

Background: Hyaluronan (HA), an extracellular glycosaminoglycan, is normally produced by three HA synthase (Has) enzymes.
Results: Skin fibroblasts from Has1/Has3 double knock-out mice have higher Has2 expression and HA levels and are resistant to cell death after UVB exposure or serum starvation.
Conclusion: HA modulates injury-induced apoptotic responses in fibroblasts.
Significance: HA has an important role in cell death responses.
Keywords: Apoptosis, Caspase, Extracellular Matrix, Fibroblast, Hyaluronan, Skin, Stress
Abstract
A balanced turnover of dermal fibroblasts is crucial for structural integrity and normal function of the skin. During recovery from environmental injury (such as UV exposure and physical wounding), apoptosis is an important mechanism regulating fibroblast turnover. We are interested in the role that hyaluronan (HA), an extracellular matrix molecule synthesized by HA synthase enzymes (Has), plays in regulating apoptosis in fibroblasts. We previously reported that Has1 and Has3 double knock-out (Has1/3 null) mice show accelerated wound closure and increased numbers of fibroblasts in the dermis. In the present study, we report that HA levels and Has2 mRNA expression are higher in cultured Has1/3 null primary skin fibroblasts than in wild type (WT) cells. Apoptosis induced by two different environmental stressors, UV exposure and serum starvation (SS), was reduced in the Has1/3 null cells. Hyaluronidase, added to cultures to remove extracellular HA, surprisingly had no effect upon apoptotic susceptibility to UVB or SS. However, cells treated with 4-methylumbelliferone to inhibit HA synthesis were sensitized to apoptosis induced by SS or UVB. When fibroblasts were transfected with Has2-specific siRNA that lowered Has2 mRNA and HA levels by 90%, both Has1/3 null and WT cells became significantly more sensitive to apoptosis. The exogenous addition of high molecular weight HA failed to reverse this effect. We conclude that Has1/3 null skin fibroblasts (which have higher levels of Has2 gene expression) are resistant to stress-induced apoptosis.
Introduction
Dermal fibroblasts are crucial cellular components for the structural integrity of the skin. They serve as the primary producer of the extracellular matrix scaffold within the dermis (1–3), participate in a dynamic interplay with other cells, such as keratinocytes, and generate a diverse array of cytokines that bind to the extracellular matrix as well as to cell surface receptors that coordinate cellular activities within the tissue. Abnormal turnover of fibroblasts is a feature of various cutaneous pathologies. For example, in systemic sclerosis or scleroderma, fibroblasts from affected skin are abnormally resistant to Fas-mediated apoptosis (4), and the activity of the antiapoptotic protein kinase, Akt, is up-regulated in these fibroblasts (5). In addition, profibrotic signaling pathways (i.e. TGF-β) are hyperactive in the scleroderma-derived fibroblasts (6, 7). Decreased susceptibility to apoptosis is also observed in fibroblasts isolated from hypertrophic scars and keloidal tissues (8, 9) and from patients with idiopathic pulmonary fibrosis (10, 11). In contrast, an increase in susceptibility to apoptosis appears to contribute to other pathological conditions of the skin. Alikhani et al. (12) demonstrated that the advanced glycation end products generated in the skin of diabetic patients could enhance proapoptotic gene expression and stimulate fibroblast apoptosis, which could be potential contributors to delayed wound healing and chronic skin ulcers in these patients. In people with fair-skinned complexions (due to few active melanocytes), UV exposure from the sun causes considerable apoptosis of dermal fibroblasts (photodamage) and contributes to the development of photoaging (13).
Hyaluronan (HA)2 is a negatively charged, unbranched, non-sulfated glycosaminoglycan consisting of alternating disaccharide units of d-glucuronic acid and N-acetylglucosamine. HA is synthesized by hyaluronan synthases (HAS1, HAS2, and HAS3), which alternately add UDP-glucosamine and UDP-glucuronic acid residues to a growing HA polymer chain that is extruded out of the plasma membrane into the extracellular space. Among the three isoenzymes, HAS1 and HAS2 synthesize very large HA chains (average molecular mass, 2 × 105 to 2 × 106 Da), whereas HAS3 produces a smaller HA size range (1 × 105 to 1 × 106 Da) (14). Studies from different groups have shown that HAS2 plays a predominant role in inducible HA synthesis in fibroblasts exposed to growth factors (15, 16), including fibroblasts undergoing transformation to myofibroblasts (17). Furthermore, down-regulation of Has2 gene expression is responsible for the inhibition of HA synthesis in skin fibroblasts in response to certain environmental stressors, such as UVB irradiation (18). The activity of HAS enzymes can be regulated at the transcriptional level (15, 16, 19, 20) or at the posttranslational level by phosphorylation (21), O-GlcNAcylation (22), or ubiquitination (23). The cellular content of UDP-sugar substrates (UDP-GlcUA and UDP-GlcNAc) also acts as a limiting factor in HA synthesis (24). In addition, HAS2 can form homodimers as well as heterodimers with HAS3 in the plasma membrane; dimerization between two HAS enzymes may potentially regulate overall enzymatic activity. Karousou et al. (23) have shown that the activity of wild type HAS2 can be quenched by heterodimerization with a loss-of-function HAS2 mutant. Overall, HAS2 appears to be the predominant HA synthase in fibroblasts, but many open questions about the regulation of its activity remain.
Hyaluronan is the major component of the extracellular matrix in many tissues, and this is particularly true in skin (25). We are interested in the role of HA in regulating fibrosis, a process in which fibroblasts undergo transformation to myofibroblasts (the cells that actually produce new extracellular matrix in the injured tissue). Several studies have shown that HA helps to regulate the TGF-β-driven transformation of human lung fibroblasts into myofibroblasts (26) by facilitating the co-localization of epidermal growth factor receptor and CD44 (the major HA receptor) in lipid rafts within plasma membranes (17, 27). However, a role for HA in regulating fibroblast viability and susceptibility to apoptosis has not been reported. Previous work from our laboratory showed that in mice lacking Has1 and Has3, healing skin wounds contain more fibroblasts and myofibroblasts at 5 days after excisional wounding relative to WT wounds (28). This suggests that a change in the level of HAS enzyme expression and HA synthesis in vivo leads to a change in the equilibrium number of fibroblasts, either through an increase in proliferation, a decrease in apoptosis, or both.
The present study was designed to investigate how HA regulates fibroblast viability, either under normal physiological conditions or during stimulation by an environmental stress. Our data show that HA, especially the HA produced by HAS2 in fibroblasts, regulates cellular susceptibility to apoptosis through modulation of caspase-9 cleavage and downstream apoptotic pathway activation.
EXPERIMENTAL PROCEDURES
Primary Cell Culture
Primary mouse dermal fibroblasts were isolated from the skin of WT or Has1/3 null mouse pups at 2–3 days of age. C57BL/6J WT mice were obtained from JAX Laboratories (Bar Harbor, ME). Mice deficient in Has1 and Has3 (Has1/3 null) were generated as described previously (28). Pups were euthanized in ice, and the entire trunk skin was removed and incubated overnight in 0.25% trypsin without EDTA, followed by mechanical separation of epidermis from dermis. To isolate fibroblasts, the dermis was finely diced and incubated with 400 units/ml collagenase type I (Worthington) for 30 min at 37 °C and with DNase for 10 min at 37 °C. The digested tissue suspension was passed through a 100-μm cell strainer to eliminate large undigested tissue chunks. The hair follicles were removed by centrifugation in a 50-ml conical tube at 20 × g for 3 min. Fibroblasts were collected and cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin mixture, at 37 °C in a humidified 5% CO2 incubator. Fresh growth medium was added every other day. Experiments were conducted after 1–2 cell passages. In some experiments, primary fibroblasts were grown in the presence of 25 μg/ml HMW-HA (∼2 × 106 Da; Lifecore Biomedical).
Proliferation and Inhibition Assays
For proliferation assays, 5 × 104 cells were plated in a 60-mm dish at day 0, and the cells were refed with fresh growth media daily. Every 24 h out to day 5, triplicate dishes of WT cells or Has1/3 null cells were trypsinized and resuspended in culture media, and total cell counts were obtained. For serum starvation experiments, cells were cultured in serum-free DMEM. For HA-inhibitory assays, 4-methylumbelliferone (4-MU) was obtained from Sigma-Aldrich, dissolved in DMSO at a concentration of 1 m, and subsequently diluted in medium to the desired concentration. Hyaluronidase from Streptomyces dysgalactiae was obtained from Calbiochem.
UV Source and Irradiation Protocol
UV light, with ∼75% in the UVB (280–320 nm) range, was delivered from a bank of UV lamps (RPR-3000A, Southern New England Ultraviolet, Branford, CT). With these bulbs, the percentages of total irradiance in the UVA/UVB/UVC spectral range were 16/74/10%, respectively; the UVC component was filtered out by a UVC filter (Kodacel, Eastman Kodak Co.). Exposure times for proper dose delivery were determined by measuring the energy output with a power meter (IL-1700, International Lights, Newburyport, MA). Murine fibroblasts in 60-mm dishes in DMEM lacking phenol red dye were exposed to UV light at 32 mJ/cm2, the previously reported LD50 for primary murine keratinocytes when using this light source (29), and harvested at the times indicated in the figures.
Inhibition of HAS2 Gene Expression by RNA Interference (RNAi)
Reverse transfection of primary mouse skin fibroblasts was performed with SMARTpool small interfering RNA (siRNA), targeted to Has2 mRNA or non-targeted scrambled siRNA (Thermo Scientific, Freemont, CA). For a 6-well tissue culture plate, 30 pmol of siRNA was mixed with 8 μl of Lipofectamine RNAiMAX (Invitrogen) in 500 ml of Opti-MEM. The final duplex concentration was 100 nm. The siRNA/transfection reagent mixture was added to the plates first, followed by fibroblasts seeded at a density of 2.2 × 105/well. The cells were refed with fresh, antibiotic-free medium at 24 h after transfection. The efficiency of Has2 gene expression knockdown by RNAi was confirmed by quantitative real-time PCR.
Western Blot Analysis
The primary antibodies for Western analyses were polyclonal rabbit anti-cleaved caspase-9 antibody (Cell Signaling Technology, catalogue no. 9509), polyclonal rabbit anti-active caspase-3 antibody (BioVision, catalog no. 3015-100), polyclonal rabbit anti-PARP antibody (Cell Signaling Technology, catalog no. 9542), and polyclonal rabbit anti-GAPDH antibody (Santa Cruz Biotechnology, Inc., catalog no. sc-25778). The secondary antibodies, goat anti-rabbit or goat anti-mouse IgG conjugated with horseradish peroxidase (HRP), were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA). At the end of treatments, cells were scraped off of the culture plates, lysed in radioimmune precipitation assay buffer supplemented with protease inhibitor mixture (Millipore), diluted in 4× NuPAGE LDS sample buffer (Invitrogen), and boiled at 70 °C for 10 min. Equal amounts of protein were loaded in a 4–12% gradient polyacrylamide gel (Invitrogen), separated by electrophoresis, and transferred to a PVDF membrane (Immobilon-P, Millipore). The membranes were blocked in 5% nonfat milk dissolved in Tris-buffered saline with 0.05% Tween 20 (TBS-T) for 1 h at room temperature before being probed with primary antibodies overnight at 4 °C, followed by incubation of the appropriate secondary antibody for 1 h at room temperature. The resulting signals were developed using an ECL Western blotting detection reagent kit (GE Healthcare). Digital records were obtained from each blot and the protein bands of interest were quantified using one-dimensional analysis software (Gel Logic, Carestream). Membranes were treated with stripping buffer (Re-Blot Plus Strong solution, catalogue number no. 2504, Millipore) and probed again using GAPDH as a loading control.
Particle Exclusion Assay
Exclusion of fixed red blood cells (RBCs) was used to visualize fibroblast pericellular HA coats. Fibroblasts were plated at a density of 30,000 cells/35-mm dish 24 h before the assay. To visualize the cell bodies, fibroblasts were incubated with calcein AM (Invitrogen; final concentration, 1 μm) for 30 min just prior to the assay and then washed three times with DMEM. Formalin-fixed RBCs were purchased from Sigma and washed in PBS several times, and the pellet was resuspended in PBS at a concentration of 5.0 × 107 cells/ml. The well mixed RBC suspension (100 μl) was added to the 35-mm dish and incubated at 37 °C for 10 min to allow the RBC to settle. Images were acquired using a Leica DMIRB inverted microscope (Leica Microsystems, GmbH, Wetzlar, Germany) with a Retiga SRV cooled CCD camera (QImaging, Surrey, Canada) and ImagePro Plus software (Media Cybernetics, Rockville, MD). Quantification of RBC-free pericellular areas (HA coats) was done using segmentation analysis with IP Lab imaging software (Scanalytics, Fairfax, VA).
Quantitation of HA and Chondroitin Sulfate (CS) by Fluorophore-assisted Carbohydrate Electrophoresis (FACE)
The HA and CS content of skin fibroblast cultures (in cell layers and in conditioned media) was measured as described previously (28). Briefly, after proteolytic digestion and ethanol precipitation/purification steps, HA in the samples was digested into disaccharides using hyaluronidase from S. dysgalactiae (2.5 milliunits/μl; 100741-1A, Seikagaku America Inc.), and CS in the samples was digested into disaccharides using chondroitinase ABC (25 milliunits/μl; 100330-1A, Seikagaku America) and then labeled with 2-aminoacridone (Invitrogen). The labeled HA and CS disaccharides were electrophoresed on a Bio-Rad mini-PROTEAN Tetra system, in a gel composed of 20% acrylamide, 40 mm Tris acetate (pH 7.0), 2.5% glycerol, 10% ammonium persulfate, and 0.1% TEMED, at constant voltage (500 V) at 4 °C for 50 min. Gels were imaged on a UV transilluminator at 365 nm using a CCD camera. The HA or CS disaccharide bands were quantified using ImageJ software, version 1.47V (National Institutes of Health).
RNA Isolation and Quantitative Real-time PCR
RNA was prepared from fibroblasts using TRIzol LS reagent (Invitrogen), following the manufacturer's instructions. The extracted RNA was then reverse-transcribed into cDNA using a random primer and Superscript III reverse transcriptase (Invitrogen). For quantitative real-time PCR (qPCR), TaqMan gene expression probes for Has1, -2, and -3 and Hyal1 and -2 and for 18 S ribosomal RNA as an endogenous control were purchased from Applied Biosystems (Foster City, CA). The above gene expression levels were measured in triplicate in both WT and Has1/3 null cell using the 2−ΔΔCt method. The mRNA levels were presented as a -fold difference relative to the untreated normal control.
Immunofluorescence Microscopy
Fibroblasts, grown in 35-mm cell culture dishes, were fixed in 4% paraformaldehyde, and the cells were permeabilized with 0.1% Triton X-100/PBS prior to staining (to visualize total cellular HA, both intracellular and extracellular). To visualize extracellular HA only, staining was done without prior permeabilization. Cells were then incubated with a biotinylated HA-binding protein (bHABP; Millipore) at 4 °C overnight, followed by incubation with Cy3-conjugated streptavidin (Jackson ImmunoResearch Laboratories) at room temperature for 1 h. For Ki-67 staining, a rabbit monoclonal antibody (Thermo Scientific) was used, followed by incubation with Cy3-conjugated donkey anti-rabbit IgG (Jackson ImmunoResearch Laboratories). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Images were acquired using a Leica DM5500B upright microscope (Leica Microsystems) with a Retiga SRV cooled CCD camera (QImaging) and ImagePro Plus software (Media Cybernetics).
Cell Viability Assays
At the specified time (either 12 h after UVB irradiation or after 4 h of serum starvation), the cells were washed with PBS, and a mixture of fluorescein diacetate (FDA; 50 μg/ml) and ethidium bromide (EB; 4 μg/ml) in DMEM was added to the cells as described previously (29). After 3 min of incubation, cells were visualized under a fluorescence microscope using an FITC/TRITC filter. For each field, green (live) and orange (dead) cells were counted using IPLab imaging software and reported as a percentage of total cells. As another assay of apoptosis, DNA fragmentation was assessed in fibroblasts using a cell death detection ELISA kit (Roche Applied Science), which measures cytosolic nucleosomes at an absorbance of 405 nm. Data were normalized to the protein concentration of the sample.
Statistical Analysis
Statistical analyses were performed using a two-sided Student's t test. p < 0.05 was considered statistically significant.
RESULTS
Has1/3 Null Skin Fibroblasts Contain Higher Levels of HA and Has2 mRNA than Normal
In order to investigate the role of HAS2 in regulating cellular behavior of mouse skin fibroblasts, primary fibroblasts were isolated from the dermis of WT mice or Has1/3 null mice (the latter having only the Has2 gene). The size of the HA-mediated pericellular coat was measured in both WT and Has1/3 null cells using a particle exclusion assay, which revealed that Has1/3 null fibroblasts form bigger pericellular HA coats than do the WT cells (Fig. 1A). In addition, the levels of HA in both the cell layer and the media of fibroblast cultures were analyzed by FACE and found to be significantly higher in the cell layer of Has1/3 null cells when compared with WT cells (Fig. 1B). Furthermore, Has2 mRNA levels were significantly higher in Has1/3 null cells than in the WT cells, as determined by qPCR (Fig. 1C). Further analyses of mRNA levels of Has1, Has2, and Has3 in WT fibroblasts by qPCR revealed that Has2 is the predominant species (92.7%) in terms of gene expression level among the three isoforms in WT primary mouse skin fibroblasts (Fig. 1D). On the other hand, there appears to be no difference in mRNA levels of hyaluronidase 1 and 2 (Hyal1 and Hyal2), the hyaluronan-degrading enzymes, between WT and Has1/3 null fibroblasts (Fig. 1E). As part of our phenotypic characterization of these two types of cells, we examined proliferation rates through the use of growth curves and expression of Ki-67 (an established proliferation marker); however, no differences between Has1/3 null and WT cells could be detected in multiple experiments (data not shown).
FIGURE 1.
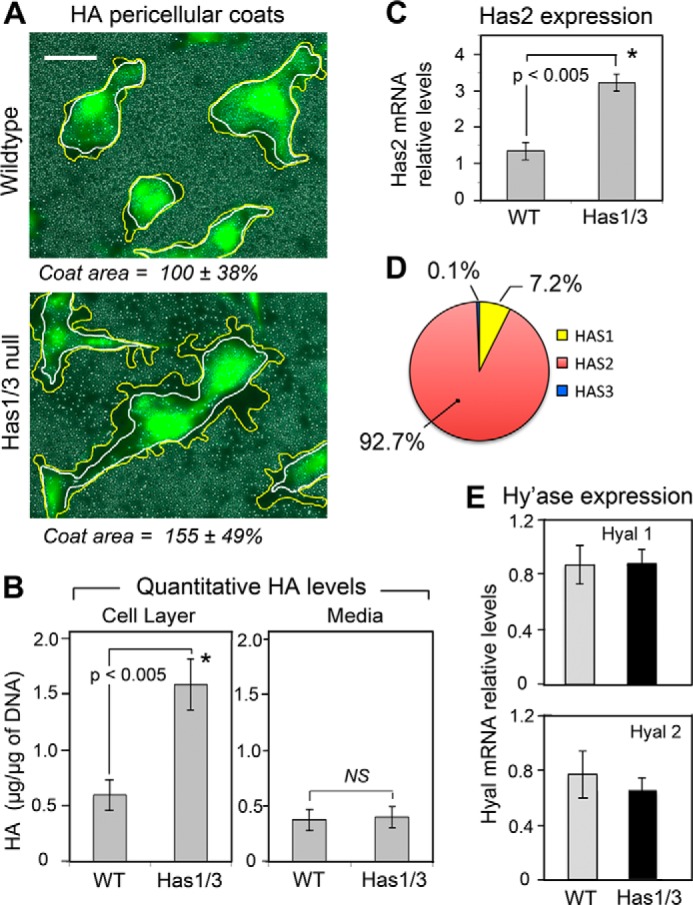
Hyaluronan levels are increased and Has2 gene expression is up-regulated in Has1/3 null skin fibroblasts. A, living fibroblasts were labeled with calcein to visualize cell bodies, and the area occupied by the pericellular HA coat was assessed by the exclusion of RBCs from the cell perimeter. Yellow lines and white lines were artificially drawn to delineate the margin of the HA coat and cell membrane, respectively. Scale bar, 50 μm. Numbers below each panel are the mean value ± S.D. of three experiments (15 cells analyzed per experiment). B, the amount of HA derived from the cell layer (left) or media (right) of WT or Has1/3 null fibroblast cultures were analyzed by FACE and normalized by DNA content of each sample. Each data point is the mean ± S.E. (error bars) of four independent experiments. C, Has2 mRNA levels in WT and Has1/3 null cells, measured by qPCR. D, pie chart of relative mRNA levels of the Has isoforms in WT skin fibroblasts, measured by qPCR using the 2−ΔΔCt method and displayed as a percentage of total Has expression. E, hyaluronidase 1 and 2 (Hyal1 and Hyal2) mRNA levels in WT and Has1/3 null cells, as determined by qPCR. In C and E, each data point represents the mean ± S.E. of three independent experiments. *, significantly different from WT by Student's t test.
Levels of HA and Has2 mRNA in Has1/3 Null Skin Fibroblasts Are More Responsive to Environmental Stress than Are WT Cells
To explore the effect of UVB irradiation on HA production in mouse skin fibroblasts, we treated the cells with UVB at a moderate dose (32 mJ/cm2, which causes less than 50% lethality) and harvested the cells at different time points for analysis of HA levels by FACE. As shown in Fig. 2A, cell-associated HA in both WT and Has1/3 null cells increased gradually during the 24 h after UVB irradiation, and the phenotypic difference in HA level between Has1/3 null and WT cells remained consistent at most time points except for the last time point, 24 h after UVB irradiation, when there was no difference in HA level in the cell layer of both types of cells. On the other hand, HA levels in the conditioned media from both types of cells did not show any difference at most time points after UVB exposure until the last time point (24 h), when a higher level of HA was detected from the media of WT cells compared with that from Has1/3 null cells (Fig. 2B). We then looked at changes in mRNA expression of the three Has isoforms in skin fibroblasts in response to UVB. First, as shown in Fig. 2C, Has2 mRNA levels in both WT and Has1/3 null skin fibroblasts decreased to about half of their initial values during the first 2 h post-UVB but then gradually recovered and exceeded baseline levels by 2–3-fold at 24 h. In addition, both Has1 and Has3 mRNA levels in WT cells are responsive to UVB exposure; Has1 mRNA showed a quick response right after UVB exposure and a steady increase to about 16-fold over baseline at 24 h (Fig. 2D); Has3 mRNA showed a delayed but robust increase, which started 8 h after UVB exposure and reached 250-fold at 24 h (Fig. 2E).
FIGURE 2.

Changes in HA levels and Has mRNA levels in WT or Has1/3 null skin fibroblasts in response to UVB exposure. A and B, time courses are shown for HA levels in the cell layer (A) or media (B) of cultures after 32 mJ/cm2 UVB exposure, with quantification by FACE analysis. Each data point is the mean ± S.D. (error bars) of four independent experiments. Changes in gene expression of the HA synthases in WT and Has1/3 cells were determined by qPCR (mean ± half-range of two experiments) for Has2 (C), Has1 (D), and Has3 (E).
To explore HA responses in fibroblasts subjected to other types of environmental stress, we treated cells with serum starvation, which causes endoplasmic reticulum stress in the cells (30, 31). As shown in the time course in Fig. 3A, HA levels in the cell layer of Has1/3 null cells decreased significantly over the first 8 h of serum starvation and then began to recover, whereas no dramatic initial decline in HA was seen in WT controls. On the other hand, HA levels in the media of both WT and Has1/3 null skin fibroblasts gradually rose, beginning at ∼6 h of serum starvation, exceeding baseline levels by 3–4-fold at 24 h (Fig. 3B). Consistent with these observations, Has2 mRNA decreased quickly after the start of serum starvation, reached a nadir around 2 h, and then recovered in a time-dependent manner (Fig. 3C). In addition, Has1 mRNA levels in WT cells rapidly decreased to about 7% of initial levels and stayed below 21% of initial levels during the remainder of serum starvation (Fig. 3D). The Has3 mRNA level in WT cells was not significantly affected by serum starvation (Fig. 3E). Overall, these results are consistent with a high dependence of Has2 expression upon environmental stressors; the latter appear to affect relative HA production more profoundly in the Has1/3 null cells, which only express Has2.
FIGURE 3.

Changes in HA levels and Has mRNA levels in WT or Has1/3 null skin fibroblasts in response to serum starvation. Time courses of HA levels in the cell layer (A) or media (B) during 24-h serum starvation are shown, measured by FACE analysis; each data point is the mean ± S.D. (error bars) of four experiments. Changes in gene expression of the HA synthases during serum starvation of WT and Has1/3 cells were determined by qPCR (mean ± half-range of two experiments) for Has2 (C), Has1 (D), and Has3 (E).
Has1/3 Null Skin Fibroblasts Are Resistant to Apoptosis Induced by either UVB Irradiation or Serum Starvation
Next, we compared the sensitivity of WT and Has1/3 null skin fibroblasts to environmental stress-induced apoptosis. Cells were treated with either UVB irradiation or serum starvation and then analyzed for cell death using a live/dead assay (FDA/EB staining). As shown in Fig. 4 (A, A′, and B), there were fewer ethidium bromide-positive dead cells in Has1/3 null cultures than in WT cultures at 12 h after UVB irradiation. Likewise, after serum starvation (4 h), fewer dead Has1/3 null fibroblasts were observed; see Fig. 4 (C, C′, and D).
FIGURE 4.
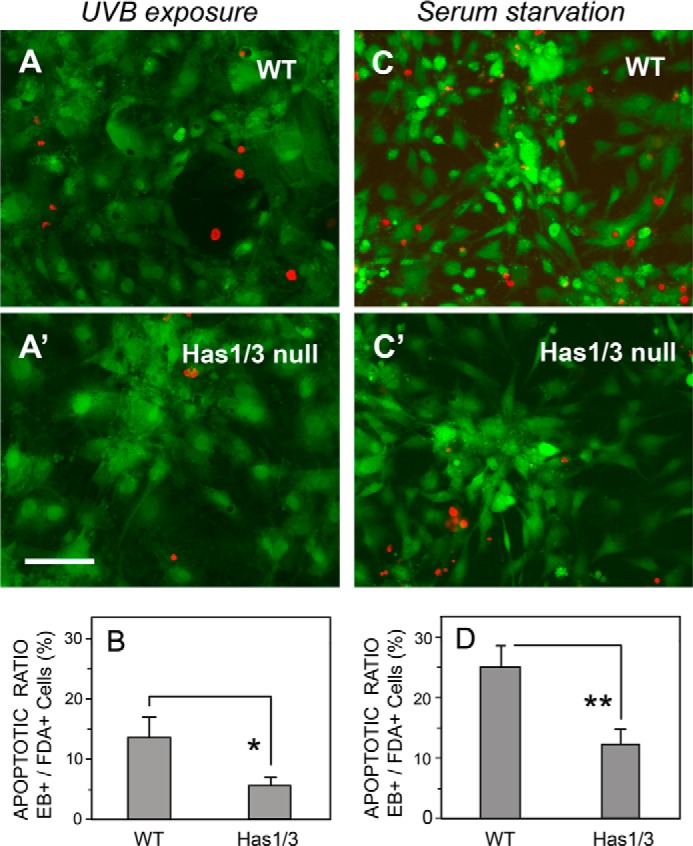
Has1/3 null fibroblasts are relatively resistant to apoptosis. Panels on left, FDA/EB “live/dead” staining of cells 12 h after UVB irradiation (32 mJ/cm2). Representative fluorescent microscopy images of WT cells (A) and Has1/3 null cells (A′) are shown above; quantitation of the ratio of dead (EB+) to live (FDA+) cells is shown below in (B). Panels on right, FDA/EB staining of cells after 4 h of serum starvation. Representative images (C and C′) and quantitation of the EB/FDA ratio (graph, D) are shown. Scale bar, 100 μm. Each bar in the graphs is the mean ± S.D. (error bars) of three independent experiments. Comparison was by Student's t test: *, p < 0.025; **, p < 0.001.
To further delineate the difference in apoptotic responses between the two types of cells, we treated fibroblasts with UVB irradiation or serum starvation and harvested the cells at different time points to investigate the activation of caspase enzymes. We analyzed the cleavage of procaspase-9 and procaspase-3 into their corresponding activated forms and also the cleavage of PARP (an established substrate for the activated caspases) by Western blotting. As shown in Fig. 5A, activation of caspase-9 and caspase-3 and cleavage of PARP were triggered in both WT and Has1/3 null cells at 12 h after UVB irradiation. However, caspase activation and PARP cleavage were significantly less in Has1/3 null cells than in WT cells at 12 h. In comparison with UVB, serum starvation triggered a relatively rapid activation of caspases and cleavage of PARP at 2 h, which peaked at 6 h after the start of serum starvation. Here again, the amount of procaspase and PARP cleavage was less in Has1/3 null cells than in WT cells (at 2 and 4 h) (Fig. 5B), but the difference disappeared when apoptosis reached its peak around 6-h serum starvation in both types of cells. Despite differences in the time course of apoptosis following serum starvation versus UVB, the reduced susceptibility of Has1/3 null fibroblasts to caspase activation was a remarkable and consistent feature.
FIGURE 5.

Attenuated apoptotic responses in Has1/3 null fibroblasts involve reduced caspase activation. A, representative Western blots of cleaved caspase-9, cleaved caspase-3, and full-length and cleaved PARP in skin fibroblasts at different times after UVB exposure (32 mJ/cm2). GAPDH was used as a loading control. B, Western blots as in A, from fibroblasts harvested at different times after serum starvation. Data in the table below these blots are the mean from densitometric scans in the corresponding lane, averaged from two independent experiments; the variance was less than 20% of the mean in all cases.
For completeness, we also looked for changes in caspase-8 (the main sensor of plasma membrane damage), in addition to caspase-9 (which responds to mitochondrial damage) and caspase-3 (the common downstream effector) (32). However, no caspase-8 was detectable in these primary murine fibroblasts, even under conditions that triggered caspase-9 and PARP cleavage (data not shown).
Removal of HA from the Extracellular Matrix Does Not Alter the Relative Resistance of Has1/3 Null Cells to Apoptosis
Many studies have demonstrated that HA in the extracellular matrix plays an important role in regulating various cellular behaviors (17, 33, 34). Our observation that Has1/3 null fibroblasts have a relatively larger extracellular HA coat and are also more resistant to apoptosis led to the obvious question of whether apoptosis resistance might depend upon the size of the HA coat. To address this question, we enzymatically removed the extracellular HA using hyaluronidase; the efficient digestion of HA was confirmed by staining non-permeabilized cells with bHABP (Fig. 6A). Fibroblasts with or without preincubation with hyaluronidase were treated with UVB irradiation (Fig. 6B) or serum starvation (Fig. 6C) to induce apoptosis. To our surprise, degradation of extracellular HA did not trigger caspase activation; nor did it have any effect upon caspase activation in cells treated with UVB (Fig. 6B) or serum starvation (Fig. 6C). This result indicates that the increased resistance to apoptosis in Has1/3 null fibroblasts is not mediated by HA in the extracellular matrix.
FIGURE 6.
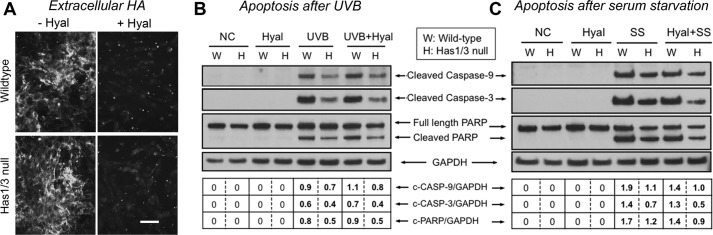
Apoptosis resistance of skin fibroblasts is not mediated by HA in the extracellular matrix. A, extracellular HA in skin fibroblast cultures treated with S. dysgalactiae hyaluronidase (0.3 unit/ml, 10 min at 37 °C) (+Hyal) or in untreated control cultures (−Hyal) and then immunostained with bHABP/streptavidin-Alexa Fluor 488. Scale bar, 100 μm. B, representative Western blots of cleaved caspase-9 and -3 and cleaved PARP in skin fibroblasts treated with either the hyaluronidase (Hyal) or UVB irradiation (UVB) or both (UVB + Hyal), versus untreated normal controls (NC). C, Western blots of cleaved caspase-9 and -3 and cleaved PARP in skin fibroblasts treated with either Hyal or serum starvation (SS) or both (Hyal + SS), versus untreated normal controls (NC). GAPDH was examined as a loading control.
Pretreatment with 4-MU Decreases Intracellular Levels of HA and Has2 mRNA and Sensitizes the Cells to Apoptosis
To further explore the role of cellular HA and Has2 in mediating susceptibility to apoptosis, we treated fibroblasts with 4-MU, a well recognized inhibitor of HA synthesis (35, 36). As shown in Fig. 7, 4-MU treatment at a dosage of 1 mm for 6 and 12 h effectively inhibited overall HA synthesis (measured by FACE) in WT and Has1/3 null cells (Fig. 7, A and B), with no effect upon an unrelated glycosaminoglycan, CS (data not shown). Inhibition of HA synthesis was relatively greater in the Has1/3 null cells, as evidenced in both the cell layer (Fig. 7A) and the media (Fig. 7B). At the same time, a comparison of extracellular HA (by HABP staining of non-permeabilized cells) versus intracellular HA (by bHABP staining performed after hyaluronidase digestion and subsequent permeabilization) revealed that intracellular HA levels are proportionally more affected than extracellular HA levels after a 6-h treatment with 4-MU (Fig. 7C). The 6-h 4-MU treatment also significantly inhibited Has2 gene expression in both types of cells (Fig. 7D) and Has1 gene expression in WT cells (Fig. 7E), as determined by qPCR analysis. This result is consistent with observations from other groups (35, 36). Importantly, the 6-h treatment with 4-MU alone was not cytotoxic because it did not trigger apoptosis, yet it clearly sensitized the cells to apoptosis induced by either UVB (Fig. 7F) or serum starvation (Fig. 7G), as can be seen by examining the last two lanes in each set of Western blots.
FIGURE 7.

4-MU treatment sensitizes skin fibroblasts to apoptosis. A, HA levels in the cell layer (A) or culture media (B) of fibroblasts treated with 4-MU (1 mm) for either 6 or 12 h were quantified by FACE analysis. C, quantitation of fluorescence intensity of extracellular HA (left) and intracellular HA (right) staining with bHABP, in skin fibroblasts treated with 4-MU for 6 h and analyzed by image processing. Gray bars, no 4-MU; black bars, +4-MU. *, p < 0.05 by Student's t test (significant difference from vehicle-treated cells). Changes in Has2 mRNA levels in both types of fibroblasts (D) or in Has1 and Has3 mRNA levels in WT fibroblasts (E), treated with 4-MU (1 mm) or vehicle alone for 6 h, were analyzed by qPCR. Each data point is the mean ± S.D. of two independent experiments. F, representative Western blots of cleaved caspase-9 and -3 and cleaved PARP in skin fibroblasts treated with 4-MU or vehicle control for 6 h, followed by 32 mJ/cm2 UVB (4-MU + UVB and Veh + UVB). G, Western blots of cleaved caspase-9 and -3 and cleaved PARP in skin fibroblasts treated with either 4-MU or vehicle control for 6 h followed by 4 h of serum starvation (4-MU + SS and Veh + SS). GAPDH, loading control.
Knockdown of Has2 Gene Expression Using Targeted siRNA Sensitizes the Cells to Apoptosis
Seeking a way to inhibit HA synthesis more specifically, we reduced Has2 gene expression via RNAi using Has2-targeted siRNA transfection. Efficient knockdown of Has2 gene expression was confirmed by qPCR (Fig. 8A). Interestingly, a 2-fold increase in Has1 gene expression in WT cells was observed in response to Has2 knockdown (Fig. 8B), whereas Has3 was unchanged (Fig. 8C). This up-regulated Has1 gene expression was not sufficient to compensate for the loss of HA synthesis capacity in the WT cells caused by Has2 gene knockdown, because an effective inhibition of HA accumulation in both the cell layer and media (Fig. 8D) of both WT and Has1/3 null cells was detected by FACE analysis. After transfection with either Has2-targeted or non-targeted scrambled siRNA, fibroblasts were challenged with UVB or with serum starvation to trigger apoptosis. Has2 RNAi did not trigger caspase activation on its own (Fig. 8F, lanes C and D) but did enhance caspase activation in cells exposed to UVB irradiation (Fig. 8E, lanes 7 and 8) or serum starvation (Fig. 8F, lanes 7 and 8), indicating that Has2 RNAi sensitizes fibroblasts to stress-induced apoptosis. Furthermore, Has2 RNAi diminished the phenotypic difference in apoptosis susceptibility between Has1/3 null and WT cells. For example, in Fig. 8E, compare the value of c-CASP-3/GAPDH in lanes 7 and 8 (equivalent intensities) with that in lanes 5 and 6 (different intensities), or in Fig. 8F, compare the value of c-CASP-3/GAPDH in lanes 7 and 8 (equivalent intensities) with that in lanes 5 and 6 (different intensities).
FIGURE 8.

Knockdown of Has2 gene expression by Has2 siRNA transfection sensitizes skin fibroblasts to apoptosis. mRNA expression levels of Has2 (A), Has1 (B), and Has3 (C) in either WT or Has1/3 null fibroblasts, analyzed by qPCR. No Rx control, untreated normal control; Scrambled siRNA, transfected with non-targeted scrambled siRNA; Has2 siRNA, transfected with Has2 siRNA. Each bar represents the mean ± S.D. (error bars) of two independent experiments. D, HA levels in the cell layer (left) or media (right) from fibroblast cultures, treated as described for A–C and analyzed by FACE. Each bar is the mean ± S.D. of two independent experiments. E and F, Western blots of skin fibroblasts after UVB (32 mJ/cm2) or serum starvation (4 h), respectively, showing effects upon the expression of cleaved caspase-9 and -3, relative to GAPDH loading controls, in cells transfected with siRNA against Has2 (Has2 siRNA), non-targeted scrambled siRNA (Scrambled), or untreated normal control (NC).
The Addition of High Molecular Weight HA Failed to Abrogate the Apoptosis Sensitization Caused by Has2 RNAi
Vigetti et al. (37) reported that HMW-HA effectively prevents the apoptosis of human aortic smooth muscle cells caused by 4-MU treatment. In order to test whether HMW-HA could protect skin fibroblasts against apoptosis, HA synthesis in fibroblasts was first inhibited by knockdown of Has2 gene expression with RNA interference, followed by the addition of HMW-HA to the culture media during serum starvation or immediately following UVB exposure. As shown in Fig. 9 (A and B), adding HMW-HA back to the media failed to protect the fibroblasts against cell death induced by either UVB or serum starvation (compare lane 3 with lane 2 in the corresponding figure panels). Nor did HMW-HA rescue the Has2 RNAi-induced sensitization to apoptosis (compare lane 7 with lane 6), as determined by a cell death ELISA.
FIGURE 9.
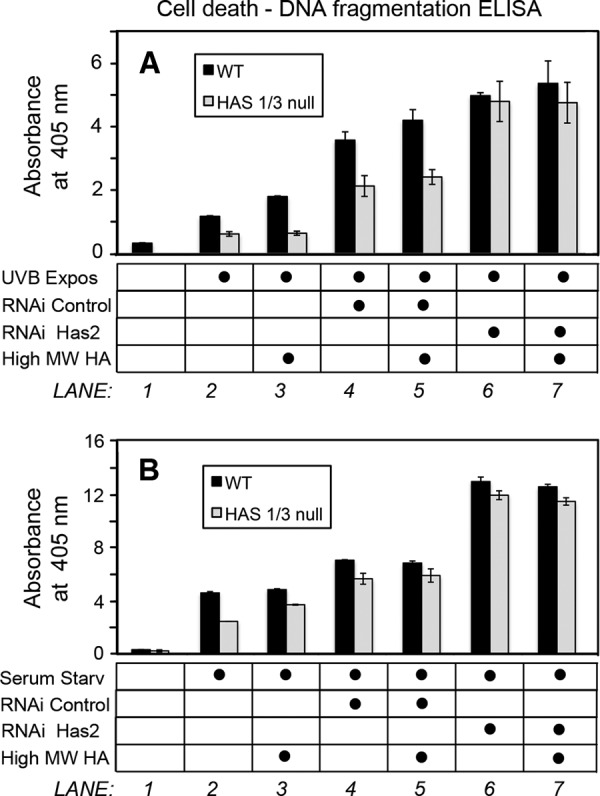
External addition of high molecular weight HA fails to rescue the apoptotic sensitization caused by Has2 RNAi. Shown is cell death/DNA fragmentation in fibroblasts transfected with Has2 siRNA (RNAi Has2) or with scrambled siRNA (RNAi Control), prior to a UVB 32 mJ/cm2 exposure (A) or 4-h serum starvation (B), in the presence or absence of high molecular weight HA (High MW HA). Apoptosis was measured with a cell death detection ELISA kit.
DISCUSSION
In this work, we have examined how the loss of HAS1 and HAS3 (two of the three known HAS enzymes) can affect HA synthesis, cell growth, and cell survival in primary skin fibroblasts. We found that Has2 gene expression and levels of HA are higher at baseline in Has1/3 null skin fibroblasts than in WT cells. Under stressful conditions, either UVB exposure or serum starvation, Has2 expression initially declines but then fully recovers in both the WT and the Has1/3 null fibroblasts, suggesting that inducible HAS2 enzyme activity plays a major role in both types of cells. Furthermore, we found that Has1/3 null cells are more resistant to stress-induced apoptosis than are WT cells. This resistance is not mediated by HA in the extracellular matrix, because removal of the pericellular HA coat does not abrogate resistance and because the addition of exogenous HA does not confer resistance to environmental stress. On the other hand, either 4-MU or Has2 RNAi treatment successfully rendered the cells more sensitive to apoptosis, suggesting that the apoptosis resistance in fibroblasts is mediated either by intracellular HA synthesized by HAS2 or by some unknown HA-independent function of HAS2. Interestingly, a recent study from the Heldin group (38) showed that it was HAS2 and not extracellular HA that plays a crucial role in regulating TGF-β-induced epithelial-mesenchymal transition and cell migration, which suggests an HA-independent role of HAS2 as a signaling molecule. Overall, the findings suggest that either HA synthesis driven by HAS2 or HAS2 itself is crucial to maintain cell viability, especially under stressful or diseased conditions. These findings will now be discussed in more detail.
Increased Hyaluronan Synthesis in Has1/3 Null Skin Fibroblasts
Prior to analyzing the HA content in Has1/3 null cells, we had anticipated that HA synthesis in the cells might be decreased due to the loss of both Has1 and Has3. But contrary to expectations, HA levels and Has2 gene expression were observed to be higher in Has1/3 null fibroblasts than in WT cells. Several mechanisms could contribute to increased HA accumulation in Has1/3 null fibroblasts. First, up-regulated Has2 gene expression could be at least partially responsible for the higher amount of HA observed in the Has1/3 null cells. Silencing of the two enzymes, HAS1 and HAS3, apparently contributes to increased expression of Has2 mRNA, suggesting that expression of Has1, Has3, or both has a negative regulatory effect on Has2 gene expression at the transcriptional level. Growth factors and cytokines have been shown to either induce or repress Has2 gene expression by regulating transcription factors that bind to response elements in the Has2 promoter (24). The promoters for Has1 and Has3 are less well studied, but one can speculate that deletion of the Has1 and Has3 genes might increase the availability of certain transcription factors to stimulate Has2 transcription. A second reason that HA synthesis might be increased in Has1/3 null fibroblasts is an enhancement of HAS2 protein stability and enzyme activity. HAS2 undergoes regulation at the post-translational level; Vigetti et al. (22) reported that HAS2 can be O-GlcNAcylated on serine 221, resulting in a significant enhancement of both the stability and activity of HAS2, and this O-GlcNAcylation is regulated by cytosolic availability of UDP-GlcNAc. It would be interesting if one or more post-translational changes occur that stabilize the HAS2 enzyme and increase its enzymatic activity in cells lacking Has1 and Has3. We hope to pursue this possibility in subsequent work. A third potential mechanism involves heterodimerization of HAS proteins. Karousou et al. (23) showed that HAS2 and HAS3 can dimerize with each other, and a loss-of-function mutation in one unit of the dimer significantly decreases the overall enzymatic activity. This observation suggests that in WT fibroblasts, HAS2 might be dimerized to some extent with other HAS proteins, whereas in the Has1/3 null cells, HAS2 can only form homodimers. Increased HA synthesis in the Has1/3 null cells would be consistent with loss of an inhibitory effect of heterodimerization between HAS2 and HAS3 or between HAS2 and HAS1. A fourth possibility to explain up-regulation of HA synthesis in the Has1/3 null cells would be a significant difference in affinity of the various HAS enzymes for sugar substrates (e.g. a higher affinity and/or slower reaction kinetics with HAS1 and/or HAS3 as compared with HAS2). The Tammi group (39) recently presented evidence that HAS1 is relatively less active than HAS2 or HAS3 in a transfected cos-1 cell system; if the situation is similar in fibroblasts, this might argue against a substrate affinity-based mechanism contributing to the HA changes that we observe. A fifth possibility (i.e. that an increase in accumulation of HA is due to a relative decrease in hyaluronan catabolism in the Has1/3 null cells) appears to be ruled out because Hyal1 and Hyal2 are unchanged in the Has1/3 null cells (Fig. 1E).
Alteration of HA Synthesis and Has2 Gene Expression in Fibroblasts in Response to Environmental Stress
As mentioned in the Introduction, fibroblast proliferation and survival is an essential component of repair processes in the skin following injury from external factors, such as UV radiation and physical wounding. For example, the skin is the organ most susceptible to UV-induced damage from the sun (13). Various groups have investigated UVB-induced changes in HA production either in vivo or in vitro. Averbeck et al. (40) reported that HA production decreased at 3 h and then increased 24 h after UVB exposure in HaCaT human keratinocyte cell cultures but remained suppressed in cultured human fibroblasts. Further study of human skin in vivo yielded results consistent with the in vitro findings (40). Rauhala et al. (41) showed that a single, acute UVB exposure of rat epidermal keratinocytes in monolayer and organotypic cultures induced a persistent elevation of Has2 gene expression at 8 h after exposure, along with an accumulation of HA, and that these effects are mediated by p38 and Ca2+/calmodulin-dependent protein kinase II. On the other hand, a series of reports from the Fischer group (42) demonstrated that chronic UVB exposure caused a significant loss of HA from the dermis of mouse skin and decreased gene expression of all three HAS enzymes in the dermis. The UVB-induced down-regulation of Has2 was mediated by the activation of αVβ3-integrin induced by collagen fragments (18). Another group reported that a single UVB exposure caused an increase in chondroitin sulfate but not in other glycosaminoglycans, including HA, in human skin (43). This illustrates that results from different studies vary substantially, depending upon tissue type, cell type, wavelength, and dosage of UV radiation as well as the timing of observations. In the present study, primary skin fibroblasts were exposed to a single dose of 32 mJ/cm2 UVB, which induced a time-dependent increase of both Has2 gene expression and HA synthesis in skin fibroblasts observable mainly at 12 and 24 h (Fig. 2, A–C). The phenotypic difference between Has1/3 null and WT cells, in which HA and Has2 were higher in the Has1/3 null cells at most time points after UVB exposure, attests to a predominant role of Has2 in this system, especially at baseline level or normal physiological conditions. However, under stressful conditions, Has1 and Has3 in WT cells could be mobilized and play a more important role in generating HA, especially when Has2 function is either suppressed or saturated at this condition. In our study, at the late time point (24 h) after UVB irradiation, HA levels in WT cells caught up to the HA levels in Has1/Has3 null cells (Fig. 2, A and B), which may be explained by the induction of Has1 and Has3 expression observed in the WT cells at 24 h after UVB (Fig. 2, D and E).
To emulate some aspects of the stress that occurs in wounded or ischemic tissues, we subjected fibroblasts to serum starvation. In the Has1/3 null cells, Has2 mRNA expression declined rapidly (down ∼70% at 2 h), perhaps due to withdrawal of the serum growth factors known to sustain Has2 transcription (24); Has2 then recovered to control levels by 12 h (Fig. 3C). In contrast, Has1 mRNA was significantly suppressed (Fig. 3D), and Has3 mRNA levels were relatively unaffected (Fig. 3E) by serum starvation. Levels of cell-associated HA levels lagged behind the changes in Has2, as might be expected (Fig. 3A). Interestingly, in contrast to the changes in cell-associated HA in Has1/3 null cells, HA levels in the cell layer of WT cells remained essentially constant during 24 h of serum starvation (Fig. 3A). HA levels in the media from both types of cells showed a moderate increase during the course of serum starvation (Fig. 3B), probably due to the HA released into the media from the apoptotic cells. Overall, these data suggest that the presence of all three Has isoforms acts to maintain HA synthesis at a relatively constant level under stressful conditions.
Hyaluronan and Has2-mediated Resistance to Apoptosis in Skin Fibroblasts
HA and HA-dependent extracellular matrices are already known to play an important role in regulating various cellular behaviors, including proliferation, migration, and cellular adhesion in different tissues (34), and in regulating TGFβ-1 induced transformation of fibroblasts into myofibroblasts during wound healing processes (17). However, the role of HA in regulating cell death and survival has proven to be very complicated. HA is reported to protect against apoptosis and to promote survival in various types of cells, including granulosa cells (44), articular chondrocytes (45, 46), and human aortic smooth muscle cells (37). In contrast, other studies showed that HA treatment induces cell death in cultured human gingival fibroblasts (47) and lymphoma cell lines (48). Thus, the regulatory effect of HA upon cell viability appears to depend upon cell context, even when controlling for the size of HA, because high molecular weight HA has been shown to either promote cellular survival (37) or induce cell death (49) in different cell types.
In our current study, which showed that Has1/3 null fibroblasts overexpress HA, produce large extracellular HA matrices, and are resistant to stress-induced apoptosis, we initially expected that the resistance to apoptosis would involve pericellular (extracellular) HA. This preconception was based upon published studies, done mainly in cancer cells, showing that cell death, growth, and survival can be regulated by interactions of HA with HA receptors (mainly CD44) on the plasma membrane (21, 33, 49, 50). Our finding that removal of the pericellular HA coat neither affects cell viability nor sensitizes the cells to apoptosis came as a surprise and motivated us to confirm the observation in a number of ways. First we employed 4-MU, a well established HA synthesis inhibitor that is proposed to work by depleting the GlcUA substrates and by repressing Has2 and Has3 gene expression (35, 36). Interestingly, 4-MU has also attracted attention recently as a potential chemotherapeutic agent (51–53). We chose the dose of 4-MU (1 mm) based upon work by Vigetti et al. (36) in human aortic smooth muscle cells (myocytes). We reasoned that fibroblasts (which undergo transformation to myofibroblasts in wounds) might be comparable with myocytes, and indeed, a 4-MU (1 mm) treatment of 6 h was sufficient to suppress Has2 gene expression and to reduce HA production (markedly for secreted HA, less so for cell-associated HA; Fig. 7, A and B). Importantly, despite a minimal effect of 4-MU upon cell-associated HA (the pericellular coat), 4-MU was able to completely reverse the resistance to apoptosis in Has1/3 null fibroblasts and to sensitize the WT cells to apoptosis (Fig. 7, F and G). To corroborate the results of the 4-MU treatment, we specifically inhibited Has2 gene expression using siRNA transfection and showed a similar result (Fig. 8). As a third form of validation, we performed experiments in which high molecular weight HA was added back to cultures in which HA synthesis had been inhibited by Has2 siRNA (Fig. 9). The results showed that exogenous HA had no protective effect upon UVB-induced or serum starvation-induced apoptosis. This contrasts with findings by Vigetti et al. (37), in which the addition of exogenous HA was shown to protect human aortic smooth muscle cells from cell death caused by 4-MU treatment; major differences in cell type, culture conditions, and inhibitors used could easily explain the different outcomes.
Another observation worthy of mention is the fact that differences in apoptotic resistance between WT and Has1/3 null cells only occurred at early time points during stress (before 12 h after UVB exposure and before 4 h after serum starvation) but then became equivalent. We attribute this to the highly likely possibility that other compensatory mechanisms are activated over time, ultimately masking the initial differences between cell-specific responses to the stressful conditions.
Our investigation has provided results that suggest involvement of Has2 in classical apoptotic pathways. Environmental stress was shown to trigger cleavage of caspase-9 (and of its downstream targets, caspase-3 and PARP) in the fibroblasts. Caspase-9 cleavage was less in the Has1/3 null fibroblasts, suggesting that HA or Has2 may affect processes occurring in mitochondria, the source of signals that activate caspase-9 (32). The other major apoptotic pathway, mediated by caspase-8 activation (triggered by signals from proapoptotic receptors, such as Fas and TNFα, in the plasma membrane) is unlikely to play a role here because caspase-8 is not detectable in the murine skin fibroblasts (data not shown). The mechanism by which HA or the activity of HAS2 affects caspase-9 cleavage and whether this involves modulation of mitochondrial membrane potential or mitochondrial proteins involved in apoptosis will be a focus of ongoing and future studies.
In conclusion, the present study shows that the ability of skin-derived fibroblasts to generate HA and to form a pericellular HA coat is significantly enhanced in Has1/3 null cells. In addition, Has2 gene expression is significantly up-regulated in the Has1/3 null fibroblasts, suggesting that the other two HA synthase isoforms (Has1 and Has3) might exert negative regulation on Has2 gene expression. Has1/3 null skin fibroblasts are more resistant to apoptosis induced by environmental stress, and this enhanced apoptosis resistance in Has1/3 null cells is not mediated by the pericellular HA coat but is dependent on Has2 gene expression and HA synthetic activity in the cells. This suggests that HA synthesis driven by Has2 or an HA-independent function of Has2 plays an indispensable role in protecting the cells against apoptosis. These findings provide novel mechanistic insight into how Has2 and hyaluronan synthesis regulate the turnover and function of fibroblasts under normal physiological and certain pathological conditions.
Acknowledgments
We sincerely thank Valbona Cali for help in generating the FACE data and Dr. Judy Drazba and Eric Diskin for assistance with cell imaging. We would also like to thank Dr. Vincent Hascall for his mentorship and support.
This work was supported, in whole or in part, by National Institutes of Health Grant P01 HL107147.
- HA
- hyaluronan
- HAS
- hyaluronan synthase
- HMW-HA
- high molecular weight HA
- 4-MU
- 4-methylumbelliferone
- RBC
- red blood cell
- CS
- chondroitin sulfate
- FACE
- fluorophore-assisted carbohydrate electrophoresis
- TEMED
- N,N,N′,N′-tetramethylethylenediamine
- qPCR
- quantitative real-time PCR
- FDA
- fluorescein diacetate
- EB
- ethidium bromide
- TRITC
- tetramethylrhodamine isothiocyanate
- PARP
- poly(ADP-ribose) polymerase
- bHABP
- biotinylated HA-binding protein.
REFERENCES
- 1. Kubo K., Kuroyanagi Y. (2005) A study of cytokines released from fibroblasts in cultured dermal substitute. Artif. Organs 29, 845–849 [DOI] [PubMed] [Google Scholar]
- 2. Wong T., McGrath J. A., Navsaria H. (2007) The role of fibroblasts in tissue engineering and regeneration. Br. J. Dermatol. 156, 1149–1155 [DOI] [PubMed] [Google Scholar]
- 3. Werner S., Krieg T., Smola H. (2007) Keratinocyte-fibroblast interactions in wound healing. J. Invest. Dermatol. 127, 998–1008 [DOI] [PubMed] [Google Scholar]
- 4. Jelaska A., Korn J. H. (2000) Role of apoptosis and transforming growth factor β1 in fibroblast selection and activation in systemic sclerosis. Arthritis Rheum. 43, 2230–2239 [DOI] [PubMed] [Google Scholar]
- 5. Jun J. B., Kuechle M., Min J., Shim S. C., Kim G., Montenegro V., Korn J. H., Elkon K. B. (2005) Scleroderma fibroblasts demonstrate enhanced activation of Akt (protein kinase B) in situ. J. Invest. Dermatol. 124, 298–303 [DOI] [PubMed] [Google Scholar]
- 6. Takagawa S., Lakos G., Mori Y., Yamamoto T., Nishioka K., Varga J. (2003) Sustained activation of fibroblast transforming growth factor-β/Smad signaling in a murine model of scleroderma. J. Invest. Dermatol. 121, 41–50 [DOI] [PubMed] [Google Scholar]
- 7. Pannu J., Trojanowska M. (2004) Recent advances in fibroblast signaling and biology in scleroderma. Curr. Opin. Rheumatol. 16, 739–745 [DOI] [PubMed] [Google Scholar]
- 8. Luo S., Benathan M., Raffoul W., Panizzon R. G., Egloff D. V. (2001) Abnormal balance between proliferation and apoptotic cell death in fibroblasts derived from keloid lesions. Plast. Reconstr. Surg. 107, 87–96 [DOI] [PubMed] [Google Scholar]
- 9. Ishihara H., Yoshimoto H., Fujioka M., Murakami R., Hirano A., Fujii T., Ohtsuru A., Namba H., Yamashita S. (2000) Keloid fibroblasts resist ceramide-induced apoptosis by overexpression of insulin-like growth factor I receptor. J. Invest. Dermatol. 115, 1065–1071 [DOI] [PubMed] [Google Scholar]
- 10. Moodley Y. P., Caterina P., Scaffidi A. K., Misso N. L., Papadimitriou J. M., McAnulty R. J., Laurent G. J., Thompson P. J., Knight D. A. (2004) Comparison of the morphological and biochemical changes in normal human lung fibroblasts and fibroblasts derived from lungs of patients with idiopathic pulmonary fibrosis during FasL-induced apoptosis. J. Pathol. 202, 486–495 [DOI] [PubMed] [Google Scholar]
- 11. Bühling F., Wille A., Röcken C., Wiesner O., Baier A., Meinecke I., Welte T., Pap T. (2005) Altered expression of membrane-bound and soluble CD95/Fas contributes to the resistance of fibrotic lung fibroblasts to FasL induced apoptosis. Respir. Res. 6, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alikhani Z., Alikhani M., Boyd C. M., Nagao K., Trackman P. C., Graves D. T. (2005) Advanced glycation end products enhance expression of pro-apoptotic genes and stimulate fibroblast apoptosis through cytoplasmic and mitochondrial pathways. J. Biol. Chem. 280, 12087–12095 [DOI] [PubMed] [Google Scholar]
- 13. Fisher G. J., Kang S., Varani J., Bata-Csorgo Z., Wan Y., Datta S., Voorhees J. J. (2002) Mechanisms of photoaging and chronological skin aging. Arch. Dermatol. 138, 1462–1470 [DOI] [PubMed] [Google Scholar]
- 14. Itano N., Sawai T., Yoshida M., Lenas P., Yamada Y., Imagawa M., Shinomura T., Hamaguchi M., Yoshida Y., Ohnuki Y., Miyauchi S., Spicer A. P., McDonald J. A., Kimata K. (1999) Three isoforms of mammalian hyaluronan synthases have distinct enzymatic properties. J. Biol. Chem. 274, 25085–25092 [DOI] [PubMed] [Google Scholar]
- 15. Jacobson A., Brinck J., Briskin M. J., Spicer A. P., Heldin P. (2000) Expression of human hyaluronan synthases in response to external stimuli. Biochem. J. 348, 29–35 [PMC free article] [PubMed] [Google Scholar]
- 16. Recklies A. D., White C., Melching L., Roughley P. J. (2001) Differential regulation and expression of hyaluronan synthases in human articular chondrocytes, synovial cells and osteosarcoma cells. Biochem. J. 354, 17–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Midgley A. C., Rogers M., Hallett M. B., Clayton A., Bowen T., Phillips A. O., Steadman R. (2013) Transforming growth factor-β1 (TGF-β1)-stimulated fibroblast to myofibroblast differentiation is mediated by hyaluronan (HA)-facilitated epidermal growth factor receptor (EGFR) and CD44 co-localization in lipid rafts. J. Biol. Chem. 288, 14824–14838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Röck K., Grandoch M., Majora M., Krutmann J., Fischer J. W. (2011) Collagen fragments inhibit hyaluronan synthesis in skin fibroblasts in response to ultraviolet B (UVB): new insights into mechanisms of matrix remodeling. J. Biol. Chem. 286, 18268–18276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stuhlmeier K. M., Pollaschek C. (2004) Differential effect of transforming growth factor β (TGF-β) on the genes encoding hyaluronan synthases and utilization of the p38 MAPK pathway in TGF-β-induced hyaluronan synthase 1 activation. J. Biol. Chem. 279, 8753–8760 [DOI] [PubMed] [Google Scholar]
- 20. Michael D. R., Phillips A. O., Krupa A., Martin J., Redman J. E., Altaher A., Neville R. D., Webber J., Kim M. Y., Bowen T. (2011) The human hyaluronan synthase 2 (HAS2) gene and its natural antisense RNA exhibit coordinated expression in the renal proximal tubular epithelial cell. J. Biol. Chem. 286, 19523–19532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bourguignon L. Y., Gilad E., Peyrollier K. (2007) Heregulin-mediated ErbB2-ERK signaling activates hyaluronan synthases leading to CD44-dependent ovarian tumor cell growth and migration. J. Biol. Chem. 282, 19426–19441 [DOI] [PubMed] [Google Scholar]
- 22. Vigetti D., Deleonibus S., Moretto P., Karousou E., Viola M., Bartolini B., Hascall V. C., Tammi M., De Luca G., Passi A. (2012) Role of UDP-N-acetylglucosamine (GlcNAc) and O-GlcNAcylation of hyaluronan synthase 2 in the control of chondroitin sulfate and hyaluronan synthesis. J. Biol. Chem. 287, 35544–35555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Karousou E., Kamiryo M., Skandalis S. S., Ruusala A., Asteriou T., Passi A., Yamashita H., Hellman U., Heldin C. H., Heldin P. (2010) The activity of hyaluronan synthase 2 is regulated by dimerization and ubiquitination. J. Biol. Chem. 285, 23647–23654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tammi R. H., Passi A. G., Rilla K., Karousou E., Vigetti D., Makkonen K., Tammi M. I. (2011) Transcriptional and post-translational regulation of hyaluronan synthesis. FEBS J. 278, 1419–1428 [DOI] [PubMed] [Google Scholar]
- 25. Manuskiatti W., Maibach H. I. (1996) Hyaluronic acid and skin: wound healing and aging. Int. J. Dermatol. 35, 539–544 [DOI] [PubMed] [Google Scholar]
- 26. Webber J., Meran S., Steadman R., Phillips A. (2009) Hyaluronan orchestrates transforming growth factor-β1-dependent maintenance of myofibroblast phenotype. J. Biol. Chem. 284, 9083–9092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Meran S., Thomas D., Stephens P., Martin J., Bowen T., Phillips A., Steadman R. (2007) Involvement of hyaluronan in regulation of fibroblast phenotype. J. Biol. Chem. 282, 25687–25697 [DOI] [PubMed] [Google Scholar]
- 28. Mack J. A., Feldman R. J., Itano N., Kimata K., Lauer M., Hascall V. C., Maytin E. V. (2012) Enhanced inflammation and accelerated wound closure following tetraphorbol ester application or full-thickness wounding in mice lacking hyaluronan synthases Has1 and Has3. J. Invest. Dermatol. 132, 198–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Anand S., Chakrabarti E., Kawamura H., Taylor C. R., Maytin E. V. (2005) Ultraviolet light (UVB and UVA) induces the damage-responsive transcription factor CHOP/gadd153 in murine and human epidermis: evidence for a mechanism specific to intact skin. J. Invest. Dermatol. 125, 323–333 [DOI] [PubMed] [Google Scholar]
- 30. Bennett H. L., Fleming J. T., O'Prey J., Ryan K. M., Leung H. Y. (2010) Androgens modulate autophagy and cell death via regulation of the endoplasmic reticulum chaperone glucose-regulated protein 78/BiP in prostate cancer cells. Cell Death Dis. 1, e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li Z., Wei H., Liu X., Hu S., Cong X., Chen X. (2010) LPA rescues ER stress-associated apoptosis in hypoxia and serum deprivation-stimulated mesenchymal stem cells. J. Cell Biochem. 111, 811–820 [DOI] [PubMed] [Google Scholar]
- 32. Budihardjo I., Oliver H., Lutter M., Luo X., Wang X. (1999) Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 15, 269–290 [DOI] [PubMed] [Google Scholar]
- 33. Toole B. P. (2004) Hyaluronan: from extracellular glue to pericellular cue. Nat. Rev. Cancer 4, 528–539 [DOI] [PubMed] [Google Scholar]
- 34. Evanko S. P., Tammi M. I., Tammi R. H., Wight T. N. (2007) Hyaluronan-dependent pericellular matrix. Adv. Drug Deliv. Rev. 59, 1351–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kultti A., Pasonen-Seppänen S., Jauhiainen M., Rilla K. J., Kärnä R., Pyöriä E., Tammi R. H., Tammi M. I. (2009) 4-Methylumbelliferone inhibits hyaluronan synthesis by depletion of cellular UDP-glucuronic acid and downregulation of hyaluronan synthase 2 and 3. Exp. Cell Res. 315, 1914–1923 [DOI] [PubMed] [Google Scholar]
- 36. Vigetti D., Rizzi M., Viola M., Karousou E., Genasetti A., Clerici M., Bartolini B., Hascall V. C., De Luca G., Passi A. (2009) The effects of 4-methylumbelliferone on hyaluronan synthesis, MMP2 activity, proliferation, and motility of human aortic smooth muscle cells. Glycobiology 19, 537–546 [DOI] [PubMed] [Google Scholar]
- 37. Vigetti D., Rizzi M., Moretto P., Deleonibus S., Dreyfuss J. M., Karousou E., Viola M., Clerici M., Hascall V. C., Ramoni M. F., De Luca G., Passi A. (2011) Glycosaminoglycans and glucose prevent apoptosis in 4-methylumbelliferone-treated human aortic smooth muscle cells. J. Biol. Chem. 286, 34497–34503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Porsch H., Bernert B., Mehić M., Theocharis A. D., Heldin C. H., Heldin P. (2013) Efficient TGFβ-induced epithelial-mesenchymal transition depends on hyaluronan synthase HAS2. Oncogene 32, 4355–4365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rilla K., Oikari S., Jokela T. A., Hyttinen J. M., Kärnä R., Tammi R. H., Tammi M. I. (2013) Hyaluronan synthase 1 (HAS1) requires higher cellular UDP-GlcNAc concentration than HAS2 and HAS3. J. Biol. Chem. 288, 5973–5983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Averbeck M., Gebhardt C. A., Voigt S., Beilharz S., Anderegg U., Termeer C. C., Sleeman J. P., Simon J. C. (2007) Differential regulation of hyaluronan metabolism in the epidermal and dermal compartments of human skin by UVB irradiation. J. Invest. Dermatol. 127, 687–697 [DOI] [PubMed] [Google Scholar]
- 41. Rauhala L., Hämäläinen L., Salonen P., Bart G., Tammi M., Pasonen-Seppänen S., Tammi R. (2013) Low dose ultraviolet B irradiation increases hyaluronan synthesis in epidermal keratinocytes via sequential induction of hyaluronan synthases Has1–3 mediated by p38 and Ca2+/calmodulin-dependent protein kinase II (CaMKII) signaling. J. Biol. Chem. 288, 17999–18012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dai G., Freudenberger T., Zipper P., Melchior A., Grether-Beck S., Rabausch B., de Groot J., Twarock S., Hanenberg H., Homey B., Krutmann J., Reifenberger J., Fischer J. W. (2007) Chronic ultraviolet B irradiation causes loss of hyaluronic acid from mouse dermis because of down-regulation of hyaluronic acid synthases. Am. J. Pathol. 171, 1451–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Werth B. B., Bashir M., Chang L., Werth V. P. (2011) Ultraviolet irradiation induces the accumulation of chondroitin sulfate, but not other glycosaminoglycans, in human skin. PLoS One 6, e14830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kaneko T., Saito H., Toya M., Satio T., Nakahara K., Hiroi M. (2000) Hyaluronic acid inhibits apoptosis in granulosa cells via CD44. J. Assist. Reprod. Genet. 17, 162–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Peng H., Zhou J. L., Liu S. Q., Hu Q. J., Ming J. H., Qiu B. (2010) Hyaluronic acid inhibits nitric oxide-induced apoptosis and dedifferentiation of articular chondrocytes in vitro. Inflamm. Res. 59, 519–530 [DOI] [PubMed] [Google Scholar]
- 46. Liu S., Zhang Q. S., Hester W., O'Brien M. J., Savoie F. H., You Z. (2012) Hyaluronan protects bovine articular chondrocytes against cell death induced by bupivacaine at supraphysiologic temperatures. Am. J. Sports Med. 40, 1375–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yanagida-Suekawa T., Tanimoto K., Tanne Y., Mitsuyoshi T., Hirose N., Su S., Tanne K., Tanaka E. (2013) Synthesis of hyaluronan and superficial zone protein in synovial membrane cells modulated by fluid flow. Eur. J. Oral. Sci. 121, 566–572 [DOI] [PubMed] [Google Scholar]
- 48. Alaniz L., García M. G., Gallo-Rodriguez C., Agusti R., Sterín-Speziale N., Hajos S. E., Alvarez E. (2006) Hyaluronan oligosaccharides induce cell death through PI3-K/Akt pathway independently of NF-κB transcription factor. Glycobiology 16, 359–367 [DOI] [PubMed] [Google Scholar]
- 49. Ruffell B., Johnson P. (2008) Hyaluronan induces cell death in activated T cells through CD44. J. Immunol. 181, 7044–7054 [DOI] [PubMed] [Google Scholar]
- 50. Chen L., Bourguignon L. Y. (2014) Hyaluronan-CD44 interaction promotes c-Jun signaling and miRNA21 expression leading to Bcl-2 expression and chemoresistance in breast cancer cells. Mol. Cancer 13, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lokeshwar V. B., Lopez L. E., Munoz D., Chi A., Shirodkar S. P., Lokeshwar S. D., Escudero D. O., Dhir N., Altman N. (2010) Antitumor activity of hyaluronic acid synthesis inhibitor 4-methylumbelliferone in prostate cancer cells. Cancer Res. 70, 2613–2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Saito T., Dai T., Asano R. (2013) The hyaluronan synthesis inhibitor 4-methylumbelliferone exhibits antitumor effects against mesenchymal-like canine mammary tumor cells. Oncol. Lett. 5, 1068–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Uchakina O. N., Ban H., McKallip R. J. (2013) Targeting hyaluronic acid production for the treatment of leukemia: treatment with 4-methylumbelliferone leads to induction of MAPK-mediated apoptosis in K562 leukemia. Leuk. Res. 37, 1294–1301 [DOI] [PubMed] [Google Scholar]