

Background: The cysteine peptidase gingipain K is a major proteolytic virulence factor of Porphyromonas gingivalis.
Results: The structure of the catalytic and immunoglobulin-type domains has been solved in complex with a covalent inhibitor.
Conclusion: A distinct S1 pocket explains its high specificity for lysines.
Significance: The structural details reveal the working mechanism and may lead to the design of drugs to selectively treat periodontitis.
Keywords: Cysteine Protease, Enzyme Catalysis, Microbial Pathogenesis, Protease Inhibitor, X-ray Crystallography, Oral Pathogen
Abstract
Cysteine peptidases are key proteolytic virulence factors of the periodontopathogen Porphyromonas gingivalis, which causes chronic periodontitis, the most prevalent dysbiosis-driven disease in humans. Two peptidases, gingipain K (Kgp) and R (RgpA and RgpB), which differ in their selectivity after lysines and arginines, respectively, collectively account for 85% of the extracellular proteolytic activity of P. gingivalis at the site of infection. Therefore, they are promising targets for the design of specific inhibitors. Although the structure of the catalytic domain of RgpB is known, little is known about Kgp, which shares only 27% sequence identity. We report the high resolution crystal structure of a competent fragment of Kgp encompassing the catalytic cysteine peptidase domain and a downstream immunoglobulin superfamily-like domain, which is required for folding and secretion of Kgp in vivo. The structure, which strikingly resembles a tooth, was serendipitously trapped with a fragment of a covalent inhibitor targeting the catalytic cysteine. This provided accurate insight into the active site and suggested that catalysis may require a catalytic triad, Cys477-His444-Asp388, rather than the cysteine-histidine dyad normally found in cysteine peptidases. In addition, a 20-Å-long solvent-filled interior channel traverses the molecule and links the bottom of the specificity pocket with the molecular surface opposite the active site cleft. This channel, absent in RgpB, may enhance the plasticity of the enzyme, which would explain the much lower activity in vitro toward comparable specific synthetic substrates. Overall, the present results report the architecture and molecular determinants of the working mechanism of Kgp, including interaction with its substrates.
Introduction
Bacteria are normally part of the commensal flora that is generally beneficial for human health (1). However, in a susceptible host, some are pathogenic and invade cells and tissues, causing infection and disease. Moreover, the emergence of resistant strains, which are currently responsible for half of all infections, has exacerbated the danger (2). In the United States alone, resistant pathogens infect at least 2 million people every year, which makes such infections more common than cancer, and they cause 23,000 deaths (2). The only way to keep pace with these extremely adaptive pathogens is via continuous effort in the development of new antimicrobials. Inexplicably, however, the pharmaceutical industry has neglected the development of new antibiotics in recent decades: only four new drug applications were approved by the United States Food and Drug Administration in 2005–2012 (3). Responsibly, academic research must fill the gap.
Among the most prevalent human bacterial commensals turned into pathogens is Porphyromonas gingivalis, a Gram-negative oral anaerobe that causes periodontitis, an inflammatory disease that afflicts half the adult population in the United States, destroys the gums, and leads to tooth loss (4). It was even detected in the 5,300-year-old mummy of the Tyrolean Iceman “Ötzi” in what may well be the earliest report of gingival infection in Homo sapiens (5). P. gingivalis invades periodontal tissues by colonizing the gingival sulcus and proliferating in the subgingival plaque. It evades the host defense mechanisms through a panel of virulence factors that deregulate innate immune and inflammatory responses. In addition, bacteria and their products can enter the circulation, contributing to development and severity of systemic diseases at distal sites, such as cardiovascular diseases (6), rheumatoid arthritis (7), diabetes (8), and preterm delivery (9). Currently, specific treatment of severe periodontitis consists only of curettage of the affected area, which is time-consuming, painful, and needs frequent repetition (10), and the adjunct doxycycline hyclate (Periostat), which targets matrix metalloproteinases and was approved by the Food and Drug Administration in 1988 (11). Consequently, there is an urgent need for the development of novel therapeutic approaches.
Peptidases are a substantial part of the infective armamentarium of P. gingivalis (12–14). Most are cysteine peptidases, and the best characterized are the gingipains K (alias Kgp)4 and R (RgpA and RgpB) (4, 15), which are major virulence factors of the pathogen (16). Gingipains are cell surface-anchored or soluble and responsible for up to 85% of the total extracellular proteolytic activity of P. gingivalis (17). This activity yields nutrient acquisition, cleavage of host cell surface receptors, signaling via protease-activated receptors, and inactivation of cytokines and components of the complement system. The pathogen thus keeps host bactericidal activity in check and maintains chronic inflammation (4). In particular, Kgp cleaves many constituents of human connective tissue and plasma, including immunoglobulins; fibronectin; plasma kallikrein; fibrinogen; iron-, heme-, and hemoglobin-transporting proteins; and peptidase inhibitors, thus contributing to bleeding and vascular permeability as well as to heme and iron uptake by the bacterium (18, 19). Further pathophysiologically relevant substrates of Kgp include cadherins at the cell adherence junction, membrane TNFα, interleukin-8, the interleukin-6 receptor, thrombomodulin, complement regulatory protein CD46, and osteoprotegerin (18). Kgp thus contributes far more to the pathogenicity of P. gingivalis than any other peptidase (20), and so it is essential for bacterial survival and the pathological outcome of periodontitis (21). This was further confirmed by the reduction of bacterial virulence observed in a mouse model of infection upon specific inhibition of Kgp (22). Accordingly, Kgp, like RgpA and RgpB, is a promising target for the development of therapeutic inhibitors to treat periodontitis (18, 23).
Functionally, RgpA and RgpB specifically cleave bonds after arginines, whereas Kgp cleaves after lysines (21, 24). Structurally, these enzymes are translated as multidomain proteins made up of at least a signal peptide, a prodomain, a catalytic domain (CD), an immunoglobulin-superfamily domain (IgSF), and a C-terminal domain. RgpB shows just this minimal configuration (21). RgpA has four additional hemagglutinin/adhesion domains (termed RgpAA1–RgpAA4) inserted between the IgSF and the C-terminal domains. Kgp in turn has between three and five such domains (termed KgpAA1–KgpAA5), depending on the bacterial strain, thus spanning up to 1,723–1,732 residues (21). Both Kgp and RgpA are subjected to extensive post-translational proteolytic processing and are secreted as non-covalent but very tight complexes of the catalytic and hemagglutinin/adhesion domains, which are held together through oligomerization motifs (25).
Detailed structural and functional knowledge of target virulence factors at the molecular level can lead to the development of new drugs following rational drug design strategies (26). Atomic structural data are available for the catalytic and IgSF domains of RgpB, for both a zymogen complex and the active form (27, 28), and for the ancillary hemagglutinin/adhesion domains KgpAA1, KgpAA2, and KgpAA3 of Kgp (29, 30). The latter, however, do not provide insight into the proteolytic function and mechanism of Kgp. Given the importance of the distinct but complementary cleavage specificities of RgpB and Kgp, which may be related to the differences between their respective CD+IgSF moieties (27% identity; see Fig. 1), we analyzed the three-dimensional structure of a catalytically competent 455-residue fragment of Kgp from P. gingivalis strain W83 (hereafter Kgp(CD+IgSF)) and assessed its molecular determinants of action and specificity.
FIGURE 1.

Gingipain sequences. Structure-guided sequence alignment of the CD and IgSF moieties (separately framed) of Kgp from P. gingivalis strain W83 (UniProt accession number Q51817; top rows) and RgpB from P. gingivalis strain HG66 (GenBank accession number AAB41892; Protein Data Bank code 1CVR (28); bottom rows) is shown. The sequence of the latter differs from that of the ortholog from strain W83 at 12 positions (UniProt accession number 95493; Protein Data Bank code 4IEF (27)). The amino acid numbering and the regular secondary structure elements (strands as black arrows labeled β1–β21 and helices as loops labeled α1–α14) above the alignment correspond to the Kgp(CD+IgSF) structure (this study); those below the alignment correspond to RgpB(CD+IgSF) (taken from Fig. 2c of Ref. 28). Identical residues are in bold white over black background, similar residues are in bold black over gray background, and the overall sequence identity is 27%. (Potential) catalytic residues of Kgp CD are pinpointed by an open rhombus; residues framing the S1 pocket are indicated by an arrow.
EXPERIMENTAL PROCEDURES
Protein Production
Kgp(CD+IgSF) of P. gingivalis strain W83 (sequence Asp229–Pro683; see UniProt database accession number Q51817) plus a C-terminal hexahistidine tag was purified by affinity chromatography on nickel-Sepharose beads from culture medium of P. gingivalis mutant strain ABM1 expressing recombinant Kgp with one oligomerization motif disrupted by hexahistidine insertion (25, 31). In contrast to wild-type strain W83, which secretes only heterooligomeric complexes of Kgp and RgpA, strain ABM1 can release soluble and functional Kgp fragments into the medium (25). This facilitates purification of a protein variant that is compatible with crystallization. To avoid autoproteolysis, the sample was incubated with Nα-tosyl-l-lysinylchloromethane (TLCK; Sigma) prior to elution from the beads.
Crystallization and Diffraction Data Collection
Crystallization assays were performed by the sitting drop vapor diffusion method. Reservoir solutions were prepared by a Tecan robot, and 100-nl crystallization drops were dispensed on 96 × 2-well MRC plates (Innovadyne) by a Phoenix nanodrop robot (Art Robbins) or a Cartesian Microsys 4000 XL robot (Genomic Solutions) at the Automated Crystallography Platform at Barcelona Science Park. Plates were stored in Bruker steady-temperature crystal farms at 4 and 20 °C. Successful conditions were scaled up to the microliter range in 24-well Cryschem crystallization dishes (Hampton Research). The best crystals were obtained at 20 °C with protein solution (at 5.7 mg/ml in 5 mm Tris·HCl, pH 7.4, 0.02% sodium azide) and 22% polyethylene glycol 8000, 0.1 m sodium cacodylate, pH 6.5, 0.2 m calcium acetate as reservoir solution from 2:1-μl drops. Crystals were cryoprotected by immersion in harvesting solution (18% polyethylene glycol 8000, 0.08 m sodium cacodylate, pH 6.5, 0.16 m calcium acetate, 20% (v/v) glycerol). A complete diffraction data set was collected from a liquid N2 flash cryocooled crystal at 100 K (cooled by an Oxford Cryosystems 700 series cryostream) on an ADSC Q315R charge-coupled device detector at beam line ID14-4 of the European Synchrotron Radiation Facility (Grenoble, France) within the Block Allocation Group “BAG Barcelona.” This crystal was orthorhombic and contained one Kgp(CD+IgSF) moiety per asymmetric unit (VM = 2.2 Å3/Da; 44% solvent content (32)) and tightly packed (for comparison, see e.g. Ref. 33). Diffraction data were integrated, scaled, merged, and reduced with programs XDS and XSCALE (34) (see Table 1).
TABLE 1.
Crystallographic data
Values in parentheses refer to the outermost resolution shell. r.m.s.d, root mean square deviation.
| Space group/cell constants a, b, and c (Å) | P212121/56.64, 58.81, 135.50 |
| Wavelength (Å) | 0.9393 |
| No. of measurements/unique reflections | 303,645/46,309 |
| Resolution range (outermost shell) (Å) | 45.2–1.75 (1.79–1.75) |
| Completeness (%) | 99.5 (93.9) |
| Rmergea | 0.100 (0.692) |
| Rr.i.m. (=Rmeas)a/CC(1/2)b | 0.108 (0.821)/0.998 (0.656) |
| Average intensity over S.D. ([I/σ(I)]) | 16.5 (1.9) |
| B-factor (Wilson) (Å2)/average multiplicity | 22.0/6.6 (3.4) |
| Resolution range used for refinement (Å) | 44.1–1.75 |
| No. of reflections in working set/in test set | 45,532/767 |
| Crystallographic Rfactor (free Rfactor)c | 0.149 (0.172) |
| No. of protein atoms/solvent molecules/ions/ligands | 3,534/533/2 Ca2+, 1 Ni2+, 3 Na+/ 1 histidine, 2 acetate, 3 azide, 4 glycerol |
| r.m.s.d. from target values bond lengths (Å)/bond angles (°) | 0.010/1.00 |
| Average B-factor (Å2) protein atoms/CD only/IgSF only | 17.1/13.6/34.5 |
| Main-chain conformational angle analysisd | |
| Residues in favored regions/outliers/all residues | 436/1/449 |
a Rmerge = ΣhklΣi|Ii(hkl) − I(hkl)|/ΣhklΣiIi(hkl); Rr.i.m. = Σhkl(nhkl/[nhkl − 1])1/2Σi|Ii(hkl) − I(hkl)|/ΣhklΣiIi(hkl); Rp.i.m. = Σhkl(1/[nhkl − 1])1/2Σi|Ii(hkl) − I(hkl)|/ΣhklΣiIi(hkl) where Ii(hkl) is the ith intensity measurement and nhkl is the redundancy of reflection hkl, including symmetry-related reflections, and I(hkl) is its average intensity. Rr.i.m. (alias, Rmeas) and Rp.i.m. are improved multiplicity-weighted indicators of the quality of the data, the redundancy-independent merging R factor and the precision-indicating merging R factor. The latter is computed after averaging over multiple measurements (for details, see Ref. 87).
b According to Karplus and Diederichs (88).
c Crystallographic Rfactor = Σhkl‖Fobs| − k|Fcalc‖/Σhkl|Fobs| where k is a scaling factor and Fobs and Fcalc are the observed and calculated structure factor amplitudes, respectively. This factor is calculated for the working set reflections; free Rfactor is the same but for a test set of reflections (>500) not used during refinement.
d According to MolProbity (47).
Structure Solution and Refinement
The structure of Kgp(CD+IgSF) was solved by likelihood-scoring molecular replacement with the program Phaser (35) using the protein part of the structure of RgpB(CD+IgSF) of P. gingivalis strain HG66 (GenBankTM accession number AAB41892; Protein Data Bank code 1CVR (28)) as a searching model. The side chains were trimmed from the model with the program CHAINSAW within the CCP4 suite (36) based on sequence alignment performed with MULTALIN (37). A two-body search was performed with the CD and the IgSF separately to obtain suitable phases. These calculations rendered two unambiguous solutions at final Eulerian angles (α, β, and γ) of 225.7°, 87.3°, and 30.7° and 226.2°, 92.0°, and 34.2° and fractional cell coordinates (x, y, and z) of 0.019, 0.265, 0.096 and 0.959, 0.259, 0.090, respectively. The initial values for the rotation/translation function Z-scores were 7.9/10.1 and 5.4/7.2, respectively, which confirmed P212121 as the correct space group. A Fourier map calculated with the appropriately rotated and translated model was then subjected to density modification and model extension with ARP/wARP (38). The model obtained was completed through successive rounds of manual model building using programs TURBO-FRODO (39) and Coot (40) and crystallographic refinement with program BUSTER-TNT (41), which included translation/libration/screw refinement (one translation/libration/screw group for each domain) until completion of the model. The final model contained Kgp residues Asp229–Val680 (the last three residues and the hexahistidine tag were not visible in the final map) plus four glycerol, one stand-alone histidine, two acetate, and three azide molecules in addition to 533 solvent molecules and six (tentatively assigned) cations, two calciums, three sodiums, and one nickel (see Table 1). Of these, the two calcium ions (numbered Ca999 and Ca998) and two of the sodium ions (numbered Na997 and Na996) were intrinsic parts of the structure and are described under “Results and Discussion.” The nickel ion in turn was observed at the interface between two symmetric molecules, tetrahedrally coordinated by the carboxylate oxygens of Glu379, a symmetric Glu355, and an acetate from the mother liquor as well as by the Nϵ2 atom of an isolated histidine, possibly resulting from digestion of the C-terminal hexahistidine tag during purification. The third sodium ion was bound 10 Å away from the nickel by the main-chain carbonyl of Thr415, four solvent molecules, and the carboxylate side chain of a symmetric Glu498 residue. We attribute these two metal sites to purification and crystallization artifacts.
Although the main chain and most of the side chains of the entire molecule were fully defined in the final Fourier map because of the high resolution and quality of the diffraction data, the CD moiety was more rigid and better defined than the downstream IgSF as indicated by the lower average thermal displacement parameter (13.6 versus 34.5Å2; see Table 1). This is reminiscent of the structure of RgpB(CD+IgSF) in complex with its prodomain (27). The side chain of the catalytic cysteine residue (Cys477) showed additional density (see “Results and Discussion”). Moreover, Met594 showed alternate occupancy for its side chain. All other sulfur-containing side chains (three cysteines and 10 methionines) were apparently unaltered according to the final Fourier map. The only Ramachandran outlier of the structure was Ala443 (Table 1), which, however, was unambiguously defined by the final Fourier map and had similar main-chain angles in the RgpB structures (27, 28). Three proline residues were found in cis conformation (Pro241, Pro424, and Pro453).
Miscellaneous
The structure-based sequence alignment in Fig. 1 was performed with the program EXPRESSO within T-COFFEE version 10.0 (42) and represented with program ESPRipt 3.0 (43). Ideal coordinates and parameters for crystallographic refinement of non-standard ligands were obtained from the PRODRG server (44). Structural similarity searches were performed with Dali (45), and structure figures were prepared with programs Coot and Chimera (46). The model was validated with MolProbity (47). The final coordinates of P. gingivalis Kgp(CD+IgSF) have been deposited in the Protein Data Bank under code 4RBM.
RESULTS AND DISCUSSION
Overall Structure of Kgp Catalytic Domain
The structure of Kgp(CD+IgSF) is elongated with approximate maximal dimensions 75 × 50 × 45 Å (Fig. 2A). Curiously, it resembles a tooth with CD featuring the crown and IgSF the root (Fig. 2B). The neck is the interface between the two domains, and the active site is at the cusp, on the grinding surface (see below).
FIGURE 2.

General architecture of KgpB. A, ribbon-type plot of Kgp(CD+IgSF) showing the regular secondary structure elements (CD, α-helices as orange ribbons, β-strand as yellow arrows, and coils in tan; IgSF, β-strand as blue arrows labeled β16–β21 and coils in lilac). The N and C termini and the four structural cations (calciums Ca999 and Ca998 in red and sodiums Na997 and Na996 in blue) are depicted. The (putative) catalytic triad, Cys477 (covalently modified at Sγ with an l-lysinylmethyl group; carbons in lilac), His444, and Asp388, is further shown as sticks. Black arrows point to the solvent channel (see Fig. 3D). B, picture of a tooth with its parts labeled. C, (2mFobs − DFcalc)-type Fourier map of the region around the catalytic cysteine Cys477 obtained with diffraction data to 1.75-Å resolution and contoured at 1σ above threshold. D, structure of Kgp(CD+IgSF) in cross-eye stereo in standard orientation (28, 79), which corresponds to a horizontal 90° rotation of the view in A, i.e. viewing the CD cusp region. Regular secondary structure elements of the CD (helices α1–α14 and strands β1–β15) are labeled. The NSD is on top, and the CSD is at the bottom (see also Fig. 5D). E, close-up of D in stereo centered on the non-primed side of the active site. Residues framing the specificity pocket S1 and pocket S2 are labeled. Small green spheres represent solvent molecules.
The globular CD (Asp229–Pro600; see Figs. 1 and 2A) is a competent cysteine peptidase domain and conforms to the α/β-hydrolase or PLEES fold (48, 49). It contains four cation-binding sites (two sodium and two calcium ions; Fig. 3, A–C), which generally contribute to tertiary structure integrity (50). It is subdivided into a smaller N-terminal (or A) subdomain (NSD; Asp229–Lys375) and a larger C-terminal (or B) subdomain (CSD; Ser376–Pro600). The NSD starts on the left of the molecule (orientation hereafter according to Fig. 2A) with a small helical segment (α1; for regular secondary structure elements, see Figs. 1 and 2, A and D), and the polypeptide chain follows an extended trace downward along the surface. At Pro241, the chain makes a sharp turn upward and enters a four-stranded parallel pleated β-sheet (sheet I) through the second strand from the right (β1). This sheet (from left to right: β6-β3-β1-β2) has connectivity +1x,−2x,−1x according to Richardson (51) and is twisted by ∼40° but not arched or curved. The NSD is a three-layer (αβα) sandwich; thus sheet I is flanked by two almost parallel helices on its right (α2 and α5) and two more (α3 and α4) on its left. Although in general the regular secondary structure elements are connected by tight loops, the one connecting β3 with β6 (Lβ3β6) exceptionally spans 30 residues and contains a β-ribbon (β4β5) and a calcium-binding site (Ca998) in addition to helix α3. This cation is oxygen-coordinated in an octahedral manner as is usual for calcium (52) by Asp330 Oδ1, two solvent molecules, and bidentately the carboxylate oxygens of Glu343 in a plane with the ion. Asp337 Oδ1 and Phe341 oxygen are found at the apical positions (Fig. 3B). Therefore, one in-plane position of the octahedron is split into two ligands, and the binding distances range between 2.31 and 2.55 Å, which are typical values for calcium (2.36–2.39 Å (53)).
FIGURE 3.

Ion sites and solvent channel. A, detail of the Kgp(CD+IgSF) structure around Ca999 shown as a red sphere. Some residues are labeled for reference; solvents are depicted as green spheres. Liganding atoms are linked with a solid line, and the respective binding distances (in Å) are indicated. B, same as A but for Ca998. C, same as A but for the two sodium sites Na997 and Na996 (blue spheres). D, detail in stereo of the semitransparent surface of Kgp CD (solvent radius, 1.4 Å) and the solvent channel ranging from the bottom of the specificity pocket (pinpointed by the tip of the l-lysinylmethyl group attached to the catalytic cysteine C477) to the opposite surface of the molecule (pinpointed by a solvent molecule in red). The solvent molecules found in the structure are depicted as green spheres. Protein atoms have been omitted for clarity. The orientation displayed corresponds to that of Fig. 2D after a vertical clockwise rotation of 120° and a counterclockwise 90° rotation in the plane.
After α5, at the front neck surface, the polypeptide enters the CSD with helix α6, which in turn leads to a central six-stranded twisted (∼40°), but not arched or curved, pleated β-sheet (sheet II; β8-β7-β9-β13-β14-β15 from left to right). The chain enters the sheet with the second strand from the left and has connectivity −1x,+2x,+1x,+1x,+1; i.e. all strands are parallel and run upward except for the rightmost one, β15, which runs downward. The latter creates the junction with the NSD and runs parallel to the leftmost strand of sheet I but is horizontally rotated ∼60° away, giving rise to a pseudo-continuous 10-stranded supersheet. Like NSD, CSD is a three-layer (αβα) sandwich, which contains the active site cleft (see below). Five helices (α7, α11, α12, α13, and α14) are found on the left side of the sheet, and three more (α8, α9, and α10) are on the right flank. Interestingly, helices α11 and α12 are aligned with respect to their axes and almost in phase. Such interrupted helices are exceedingly rare in protein structures (51), and only segment Gly522–Gly539 (like prolines, flanking glycines are observed in hinges (54)) prevents these helices from being a single continuous unit. This intercalated segment gives rise to an extended loop that protrudes from the molecular surface and folds back to cover helix α7 like a cape, and it contains a sodium-binding site (Na997). This cation is octahedrally coordinated by six oxygen ligands, the most common coordination number for sodium (55), through Thr536 Oγ, Val521 oxygen, Asp542 Oδ2, and a solvent molecule coplanar with the metal and through Ser537 oxygen and Tyr402 Oη at the apical positions. Coordinating distances span 2.31–2.59 Å (Fig. 3C), which is consistent with most common distances for sodium (2.38–2.41 Å (53)). Nearby, Lα12α13 contains a second octahedral oxygen-liganded sodium site (Na996) 13.4 Å from the former. This ion is bound at distances of 2.36–2.69 Å by Tyr550 oxygen, Asn551 Oδ1, Ala543 oxygen, and Leu546 oxygen in the plane and apically by Thr544 oxygen and Ser549 oxygen (Fig. 3C). As found for the segment connecting strands β3 and β6 in NSD sheet I, the segment connecting β9 and β13 in the CSD is elaborate and includes a small three-stranded antiparallel β-sheet (sheet III; β10, β11, and β12), which is almost perpendicular to sheet II. The downstream loop, Lβ13α10, contributes together with Lβ3β4 of the NSD to a second calcium site (Ca999), which may thus have a role in maintaining the structural integrity of the NSD-CSD interface. This calcium is bound by Phe482 oxygen, two solvent molecules and bidentately the carboxylate oxygens of Glu491 in a plane with the ion. A further solvent molecule and bidentately the carboxylate oxygens of Asp313 are in the respective apical positions (Fig. 3A). Therefore, two positions of the octahedron are split into two ligands, and the binding distances range from 2.33 to 2.55 Å (Fig. 3A). After strand β15, at segment Asp590–Ser592, the polypeptide abruptly changes direction and runs horizontally outward from the interface with the NSD to the left molecular surface. At Ala598, the chain turns abruptly downward and leads to the interface between CD and IgSF at Pro600–Lys601.
Finally, an internal channel is found within the CSD, vertically traversing the molecule over 20 Å from the bottom of the specificity pocket in the active site (see below) to the lower outer surface of the subdomain (Fig. 3D) where it emerges through a crater surrounded by Pro595, Asn551, and Arg597. It is filled with 13 solvent molecules, which are well defined in the final Fourier map (average thermal displacement parameter, 17.0 Å2; for comparison, the overall value for the CD is 13.6 Å2, and that of all solvent molecules is 29.8 Å2). The outermost solvent molecule of the channel at the domain surface is bound by Ser669 Oγ from the downstream domain IgSF. This channel is embraced by sheet II and helices α7, α11+α12, and α13. Such extended solvent channels traversing the inner core of proteins are rare, and here its role, if any, is unknown. The channel is not wide enough to evacuate reaction products, as e.g. in catalase (56) or the ribosome (57), and is too far away from the active site cysteine to serve as supplier of solvent for the deacylation step in catalysis. In addition, in the structurally related RgpB, this channel is replaced by a compact hydrophobic core (see Protein Data Bank code 1CVR (28)).
Solvent molecules buried in internal cavities, which are integral structural components of proteins, interchange with the external bulk solvent (58), thus conferring a “breathing” motion to a protein. By serving as mobile hydrogen bonding donors or acceptors, internal waters may facilitate transition and structural rearrangement between different functional states (59), and they cluster at internal cavities of functional importance such as hinge regions or along channels (60). However, the generation of cavities inside a protein at places where a compact hydrophobic core is found in close structural relatives usually reduces stability (61), although water-filled cavities destabilize less than empty cavities: the water molecules may still interact favorably with neighboring protein residues (62). Conversely, the hydrogen bonding potential of a water molecule inside a protein structure is less exploited than in the aqueous phase, and moving a solvent molecule from bulk solvent to the interior of a protein entails entropic costs (63) and energy costs for hydration of the cavity (64), which in turn destabilize protein structures. In Kgp(CD+IgSF), the extended solvent channel could contribute more to the overall plasticity and flexibility of the enzyme than in the compact hydrophobic core of RgpB. Although certain flexibility (at least around the active site) is a prerequisite for efficient catalysis (65), destabilization of the overall enzyme moiety contravenes the axiom that proteins must adopt a stable tertiary fold to be wholly functional (66). This would be consistent with much lower activity of Kgp in vitro than RgpB against comparable synthetic substrates mimicking their respective specificities (24, 67). This in turn would apparently contract the superior role of Kgp as a proteolytic virulence factor (20). It must be kept in mind, however, that native Kgp occurs as a complex of the catalytic and hemagglutinin adhesion domains, which work as a homing device to deliver Kgp to its targets and exert essential functions for P. gingivalis such as agglutination of red blood cells, acquisition of heme, and binding to the extracellular matrix (4, 18, 19).
Overall Structure and Similarity of Kgp Immunoglobulin Superfamily Domain
With Lys601, the polypeptide chain enters the IgSF, which is essential for folding of Kgp: no properly folded CD is detected by specific monoclonal antibodies if IgSF is ablated despite the truncated kgp gene being transcribed (68). In addition, only residual Kgp-specific activity is detected in such deletion mutants (68). Structurally, IgSF consists of a six-stranded antiparallel open β-barrel adopting a Greek key topology for its first four strands (β16-β19-β18-β17) followed by a final β-ribbon structure (β20β21). The initial segment of IgSF (Lys601–Pro608) runs in rather extended conformation and partially closes the open side of the barrel, but because of a bulge at Pro608–Pro612, it only interacts through one hydrogen bond with both neighboring strands β16 (Thr606 oxygen-Gln621 oxygen; 2.99 Å) and β21 (Lys601 oxygen-Leu672 nitrogen; 2.86 Å), so strictly speaking, it cannot be considered a proper β-strand (Fig. 2A). Overall, the IgSF fold corresponds to classic immunoglobulin-like domains, which usually function as cell adhesion molecules (69). In particular, it best fits into the C2 set represented by the second domain of vascular cell adhesion molecule-1 (Protein Data Bank code 1VCA). Consistently, structural similarity searches identified, in addition to the homologous domain from RgpB (Protein Data Bank codes 1CVR and 4IEF; Dali Z-score = 14.4; root mean square deviation, 1.2 Å; length of alignment, 79; sequence identity, 23%; see also Fig. 1), the N-terminal immunoglobulin-like domains of complement components C3 (Protein Data Bank code 2WII; 8.1; 2.4 Å; 75; 15%) and C5 (Protein Data Bank code 3PRX; 7.4; 2.3 Å; 73; 12%) as related. Interestingly, these proteins themselves are degraded by Kgp (70). In addition, the macroglobulin-like MG domains are also similar (71).
The IgSF contacts the bottom of the CD through an interface that generates the neck of the tooth and involves Lα1β1, α2, Lα2β2, and the end of α5 of CD, which fit into the concave outer surface of the IgSF barrel. In turn, Lβ20β21 of IgSF inserts like a wedge between CD C-terminal segment Ser592–Pro600 and the Na996-stabilized loop Lα12α13.
Catalytic Site and Active Site Cleft
Catalysis in Kgp occurs at the cusp of the tooth through binding of peptide substrates to the active site cleft (Fig. 2, A, D, and E). As occurs in α/β-hydrolase or PLEES fold enzymes, active site residues are provided by loops connecting strands at the C-terminal edge of the central β-sheet (here sheet II of the CSD). We serendipitously trapped the structure of Kgp(CD+IgSF) in a covalent reaction intermediate mimic, which was interpreted, based on the excellent quality of the Fourier map (Fig. 2C), as an l-lysinylmethyl (LM) moiety attached to the Sγ atom of the catalytic cysteine Cys477 (provided by Lβ13α10). The latter was identified as such by active site labeling and confirmed by mutagenesis (72). LM introduces an extra methylene group between Cys477 Sγ and the carbonyl, mimicking the scissile carbonyl (Fig. 2C), so it does not yield a thioester and cannot be hydrolyzed. This covalent modification resulted from the irreversible cysteine peptidase chloromethyl ketone inhibitor TLCK used during purification (see “Experimental Procedures”). Inhibition of cysteine peptidases by TLCK was first reported by Cohen and co-workers (73), and such chloromethyl ketones are routinely used during protein purification to prevent degradation (74). Noteworthily, although these compounds target the catalytic cysteine in cysteine peptidases (75, 76), they also inhibit serine peptidases but by covalent attachment to the aromatic Nϵ2 of the respective catalytic histidines instead (77). In a second step, the nearby catalytic serine of serine peptidases may (78) or may not (77) attack the carbonyl of the Nϵ2-attached carboxymethyl moiety and yield a tetrahedral reaction intermediate-like product covalently bound to both serine Oγ and histidine Nϵ2.
The LM moiety enabled us to identify the active site cleft (Figs. 2, D and E, and 4). When viewed in the standard orientation of cysteine peptidases (28, 79), i.e. with a substrate binding horizontally from left (non-primed side) to right (primed side), the cleft is rather flat and delimited by His444–Glu447 from β10, Asp388–Val395 from Lβ7α7, and Phe527 at its bottom; Cys476–Ile478 from Lβ13α10 at its back; and Pro509–Trp513 from Lβ14α11 and Ile573–His575 from Lα13α14 at its top. Lβ9β10 contains His444, which strongly hydrogen bonds through its Nδ1 atom the carbonyl group oxygen of the LM moiety (2.61 Å apart; Figs. 2, D and E, and 4). This oxygen is close to where the scissile carbonyl oxygen is expected to be in a true substrate after the acylation step of catalysis. This is consistent with His444 playing a major role in catalysis, potentially as part of a charge relay system together with Cys477, as suggested previously (18).
FIGURE 4.

Interactions in the S1 pocket. The scheme depicts relevant interactions (as curved lines) of the l-lysinylmethyl group (bold trace) in the specificity pocket. The three solvent molecules in the pocket are shown as white spheres, and the first solvent of the large inner solvent channel is shown as a gray sphere (see also Fig. 3D).
Although most cysteine peptidases comprise a cysteine-histidine dyad as catalytic residues (80, 81), the position and proximity of one of the carboxylate oxygens of Asp388 from Lβ7α7 to His444 Nϵ2 in the present structure (2.68 Å apart; Fig. 2, D and E) strongly suggests a role for this aspartate in catalysis in Kgp as already described in the foot-and-mouth disease virus leader cysteine peptidase (82) and as postulated for RgpB. In the latter, however, Glu381 (RgpB of P. gingivalis strain HG66 numbering in italics according to GenBank accession number AAB41892; see Fig. 1; subtract 229 for the protein numbering used in Protein Data Bank code 1CVR and Ref. 28) is found instead of an aspartate (27, 28). This hypothesis would entail that Kgp had a catalytic triad spanning Cys477-His444-Asp388 and that the cleavage mechanism would include a thiolate-imidazolium ion pair making an oxyanion hole-assisted nucleophilic attack on the scissile peptide carbonyl in the acylation step (81). Alternatively, the imidazole may also function as a general base and abstract a proton from the cysteine thiol group (81). In either case, the histidine imidazolium would thereafter transfer a proton to the leaving α-amino group of the downstream cleavage product, and the upstream part of the substrate would remain covalently bound as a thioester to the catalytic cysteine. We hypothesize that the aspartate would also have a role in protonation and thus side-chain orientation of the histidine imidazole/ium during catalysis as in serine peptidases. Unfortunately, site-specific mutagenesis failed to demonstrated the catalytic efficacy of third residues in other cysteine peptidases such as papain (81), so this hypothesis remains to be verified by other methods.
The complex of Kgp(CD+IgSF) with LM also revealed that the specificity pocket in S1 can accommodate a lysine side chain whose ϵ-amino group is tetrahedrally bound by one of the carboxylate atoms of Asp516 (2.76 Å apart), Thr442 Oγ (2.99 Å), Asn475 oxygen (3.06 Å), and one of three solvent molecules forming a buried solvent cluster at the pocket bottom (2.69 Å; Figs. 2E and 4). This solvent molecule is further bound to the second water of the cluster (2.89 Å), Trp513 oxygen (2.91 Å), Asp516 Cβ (3.16 Å), and Val395 Cγ1 (2.75 Å) in a distorted square-based pyramidal manner. The second solvent in turn is further bound to Thr399 Oγ1 (2.89 Å), Thr442 Oγ1 (2.89 Å), and the third solvent molecule (2.61 Å) in a tetrahedral manner. Finally, the latter solvent is further bound to Asp516 oxygen (2.79 Å) and Ser520 Oγ (2.73 Å). The latter oxygen bridges the three-solvent cluster at the bottom of the pocket with the internal solvent channel (see above and Figs. 3D and 4). The aliphatic part of the lysine side chain of LM in turn is sandwiched among the side chain of Trp513, the main chain at Asn475–Cys477, and Ser511 Oγ. Replacement of lysine in P1 with an arginine, which would match the specificity of RgpB, would entail rupture of the salt bridge with Asp516 and the hydrogen bonds with Asn475 oxygen and Thr442 Oγ and possibly clashes with the latter atom, thus explaining why Kgp is specific for lysines and not arginines. This lysine specificity resembles trypsin, and like several trypsin-like serine peptidases, Kgp has been shown to cleave proteins involved in the blood coagulation/fibrinolysis cascade (24). In contrast to these serine peptidases, however, Kgp needs an anaerobic environment as found in the periodontal pockets of infected patients. Finally, upstream of S1, S2 is shallow and small, framed by Tyr512, His575, Trp513, and Trp391 (Figs. 2, D and E, and 4). This explains why Kgp substrates do not have arginines or lysines at position P2 (18).
Structural Kinship of the CD
Despite low overall sequence identity, the closest structural similarity of Kgp CD was found with the corresponding domain of the two essentially identical RgpB strain variants studied to date (strains HG66 and W83; Protein Data Bank codes 1CVR and 4IEF; Dali Z-score, 44.6 and 43.2; root mean square deviation, 2.1 and 2.0 Å; length of alignment, 336 and 328; sequence identity, 24 and 26%; see also Fig. 1). Although both Kgp and RgpB CDs generally fit well on top of each other, in particular at the regular secondary structure elements (Fig. 5, A and B), large differences are found in a number of loops as well as due to the long solvent-filled internal channel of Kgp, which is absent in RgpB. Notable insertions or deletions or substantially different chain traces are found at Kgp elements Lβ2α3, Lβ4β5, Lα5α6 and the interface between NSD and CSD, Lβ7α7, Lα7β8, Lβ8α8, Lα10β14, Lα11α12, Lα12α13, and Lα13α14. In particular, Lβ7α7 contains a long flap, Gly382–Ser391 in RgpB (absent in Kgp) that partially replaces the elongated cap Gly522–Gly539 of Kgp (in turn missing in RgpB). Consistently, the two Kgp helices α11 and α12 are a continuous single helix in RgpB. Overall, these differences also entail that, although both structures share the two calcium sites of Kgp but none of its sodium sites, an extra calcium site is found in RgpB at the region flanked by α7 and α11-Lα11α12-α12 in the CSD of the CD. Noteworthily, although catalytic activity of RgpB is ablated by the calcium chelator EDTA (83), Kgp remains unaffected (24). This supports that it is actually the extra calcium site of RgpB that is targeted by the chelator, whereas the two common calcium sites likely remain bound to the respective protein moieties. These differences also have a direct consequence for Lβ7α7, which provides the third putative catalytic acidic residue, Asp388 (see above). This loop protrudes slightly more in Kgp, thus explaining why an aspartate suffices in the latter to approach the catalytic histidine, whereas a glutamate is required in RgpB (see Fig. 5, B and C), as the positions of the catalytic cysteines and histidines nicely coincide in both structures (Fig. 5C).
FIGURE 5.
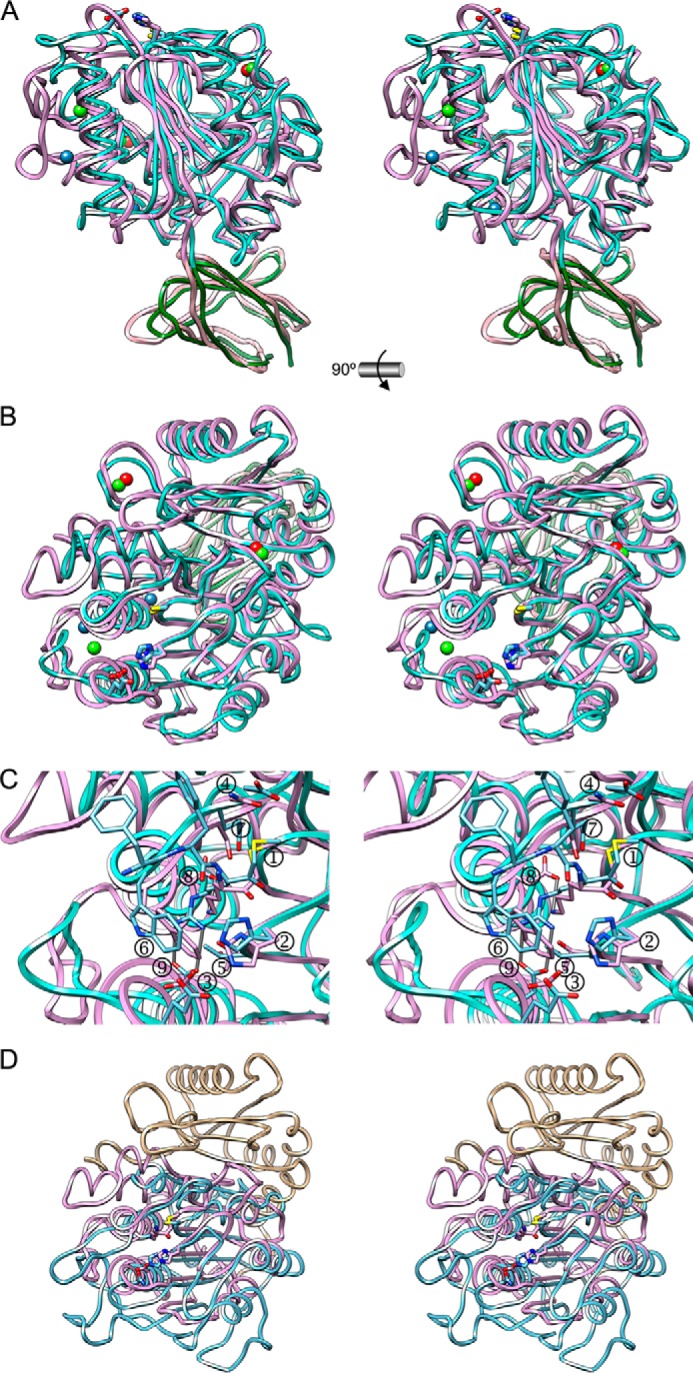
Structural similarities. A, ribbon plot in stereo showing the superposition of Kgp(CD+IgSF) (lilac + light pink) and RgpB(CD+IgSF) of strain HG66 (cyan + green; Protein Data Bank code 1CVR (28)). The two calcium and two sodium ions of Kgp are depicted as red and blue spheres, respectively, and the three calcium ions of RgpB are depicted as green spheres. The catalytic active site residues are shown as sticks for each structure with carbons colored in the respective ribbon colors. B, superposition as in A but in standard orientation. C, close-up of B focusing into the active sites. RgpB is covalently modified at its catalytic cysteine Sγ atom by a Phe-Phe-Arg-CH2- moiety. Shown are the catalytic cysteines (➀; Cys477 in Kgp and Cys473 in RgpB) and histidines (➁; His444 in Kgp and His473 in RgpB) and the putative histidine-polarizing acidic residues (➂; Asp388 in Kgp and Glu381 in RgpB) as well as some residues engaged in P1 pocket framing (➃, Asn510 in Kgp and Asp510 in RgpB; ➄, Thr442 in Kgp and Thr438 in RgpB; ➅, Trp513 in Kgp and Trp513 in RgpB; ➆, Ser511 in Kgp and Gln511 in RgpB), and the aspartates salt bridging the basic residues in P1 (➇, Asp516 in Kgp; ➈, Asp392 in RgpB). D, superposition in stereo of Kgp CD (NSD in tan and CSD in lilac) onto legumain (in turquoise; Protein Data Bank code 4AWA (86)) in standard orientation.
Substantial differences are also found in the specificity pockets. Interestingly, although in both structures an aspartate salt bridges the tip of the specific lysine or arginine (Asp516 in Kgp or Asp392 in RgpB, respectively), none of them is placed at the bottom of the pocket but rather on the side although on opposite walls of the pocket (Asp392 is close to Gly396, and Asp516 is close to Pro516; see Fig. 5C), so the Cγ atoms of these aspartates are ∼8 Å apart. This entails that, although the lysine is bound in extended conformation in Kgp, in RgpB the arginine side chain is rotated clockwise around Cδ–Nϵ by ∼50° to meet Asp392 by means of a double salt bridge through its Nη1 and Nη2 atoms. This is enabled by the presence of Val471 and Met517 and by a rearrangement of RgpB region Ser507–Gln520 (Ser507–Ser520 in Kgp), which relocates Trp513 and thus avoids a clash with the arginine (Fig. 5C). In summary, although the CD structures of Kgp and RgpB are quite similar, structural peculiarities account for their varying specificities and help to explain distinct catalytic efficiencies against comparable substrates (68).
Mechanistically, the CDs of gingipains belong to MEROPS database family C25 (80, 84). They have been grouped together with families C11 (clostripain), C13 (legumain), C14 (caspases), C50 (separases), C80 (RTX self-cleaving toxin from Vibrio cholerae), and C84 (PrtH peptidase from Tannerella forsythia) into clan CD. All these families share the following properties (80): they are broadly distributed across all kingdoms of life, the catalytic histidine is found in a histidine-glycine motif preceded by a block of four hydrophobic residues (motif II according to Aravind and Koonin (85)), and the catalytic cysteine is found in an alanine-cysteine motif (exceptionally cysteine-cysteine in Kgp) preceded by a second block of four hydrophobic residues (motif III according to Aravind and Koonin (85)). In addition, they are specific for residues in position P1 of substrates (arginine, lysine, asparagine, or aspartate for the distinct families) and resistant to the broad spectrum cysteine peptidase inhibitor E-64 but susceptible to chloromethyl ketone inhibitors (80). Structurally, despite being cysteine peptidases, Kgp and RgpB CDs are unlike any other clans and proteins structurally characterized to date. A structure-based search identified legumain as the closest structural relative of Kgp and RgpB (Fig. 5D), but it only matches the CSD and shows negligible sequence identity (Protein Data Bank codes 4AW9, 4AWA, and 4AWB; Dali Z-score, 13.5; root mean square deviation, 2.8 Å; length of alignment, 173; sequence identity, 14%). A certain similarity of the entire CD of Kgp and RgpB is also found with 2 + 2 heterotetramers of caspases (see Fig. 6 in Ref. 28).
Conclusion
The present detailed structural analysis of an essential virulence factor of a major human periodontopathogen has revealed the molecular determinants of its mode of action and specificity. This may lay the foundations for the rational design of specific inhibitors (complementary to but distinct from those against RgpB) that may curtail the survival of the pathogen and palliate the effects of periodontal disease and its associated systemic disorders. This approach is complementary to the one aimed at developing inhibitors simultaneously targeting both types of gingipains (23).
Acknowledgments
We are grateful to the joint Molecular Biology Institute of Barcelona/Institute for Research in Biomedicine Automated Crystallography Platform for assistance during crystallization experiments and to Robin Rycroft for very helpful contributions to the manuscript. We acknowledge the help provided by local contacts at the European Synchrotron Radiation Facility.
This work was supported, in whole or in part, by National Institutes of Health Grant DE09761/DE/NIDCR. This work was also supported by Polish National Science Center Grant UMO-2012/04/A/NZ1/00051; Ministry of Science and Higher Education of Poland Grant 2137/7.PR-EU/2011/2; European Union Grants FP7-HEALTH-2010-261460 “Gums&Joints,” FP7-PEOPLE-2011-ITN-290246 “RAPID,” FP7-HEALTH-2012-306029-2 “TRIGGER,” and POIG.02.01.00-12-064/08; Spanish Ministry of Economy and Competitiveness Grants BFU2012-32862 and BIO2013-49320-EXP; Spanish Ministry of Science and Education Grant CSD2006-00015; and National Government of Catalonia Grant 2014SGR9. Funding for data collection was provided in part by the European Synchrotron Radiation Facility (Grenoble, France).
The atomic coordinates and structure factors (code 4RBM) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- Kgp
- gingipain K
- CD
- catalytic domain
- CSD
- C-terminal subdomain
- IgSF
- immunoglobulin superfamily-like domain
- KgpAA1–KgpAA5
- Kgp hemagglutinin/adhesion domains A1–A5
- LM
- l-lysinylmethyl
- NSD
- N-terminal subdomain
- Rgp
- gingipain R
- RgpAA1–RgpAA4
- RgpA hemagglutinin/adhesion domains A1–A4
- TLCK
- Nα-tosyl-l-lysinylchloromethane.
REFERENCES
- 1. Le Chatelier E., Nielsen T., Qin J., Prifti E., Hildebrand F., Falony G., Almeida M., Arumugam M., Batto J. M., Kennedy S., Leonard P., Li J., Burgdorf K., Grarup N., Jørgensen T., Brandslund I., Nielsen H. B., Juncker A. S., Bertalan M., Levenez F., Pons N., Rasmussen S., Sunagawa S., Tap J., Tims S., Zoetendal E. G., Brunak S., Clément K., Doré J., Kleerebezem M., Kristiansen K., Renault P., Sicheritz-Ponten T., de Vos W. M., Zucker J. D., Raes J., Hansen T., MetaHIT consortium, Bork P., Wang J., Ehrlich S. D., Pedersen O. (2013) Richness of human gut microbiome correlates with metabolic markers. Nature 500, 541–546 [DOI] [PubMed] [Google Scholar]
- 2. May M. (2014) Drug development: time for teamwork. Nature 509, S4–S5 [DOI] [PubMed] [Google Scholar]
- 3. Hede K. (2014) Antibiotic resistance: an infectious arms race. Nature 509, S2–S3 [DOI] [PubMed] [Google Scholar]
- 4. Bostanci N., Belibasakis G. N. (2012) Porphyromonas gingivalis: an invasive and evasive opportunistic oral pathogen. FEMS Microbiol. Lett. 333, 1–9 [DOI] [PubMed] [Google Scholar]
- 5. Maixner F., Thomma A., Cipollini G., Widder S., Rattei T., Zink A. (2014) Metagenomic analysis reveals presence of Treponema denticola in a tissue biopsy of the Iceman. PLoS One 9, e99994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kurita-Ochiai T., Yamamoto M. (2014) Periodontal pathogens and atherosclerosis: implications of inflammation and oxidative modification of LDL. Biomed. Res. Int. 2014, 595981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maresz K. J., Hellvard A., Sroka A., Adamowicz K., Bielecka E., Koziel J., Gawron K., Mizgalska D., Marcinska K. A., Benedyk M., Pyrc K., Quirke A. M., Jonsson R., Alzabin S., Venables P. J., Nguyen K. A., Mydel P., Potempa J. (2013) Porphyromonas gingivalis facilitates the development and progression of destructive arthritis through its unique bacterial peptidylarginine deiminase (PAD). PLoS Pathog. 9, e1003627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tilakaratne A., Soory M. (2014) Anti-inflammatory actions of adjunctive tetracyclines and other agents in periodontitis and associated comorbidities. Open Dent. J. 8, 109–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Page R. C. (1998) The pathobiology of periodontal diseases may affect systemic diseases: inversion of a paradigm. Ann. Periodontol. 3, 108–120 [DOI] [PubMed] [Google Scholar]
- 10. Haffajee A. D., Socransky S. S., Gunsolley J. C. (2003) Systemic anti-infective periodontal therapy. A systematic review. Ann. Periodontol. 8, 115–181 [DOI] [PubMed] [Google Scholar]
- 11. Matesanz-Pérez P., García-Gargallo M., Figuero E., Bascones-Martínez A., Sanz M., Herrera D. (2013) A systematic review on the effects of local antimicrobials as adjuncts to subgingival debridement, compared with subgingival debridement alone, in the treatment of chronic periodontitis. J. Clin. Periodontol. 40, 227–241 [DOI] [PubMed] [Google Scholar]
- 12. Potempa J., Pike R. N. (2005) Bacterial peptidases. Contrib. Microbiol. 12, 132–180 [DOI] [PubMed] [Google Scholar]
- 13. Mallorquí-Fernández N., Manandhar S. P., Mallorquí-Fernández G., Usón I., Wawrzonek K., Kantyka T., Solà M., Thøgersen I. B., Enghild J. J., Potempa J., Gomis-Rüth F. X. (2008) A new autocatalytic activation mechanism for cysteine proteases revealed by Prevotella intermedia interpain A. J. Biol. Chem. 283, 2871–2882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dubin G., Koziel J., Pyrc K., Wladyka B., Potempa J. (2013) Bacterial proteases in disease—role in intracellular survival, evasion of coagulation/ fibrinolysis innate defenses, toxicoses and viral infections. Curr. Pharm. Des. 19, 1090–1113 [DOI] [PubMed] [Google Scholar]
- 15. Lamont R. J., Jenkinson H. F. (1998) Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis. Microbiol. Mol. Biol. Rev. 62, 1244–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Potempa J., Travis J. (1996) Porphyromonas gingivalis proteinases in periodontitis, a review. Acta Biochim. Pol. 43, 455–465 [PubMed] [Google Scholar]
- 17. Potempa J., Pike R., Travis J. (1997) Titration and mapping of the active site of cysteine proteinases from Porphyromonas gingivalis (gingipains) using peptidyl chloromethanes. Biol. Chem. 378, 223–230 [DOI] [PubMed] [Google Scholar]
- 18. Pike R. N., Potempa J. (2013) in Handbook of Proteolytic Enzymes (Rawlings N. D., Salvesen G., eds) pp. 2337–2344, Academic Press, Oxford [Google Scholar]
- 19. Brochu V., Grenier D., Nakayama K., Mayrand D. (2001) Acquisition of iron from human transferrin by Porphyromonas gingivalis: a role for Arg- and Lys-gingipain activities. Oral Microbiol. Immunol. 16, 79–87 [DOI] [PubMed] [Google Scholar]
- 20. Pathirana R. D., O'Brien-Simpson N. M., Brammar G. C., Slakeski N., Reynolds E. C. (2007) Kgp and RgpB, but not RgpA, are important for Porphyromonas gingivalis virulence in the murine periodontitis model. Infect. Immun. 75, 1436–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yongqing T., Potempa J., Pike R. N., Wijeyewickrema L. C. (2011) The lysine-specific gingipain of Porphyromonas gingivalis: importance to pathogenicity and potential strategies for inhibition. Adv. Exp. Med. Biol. 712, 15–29 [DOI] [PubMed] [Google Scholar]
- 22. Curtis M. A., Aduse Opoku J., Rangarajan M., Gallagher A., Sterne J. A., Reid C. R., Evans H. E., Samuelsson B. (2002) Attenuation of the virulence of Porphyromonas gingivalis by using a specific synthetic Kgp protease inhibitor. Infect. Immun. 70, 6968–6975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kataoka S., Baba A., Suda Y., Takii R., Hashimoto M., Kawakubo T., Asao T., Kadowaki T., Yamamoto K. (2014) A novel, potent dual inhibitor of Arg-gingipains and Lys-gingipain as a promising agent for periodontal disease therapy. FASEB J. 28, 3564–3578 [DOI] [PubMed] [Google Scholar]
- 24. Pike R., McGraw W., Potempa J., Travis J. (1994) Lysine- and arginine-specific proteinases from Porphyromonas gingivalis. Isolation, characterization, and evidence for the existence of complexes with hemagglutinins. J. Biol. Chem. 269, 406–411 [PubMed] [Google Scholar]
- 25. Sztukowska M., Veillard F., Potempa B., Bogyo M., Enghild J. J., Thogersen I. B., Nguyen K. A., Potempa J. (2012) Disruption of gingipain oligomerization into non-covalent cell-surface attached complexes. Biol. Chem. 393, 971–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mittl P. R., Grütter M. G. (2006) Opportunities for structure-based design of protease-directed drugs. Curr. Opin. Struct. Biol. 16, 769–775 [DOI] [PubMed] [Google Scholar]
- 27. de Diego I., Veillard F. T., Guevara T., Potempa B., Sztukowska M., Potempa J., Gomis-Rüth F. X. (2013) Porphyromonas gingivalis virulence factor gingipain RgpB shows a unique zymogenic mechanism for cysteine peptidases. J. Biol. Chem. 288, 14287–14296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eichinger A., Beisel H. G., Jacob U., Huber R., Medrano F. J., Banbula A., Potempa J., Travis J., Bode W. (1999) Crystal structure of gingipain R: an Arg-specific bacterial cysteine proteinase with a caspase-like fold. EMBO J. 18, 5453–5462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li N., Yun P., Jeffries C. M., Langley D., Gamsjaeger R., Church W. B., Hunter N., Collyer C. A. (2011) The modular structure of haemagglutinin/adhesin regions in gingipains of Porphyromonas gingivalis. Mol. Microbiol. 81, 1358–1373 [DOI] [PubMed] [Google Scholar]
- 30. Li N., Yun P., Nadkarni M. A., Ghadikolaee N. B., Nguyen K. A., Lee M., Hunter N., Collyer C. A. (2010) Structure determination and analysis of a haemolytic gingipain adhesin domain from Porphyromonas gingivalis. Mol. Microbiol. 76, 861–873 [DOI] [PubMed] [Google Scholar]
- 31. Potempa J., Nguyen K. A. (2007) Purification and characterization of gingipains. Curr. Protoc. Protein Sci. Chapter 21, Unit 21.20 [DOI] [PubMed] [Google Scholar]
- 32. Matthews B. W. (1968) Solvent content of protein crystals. J. Mol. Biol. 33, 491–497 [DOI] [PubMed] [Google Scholar]
- 33. Trillo-Muyo S., Jasilionis A., Domagalski M. J., Chruszcz M., Minor W., Kuisiene N., Arolas J. L., Solà M., Gomis-Rüth F. X. (2013) Ultratight crystal packing of a 10-kDa protein. Acta Crystallogr. D Biol. Crystallogr. 69, 464–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Corpet F. (1988) Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16, 10881–10890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Langer G., Cohen S. X., Lamzin V. S., Perrakis A. (2008) Automated macromolecular model building for x-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Carranza C., Inisan A.-G., Mouthuy-Knoops E., Cambillau C., Roussel A. (1999) in AFMB Activity Report 1996–1999, pp. 89–90, CNRS-UPR 9039, Marseille, France [Google Scholar]
- 40. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Blanc E., Roversi P., Vonrhein C., Flensburg C., Lea S. M., Bricogne G. (2004) Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr. D Biol. Crystallogr. 60, 2210–2221 [DOI] [PubMed] [Google Scholar]
- 42. Di Tommaso P., Moretti S., Xenarios I., Orobitg M., Montanyola A., Chang J. M., Taly J. F., Notredame C. (2011) T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 39, W13–W17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Robert X., Gouet P. (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schüttelkopf A. W., van Aalten D. M. (2004) PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60, 1355–1363 [DOI] [PubMed] [Google Scholar]
- 45. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 47. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ollis D. L., Cheah E., Cygler M., Dijkstra B., Frolow F., Franken S. M., Harel M., Remington S. J., Silman I., Schrag J., Sussman J. L., Verschueren K. H., Goldman A. (1992) The a/b hydrolase fold. Protein Eng. 5, 197–211 [DOI] [PubMed] [Google Scholar]
- 49. Puente X. S., López-Otín C. (1997) The PLEES proteins—a family of structurally related enzymes widely distributed from bacteria to humans. Biochem. J. 322, 947–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fraústo da Silva J. J. R., Williams R. J. P. (2001) The Biological Chemistry of the Elements: the Inorganic Chemistry of Life, 2nd Ed., Oxford University Press Inc., New York [Google Scholar]
- 51. Richardson J. S. (1981) The anatomy and taxonomy of protein structure. Adv. Protein Chem. 34, 167–339 [DOI] [PubMed] [Google Scholar]
- 52. Harding M. M. (2000) The geometry of metal-ligand interactions relevant to proteins. II. Angles at the metal atom, additional weak metal-donor interactions. Acta Crystallogr. D Biol. Crystallogr. 56, 857–867 [DOI] [PubMed] [Google Scholar]
- 53. Harding M. M. (2006) Small revisions to predicted distances around metal sites in proteins. Acta Crystallogr. D Biol. Crystallogr. 62, 678–682 [DOI] [PubMed] [Google Scholar]
- 54. Richardson J. S., Richardson D. C. (1989) in Prediction of Protein Structure and the Principles of Protein Conformation (Fasman G. D., ed.) pp. 1–98, Plenum Press, New York [Google Scholar]
- 55. Harding M. M. (2002) Metal-ligand geometry relevant to proteins and in proteins: sodium and potassium. Acta Crystallogr. D Biol. Crystallogr. 58, 872–874 [DOI] [PubMed] [Google Scholar]
- 56. Vidossich P., Loewen P. C., Carpena X., Fiorin G., Fita I., Rovira C. (2014) Binding of the antitubercular pro-drug isoniazid in the heme access channel of catalase-peroxidase (KatG). A combined structural and metadynamics investigation. J. Phys. Chem. B 118, 2924–2931 [DOI] [PubMed] [Google Scholar]
- 57. Nakatogawa H., Ito K. (2002) The ribosomal exit tunnel functions as a discriminating gate. Cell 108, 629–636 [DOI] [PubMed] [Google Scholar]
- 58. Williams M. A., Goodfellow J. M., Thornton J. M. (1994) Buried waters and internal cavities in monomeric proteins. Protein Sci. 3, 1224–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rose A., Theune D., Goede A., Hildebrand P. W. (2014) MP:PD—a data base of internal packing densities, internal packing defects and internal waters of helical membrane proteins. Nucleic Acids Res. 42, D347–D351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hildebrand P. W., Günther S., Goede A., Forrest L., Frömmel C., Preissner R. (2008) Hydrogen-bonding and packing features of membrane proteins: functional implications. Biophys. J. 94, 1945–1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lee C., Park S. H., Lee M. Y., Yu M. H. (2000) Regulation of protein function by native metastability. Proc. Natl. Acad. Sci. U.S.A. 97, 7727–7731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Takano K., Yamagata Y., Yutani K. (2003) Buried water molecules contribute to the conformational stability of a protein. Protein Eng. 16, 5–9 [DOI] [PubMed] [Google Scholar]
- 63. Dunitz J. D. (1994) The entropic cost of bound water in crystals and biomolecules. Science 264, 670–670 [DOI] [PubMed] [Google Scholar]
- 64. Zhang L., Hermans J. (1996) Hydrophilicity of cavities in proteins. Proteins 24, 433–438 [DOI] [PubMed] [Google Scholar]
- 65. Kokkinidis M., Glykos N. M., Fadouloglou V. E. (2012) Protein flexibility and enzymatic catalysis. Adv. Protein Chem. Struct. Biol. 87, 181–218 [DOI] [PubMed] [Google Scholar]
- 66. Schulenburg C., Hilvert D. (2013) Protein conformational disorder and enzyme catalysis. Top Curr. Chem. 337, 41–67 [DOI] [PubMed] [Google Scholar]
- 67. Potempa J., Pike R., Travis J. (1995) The multiple forms of trypsin-like activity present in various strains of Porphyromonas gingivalis are due to the presence of either Arg-gingipain or Lys-gingipain. Infect. Immun. 63, 1176–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sztukowska M., Sroka A., Bugno M., Banbula A., Takahashi Y., Pike R. N., Genco C. A., Travis J., Potempa J. (2004) The C-terminal domains of the gingipain K polyprotein are necessary for assembly of the active enzyme and expression of associated activities. Mol. Microbiol. 54, 1393–1408 [DOI] [PubMed] [Google Scholar]
- 69. Chothia C., Jones E. Y. (1997) The molecular structure of cell adhesion molecules. Annu. Rev. Biochem. 66, 823–862 [DOI] [PubMed] [Google Scholar]
- 70. Discipio R. G., Daffern P. J., Kawahara M., Pike R., Travis J., Hugli T. E., Potempa J. (1996) Cleavage of human complement component C5 by cysteine proteinases from Porphyromonas (Bacteroides) gingivalis. Prior oxidation of C5 augments proteinase digestion of C5. Immunology 87, 660–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Marrero A., Duquerroy S., Trapani S., Goulas T., Guevara T., Andersen G. R., Navaza J., Sottrup-Jensen L., Gomis-Rüth F. X. (2012) The crystal structure of human α2-macroglobulin reveals a unique molecular cage. Angew. Chem. Int. Ed. Engl. 51, 3340–3344 [DOI] [PubMed] [Google Scholar]
- 72. Ishida Y., Hu J., Sakai E., Kadowaki T., Yamamoto K., Tsukuba T., Kato Y., Nakayama K., Okamoto K. (2008) Determination of active site of lysine-specific cysteine proteinase (Lys-gingipain) by use of a Porphyromonas gingivalis plasmid system. Arch. Oral Biol. 53, 538–544 [DOI] [PubMed] [Google Scholar]
- 73. Shaw E., Mares-Guia M., Cohen W. (1965) Evidence for an active-center histidine in trypsin through use of a specific reagent, 1-chloro-3-tosylamido-7-amino-2-heptanone, the chloromethyl ketone derived from Nα-tosyl-L-lysine. Biochemistry 4, 2219–2224 [Google Scholar]
- 74. Frydrych I., Mlejnek P. (2008) Serine protease inhibitors N-α-tosyl-l-lysinyl-chloromethylketone (TLCK) and N-tosyl-l-phenylalaninyl-chloromethylketone (TPCK) are potent inhibitors of activated caspase proteases. J. Cell. Biochem. 103, 1646–1656 [DOI] [PubMed] [Google Scholar]
- 75. Drenth J., Kalk K. H., Swen H. M. (1976) Binding of chloromethyl ketone substrate analogues to crystalline papain. Biochemistry 15, 3731–3738 [DOI] [PubMed] [Google Scholar]
- 76. Jia Z., Hasnain S., Hirama T., Lee X., Mort J. S., To R., Huber C. P. (1995) Crystal structures of recombinant rat cathepsin B and a cathepsin B-inhibitor complex. Implications for structure-based inhibitor design. J. Biol. Chem. 270, 5527–5533 [DOI] [PubMed] [Google Scholar]
- 77. Robertus J. D., Alden R. A., Birktoft J. J., Kraut J., Powers J. C., Wilcox P. E. (1972) An x-ray crystallographic study of the binding of peptide chloromethyl ketone inhibitors to subtilisin BPN′. Biochemistry 11, 2439–2449 [DOI] [PubMed] [Google Scholar]
- 78. Bode W., Mayr I., Baumann U., Huber R., Stone S. R., Hofsteenge J. (1989) The refined 1.9 Å crystal structure of human α-thrombin: interaction with D-Phe-Pro-Arg chloromethylketone and significance of the Tyr-Pro-Pro-Trp insertion segment. EMBO J. 8, 3467–3475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Schechter I., Berger A. (1967) On the size of active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 27, 157–162 [DOI] [PubMed] [Google Scholar]
- 80. Chen J.-M., Rawlings N. D., Stevens R. A., Barrett A. J. (1998) Identification of the active site of legumain links it to caspases, clostripain and gingipains in a new clan of cysteine endopeptidases. FEBS Lett. 441, 361–365 [DOI] [PubMed] [Google Scholar]
- 81. Polgár L. (2013) in Handbook of Proteolytic Enzymes (Rawlings N. D., Salvesen G. S., eds) 3rd Ed., pp. 1773–1784, Academic Press, Oxford [Google Scholar]
- 82. Guarné A., Hampoelz B., Glaser W., Carpena X., Tormo J., Fita I., Skern T. (2000) Structural and biochemical features distinguish the foot-and-mouth disease virus leader proteinase from other papain-like enzymes. J. Mol. Biol. 302, 1227–1240 [DOI] [PubMed] [Google Scholar]
- 83. Chen Z., Potempa J., Polanowski A., Wikstrom M., Travis J. (1992) Purification and characterization of a 50-kDa cysteine proteinase (gingipain) from Porphyromonas gingivalis. J. Biol. Chem. 267, 18896–18901 [PubMed] [Google Scholar]
- 84. Rawlings N. D., Waller M., Barrett A. J., Bateman A. (2014) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 42, D503–D509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Aravind L., Koonin E. V. (2002) Classification of the caspase-hemoglobinase fold: detection of new families and implications for the origin of the eukaryotic separins. Proteins 46, 355–367 [DOI] [PubMed] [Google Scholar]
- 86. Dall E., Brandstetter H. (2013) Mechanistic and structural studies on legumain explain its zymogenicity, distinct activation pathways, and regulation. Proc. Natl. Acad. Sci. U.S.A. 110, 10940–10945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Weiss M. S. (2001) Global indicators of x-ray quality. J. Appl. Crystallogr. 34, 130–135 [Google Scholar]
- 88. Karplus P. A., Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]