


Abstract
A new functional gene database, FOAM (Functional Ontology Assignments for Metagenomes), was developed to screen environmental metagenomic sequence datasets. FOAM provides a new functional ontology dedicated to classify gene functions relevant to environmental microorganisms based on Hidden Markov Models (HMMs). Sets of aligned protein sequences (i.e. ‘profiles’) were tailored to a large group of target KEGG Orthologs (KOs) from which HMMs were trained. The alignments were checked and curated to make them specific to the targeted KO. Within this process, sequence profiles were enriched with the most abundant sequences available to maximize the yield of accurate classifier models. An associated functional ontology was built to describe the functional groups and hierarchy. FOAM allows the user to select the target search space before HMM-based comparison steps and to easily organize the results into different functional categories and subcategories. FOAM is publicly available at http://portal.nersc.gov/project/m1317/FOAM/.
INTRODUCTION
Continuous evolution of next generation sequencing (NGS) permits acquisition of increasingly large DNA and RNA sequence datasets at a relatively low cost. These NGS approaches have resulted in tremendous breakthroughs in the study of human-associated microbial communities (1,2). The environmental microbiology scientific community is also applying NGS to study the composition of phylogenetic and functional genes in microbial communities (metagenomics) and to study which genes are expressed (metatranscriptomics). Recent examples include understanding of the impact of climate change on carbon cycling and greenhouse gas emissions in soil and sediment microbial communities (3–6), and mining of metagenomes for genes that encode novel enzymes of interest for biotechnology applications and biofuel production (7,8). However, a current bottleneck in meta-omic analyses of environmental microbial communities is the lack of tools to accurately assess functional information without excessive computational time.
In particular, soil has been considered the ‘grand challenge’ for metagenomics sequencing projects for several reasons (9). First, the majority of the microorganisms in soil have never been cultivated and their functions are not known. Second, the high diversity and complexity of soil microbial communities is a challenge for metagenome assembly (10). Third, the annotations suffer from lacking functional assignments (as the microorganisms from which they originate have not yet been cultivated and no genome sequence data exists), and there are several genes that have more than one functional assignment in existing databases making correct gene assignments difficult. This deficiency is exemplified when screening for genes involved in specific biochemical cycles, such as methane oxidation or methanogenesis in KEGG (Kyoto Encyclopedia of Genes and Genomes), which are currently classified by Gene Ontology (11) within five categories: ‘biological process’, ‘metabolic process’, ‘biosynthetic process’, ‘cellular biosynthetic process’ and ‘alkane biosynthetic process’. Another example, KEGG Brite (12), classifies methanogenesis at the fourth functional sublevel (as ‘methane metabolism’). Finally, there remain computational challenges when screening big datasets, such as those represented by gigabases (Gb) to terabases (Tb) of sequence data, due to the requirement for access to super computers and excessive computational time (13).
MATERIALS AND METHODS
To address these challenges we aimed to build a manually curated and validated database for screening of environmental metagenomic and metatranscriptomic sequence datasets for functional genes. We focused on biochemical functions and metabolic pathways important in environmental microbial ecology, including global carbon and nitrogen cycles, by manually selecting and organizing functional gene information into a database here called ‘FOAM’ (Functional Ontology Assignments for Metagenomes).
First, KEGG orthologs (KOs) (12) were retrieved to fit within a hierarchical organization from general features to specific pathways (such as denitrification, methanogenesis, etc.). KEGG KO (a reference set of homologous genes, consistent in known functions) benefits from stability, good maintenance, curation, and third party annotation. The KEGG KO was chosen as the FOAM ‘unit’ because it is a qualitative and dynamically maintained knowledge base associated with a rich tool environment that is available within or outside of KEGG. Additionally, using KEGG KO permits the use of all visualization KEGG tools or third party software that have been released [e.g. Cytoscape (14), Glamm (15), Voronto (16), iPATH (17), bioconductor Pathview package (18)]. KEGG KO lists the genes defined in KEGG that belong to each functional and homologous family and, as a consequence, these can be multi-domain and multi-functional. Here, to provide accurate functional annotation, each FOAM module was constructed to ideally target one function.
The reduced size of the resultant FOAM database, compared to non-specific sequence databases, was a first step towards significant improvement in the speed and specificity of similarity searches. In addition, to improve upon the sensitivity of conventional heuristic alignment programs, we turned each KO set into Hidden Markov Models (HMMs; 19) by fetching their corresponding protein family (Pfam) profiles (20) as described in Figure 1. This step generated a sizeable number of conflicts (several Pfam per KO and vice versa) that were automatically resolved by functional assignments to KO. For the few remaining unresolved assignations, the corresponding set of sequences was manually split according to the topology of their phylogenetic trees. At this point the HMMs were re-trained from the new pool of sequences.
Figure 1.
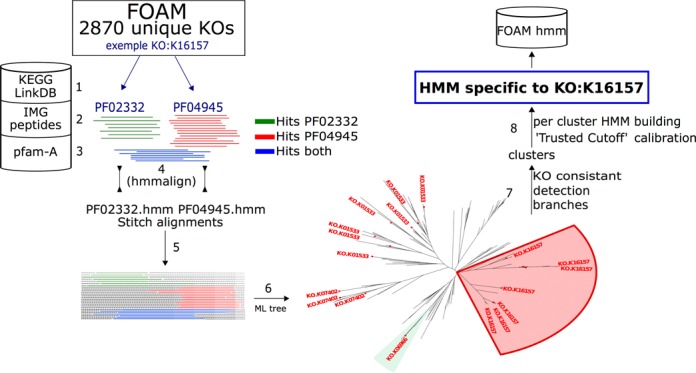
HMM building pipeline: example with KO:K16157 (methane monooxygenase). Step 1—find Pfam(s) combination assigned to the KO of interest (a) and (b) check for redundancy. Step 2—fetch IMG peptide sequences which hit the retrieved Pfam(s). Step 3—fetch from Pfam-A database the HMM of interest. Step 4—alignment (hmmalign) and filter each Pfam from extra sequences obtained in IMG. Step 5—stitch filtered alignments. Step 6—draw a Maximum Likelihood tree (fasttree). Step 7—find clusters in tree with same KO. Step 8—split alignment (step 5 output) by cluster (step 7 output) and build HMM for each, and process the ‘Trusted Cutoff’ computation.
By retrieving the sequences of the corresponding Pfam of each selected KO, in addition to the sequences already present in the FOAM database, we ensured precise detection of functions from potentially distant homologs. With this method, ∼74 000 peptide sequence profiles were specifically tailored and trained to predict functions as defined in KEGG KO. This profile-based searching approach enabled identification of less conserved regions along sequence alignments. Thus this method is applicable for searching for more distant homologs, similar to the approach used by Pfam (20) and TIGRFAM (21). However, we found that most Pfam and TIGRFAM models provide multiple KEGG KO assignments and did not serve our needs for retrieval of functionally specific annotations from metagenomes. Also, Pfam and TIGRFAM do not focus on environmental processes and cover only few functions of interest for different environmental sources. Additionally, Pfam and TIGRFAM are based on a simplified alignment, called ‘SEED’, which is composed of a collection of sequences representative of a protein family, whereas our aim was a more comprehensive recruitment of more distant homologs. Recently, FunGene (22) was published as a new toolkit specialized to process amplicon data for functional genes, focusing on marker genes (∼100 currently available). FunGene provides users with HMMs for their marker genes of interest as a tool to test primers and probes. Moreover, FunGene allows users to build and submit new HMMs. FOAM is complementary to FunGene: it includes ∼3000 custom protein models obtained by enriching Pfams relevant to environmental microbiology with more protein sequences. An additional attribute of FOAM is that KO assignments were screened during the manual calibration to ensure that the Pfam alignments all targeted the same KO. If parts of the alignments targeted other KOs they were omitted from building the models or manually reassigned. Importantly, FOAM is a database that can be complemented with input from the user community. The FOAM database is by no means complete and we encourage recommendations from future users for additional categories to input into FOAM.
Ontology definition
The ontology was defined according to following constraints: (i) cover major biochemical functions and pathways relevant to environmental microbiology; (ii) organize functional classes hierarchically to simplify functional group selections before or after the search step; (iii) use KEGG KO‘s to refine the functional classification. FOAM therefore benefits from the quality of KEGG annotation and all the tools that make use of KO IDs. Several KOs are present in several functional classes to keep the pathway complete at the higher level.
To address the first two constraints, we mainly drew from a comprehensive bacterial physiology and metabolism reference text (23) that was completed by KEGG BRITE ontology (12) information. The resulting FOAM hierarchy is limited to five functional levels: level 1 is the most general function group definition and level 5 the most refined, with level 5 corresponding to KO levels. The resulting FOAM ontology is distributed in the form of a tabulated file on the FOAM website. For example, the level 1 FOAM entry #11 refers to the nitrogen cycle (Table 1; http://portal.nersc.gov/project/m1317/FOAM/data/release_1/) and contains 52 KOs. There are four functional groups at level 2 of the database: ‘reduction of nitrogenous compounds’ (i.e. reductive reactions involving nitrogen compounds), ‘nitrogen fixation’, ‘ammonia assimilation’ and ‘nitrification’. The FOAM level 2 group, ‘reduction of nitrogenous compounds’, is further subdivided into four categories: ‘denitrification’, ‘assimilatory nitrate reduction’, ‘dissimilatory nitrate reduction’ and ‘hydroxylamine reduction’. Subsequently, the FOAM level 2 group of ‘nitrification’ is subdivided into the following categories: ‘ammonia to hydroxylamine’ and ‘hydroxylamine to nitrite’. This example illustrates that some KOs (e.g. hydroxylamine oxidoreductase) may be found in more than one FOAM category, as gene products may be involved in more than one biochemical pathway.
Table 1. The current FOAM database is made of 73 969 HMMs designed to target 2870 different Kos.
| #HMM | #KO | #hmm/KO | |
|---|---|---|---|
| 01_Fermentation | 1342.5 | 173 | 7.76 |
| 02_Homoacetogenesis | 336 | 118 | 2.85 |
| 03_Superpathway of thiosulfate metabolism | 36 | 7 | 5.14 |
| 04_Utililization of sugar, conversion of pentose to EMP pathway intermediates | 100.5 | 14 | 7.18 |
| 05_Fatty acid oxidation | 1179.5 | 41 | 28.77 |
| 06_Amino acid utilization biosynthesis metabolism | 7773 | 805 | 9.66 |
| 07_Nucleic acid metabolism | 2734 | 288 | 9.49 |
| 08_Hydrocarbon degradation | 1415.5 | 85 | 16.65 |
| 09_Carbohydrate Active enzyme (CAZy) | 2305.5 | 305 | 7.56 |
| 10_TCA cycle | 478.5 | 35 | 13.67 |
| 11_Nitrogen cycle | 217.5 | 52 | 4.18 |
| 12_Transporters | 0.5 | 543 | 0.00 |
| 13_Hydrogen metabolism | 194.5 | 16 | 12.16 |
| 14_Methanogenesis | 524.5 | 57 | 9.20 |
| 15_Methylotrophy | 238 | 69 | 3.45 |
| 16_Embden Meyerhof-Parnos (EMP) | 209 | 35 | 5.97 |
| 17_Gluconeogenesis | 258 | 28 | 9.21 |
| 18_Sulfur metabolism | 35.5 | 33 | 1.08 |
| 19_Synthesis of saccharides and deriviatives | 2015.5 | 419 | 4.81 |
| 20_Polymers hydrolysis | 2740 | 358 | 7.65 |
| 21_Cellular response to stress | 11647 | 825 | 14.12 |
| Total: 35781 | Different KOs: 2870 |
On average, an HMM is made from an alignment of 81 peptide sequences and about 26 HMMs are built per KO. The file size is ∼7 GB.
Database construction
Approximately 74 000 HMMs were generated and distributed into either a single file, or 21 HMMer (19) formatted files, where each HMM corresponded to the most refined functional level in the hierarchical FOAM ontology. To generate models, we developed a pipeline, triggered for each KO referenced in FOAM. Then, based on KEGG assignment information, the system retrieved all combinations of Pfams assigned to a KO. From those combinations, sequences in the IMG (version 4) protein database containing the Pfams were retrieved; including protein sequences annotated from microbial genomes and metagenomes. This process enriched preexisting Pfams with additional sequence information (particularly metagenomic sequences). The sequences were aligned with Pfam models using hmmalign (part of HMMer 3 suite) and recurring gaps were removed after this step. At this stage, we gained a number of protein profiles (defined as an alignment or model that represents a group of sequences which serves for comparison) per KO equal to retrieved Pfams. An individual Pfam usually has multiple-KO assignments, making them less specific. Therefore, profiles were grafted together into one alignment and a Maximum Likelihood tree was built through the FastTree program (24) to identify clades on the topology consistent with sequence KO annotation—here called clusters (Figure 1). Grafting alignments is relevant because some sequences may be present in several Pfams, linking them with other sequences in a different Pfam (see Figure 1 step 5 and Venn diagram in Supplementary Data 2).
Detection of KO consistent branches was done by browsing the tree from the leaves to ascendant nodes provided that the subtree gathered leaves with only the same KO assignment. Once completed, the subtree was excised and the algorithm restarted from the remaining tree. A consistent subtree was defined as the largest subtree that has a single KO annotation. Due to the nature of the KO annotation it is possible that some genes may have had more than one KO assigned to them. In that case, we still accepted them if they had the common KO of the subtree. Furthermore, each KO was mapped to EC numbers and a consistent subtree may have all genes assigned to the same EC number, or the same first three digits of the EC number. In any of the above scenarios, we reported the members of the subtree and their corresponding annotation (common KO or EC number). A caveat of this approach is that we did not consider the quality of the tree in the tree-splitting step (i.e. weakly supported branches were equally treated as strongly supported ones), producing models of different qualities. Nevertheless, we decided that the approach of rational classification is better than no classification at all. In the future, the groups could be recomputed, or split more optimally when more data become available (e.g. more KOs). From each cluster related to the KO in process, we extracted the alignment from which HMMs were eventually built.
RESULTS
Validation
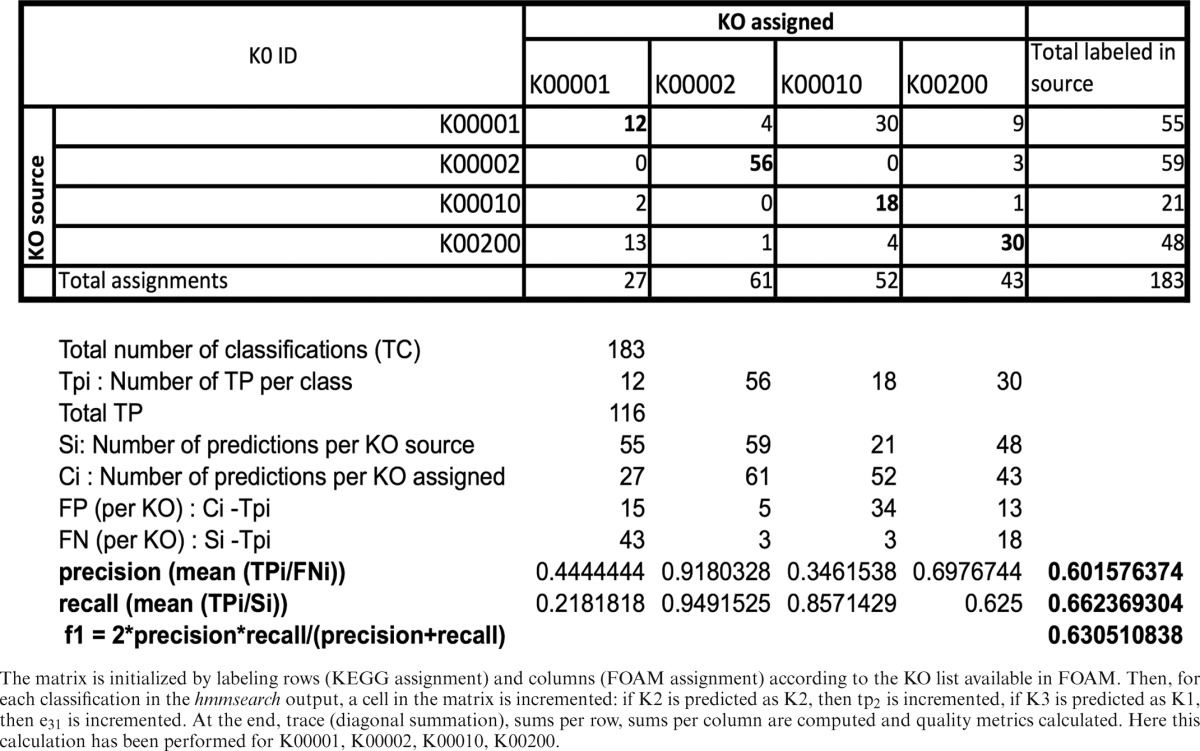
As FOAM was constructed entirely from protein sequences in Pfam and in JGI-IMG without sourcing KEGG peptides, we used KEGG peptides for validation. Because each KEGG sequence has a KO assignment, resulting comparisons were relatively straightforward. We also compared FOAM HMMs to UNIPROT—Swissprot (25), but this required searching for files generated by KEGG to make the link between KEGG and Swissprot IDs. Therefore, validation of HMMs in FOAM included all KEGG protein sequences (with genuine KO assignments) and those in Swissprot. To accomplish this, the hmmsearch program was launched and the best hits with a ‘per domain’ score >25 were kept. In order to compute quality metrics, we had to address a multi-class problem because each item (here sequence) had to be labeled with one of more than two KOs. To do so, we built a ‘confusion matrix’ from which the validation metrics were derived (Supplementary Table S1). As each FOAM HMM targets one or multiple KOs, or each classified sequence, we considered the best hit as a true positive if at least one KO assigned by KEGG corresponded to one predicted KO (an example of how a confusion matrix is filled and metrics are computed is given in Table 2).
Table 2. Example of confusion matrix construction for database validation.
Validation results are shown in Figure 2 and indicate good FOAM classification performance for all indices. Indeed, at the KO level (the most refined classification level) precision reached 92% in both tests; with 82% (Swissprot test) or 69% (KEGG peptides test) sensitivity. Notably, a majority of misclassifications at the KO level were assigned to very close KOs (in terms of ID which usually means functional similarity) explaining the jump in values when KOs are gathered in higher classes starting at FOAM ontogeny level 4 which normally gathers several very close KOs.
Figure 2.

Validation results. For each of the five functional levels available in the FOAM ontology, three metrics were computed: recall (or sensitivity), precision (known also as ‘positive predictive value’ or sometime ‘specificity’) and F1-score (the harmonic mean of both). In all cases, precision stays >92% at the KO level to reach 97% at level 1 (21 classes). Recall varies much more, from 69% at the KO level, to 98% at level 1. Levels 2, 3 and 4 gave similar performance results for both recall and precision; and their F1-score ‘mean’ within a range of 92–94%.
As a specific example, we extracted the data from the validation we performed for K02588, which contains the nifH gene, and is known to be problematic (many false positives) due to its domain architecture. The Swissprot database contains 70 proteins annotated as K02588. Running the HMMs, we were able to classify 68 of these correctly, 0 were incorrect, and 2 were false negatives; thus illustrating the power of FOAM.
DISCUSSION
In the new era of metagenomics, faced with the growth of large databases, increasing numbers of sequencing projects, and consequently large sequence datasets, we identified a need for a functional gene database that covers the diversity of known microbial metabolic processes; in particular for environmental metagenomes. The resulting FOAM database that we present here has many strengths for the user including core function models, i.e. FOAM HMMs tailored for KO assignments, and an organizational structure based on biochemical cycles with environmental relevance. The resulting HMM construction pipeline has several strengths, including: (i) improved specificity for classifying peptides or translated reads/contigs into functions because sequences were selected according to phylogenetic analyses to become KO-specific classifiers and (ii) increased sensitivity that is crucial when dealing with diverse and underexplored environments (e.g. soil). The KEGG KO was chosen as the FOAM ‘unit’ because it is a dynamically maintained knowledge base associated with a rich tool set available within or outside of KEGG (KEGG-ML, GLAMM, iPath, Voronto, etc.). That being said, FOAM does not aim to replace other existing systems, such as MG-RAST, IMG/M, KEGG genes, KO, Phylofacts (FAT-CAT) (26,27), or Pfam, for metagenome annotation. Rather, FOAM provides the community with a tool for precise functional annotations even when distant homologs have been obtained by shotgun metagenomic sequencing.
We also defined a large set of environmentally important functions for gene mining, which we organized into 5 levels of resolution. This enables the user to focus on specific functional groups by preselecting the relevant models or by organizing the results into classified functional families. As an example, we annotated two soil metagenomic datasets after quality trimming/filtering and assembly using Velvet. The coding regions were then searched from contigs using Prodigal (in mode ‘meta’) and HMMer (19) run against the HMMs from FOAM CarboActive enzyme ontology group. The first metagenome, from a prairie soil, was ∼300 Gbp, assembled in 5 901 346 contigs (N50: 609 bp), producing 7 716 071 ORFs. The second metagenome, from adjacent cultivated soil, was ∼200 Gb, assembled in 4 592 072 contigs (N50: 548 bp) with 6 059 007 ORFs. The annotation of these two datasets took ∼200 cpu hours. In another example, we used the whole FOAM database to annotate 6 657 648 Miseq reads (∼75 bp in length) from permafrost. The sequences were translated into all six reading frames and the translated file was run through hmmsearch on a Dell PowerEdge R910 with 40 cores and 1 TB of RAM. The hmmsearch devoted 5 cpus on average at any given time and this search took ∼55 h to run, i.e. ∼275 cpu hours. These two examples demonstrate that FOAM is an appropriate tool for characterization and comparison of different functional genes in large and highly complex environmental metagenome datasets.
In summary, analysis of current environmental metagenomes is often challenging due to the high diversity and large proportion of uncharacterized microbial taxa in most environmental habitats. Such data require more sensitive tools to identify distant homologs while minimizing computational cost. The FOAM database represents a useful and expedient tool for informative analyses of these data. The assignment sensitivity was increased with HMMs that were trained from sequence alignment profiles, and use of HMMer 3.0 provided model queries that were as expedient as BLAST (28,29). Additionally, FOAM allows easy narrowing of a search to specific target functional categories. As each model is trained with an average of 80 sequences, FOAM screens a dataset with a selective database ∼80× faster than BLAST to test the same content, while resulting in a higher sensitivity.
Deliveries (Results)
HMM files and ontology are available here: http://portal.nersc.gov/project/m1317/FOAM/
FOAM (the ontology)
The ontology is available as a tab-separated values file.
FOAM (the HMMs)
FOAM is currently made of 73 969 HMMs built to classify into 2870 different KO (about 25 HMMs per KO on average). A brief description of FOAM statistics is illustrated in Table 1.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Olivia Mason for her help and her advice in organizing the database, as well as Vanessa Sochat for her assistance with MetaCyc analyses and the website.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be considered as Joint First Authors.
FUNDING
DOE-BER, (US Department of Energy - Biological and Environmental Research) Biological Systems Research on the Role of Microbial Communities in Carbon Cycling Program [DE-SC0004953]; US DOE [DE/AC02-05CH11231]. Funding for open access charge: DOE-BER [DE-SC0004953].
Conflict of interest statement. None declared.
REFERENCES
- 1.Human Microbiome Jumpstart Reference Strains Consortium. Nelson K.E., Weinstock G.M., Highlander S.K., Worley K.C., Creasy H.H., Wortman J.R., Rusch D.B., Mitreva M., Sodergren E., et al. A catalog of reference genomes from the human microbiome. Science. 2010;328:994–999. doi: 10.1126/science.1183605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qin J., Li R., Raes J., Arumugam M., Burgdorf K.S., Manichanh C., Nielsen T., Pons N., Levenez F., Yamada T., et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jansson J.K. Towards ‘Tera-Terra’: Terabase sequencing of terrestrial metagenomes. Microbe. 2011;6:309–315. [Google Scholar]
- 4.Mackelprang R., Waldrop M.P., DeAngelis K.M., David M.M., Chavarria K.L., Blazewicz S.J., Rubin E.M., Jansson J.K. Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature. 2011;480:368–371. doi: 10.1038/nature10576. [DOI] [PubMed] [Google Scholar]
- 5.Luo C., Rodriguez-R L.M., Johnston E.R., Wu L., Cheng L., Xue K., Tu Q., Deng Y., He Z., Shi J.Z. Soil microbial community responses to a decade of warming as revealed by comparative metagenomics. Appl. Environ. Microbiol. 2013;37:12–13. doi: 10.1128/AEM.03712-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mason O.U., Hazen T.C., Borglin S., Chain P.S.G., Dubinsky E.A., Fortney J.L., Han J., Holman H.-Y.N., Hultman J., Lamendella R., et al. Metagenome, metatranscriptome and single-cell sequencing reveal microbial response to Deepwater Horizon oil spill. ISME J. 2012;6:1715–1727. doi: 10.1038/ismej.2012.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Elsas J.D., Costa R., Jansson J., Sjöling S., Bailey M., Nalin R., Vogel T.M., van Overbeek L. The metagenomics of disease-suppressive soils – experiences from the METACONTROL project. Trends Biotechnol. 2008;26:591–601. doi: 10.1016/j.tibtech.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 8.DeAngelis K.M., D'haeseleer P., Chivian D., Simmons B., Arkin A.P., Mavromatis K., Malfatti S., Tringe S., Hazen T.C. Metagenomes of tropical soil-derived anaerobic switchgrass-adapted consortia with and without iron. Stand. Genomic Sci. 2013;7:382–398. doi: 10.4056/sigs.3377516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jansson J.K., Neufeld J.D., Moran M.A., Gilbert J.A. Omics for understanding microbial functional dynamics. Environ. Microbiol. 2012;14:1–3. doi: 10.1111/j.1462-2920.2011.02518.x. [DOI] [PubMed] [Google Scholar]
- 10.Pell J., Hintze A., Canino-Koning R., Howe A., Tiedje J.M., Brown C.T. Scaling metagenome sequence assembly with probabilistic de Bruijn graphs. PNAS. 2011;109:13272–13277. doi: 10.1073/pnas.1121464109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ashburner M.M., Ball C.A.C., Blake J.A.J., Botstein D.D., Butler H.H., Cherry J.M.J., Davis A.P.A., Dolinski K.K., Dwight S.S.S., Eppig J.T.J., et al. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kanehisa M., Araki M., Goto S., Hattori M., Hirakawa M., Itoh M., Katayama T., Kawashima S., Okuda S., Tokimatsu T., et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:D480–D484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas T.T., Gilbert J.J., Meyer F. Metagenomics - a guide from sampling to data analysis. Microb. Inform. Exp. 2011;2:3. doi: 10.1186/2042-5783-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bates J.T., Chivian D., Arkin A.P. GLAMM: genome-linked application for metabolic maps. Nucleic Acids Res. 2011;39:W400–W405. doi: 10.1093/nar/gkr433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santamaria R., Pierre P. Voronto: mapper for expression data to ontologies. Bioinformatics. 2012;28:2281–2282. doi: 10.1093/bioinformatics/bts428. [DOI] [PubMed] [Google Scholar]
- 17.Yamada T., Letunic I., Okuda S., Kanehisa M., Bork P. iPath2.0: interactive pathway explorer. Nucleic Acids Res. 2011;39:W412–W415. doi: 10.1093/nar/gkr313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luo W., Brouwer C. Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics. 2013;29:1830–1831. doi: 10.1093/bioinformatics/btt285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eddy S.R.S. Accelerated profile HMM searches. PLoS Comput. Biol. 2011;7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finn R.D., Mistry J., Tate J., Coggill P., Heger A., Pollington J.E., Gavin O.L., Gunasekaran P., Ceric G., Forslund K. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haft D.H., Haft D.H., Selengut J.D., Selengut J.D., White O., White O. The TIGRFAMs database of protein families. Nucleic Acids Res. 2003;31:371–373. doi: 10.1093/nar/gkg128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fish J.A., Chai B., Wang Q., Sun Y., Brown C.T., Tiedje J.M., Cole J.R. FunGene: the functional gene pipeline and repository. Front. Microbiol. 2013;4:1–14. doi: 10.3389/fmicb.2013.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim B.H., Gadd G.M. Bacterial Physiology and Metabolism. Cambridge: Cambridge University Press; 2008. [Google Scholar]
- 24.Price M.N., Dehal P.S., Arkin A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.UniProt Consortium. The universal protein resource (UniProt) Nucleic Acids Res. 2008;36:D190–D195. doi: 10.1093/nar/gkm895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krishnamurthy N., Brown D.P., Kirshner D., Sjölander K. PhyloFacts: an online structural phylogenomic encyclopedia for protein functional and structural classification. Genome Biol. 2006;7:1–17. doi: 10.1186/gb-2006-7-9-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Afrasiabi C., Samad B., Dineen D., Meacham C., Sjölander K. The PhyloFacts FAT-CAT web server: ortholog identification and function prediction using fast approximate tree classification. Nucleic Acids Res. 2013;41:W242–W248. doi: 10.1093/nar/gkt399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 29.Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T.L. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:1–9. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.