

Case 1
A 61-year-old healthy man presents for a routine outpatient clinic visit. He was diagnosed with hypertension 6 years ago with blood pressure well controlled on amlodipine 5 mg daily. He is a lifelong nonsmoker and has no family history of premature coronary disease. He is employed as a business executive. Exercise is limited to 15 minutes of walking daily in transit to work. Diet includes frequent dining out with clients and occasional moderate alcohol consumption. Examination is notable for a blood pressure of 125/70 mm Hg and a body mass index of 32 kg/m2. A fasting lipid panel is notable for total cholesterol of 188 mg/dL, high-density lipoprotein-cholesterol (HDL-C) of 31 mg/dL, calculated low-density lipoprotein-cholesterol (LDL-C) of 125 mg/dL, and triglycerides of 160 mg/dL.
Case 2
A 58-year-old man with coronary artery disease presents for follow-up. When he was 50 years of age, chest discomfort led to diagnosis of a non–ST-segment elevation myocardial infarction. Cardiac catheterization revealed a 90% left anterior descending lesion, which was stented, and 40% left circumflex artery stenosis. A lipid profile was returned with total cholesterol of 180 mg/dL, LDL-C of 130 mg/dL, HDL-C of 28 mg/dL, and triglycerides of 150 mg/dL. In addition to other cardiovascular medications, the patient was treated with atorvastatin 80 mg. The follow-up LDL-C was 72 mg/dL, and HDL-C was 26 mg/dL; vital signs were well controlled. Four years ago, at 54 years of age, he re-presented with exertional chest discomfort. Stress testing led to repeat catheterization and stenting of a 90% left circumflex lesion and a 90% left anterior descending lesion distal to the previous intervention. Lipoprotein(a), checked because of a family history of premature coronary artery disease, was 110 mg/dL (upper limit of normal, 40 mg/dL). A trial of niacin was started, and titrated, which the patient tolerated. On 2 g of niacin and atorvastatin 80 mg daily, his most recent lipid profile was returned with a total cholesterol of 132 mg/dL, HDL-C of 44 mg/dL, LDL-C of 64 mg/dL, triglycerides of 120 mg/dL, and lipoprotein(a) of 44 mg/dL.
The Clinical Problem
A low level of circulating HDL-C represents a problem that is both common and clinically challenging, with a difficult-to-reconcile current database. National Cholesterol Treatment Panel guidelines suggest that a HDL-C level of <40 mg/dL be considered low.1 National Health and Nutrition Examination Survey data indicate that 31% of men and 12% of women in the community currently meet this criterion.2 The National Cholesterol Treatment Panel, in their more detailed text regarding metabolic syndrome, recognize sex-specific cutoffs of HDL-C <40 mg/dL for men and <50 mg/dL for women as undesirable. Low HDL, linked to premature coronary artery disease, is being identified with increasing frequency as lipid screening recommendations are increased.
The inverse association between plasma HDL-C and cardiovascular disease remains one of the most robust epidemiological observations ever made (Figure 1A). First described in 1977 in Framingham Heart Study data, the relationship has been replicated in multiple cohorts.3 Epidemiological data link a 1 mg/dL increase in HDL to a 2% to 3% reduction in cardiovascular events, an impact greater than that noted for LDL-C. However, pharmacological interventions that raise HDL-C have not been clearly shown to reduce adverse cardiovascular outcomes. This conundrum has forced clinicians to reconsider existing and emerging data on HDL biology and therapeutic strategies to optimize clinical outcomes in the statin era.
Figure 1.
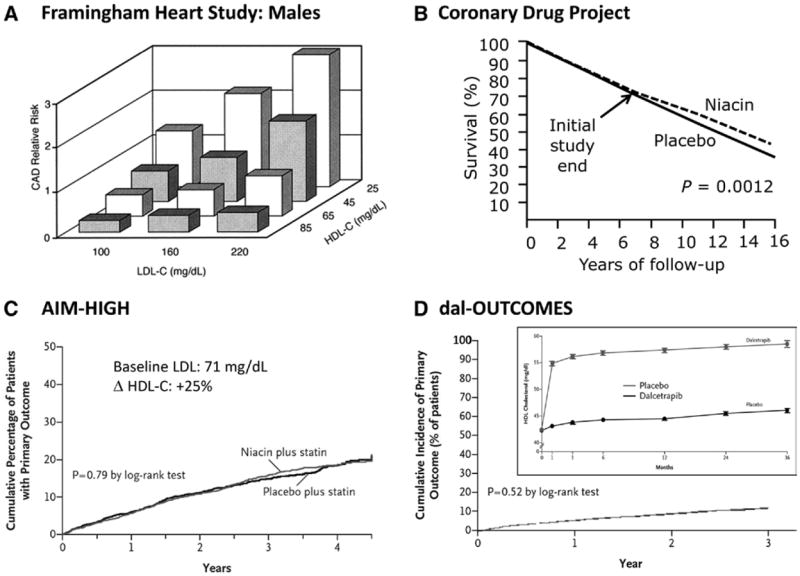
The HDL conundrum in 4 representative datasets. A, A strong epidemiological risk association. Total HDL-C levels exhibit a wellvalidated, robust inverse relationship with cardiovascular risk, even among patients with the same (including lower) LDL levels, as seen in the Framingham Heart Study. B, Encouraging prospects for cardiovascular benefit. In the Coronary Drug Project, niacin significantly decreased mortality at long-term (16-year) follow-up. C, Missing prospective event reduction? In AIM-HIGH, adding niacin to statin-treated cardiovascular disease patients with well-controlled baseline LDL levels had no impact on cardiovascular events despite significant HDL increases. D, Novel strategies, more disappointment? CETP inhibitors, like dalcetrapib, can significantly increase HDL levels, but have yet to show clinical benefit, as seen in Dalcetrapib Outcomes Study (dal-OUTCOMES) (post–acute coronary syndrome trial). AIM-HIGH indicates Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides: Impact on Global Health Outcomes; CAD, coronary artery disease; CETP, cholesteryl ester transfer protein; dal-OUTCOMES, Dalcetrapib Outcomes Study; HDL, high-density lipoprotein; HDL-C, high-density lipoprotein-cholesterol; LDL, low-density lipoprotein; and LDL-C, low-density lipoprotein-cholesterol.
The Biology
The epidemiological association between HDL-C and cardiovascular disease has stimulated research to clarify its mechanistic underpinnings and provide biological plausibility for causation. Numerous models have been proposed, based mainly on animal or in vitro models. HDL-C promotes reverse cholesterol transport, which facilitates cholesterol transport from peripheral lipid-laden macrophages to the liver for biliary excretion. In this construct, HDL is atheroprotective by removing cholesterol from pathological sites, like the vasculature, and depositing it in the liver and gastrointestinal tract. Considerable preclinical research also suggests other HDL properties that might decrease atherosclerotic complications, including anti-inflammatory effects, decreased low-density lipoprotein (LDL) oxidation, increased nitric oxide production, decreased endothelial adhesion molecule expression, decreased thrombogenicity, and decreased inflammatory-induced apoptosis (Figure 2).
Figure 2.
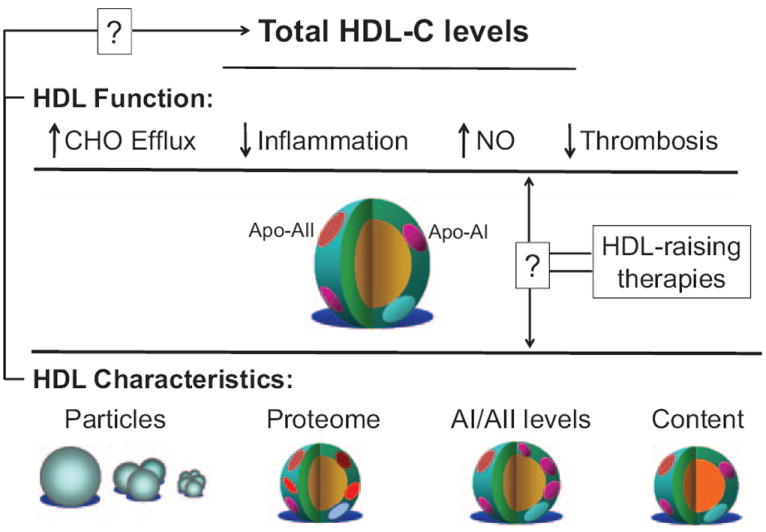
HDL-cholesterol: levels vs function? HDL, like other lipoproteins, includes a range of complex macromolecules with diverse functional effects and distinct characteristics. HDL, in part, through its major apolipoprotein, apolipoprotein A-I, promotes cholesterol efflux and may also decrease vascular inflammation, increase nitric oxide levels (NO), and decrease thrombosis. These and other pleiotropic effects may relate to variable HDL characteristics involving particles (number, size, subtypes), the proteins present, relative apolipoprotein levels, and the specific lipid content. How these properties relate to total HDL-C levels, or respond to different HDL-raising therapies, remains unknown but offers other ways to consider HDL. Apo indicates apolipoprotein; CHO, cholesterol; HDL, high-density lipoprotein; and HDL-C, high-density lipoprotein-cholesterol.
The Controversy
Recently, the HDL hypothesis—the notion that HDL-mediated atheroprotection can be modulated for therapeutic benefit—has faced multiple challenges (Figure 1). Analyses seeking to isolate the impact of HDL-C via observational epidemiology are limited by extensive confounding; multiple cardiovascular risk factors, including male sex, smoking, visceral adiposity, insulin resistance, and systemic inflammation alter HDL-C levels. Furthermore, intensive statin therapy may attenuate responses that might have otherwise been significant; null associations between on-treatment HDL-C and cardiovascular outcomes were noted in several recent clinical trials, although a risk relationship with HDL-C despite statin therapy persisted in other studies.
Although human genetics analyses have further solidified the causal relationship between LDL-C and atherosclerosis, analogous studies of patients with genetic mutations that alter HDL-C levels have yielded inconsistent results. Markedly decreased HDL concentrations in patients with monogenic disorders involving apolipoprotein A-I (ie, apolipoprotein A-IMilano), ABCA1 (Tangier disease), and LCAT (familial lecithin cholesterol acyltransferase deficiency) have not been clearly associated with increased or premature cardiovascular disease. Mendelian randomization analyses use high-throughput genotyping to try to overcome confounding in assessing causality. For example, if HDL-C is directly atheroprotective, inherited variations in HDL-C levels should change cardiovascular risk by a magnitude predicted by known epidemiology. In 1 recent analysis, a genetic score that combined 14 common polymorphisms that influence HDL-C levels had no association with cardiovascular events. However, some specific polymorphisms did correlate with reduced risk.4 This study suggests that HDL-C levels as a predictor of cardiovascular benefit may depend on how a given intervention alters HDL function (Figure 2).
With regard to therapeutics, evidence that pharmacological agents that increase HDL-C improve cardiovascular outcomes has been limited. Despite early smaller clinical trials and long-term follow-up studies of HDL-C interventions suggesting significant cardiovascular benefit, 4 recent high-profile clinical trials of HDL-raising therapies have argued otherwise: Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides: Impact on Global Health Outcomes (AIM-HIGH; secondary prevention trial adding niacin to LDL-lowering therapy)5; Heart Protection Study 2: Treatment of HDL to Reduce the Incidence of Vascular Events (HPS-2 THRIVE; secondary prevention of adding niacin/laropiprant to statin therapy)6; a Study Examining Torcetrapib/Atorvastatin And Atorvastatin Effects on Clinical CV Events in Patients With Heart Disease (ILLUMINATE, primary prevention trial in high-risk patients adding the cholesteryl ester transfer protein [CETP] inhibitor torcetrapib to atorvastatin)7; and Dalcetrapib Outcomes Study (Dal-OUTCOMES, secondary prevention trial in acute coronary syndrome patients adding the CETP inhibitor dalcetrapib to optimal therapy; Figure 1D).8 Each of these studies has complicating features that preclude definitive conclusions about the impact of the HDL changes seen. However, these negative results reinforce the need for caution and reexamination of HDL-centric therapies.
Nonpharmacological Interventions
Aerobic Exercise
A high level of exercise was previously thought necessary in order to increase HDL-C levels. However, sustained and regular moderate-intensity activity can increase HDL-C levels by 5% to 10%. Individuals with low HDL-C and elevated triglycerides and abdominal obesity, as in case 1 above, may have particularly robust HDL-C responses to exercise, potentially mediated through increased insulin sensitivity independent of diet. Observational data and randomized trials with surrogate cardiovascular end points support the potential for cardiovascular risk reduction with exercise. Therefore, adherence to current American Heart Association guidelines for physical activity in adults (at least 30 minutes most days of the week and at least 60 minutes for weight loss) remains advisable.8a
Weight Loss
Overweight and obesity are strongly associated with both low HDL-C and elevated circulating triglycerides. Acute changes in weight loss may modestly decrease HDL-C levels, likely via decreased lipoprotein lipase activity. However, on weight stabilization, HDL-C levels generally increase by 0.35 mg/dL per kilogram.9 This increase in HDL-C occurs regardless of weight loss strategy: diet, physical activity, pharmacological therapies, or surgical approaches.
Nutrition
Patients who consume very low fat diets often have lower HDL-C levels, although LDL-C is also reduced, confounding the assignment of potential risk reduction. Meanwhile, diets high in saturated or trans fats tend to increase HDL-C levels at the possible expense of impaired HDL functionality and increased LDL-C. Processed carbohydrates that rapidly increase glucose levels (ie, high glycemic index) may lower HDL-C levels, increase inflammation, and increase cardiovascular risk. An emphasis on modest portion sizes and a diet high in fresh fruits and vegetables, low in processed carbohydrates and meats, and high in polyunsaturated fatty acids remains advisable.
Smoking Cessation
The oxidant stress and inflammation induced by cigarette smoking have been linked to both decreased HDL levels and function. Smokers tend to have an HDL-C level that is 5% to 10% lower than matched controls. Importantly, smoking cessation leads to an increase in HDL-C of ≈4 mg/dL with minimal impact on other lipid parameters.10
Alcohol Intake
Alcohol consumption can increase HDL-C ≈2 mg/dL per daily alcoholic beverage.11 Moderate alcohol intake (1–2 drinks/d in men, 1 drink/d in women) has been associated with improved cardiovascular outcomes in observational studies, although via unclear mechanisms. Recommending that nondrinkers with low HDL-C initiate moderate alcohol consumption for cardiovascular risk reduction remains speculative and difficult to endorse given the adverse outcomes associated with alcohol intake. In select patients at low risk for alcohol-related problems, providing the relevant information regarding modest, responsible alcohol intake and cardiovascular risk might be considered.
Pharmacological Interventions
Niacin
Niacin, otherwise known as nicotinic acid or vitamin B3, when prescribed at a dose of 1 to 2 g per day, can increase HDL-C levels 15% to 30%, the most potent effect among currently available medications. Niacin also favorably alters LDL-C, triglyceride, and in some cases, lipoprotein (a) levels. In the Coronary Drug Project, a secondary prevention trial in men performed in the 1960s, niacin reduced myocardial infarctions by 26%12; on long-term follow-up, mortality was also significantly decreased (Figure 1B).
However, data from recent clinical outcome trials that included concomitant statin therapy have failed to show benefit with niacin. The AIM-HIGH trial, a secondary prevention trial in patients with low HDL-C and elevated triglycerides that added extended-release niacin to simvastatin (and ezetimibe as needed to achieve LDL-C goals), was stopped owing to the clinical futility for demonstrating event reduction, despite significant HDL-C increases and a significant event rate (Figure 1C).5 Potential limitations include the baseline LDL-C levels of 70 mg/dL for all subjects, the low absolute difference in on-treatment HDL-C between placebo and niacin groups (38 versus 42 mg/dL), ezetimibe use as a confounding variable, low-dose niacin use in the control group to avoid symptomatic reactions resulting in functional unblinding, and relatively short follow-up (mean, 3 years). The significantly larger HPS-2 THRIVE trial studied extended-release niacin and laropiprant (a flushing inhibitor) versus placebo added to LDL-lowering therapy in >25 000 patients with cardiovascular disease. The study sponsor recently announced a failure to achieve any reduction in the primary composite cardiovascular end point.6 Adverse events were also increased in the active treatment group with details pending peer-reviewed publication of the clinical trial results.
Tolerability also limits niacin use. Flushing is an issue, which can be attenuated with extended-release niacin formulations, taking aspirin 30 minutes before niacin, gradual dose titration, avoiding missed niacin doses, and limiting concomitant intake of drinks that promote flushing, eg, alcohol, hot liquids. Over the counter no-flush preparations may be less effective in HDL raising. Although niacin is generally regarded as safe, it can increase uric acid levels, precipitating gout, and worsen insulin sensitivity.
Niacin use will likely decrease in the future, particularly if purported adverse events in HPS-2 THRIVE are substantial. Whether there is a role for niacin therapy in certain patient subgroups, like statin intolerance, like statin intolerance, isolated low HDL-C, or elevated lipoprotein (a), as in case 2, remains unresolved.
Fibrates
Fibrate therapy lowers triglycerides while raising HDL-C ≈10% to 20%, effects thought to occur via peroxisome proliferator-activated receptor alpha (PPARα) activation. The Helsinki Heart Study (HHS, enrollment 1981–1982) and the Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial (VA-HIT, enrollment 1991–1995) demonstrated that gemfibrozil significantly reduced coronary risk (34% and 22%, respectively), although in the absence of statin use.13,14 The incremental benefit of combining fibrates with statin therapy remains unproven. In the Action to Control Cardiovascular Risk in Diabetes – Lipid arm (ACCORD-Lipid, enrollment 2001–2005), adding fenofibrate to simvastatin did not improve the primary cardiovascular end point.15 Fibrate benefits, especially in the statin era, may be restricted to patients with significantly low HDL-C and elevated triglycerides, a concept supported by post hoc subgroup analyses.16 Regarding safety, fibrates may rarely increase myopathy and rhabdomyolysis in combination with a statin (seen more often with gemfibrozil than ferofibrate) and must be used with caution in chronic kidney disease.
Statins
Statin therapy has proven risk reduction in both primary and secondary prevention of cardiovascular disease. Statins modestly increase HDL-C by 5% to 10%, with some variability among class members, especially at higher doses; rosuvastatin may have greater HDL-raising effects than other statins at their maximal doses. Statin therapy may also favorably modify the HDL proteome, limit inflammation, and offset the risk of low HDL-C levels.
Oral Antidiabetic Agents
Type 2 diabetes mellitus and insulin resistance are commonly associated with atherogenic dyslipidemia, a triad consisting of low HDL-C levels, elevated triglycerides, and small, dense LDL particles. Medications that improve glycemic control may improve low HDL-C levels either directly or indirectly, for example, by lowering triglycerides. The insulin sensitizer pioglitazone, which activates PPARγ, and may have some PPARα activity, can increase HDL-C modestly, an effect implicated in pioglitazone’s anti-atherosclerotic effects.17 Pioglitazone’s untoward effects may offset these potential benefits. Whether newer antidiabetic agents like glucagon-like peptide 1 agonists and dipeptidyl peptidase IV inhibitors improve cardiovascular outcomes through lipid changes remains to be determined.
The HDL-Directed Therapeutic Pipeline
Despite recent setbacks, multiple HDL-directed therapeutic agents remain in clinical development (Table). Pharmacological CETP inhibition, a potent mechanism for increasing HDL levels, has been under intense study. The CETP inhibitor torcetrapib significantly raised HDL in phase III studies but increased cardiovascular events, perhaps because of off-target blood pressure effects. In the Dal-OUTCOMES trial, dalcetrapib had no impact on cardiovascular events, perhaps owing to less CETP inhibition and more modest lipid effects.8 Evacetrapib increases HDL-C levels by up to 132% while reducing LDL-C by 40%; anacetrapib similarly increases HDL-C by 138% and decreases LDL-C by 40%. These more potent CETP inhibitors, appear safe in phase II trials; phase III secondary prevention trial data are eagerly awaited, eg, the anacetrapib REVEAL trial (clinicaltrials.gov identifier: NCT00658515).
Table. Emerging Therapeutic Approaches for Treating Low HDL-C.
| Therapeutic Strategy | Rationale |
|---|---|
| CETP inhibition | Increase HDL-C by inhibiting transfer of cholesteryl esters from HDL-C to Apo-B–containing particles. |
| Apo A-I/reconstituted HDL infusion | Promote reverse cholesterol transport and stabilize atherosclerotic plaque |
| Apo A-I mimetic peptides/ upregulators of Apo A-I production | Seek to mimic pleiotropic beneficial effects of Apo A-I |
| LXR agonism/ inhibition of microRNA MiR-33 | Upregulate transporters involved in efflux of cholesterol from macrophages, promoting reverse cholesterol transport |
| Dual PPARα/PPARγ agonism | Combines PPARα effect of fibrates on lipid metabolism with PPARγ effect on improved insulin resistance |
Examples of strategies for increasing levels of HDL or its components, and the rationale for these approaches are provided.
Apo indicates apolipoprotein; CETP, cholesteryl ester transfer protein; HDL, highdensity lipoprotein; HDL-C, high-density lipoprotein cholesterol; LXR, liver X receptor; PPARα, peroxisome proliferator-activated receptor alpha; and PPARγ, peroxisome proliferator-activated receptor gamma.
Beyond CETP inhibition, direct infusion of reconstituted nascent HDL or recombinant apolipoprotein A-IMilano reportedly decreases atheroma in small human proof-of-concept studies (Table). Oral therapies that increase apolipoprotein A-I production or serve as mimetic peptides improve HDL-function ex vivo. Liver X receptor agonists promote cholesterol efflux and augment intestinal HDL generation. Clinical development of liver X receptor modulators has been limited by various side effects, including hypertriglyceridemia. microRNA miR-33 inhibition may enhance expression of genes involved in cholesterol efflux, although this remains untested in humans. Aleglitazar, a dual PPARα/PPARγ agonist that combines HDL raising/triglyceride lowering with insulin sensitization/glucose lowering is under study in acute coronary syndrome patients with diabetes mellitus (clinicaltrials.gov identifier: NCT01715818).
Novel Conceptual Approaches
The complexities of HDL metabolism remain a highly active area of research. Substantial heterogeneity exists in the biological and chemical properties among circulating HDL particles and their different subtypes (Figure 2). Proteomics studies have revealed numerous distinct proteins associated with HDL, including several involved in complement regulation and the acute phase response. Intriguingly, the HDL proteome may differ between individuals with and without coronary disease. Advanced HDL phenotyping may identify better predictors of cardiovascular disease, for example, HDL particle number as assessed by nuclear magnetic resonance spectroscopy may be a better predictor of atherosclerosis than HDL-C levels in observational studies. Whether routine clinical use of advanced lipoprotein characterization improves clinical outcomes remains unclear. The notion that functional HDL properties may vary among individuals has existed for some time; ex vivo assays that quantify HDL function are in development.18 One recent study demonstrated that cholesterol efflux capacity, as measured by HDL-mediated efflux of cholesterol from macrophages, strongly correlated with atherosclerotic burden, even after adjusting for HDL-C levels.19
Management Strategy
Low HDL levels should prompt testing for secondary causes, some of which are reversible or artifactual. For example, circulating paraproteins, as seen in hematologic malignancies like multiple myeloma, can rarely interfere with HDL-C quantification. Multiple genetic abnormalities are associated with very low HDL-C levels (<10 mg/dL). Various drugs may decrease HDL-C, including anabolic steroids, atypical antipsychotics, highly active antiretroviral therapy for HIV/AIDS, β-blockers, and immunosuppressive agents. Liver disease, including cirrhosis, can decrease all cholesterol subfractions by impairing apolipoprotein production. Last, low HDL-C levels are frequently noted in acute inflammatory states.
For now, statins remain the first-line therapy among individuals with low HDL-C levels and significant cardiovascular risk who warrant intervention, as defined by validated risk algorithms.20 Therapeutic lifestyle changes offer general health benefits, including the possibility of improved HDL-C levels and function. In terms of specific HDL-C–targeting therapies, fibrates may be reasonable to use in patients with significant cardiovascular risk, along with statins, if triglycerides are significantly elevated, eg, >200 mg/dL, and HDL-C is low. Niacin, if tolerated, remains another theoretical option in patients with substantially elevated lipoprotein(a), statin intolerance, or, in addition to statin therapy, in patients with isolated low HDL-C and progressive cardiovascular events.
The issues and experience around HDL as a risk predictor and a target for therapy are highlighted in the 2 cases initially presented. In case 1, although the low HDL-C may help identify the patient at increased cardiovascular risk, there is no strong clinical trial evidence to argue for niacin as first-line therapy for this patient. After discussion with the patient, a statin was initiated and well-tolerated. Lifestyle interventions were also implemented. In case 2, the patient had early-onset atherosclerotic complications and evidence for progression despite therapy. Moreover, he tolerated niacin, experienced a significant HDL-C increase, and notable lipoprotein (a) lowering. Although the impact of these changes on cardiovascular outcomes remains unproven, after discussion of the data, the patient opted to continue niacin/statin combination therapy and not forego the potential benefit of these changes.
Footnotes
Disclosures
Dr Plutzky is a consultant of Abbott, Merck, Roche/Genentech. Dr Khera reports no conflicts.
References
- 1.National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002;106:3143–421. [PubMed] [Google Scholar]
- 2.Carroll MD, Kit BK, Lacher DA. NCHS data brief no 92. Hyattsville, MD: National Center for Health Statistics; 2012. Total and high-density lipoprotein cholesterol in adults, 2009–2010. [PubMed] [Google Scholar]
- 3.Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med. 1977;62:707–714. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- 4.Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, Hindy G, Hólm H, Ding EL, Johnson T, Schunkert H, Samani NJ, Clarke R, Hopewell JC, Thompson JF, Li M, Thorleifsson G, Newton-Cheh C, Musunuru K, Pirruccello JP, Saleheen D, Chen L, Stewart A, Schillert A, Thorsteinsdottir U, Thorgeirsson G, Anand S, Engert JC, Morgan T, Spertus J, Stoll M, Berger K, Martinelli N, Girelli D, McKeown PP, Patterson CC, Epstein SE, Devaney J, Burnett MS, Mooser V, Ripatti S, Surakka I, Nieminen MS, Sinisalo J, Lokki ML, Perola M, Havulinna A, de Faire U, Gigante B, Ingelsson E, Zeller T, Wild P, de Bakker PI, Klungel OH, Maitland-van der Zee AH, Peters BJ, de Boer A, Grobbee DE, Kamphuisen PW, Deneer VH, Elbers CC, Onland-Moret NC, Hofker MH, Wijmenga C, Verschuren WM, Boer JM, van der Schouw YT, Rasheed A, Frossard P, Demissie S, Willer C, Do R, Ordovas JM, Abecasis GR, Boehnke M, Mohlke KL, Daly MJ, Guiducci C, Burtt NP, Surti A, Gonzalez E, Purcell S, Gabriel S, Marrugat J, Peden J, Erdmann J, Diemert P, Willenborg C, König IR, Fischer M, Hengstenberg C, Ziegler A, Buysschaert I, Lambrechts D, Van de Werf F, Fox KA, El Mokhtari NE, Rubin D, Schrezenmeir J, Schreiber S, Schäfer A, Danesh J, Blankenberg S, Roberts R, McPherson R, Watkins H, Hall AS, Overvad K, Rimm E, Boerwinkle E, Tybjaerg-Hansen A, Cupples LA, Reilly MP, Melander O, Mannucci PM, Ardissino D, Siscovick D, Elosua R, Stefansson K, O’Donnell CJ, Salomaa V, Rader DJ, Peltonen L, Schwartz SM, Altshuler D, Kathiresan S. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–580. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. AIM-HIGH Investigators. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–2267. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- 6. [January 11, 2013];Merck Announces HPS2-THRIVE Study of TREDAPTIVE™ (Extended-Release Niacin/Laropiprant) Did Not Achieve Primary Endpoint. [press release]. http://www.mercknewsroom.com/press-release/prescription-medicine-news/merck-announces-hps2-thrive-study-tredaptive-extended-relea.
- 7.Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, Lopez-Sendon J, Mosca L, Tardif JC, Waters DD, Shear CL, Revkin JH, Buhr KA, Fisher MR, Tall AR, Brewer B. ILLUMINATE Investigators. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–2122. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- 8.Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, Chaitman BR, Holme IM, Kallend D, Leiter LA, Leitersdorf E, McMurray JJ, Mundl H, Nicholls SJ, Shah PK, Tardif JC, Wright RS. dal-OUTCOMES Investigators. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367:2089–2099. doi: 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
- 8a.Thompson PD, Buchner D, Pina IL, Balady GJ, Williams MA, Marcus BH, Berra K, Blair SN, Costa F, Franklin B, Fletcher GF, Gordon NF, Pate RR, Rodriguez BL, Yancey AK, Wenger NK. American Heart Association Council on Clinical Cardiology Subcommittee on Exercise, Rehabilitation, and Prevention; American Heart Association Council on Nutrition, Physical Activity, and Metabolism Subcommittee on Physical Activity. Exercise and physical activity in the prevention and treatment of atherosclerotic cardiovascular disease. Circulation. 2003;107:3109–3116. doi: 10.1161/01.CIR.0000075572.40158.77. [DOI] [PubMed] [Google Scholar]
- 9.Dattilo AM, Kris-Etherton PM. Effects of weight reduction on blood lipids and lipoproteins: a meta-analysis. Am J Clin Nutr. 1992;56:320–328. doi: 10.1093/ajcn/56.2.320. [DOI] [PubMed] [Google Scholar]
- 10.Maeda K, Noguchi Y, Fukui T. The effects of cessation from cigarette smoking on the lipid and lipoprotein profiles: a meta-analysis. Prev Med. 2003;37:283–290. doi: 10.1016/s0091-7435(03)00110-5. [DOI] [PubMed] [Google Scholar]
- 11.Rimm EB, Williams P, Fosher K, Criqui M, Stampfer MJ. Moderate alcohol intake and lower risk of coronary heart disease: meta-analysis of effects on lipids and haemostatic factors. BMJ. 1999;319:1523–1528. doi: 10.1136/bmj.319.7224.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clofibrate and niacin in coronary heart disease. JAMA. 1975;231:360–381. [PubMed] [Google Scholar]
- 13.Frick MH, Elo O, Haapa K, Heinonen OP, Heinsalmi P, Helo P, Huttunen JK, Kaitaniemi P, Koskinen P, Manninen V. Helsinki Heart Study: primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. N Engl J Med. 1987;317:1237–1245. doi: 10.1056/NEJM198711123172001. [DOI] [PubMed] [Google Scholar]
- 14.Rubins HB, Robins SJ, Collins D, Fye CL, Anderson JW, Elam MB, Faas FH, Linares E, Schaefer EJ, Schectman G, Wilt TJ, Wittes J. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N Engl J Med. 1999;341:410–418. doi: 10.1056/NEJM199908053410604. [DOI] [PubMed] [Google Scholar]
- 15.Ginsberg HN, Elam MB, Lovato LC, Crouse JR, III, Leiter LA, Linz P, Friedewald WT, Buse JB, Gerstein HC, Probstfield J, Grimm RH, Ismail-Beigi F, Bigger JT, Goff DC, Jr, Cushman WC, Simons-Morton DG, Byington RP. ACCORD Study Group. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362:1563–1574. doi: 10.1056/NEJMoa1001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldfine AB, Kaul S, Hiatt WR. Fibrates in the treatment of dyslipidemias–time for a reassessment. N Engl J Med. 2011;365:481–484. doi: 10.1056/NEJMp1106688. [DOI] [PubMed] [Google Scholar]
- 17.Lincoff AM, Wolski K, Nicholls SJ, Nissen SE. Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: a meta-analysis of randomized trials. JAMA. 2007;298:1180–1188. doi: 10.1001/jama.298.10.1180. [DOI] [PubMed] [Google Scholar]
- 18.deGoma EM, deGoma RL, Rader DJ. Beyond high-density lipoprotein cholesterol levels evaluating high-density lipoprotein function as influenced by novel therapeutic approaches. J Am Coll Cardiol. 2008;51:2199–2211. doi: 10.1016/j.jacc.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, Mohler ER, Rothblat GH, Rader DJ. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cholesterol Treatment Trialists’ (CTT) Collaborators. Mihaylova B, Emberson J, Blackwell L, Keech A, Simes J, Barnes EH, Voysey M, Gray A, Collins R, Baigent C. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet. 2012;380:581–590. doi: 10.1016/S0140-6736(12)60367-5. [DOI] [PMC free article] [PubMed] [Google Scholar]