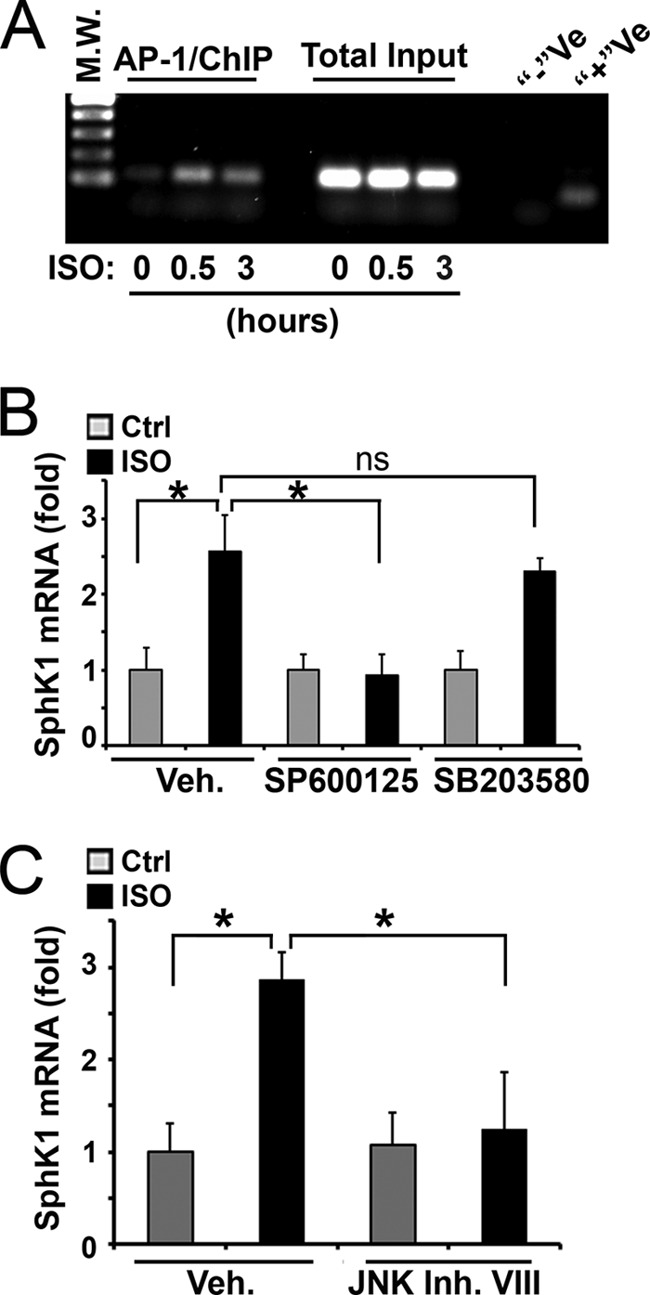
The JNK/c-Jun signaling pathway mediates the ADRB3/HSL signaling up-regulated SphK1 expression. A, differentiated 3T3-L1 adipocytes were treated with isoproterenol (10 μm) for indicated times. AP-1 ChIP assays were performed by amplifying the anti-c-Jun precipitates with primer pairs specific for AP-1 site in the SphK1 promoter region (SABiosciences, GPM1030068(−)02A). Total input, PCR amplification of total input chromatin, was used as a loading control. −Ve, negative control, immunoprecipitation was performed by using normal rabbit IgG. +Ve, GAPDH ChIP assay of anti-RNA polymerase II precipitates to ensure ChIP reaction was successful. M.W., molecular weight markers. B, 3T3-L1 cells were pretreated with inhibitor of JNK (SP600125, 10 μm) or p38 (SB203580, 10 μm) for 30 min, followed by stimulating with isoproterenol for 3 h. Levels of SphK1 were measured by qPCR analysis. C, qPCR quantitation of SphK1 in 3T3-L1 cells pretreated with JNK inhibitor VIII (JNK Inh. VIII, 10 μm, 0.5 h), followed by stimulating with isoproterenol for 3 h. Note that JNK inhibitors (SP600125 and JNK inhibitor VIII) completely abrogated the isoproterenol-increased SphK1 expression. Data are mean ± S.D. of triplicate determinations, which were repeated two times with similar results. *, p < 0.05; ns, not statistically significant; t test; Veh, vehicle; Ctrl, control.
SphK1 Plays a Critical Role in ADRB3/HSL-stimulated Expression of Proinflammatory Cytokine IL-6
In previous work, we showed that activation of ADRB3/HSL signaling in adipocytes in vivo and model adipocytes in vitro triggers acute changes in metabolism that alter patterns of gene expression, including inflammatory genes IL-6, CCL2, and PAI-1 (13). IL-6 is a key player in chronic inflammation, and levels of circulating IL-6 are elevated in several proinflammatory diseases. Moreover, plasma IL-6 concentrations are elevated in obesity (40, 41). Therefore, we examined the role of SphK1 in lipolysis-mediated IL-6 gene expression. As shown in Fig. 4, activation of ADRB3 pathway up-regulated IL-6 gene expression (Fig. 4A) and increased the appearance of IL-6 protein in adipocytes (Fig. 4B) and the release of the cytokine into media (Fig. 4C). Importantly, both ADRB3-triggered production and release of IL-6 were greatly suppressed by BAY treatment, indicating the requirement of HSL activity.
FIGURE 4.
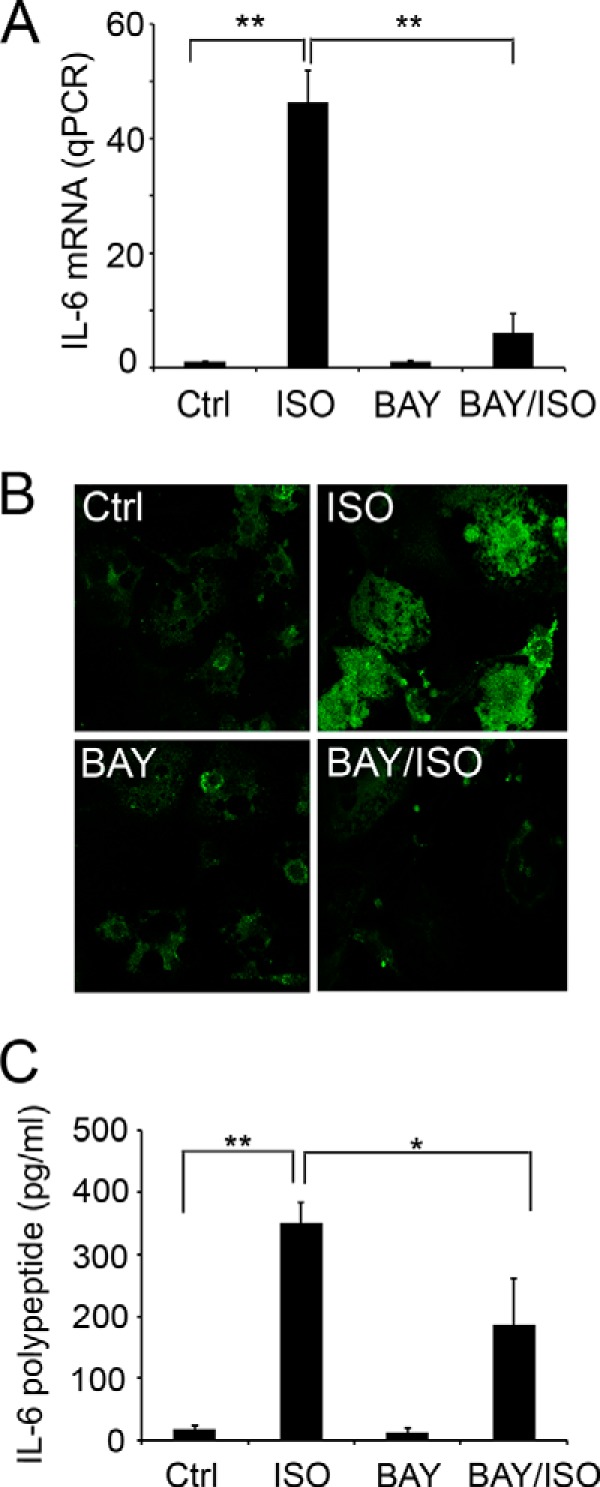
Activation of ADRB3/HSL signaling stimulates the expression of IL-6. A, 3T3-L1 cells were treated with or without isoproterenol in the presence and absence of HSL inhibitor BAY. Expression levels of IL-6 were measured by qPCR quantitation. B, immunofluorescent staining with anti-IL-6 showed that IL-6 expression was markedly increased after isoproterenol treatment and was inhibited by BAY. C, 3T3-L1 cells were treated with or without isoproterenol for 3 h, in the presence and absence of BAY. Levels of IL-6 proteins present in culture media were measured by ELISA. Note that isoproterenol treatment increased IL-6 in culture media, and BAY significantly diminished the isoproterenol-increased media IL-6. Data in A and C are mean ± S.D. of triplicate determinations (**, p < 0.01; *, p < 0.05; t test). Experiments were repeated at least two times with similar results. Ctrl, control.
We next investigated whether lipolysis-activated SphK1 is required for ADRB3/HSL signaling-induced IL-6 production in adipocytes and white adipose tissues. 3T3-L1 cells were treated with or without isoproterenol in the presence and absence of SKI-2 (4-[[4-(4-chlorophenyl)-2-thiazolyl]amino]phenol), a selective inhibitor of SphKs (27). IL-6 levels were markedly up-regulated in isoproterenol-treated adipocytes, and ∼65% of the isoproterenol-induced IL-6 was inhibited in the presence of SKI-2 (p < 0.05, n = 3) (Fig. 5A). We also examined the role of SphK1 in lipolysis-stimulated IL-6 production in mouse white adipose tissue. Mice were injected with control vehicle or SKI-2 for 1 h, followed by injection of CL or saline, and the expression level of IL-6 was measured by qPCR. As shown in Fig. 5B, IL-6 was dramatically up-regulated by CL administration in vivo (n = 8, p < 0.01). SKI-2 pretreatment inhibited ∼80% of CL-stimulated IL-6 expression (n = 4, p < 0.05) (Fig. 5B). These in vitro and animal experiments, utilizing SKI-2, strongly suggest that SphK1 plays an important role in the lipolysis-induced pro-inflammatory cytokine IL-6 expression.
FIGURE 5.
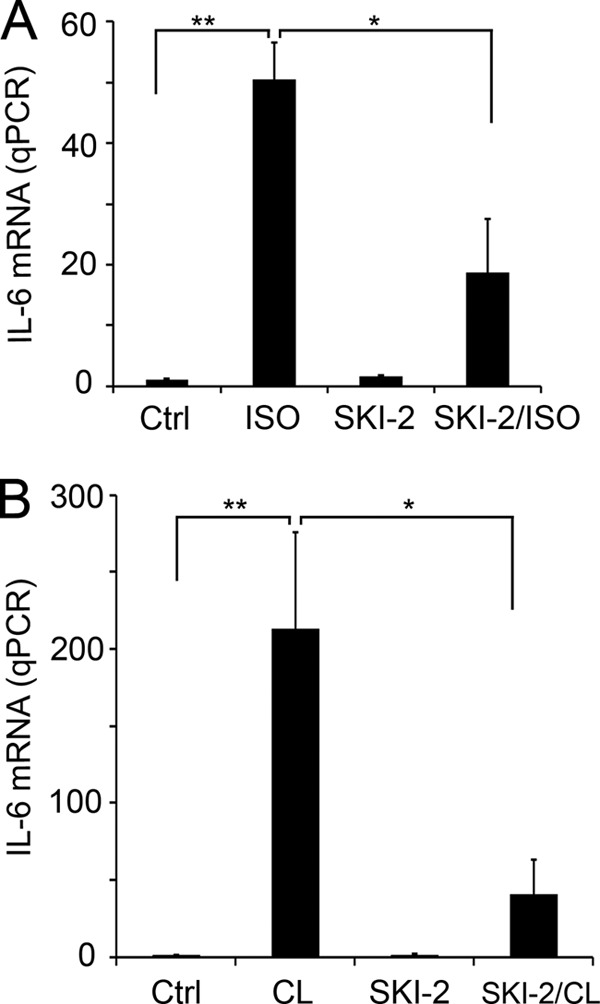
SKI-2, a pharmacological inhibitor of SphK, diminishes the lipolysis-stimulated IL-6 expression. Pretreatment of SKI-2 (5 μm, 1 h) significantly inhibited the ADRB3 activation increased expression of IL-6 mRNA in cultured adipocytes (A) and in EWAT in animals (B). **, p < 0.01; *, p < 0.05; t test. Ctrl, control.
To further validate the role of SphK1 in the adipose lipolysis-induced IL-6 expression, we examined the effects of selective SphK1 or SphK2 knockdown in 3T3-L1 adipocytes. Compared with cells transfected with non-target siRNA, SphK1 and SphK2 siRNAs selectively reduced expression of the targeted mRNAs by ∼50% under basal and stimulated conditions (Fig. 6, A and B). Knockdown of SphK1, but not SphK2, reduced induction of IL-6 mRNA and proteins by isoproterenol stimulation (Fig. 6, C and D).
FIGURE 6.
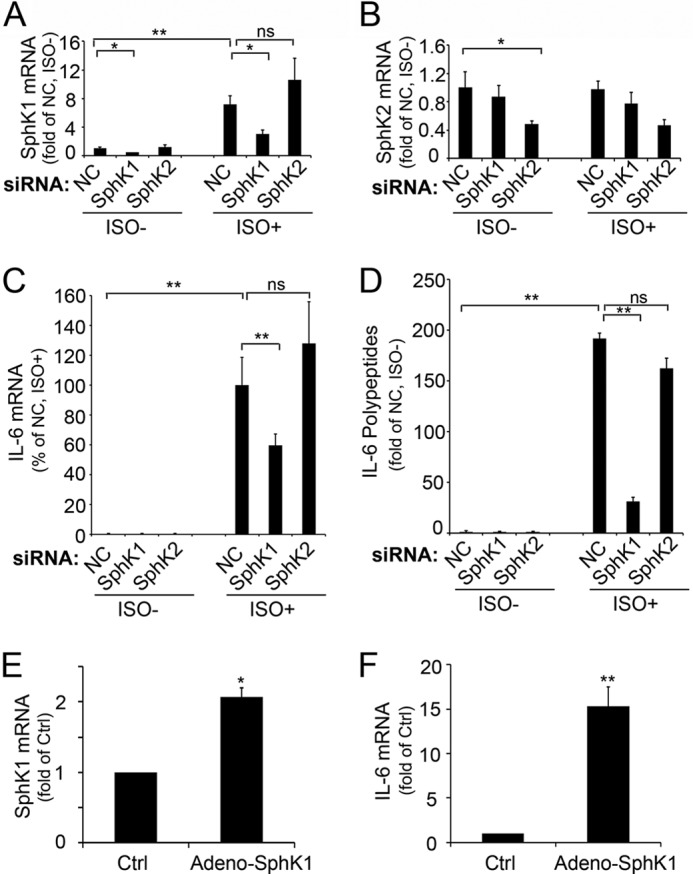
SphK1 contributes to ADRB3/HSL enhanced IL-6 expression. 3T3-L1 cells were transfected with sequence specific si-SphK1, si-SphK2, or non-targeting control (NC) siRNA oligonucleotides (50 nm, Invitrogen). Cells were stimulated with or without isoproterenol (10 μm) for 3 h at 48 h post-transfection. Cell pellets were collected for qPCR quantitation of expression levels of SphK1 (A), SphK2 (B), and IL-6 (C). Cultural media were used for IL-6 measurement by ELISA analysis (D). Note that SphK1 was profoundly knocked down in cells transfected with si-SphK1, compared with that in cells transfected with non-targeting control siRNA or si-SphK2 and that isoproterenol-induced SphK1 up-regulation was diminished specifically in cells transfected with si-SphK1 (A). Similarly, SphK2 expression was specifically knocked down in cells transfected with si-SphK2 (B). Also note that isoproterenol-induced IL-6 mRNA (C) and proteins (D) were specifically inhibited in cells transfected with si-SphK1. In contrast, transfection with si-SphK2 had no effect on isoproterenol-enhanced IL-6 expression (C and D). E and F, 3T3-L1-CAR cells were transduced with a multiplicity of 200 of adenoviral particles carry SphK1 (adeno-SphK1) or β-galactosidase (control; Ctrl). Expression levels of SphK1 (E) or IL-6 (F) were measured by qPCR analysis. Note that ectopic expression of SphK1 significantly increased IL-6 expression. Data are mean ± S.D. of triplicate determinations (**, p < 0.01; *, p < 0.05; ns, not statistically significant; t test). All experiments were repeated at least twice with similar results.
We also examined the effects of adenoviral overexpression of SpkK1 in 3T3-L1-CAR cells. SphK1 was ectopically expressed in 3T3-L1-CAR cells to a level comparable with that induced by isoproterenol treatment (Fig. 6E). As shown in Fig. 6F, ectopic expression of SphK1 alone was able to stimulate IL-6 production in the absence of isoproterenol treatment.
Finally, we examined the role of SphK1 in lipolysis-mediated IL-6 production in wild-type and SphK1 null mice (34). As shown in Fig. 7, A and B, CL treatment increased expression of SphK1 and IL-6 by 31- and 160-fold, respectively, in wild-type mice. Knock-out of SphK1 reduced CL-mediated induction of IL-6 mRNA by two-thirds (Fig. 7B) and completely eliminated up-regulation of IL-6 protein expression (Fig. 7, C and D). Collectively, these data demonstrate that SphK1 is a critical regulator of lipolysis-stimulated IL-6 production in adipocytes.
FIGURE 7.
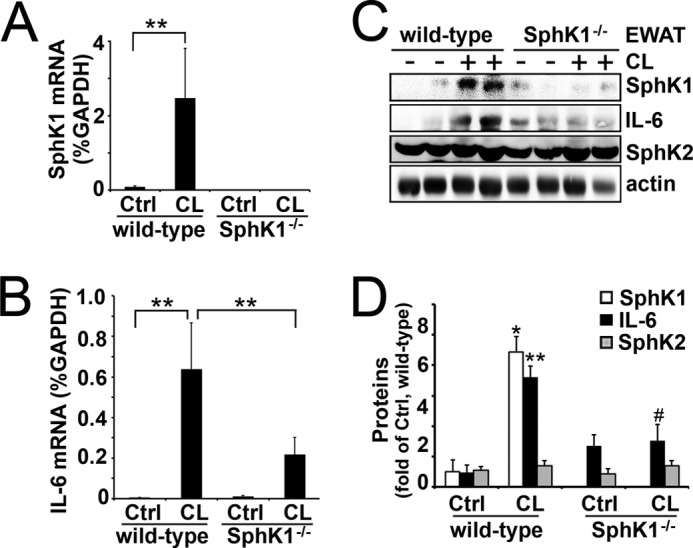
SphK1 is required for lipolysis-triggered IL-6 production in animals. Wild-type and SphK1 null mice (SphK1−/−) were injected (i.p.) with CL 316243. Three hours later, EWAT were collected, and levels of SphK1 (A) and IL-6 (B) were measured by qPCR and Western blot analysis (C and D). Note that SphK1 and IL-6 were dramatically up-regulated in wild-type mice after CL-316243 administration. Also note that the CL-316243-increased IL-6 expression was significantly diminished in SphK1−/− mice. ** and ##, p < 0.01; * and #, p < 0.05 (t test, n = 6). C, * and **, CL versus control (Ctrl), wild-type mice; # and ##, CL in SphK1−/− versus CL in wild-type mice.
DISCUSSION
Acute ADRB3 activation triggers a proinflammatory response in adipose tissue (13, 42), and this effect is mediated by lipolytic products derived from activation of HSL (13). However, the signaling events downstream of HSL leading to adipose inflammation are incompletely understood. In this report, we investigated mechanisms by which excess lipolysis produces adipose tissue inflammation. We found that ADRB3/HSL signaling selectively activated SphK1, and not SphK2, and increased S1P production in adipocytes. We previously showed that activation of ADRB3/HSL signaling in adipocytes in vivo and model adipocytes in vitro triggers acute changes in metabolism that alter patterns of gene expression, including inflammatory genes IL-6, CCL2, and PAI-1 (13). IL-6 has been shown to be a key player in chronic inflammation, and levels of circulating IL-6 are elevated in several proinflammatory diseases such as rheumatoid arthritis, systemic juvenile idiopathic arthritis, systemic lupus erythematosus, ankylosing spondylitis, psoriasis, and Crohn disease (43). Therefore, we focused on investigating the role of SphK1 activation in lipolysis-induced IL-6 gene expression in the present study. Our data demonstrate that SphK1 appears to link adrenergic activation of HSL with the production of the proinflammatory cytokine IL-6. Plasma IL-6 concentrations are elevated in obesity (40, 41). Therefore, our study implies that SphK1 activation may be functionally involved in obese associated adipocyte inflammation.
HSL-stimulated lipolysis results in the liberation of free fatty acids, including palmitate from triglycerides in adipocytes (13, 29, 44). Palmitate treatment enhanced SphK1 expression in C2C12 myotubes (45). The study of Ross et al. (46) suggested that peroxisome proliferator-activated receptor α is involved in the palmitate-induced SphK1 expression in muscle. In contrast, we found that the stress kinase JNK/AP-1 signaling cascade contributes to the HSL-triggered SphK1 up-regulation in adipocytes. These results suggest that distinct signaling pathway may mediate SphK1 up-regulation in different cell types.
It was shown that high fat diet treatment caused a SphK1-dependent up-regulation of IL-6 in muscle, whereas high fat diet had no effect on the IL-6 levels in adipose tissue (46). In contrast, Wang et al. (37) recently observed that high fat diet triggered adipose inflammation (including up-regulation of IL-6), which was dependent on SphK1 activity. The reason for these divergent results is not known, but may involve differences in levels of adiposity and basal levels of adipocyte lipolysis. In the present experiments, the rapid activation of IL-6 production by adrenergic activation allowed detailed mechanistic analysis. Our results demonstrated that lipolysis rapidly up-regulated SphK1 and increased S1P production in adipocytes, which ultimately led to enhanced expression of IL-6 proinflammatory cytokine. This pathway, which was confirmed in vitro and in vivo by pharmacological and genetic approaches, clearly demonstrates that SphK1 is a critical mediator of proinflammatory responses in adipocytes and adipose tissues. These observations suggest that targeting adipose SphK1 may provide a therapeutic avenue for treating adipose inflammation and its associated pathological disorders.
Acknowledgment
We are grateful for the adenoviral particles carrying SphK1 and SphK2 from Dr. Timothy Hla.
This work is supported by National Institutes of Health Grants HL-071071 (to M.-J. L.), DK-076229 and DK-062292 (to J. G. G.), the Intramural Research Program of the National Institutes of Health, NIDDK (to R. L. P.), and by a Pilot and Feasibility Grant from the Michigan Diabetes Research and Training Center (National Institutes of Health Grant 5P600-DK020572) (to M. L.).
- ADRB3
- β3-adrenergic receptor
- 3T3-L1-CAR
- 3T3-L1 cell line stably expressing the coxsackie and adenovirus receptor (CAR)
- BAY
- BAY 59-9435, selective HSL inhibitor
- CL
- CL-316243, specific agonist of β3-adrenergic receptor
- EWAT
- epididymal white adipose tissues
- HSL
- hormone-sensitive lipase
- IL-6
- interleukin 6
- SKI-2
- sphingosine kinase inhibitor 2, a non-lipid inhibitor of sphingosine kinases
- SphK1
- sphingosine kinase 1
- qPCR
- quantitative PCR.
REFERENCES
- 1. Eckel R. H., York D. A., Rössner S., Hubbard V., Caterson I., St Jeor S. T., Hayman L. L., Mullis R. M., Blair S. N., and American Heart Association (2004) Prevention Conference VII: Obesity, a worldwide epidemic related to heart disease and stroke: executive summary. Circulation 110, 2968–2975 [DOI] [PubMed] [Google Scholar]
- 2. Engeland A., Bjørge T., Søgaard A. J., Tverdal A. (2003) Body mass index in adolescence in relation to total mortality: 32-year follow-up of 227,000 Norwegian boys and girls. Am. J. Epidemiol. 157, 517–523 [DOI] [PubMed] [Google Scholar]
- 3. Flegal K. M., Carroll M. D., Ogden C. L., Johnson C. L. (2002) Prevalence and trends in obesity among US adults, 1999–2000. JAMA 288, 1723–1727 [DOI] [PubMed] [Google Scholar]
- 4. Flegal K. M., Carroll M. D., Kuczmarski R. J., Johnson C. L. (1998) Overweight and obesity in the United States: prevalence and trends, 1960–1994. Int. J. Obes. Relat. Metab. Disord. 22, 39–47 [DOI] [PubMed] [Google Scholar]
- 5. Poirier P., Giles T. D., Bray G. A., Hong Y., Stern J. S., Pi-Sunyer F. X., Eckel R. H. (2006) Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss. Arterioscler. Thromb. Vasc. Biol. 26, 968–976 [DOI] [PubMed] [Google Scholar]
- 6. Andersson C. X., Gustafson B., Hammarstedt A., Hedjazifar S., Smith U. (2008) Inflamed adipose tissue, insulin resistance and vascular injury. Diabetes Metab. Res. Rev. 24, 595–603 [DOI] [PubMed] [Google Scholar]
- 7. Visser M., Bouter L. M., McQuillan G. M., Wener M. H., Harris T. B. (1999) Elevated C-reactive protein levels in overweight and obese adults. JAMA 282, 2131–2135 [DOI] [PubMed] [Google Scholar]
- 8. Makki K., Froguel P., Wolowczuk I. (2013) Adipose tissue in obesity-related inflammation and insulin resistance: cells, cytokines, and chemokines. ISRN Inflamm. 2013, 139239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Esser N., Legrand-Poels S., Piette J., Scheen A. J., Paquot N. (2014) Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 105, 141–150 [DOI] [PubMed] [Google Scholar]
- 10. Das A., Mukhopadhyay S. (2011) The evil axis of obesity, inflammation and type-2 diabetes. Endocr. Metab. Immune Disord. Drug Targets 11, 23–31 [DOI] [PubMed] [Google Scholar]
- 11. Bézaire V., Langin D. (2009) Regulation of adipose tissue lipolysis revisited. Proc. Nutr. Soc. 68, 350–360 [DOI] [PubMed] [Google Scholar]
- 12. Sabio G., Das M., Mora A., Zhang Z., Jun J. Y., Ko H. J., Barrett T., Kim J. K., Davis R. J. (2008) A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 322, 1539–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mottillo E. P., Shen X. J., Granneman J. G. (2010) beta3-adrenergic receptor induction of adipocyte inflammation requires lipolytic activation of stress kinases p38 and JNK. Biochim. Biophys. Acta 1801, 1048–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hla T., Lee M. J., Ancellin N., Liu C. H., Thangada S., Thompson B. D., Kluk M. (1999) Sphingosine-1-phosphate: extracellular mediator or intracellular second messenger? Biochem. Pharmacol. 58, 201–207 [DOI] [PubMed] [Google Scholar]
- 15. Igarashi Y., Yatomi Y. (1998) Sphingosine 1-phosphate is a blood constituent released from activated platelets, possibly playing a variety of physiological and pathophysiological roles. Acta Biochim. Pol. 45, 299–309 [PubMed] [Google Scholar]
- 16. Moolenaar W. H. (1999) Bioactive lysophospholipids and their G protein-coupled receptors. Exp. Cell Res. 253, 230–238 [DOI] [PubMed] [Google Scholar]
- 17. Spiegel S. (1999) Sphingosine 1-phosphate: a prototype of a new class of second messengers. J. Leukoc. Biol. 65, 341–344 [DOI] [PubMed] [Google Scholar]
- 18. Lee M. J., Van Brocklyn J. R., Thangada S., Liu C. H., Hand A. R., Menzeleev R., Spiegel S., Hla T. (1998) Sphingosine-1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science 279, 1552–1555 [DOI] [PubMed] [Google Scholar]
- 19. Hait N. C., Allegood J., Maceyka M., Strub G. M., Harikumar K. B., Singh S. K., Luo C., Marmorstein R., Kordula T., Milstien S., Spiegel S. (2009) Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 325, 1254–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alvarez S. E., Harikumar K. B., Hait N. C., Allegood J., Strub G. M., Kim E. Y., Maceyka M., Jiang H., Luo C., Kordula T., Milstien S., Spiegel S. (2010) Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 465, 1084–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Niessen F., Schaffner F., Furlan-Freguia C., Pawlinski R., Bhattacharjee G., Chun J., Derian C. K., Andrade-Gordon P., Rosen H., Ruf W. (2008) Dendritic cell PAR1-S1P3 signalling couples coagulation and inflammation. Nature 452, 654–658 [DOI] [PubMed] [Google Scholar]
- 22. Skoura A., Michaud J., Im D. S., Thangada S., Xiong Y., Smith J. D., Hla T. (2011) Sphingosine-1-phosphate receptor-2 function in myeloid cells regulates vascular inflammation and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 31, 81–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang W., An J., Jawadi H., Siow D. L., Lee J. F., Zhao J., Gartung A., Maddipati K. R., Honn K. V., Wattenberg B. W., Lee M. J. (2013) Sphingosine-1-phosphate receptor-2 mediated NFκB activation contributes to tumor necrosis factor-α induced VCAM-1 and ICAM-1 expression in endothelial cells. Prostaglandins Other Lipid Mediat. 106, 62–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Baker D. A., Barth J., Chang R., Obeid L. M., Gilkeson G. S. (2010) Genetic sphingosine kinase 1 deficiency significantly decreases synovial inflammation and joint erosions in murine TNF-α-induced arthritis. J. Immunol. 185, 2570–2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bachmaier K., Guzman E., Kawamura T., Gao X., Malik A. B. (2012) Sphingosine kinase 1 mediation of expression of the anaphylatoxin receptor C5L2 dampens the inflammatory response to endotoxin. PLoS One 7, e30742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hsu A., Zhang W., Lee J. F., An J., Ekambaram P., Liu J., Honn K. V., Klinge C. M., Lee M. J. (2012) Sphingosine-1-phosphate receptor-3 signaling up-regulates epidermal growth factor receptor and enhances epidermal growth factor receptor-mediated carcinogenic activities in cultured lung adenocarcinoma cells. Int. J. Oncol. 40, 1619–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. French K. J., Upson J. J., Keller S. N., Zhuang Y., Yun J. K., Smith C. D. (2006) Antitumor activity of sphingosine kinase inhibitors. J. Pharmacol. Exp. Ther. 318, 596–603 [DOI] [PubMed] [Google Scholar]
- 28. Mottillo E. P., Granneman J. G. (2011) Intracellular fatty acids suppress beta-adrenergic induction of PKA-targeted gene expression in white adipocytes. Am. J. Physiol. Endocrinol. Metab. 301, E122–E131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mottillo E. P., Shen X. J., Granneman J. G. (2007) Role of hormone-sensitive lipase in β-adrenergic remodeling of white adipose tissue. Am. J. Physiol. Endocrinol. Metab. 293, E1188–E1197 [DOI] [PubMed] [Google Scholar]
- 30. Claus T. H., Lowe D. B., Liang Y., Salhanick A. I., Lubeski C. K., Yang L., Lemoine L., Zhu J., Clairmont K. B. (2005) Specific inhibition of hormone-sensitive lipase improves lipid profile while reducing plasma glucose. J. Pharmacol. Exp. Ther. 315, 1396–1402 [DOI] [PubMed] [Google Scholar]
- 31. Ebdrup S., Refsgaard H. H., Fledelius C., Jacobsen P. (2007) Synthesis and structure-activity relationship for a novel class of potent and selective carbamate-based inhibitors of hormone selective lipase with acute in vivo antilipolytic effects. J. Med. Chem. 50, 5449–5456 [DOI] [PubMed] [Google Scholar]
- 32. Jiao P., Ma J., Feng B., Zhang H., Diehl J. A., Chin Y. E., Yan W., Xu H. (2011) FFA-induced adipocyte inflammation and insulin resistance: involvement of ER stress and IKKβ pathways. Obesity 19, 483–491 [DOI] [PubMed] [Google Scholar]
- 33. Lee M. J., Thangada S., Paik J. H., Sapkota G. P., Ancellin N., Chae S. S., Wu M., Morales-Ruiz M., Sessa W. C., Alessi D. R., Hla T. (2001) Akt-mediated phosphorylation of the G protein-coupled receptor EDG-1 is required for endothelial cell chemotaxis. Mol. Cell 8, 693–704 [DOI] [PubMed] [Google Scholar]
- 34. Allende M. L., Sasaki T., Kawai H., Olivera A., Mi Y., van Echten-Deckert G., Hajdu R., Rosenbach M., Keohane C. A., Mandala S., Spiegel S., Proia R. L. (2004) Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. J. Biol. Chem. 279, 52487–52492 [DOI] [PubMed] [Google Scholar]
- 35. Muradashvili N., Khundmiri S. J., Tyagi R., Gartung A., Dean W. L., Lee M. J., Lominadze D. (2014) Sphingolipids affect fibrinogen-induced caveolar transcytosis and cerebrovascular permeability. Am. J. Physiol. Cell Physiol. 307, C169–C179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mottillo E. P., Bloch A. E., Leff T., Granneman J. G. (2012) Lipolytic products activate peroxisome proliferator-activated receptor (PPAR)α and δ in brown adipocytes to match fatty acid oxidation with supply. J. Biol. Chem. 287, 25038–25048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang J., Badeanlou L., Bielawski J., Ciaraldi T. P., Samad F. (2014) Sphingosine kinase 1 regulates adipose proinflammatory responses and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 306, E756–E768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Szczepankiewicz B. G., Kosogof C., Nelson L. T., Liu G., Liu B., Zhao H., Serby M. D., Xin Z., Liu M., Gum R. J., Haasch D. L., Wang S., Clampit J. E., Johnson E. F., Lubben T. H., Stashko M. A., Olejniczak E. T., Sun C., Dorwin S. A., Haskins K., Abad-Zapatero C., Fry E. H., Hutchins C. W., Sham H. L., Rondinone C. M., Trevillyan J. M. (2006) Aminopyridine-based c-Jun N-terminal kinase inhibitors with cellular activity and minimal cross-kinase activity. J. Med. Chem. 49, 3563–3580 [DOI] [PubMed] [Google Scholar]
- 39. Turpeinen T., Nieminen R., Moilanen E., Korhonen R. (2010) Mitogen-activated protein kinase phosphatase-1 negatively regulates the expression of interleukin-6, interleukin-8, and cyclooxygenase-2 in A549 human lung epithelial cells. J. Pharmacol. Exp. Ther. 333, 310–318 [DOI] [PubMed] [Google Scholar]
- 40. Mohamed-Ali V., Goodrick S., Rawesh A., Katz D. R., Miles J. M., Yudkin J. S., Klein S., Coppack S. W. (1997) Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-α, in vivo. J. Clin. Endocrinol. Metab. 82, 4196–4200 [DOI] [PubMed] [Google Scholar]
- 41. Bastard J. P., Jardel C., Bruckert E., Blondy P., Capeau J., Laville M., Vidal H., Hainque B. (2000) Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. J. Clin. Endocrinol. Metab. 85, 3338–3342 [DOI] [PubMed] [Google Scholar]
- 42. Granneman J. G., Li P., Zhu Z., Lu Y. (2005) Metabolic and cellular plasticity in white adipose tissue I: effects of β3-adrenergic receptor activation. Am. J. Physiol. Endocrinol. Metab. 289, E608–E616 [DOI] [PubMed] [Google Scholar]
- 43. Gabay C. (2006) Interleukin-6 and chronic inflammation. Arthritis Res. Ther. 8, S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ström K., Gundersen T. E., Hansson O., Lucas S., Fernandez C., Blomhoff R., Holm C. (2009) Hormone-sensitive lipase (HSL) is also a retinyl ester hydrolase: evidence from mice lacking HSL. FASEB J. 23, 2307–2316 [DOI] [PubMed] [Google Scholar]
- 45. Hu W., Bielawski J., Samad F., Merrill A. H., Jr., Cowart L. A. (2009) Palmitate increases sphingosine-1-phosphate in C2C12 myotubes via upregulation of sphingosine kinase message and activity. J. Lipid Res. 50, 1852–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ross J. S., Hu W., Rosen B., Snider A. J., Obeid L. M., Cowart L. A. (2013) Sphingosine kinase 1 is regulated by peroxisome proliferator-activated receptor alpha in response to free fatty acids and is essential for skeletal muscle interleukin-6 production and signaling in diet-induced obesity. J. Biol. Chem. 288, 22193–22206 [DOI] [PMC free article] [PubMed] [Google Scholar]