


Abstract
Development of single-molecule localization microscopy techniques has allowed nanometre scale localization accuracy inside cells, permitting the resolution of ultra-fine cell structure and the elucidation of crucial molecular mechanisms. Application of these methodologies to understanding processes underlying DNA replication and repair has been limited to defined in vitro biochemical analysis and prokaryotic cells. In order to expand these techniques to eukaryotic systems, we have further developed a photo-activated localization microscopy-based method to directly visualize DNA-associated proteins in unfixed eukaryotic cells. We demonstrate that motion blurring of fluorescence due to protein diffusivity can be used to selectively image the DNA-bound population of proteins. We designed and tested a simple methodology and show that it can be used to detect changes in DNA binding of a replicative helicase subunit, Mcm4, and the replication sliding clamp, PCNA, between different stages of the cell cycle and between distinct genetic backgrounds.
INTRODUCTION
The development of single-molecule localization microscopy (SMLM), in particular for super-resolution imaging, has allowed researchers to visualize biological processes occurring at a scale below the diffraction limit. Single-molecule-based super-resolution techniques such as photo-activated localization microscopy (PALM) (1), fluorescence PALM (2) and stochastic optical reconstruction microscopy (3) temporally separate the fluorescence emission of photoactivatable or photoswitchable fluorescent probes. Fluorophore density can be controlled by iterative photoswitching of spatially isolated single emitters, which allows for high precision localization of each activated fluorophore by fitting individual point-spread functions (PSFs) (4). The resulting localizations can subsequently be reconstructed into a pointillist image with a spatial resolution greater than 10 times that of wide-field microscopy [reviewed in (5)].
To date, the majority of SMLM studies have focussed on problems paralleled in structural biochemistry including: visualizing well-defined structures such as actin fibres (6), characterizing protein complexes, where the stoichiometry is already known [for example nuclear pores (7)], and estimating protein copy number in diffraction-limited foci (8). The application of single-molecule microscopy to understanding phenomena which do not possess any ordered structure has largely been confined to prokaryotes, exploiting their physical dimensions with techniques such as total internal reflection fluorescence microscopy (TIRF). Prokaryotic cells typically have a small axial size, allowing TIRF or ‘near-TIRF’ (where illumination is at a subcritical angle and provides a thin sheet of excitation light) to directly image the whole cell (9).
Fields of study that have particularly benefited from the use of SMLM in bacterial systems include DNA replication and DNA repair [reviewed in (10)]. Studies to date have largely concentrated on the visualization of individual replication events and the quantification of the numbers of specific proteins present at each fork [e.g. (11,12)] as well as residence times of DNA repair proteins on DNA during repair (13). Although the advantages of SMLM approaches are clear, research in eukaryotic cells, particularly within the replication and repair fields, is less well developed. In part, this is due to the lack of defined techniques to overcome the problems associated with a greater depth of field (e.g. the greater depth of cells results in increased background fluorescence from out of focus fluorophores). Since eukaryotic model organisms share greater homology of protein structure, organization and function with humans than prokaryotes do, it is clear that SMLM methodologies that allow exploitation of equivalent SMLM approaches to eukaryotic model systems need to be developed.
Schizosaccharomyces pombe is a tractable model eukaryote that is frequently used to characterize DNA replication and repair processes. It provides researchers with the ability to perform sophisticated genetic experiments with the relative technical ease of a single-cell organism while being more closely related to humans than prokaryotes. Therefore, we have developed SMLM methodologies to study DNA replication and repair proteins in S. pombe. Here we present a simple method using PALM to directly observe DNA-association characteristics of proteins inside cells. This methodology does not require chemical fixation and allows users to quantify relative difference in DNA association of proteins in cells during different cycle stages as well as in distinct genetic backgrounds.
MATERIALS AND METHODS
S. pombe strain construction
We used recombination-mediated cassette exchange (RMCE) (14) to introduce copies of the pcn1 [proliferating cell nuclear antigen (PCNA)] and cdc21 (Mcm4) genes fused to the photoactivatable fluorophore mEos3.1. pcn1 strains were created using the essential gene replacement strategy (14): a pcn1 ‘base strain’ was constructed by inserting the loxP site upstream of the pcn1 start codon and ura4+-loxM3 directly downstream of the pcn1 stop codon. To insert the loxP site, Phusion DNA Polymerase (New England Biolabs) [used for all subsequent polymerase chain reaction (PCR) reactions unless stated otherwise] was used to amplify the loxP-ura4+-loxP cassette from plasmid template pAW41 (14) using primers P1 and P2. The resulting PCR fragment has ura4+ flanked by loxP sites between 80-bp stretches of sequence homologous to the pcn1 locus 927 bp upstream of the pcn1 start codon. This was used to transform S. pombe strain AW310 (h− ura4-D18, leu1-32) (15). Following transformation, cells were directly plated onto amino acids adenine (EMM) supplemented with EMM and leucine (EMM+L) plates and grown at 30°C for 4–5 days until colonies appeared. Transformants were restreaked onto fresh EMM+L plates.
To remove the ura4+ marker, cells were transformed with the Cre-recombinase expressing plasmid pAW5 (14), plated onto EMM plates supplemented with uracil (EMM+U) and grown at 30°C until colonies appeared. Transformants were restreaked onto fresh EMM+U and subsequently grown in liquid YE media at 30°C overnight to saturation (∼5 × 107 cells/ml). Five hundred cells were plated on Yeast Extract Agar (YEA) containing 5-fluoroorotic acid, Formedium (5-FOA) (YEA 5-FOA) to select for uracil auxotrophic cells. Plates were replica plated onto EMM+U to identify 5-FOA resistant leu− colonies, which have lost the Cre expressing plasmid pAW5.
A second integration introducing the ura4+ marker and loxM3 site downstream of the stop codon was then performed. The ura4+-loxM3 cassette from plasmid template pAW12 (14) was amplified using primers P3 and P4. The resulting PCR fragment was flanked with 80-bp stretches homologous to sequences directly downstream of the pcn1 stop codon and was used to transform into cells. After transformation, cells were directly plated onto EMM+L+A plates and grown at 30°C for 4–5 days to allow colony growth. Transformants were restreaked onto fresh EMM+L+A plates. The resulting pcn1 base strain TJE47 (h− loxP:pcn1:ura4:loxM3, leu1-32, ura4-D18) was confirmed by PCR and sequencing. To create the mEos3.1-pcn1 construct, the pcn1 Open Reading Frame (ORF) and 927bp of upstream sequence was amplified from total S. pombe genomic DNA using primers P5 and P6. The product was then cloned into SphI/SpeI restricted plasmid pGEM5fz to create pGEM5fz-pcn1 and the insert confirmed by sequencing. In order to N-terminally tag Pcn1 with the mEos3.1 protein, we introduced a BamHI restriction site directly upstream of the pcn1 start codon using single-site mutational PCR kit (Stratagene). An mEos3.1 coding sequence was commercially synthesized that was codon-optimized for S. pombe (16), encoded a C-terminal poly-threonine-glycine-serine linker and was flanked by BamHI restriction sites (Genscript). The sequence was subcloned into BamHI restricted pGEM5fz-pcn1 to create pGEM5fz-mEos3.1-pcn1. The mEos3.1-pcn1 construct was then finally subcloned from pGEM5fz-pcn1-mEos3.1 into the Cre-expression plasmid pAW8 (14)to create pAW8-pcn1-mEos3.1.
Pcn1PCNA has been previously N-terminally tagged in S. pombe and was shown to be functional when combined with an untagged copy expressed at the same level (17). To allow comparisons with previous literature, we therefore made a similar strain expressing both tagged and untagged versions of Pcn1. To introduce an untagged copy of the pcn1 gene we used a his3 targeting plasmid (18). The promoter-pcn1 fragment from plasmid pAW8-pcn1 was PCR amplified by primers P7 and P8. The resulting fragment and plasmid pHIS3K were restricted with SphI and SalI enzymes before the fragment was ligated into the vector to create plasmid pHIS3-pcn1. Plasmid pHIS3-pcn1 was linearized with NotI and transformed into the pcn1 base strain (TJE47). Cells were grown on YEA plates before being replica plated onto YEA+hygromycin (200 μg/ml) (Melford) to select for successful transformants. Integration was further confirmed by patching cells on EMM+U+L plates to check for histidine auxotrophy. Cells that were hygromycin resistant and histidine auxotrophic generated strain TJE177 (loxP-pcn1-ura4+-loxM3, his3::pHIS3-pcn1-HPHMX6, leu1-32, ura4-D18).
To introduce the mEos3.1-pcn1 construct into cells, we performed Cre-mediated cassette exchange (14) using the plasmid pAW8-mEos3.1-pcn1 to transform strain TJE177. Following transformation, cells were plated onto EMM+U plates and grown at 30°C until colonies appeared. Colonies were restreaked onto fresh EMM+U and then grown in YE media at 30°C overnight to saturation (∼5 × 107 cells/ml) and 500 cells plated on YEA 5-FOA to select for uracil auxotrophic cells. Plates were replica plated onto EMM+U to identify 5-FOA-resistant leucine auxotrophic colonies that have lost the pAW8-mEos3.1-pcn1 plasmid.
The mEos3.1:pcn1 elg1Δ strain was created by replacing the elg1 ORF with the natMX6 cassette. The natMX6 cassette was PCR amplified from plasmid pFA6-natMX6 (19) using primers P9 and P10 and transformed into the pcn1 base strain TJE177. Successful transformants (strain TJE214) were selected for by growing cells on YEA+NAT (Nourseothricin, 100 μg/ml) plates. The mEos3.1-pcn1 construct was then introduced by Cre-mediated recombination as described earlier to generate strain (TJE214: mEos3-1:pcn1 leu1-32, ura4-D18, ade6-704 elg1::natMX6).
Cells expressing Mcm4 protein C-terminally tagged with mEos3.1 were made by C-terminally tagging RMCE method (14). The C-terminal tagging base strain was a gift from Dr Izumi Miyabe. This was transformed with plasmid pAW8ENdeI-mEos3.1. Following transformation, cells were plated onto EMM+U+A and plated on YEA 5-FOA as previously described. Cells that had lost the plasmid were streaked to single colonies to create strain TJE148 (cdc21:loxP:mEos3.1:loxM3, leu1-32, ura4-D18, ade6-704).
PALM microscope
S. pombe cells with mEos3.1-tagged proteins were imaged with a custom-built inverted microscope (Olympus IX71) fitted with a motorized stage (Prior H117E1I4), using a 561-nm imaging laser (Cobolt, Jive) and a 405-nm activation laser (LaserBoxx, Oxxius). Each laser line displayed a quarter-wave plate (Thorlabs WPQ05M-405 and -561) and a low pass filter (Semrock FF01-417/60-25 and FF01-561/14-25). Both laser beams were expanded and collimated with a custom-built beam expander constituted of two matching lenses (Thorlabs LC1975 and LA1986), and coupled using a dichroic mirror (Semrock FF552-Di02-25). The resulting beams were focused to the back focal plane of an apochromatic 1.45 NA, 60× TIRF objective (Olympus, UIS2 APON 60× OTIRF) using a coated plane convex lens (Thorlabs LA1253-A). A multi-band dichroic mirror (Semrock Di01-R405/488/561/635-25 36), a band-pass filter (Semrock FF01-580/14-25) and a longpass filter (Semrock BLP02-561R-25) were used to separate fluorescence signal from the laser emission. The emission beam was further enlarged by a 2.5 beam expander, leading to an optimized pixel size of 107 nm/pixel after projection onto the EMCCD camera (Photometrics Evolve 512).
Sample preparation
Fission yeast cells were cultured overnight at 30°C in EMM2 minimal media supplemented with adenine, leucine, histidine and uracil. To enrich for either G2 or S-phase cells, cultures were synchronized by lactose gradients to separate out G2 cells, which were either directly processed for imaging or resuspended in YE media and incubated at 30°C for 120 min to acquire S-phase cells. For mEos3.1:pcn1 strains, both G2 and S-phase samples were treated with sodium azide (final concentration of 1 mg/ml) for 5 min in order to kill cells and prevent any unwanted adenosine triphosphate (ATP)-dependent DNA loading/unloading reactions. After incubation, cells were then washed three times and resuspended in ice cold phosphate buffered saline. For mcm4-mEos3.1 strain, S-phase cells were incubated in YE + 10-mM hydroxyurea (Sigma) in order to arrest replication. Directly prior to imaging cells were placed on a 1% (w/v) agarose pad and sandwiched between two ozonated coverslips that were then sealed with paraffin wax.
Image acquisition
For experiments in Figures 1B and 2A, mEos3.1 fluorophores were activated with sequential pulses of the 405-nm laser at power densities ranging from 0.1 to 10 W/cm2, adjusted manually to ensure single-molecule activation. Each activation cycle was followed by imaging of the sample using the 561-nm laser under incident intensities of the order of 1 kW/cm2. Iterative cycling continued until no further in-focus nuclear fluorescence could be detected. Samples for Figures 3 and 4 were imaged using continuous wave illumination of 405 nm (0.1–1 W/cm2) and 561-nm (1 kW/cm2) laser light, in order to reduce experiment duration. Control experiments showed no significant difference in the total number of localization using the pulsed activation method or the continuous wave approach (data not shown). All experiments were carried out at 17.0 ± 0.5°C. The typical number of frames (350 ns): 6000–11 000.
Figure 1.
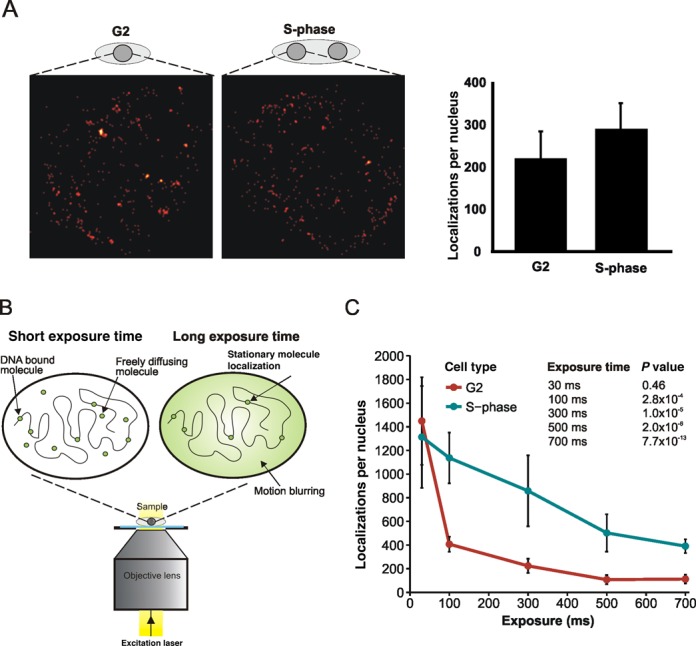
Selecting for DNA-bound proteins using long exposure times. (A) Fluorescent localization images of Mcm4-mEos3.1 fusion proteins visualized in G2 and S-phase nuclei in formaldehyde-fixed fission yeast. Cells were harvested from lactose gradient synchronization and fixed in 1% formaldehyde. There are an average of 219 localizations in G2 and 287 in S-phase cells (a 1.3-fold increase), which likely reflects the fact that replication genes are transcriptionally induced at the G1/S boundary. Differences in the number of localizations per nucleus do not reflect proteins that are DNA bound in this form of analysis. (B) Schematic representation of the effect of exposure time on detection of single molecules in unfixed cells. Left: at short exposure times, molecules from both the DNA bound and freely diffusing subpopulations are detected. Right: at long exposure times, fluorescence from diffusing molecules is dispersed due to motion blurring. Molecules bound to DNA are relatively static and fluorescence remains concentrated, allowing the molecule to be localized by Gaussian algorithms. (C) Numbers of Mcm4-mEos3.1 fluorescent localizations per nucleus decreases as a function of exposure time. G2 and S-phase cells obtained from lactose gradient synchronization were imaged on agarose pads at different exposure times. Raw data were processed with 2D-Gaussian fitting routines and the number of localizations per nucleus of in-focus nuclei was counted. A minimum of six nuclei were analysed per data point. Error bars: standard deviation. P values for each exposure time relate to the comparison between S and G2 phase.
Figure 2.
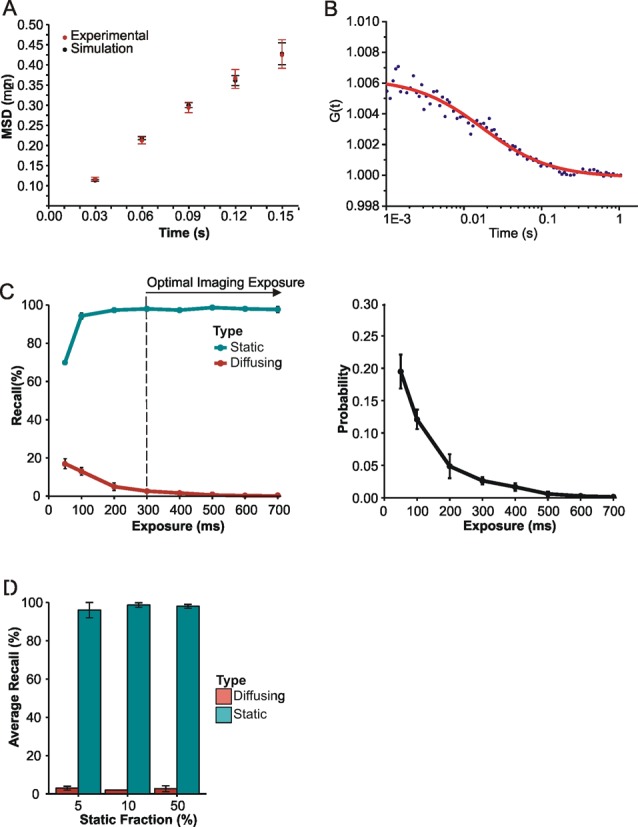
Simulating nuclear protein diffusion to optimize exposure time. (A) Mean square displacement (+/− SE) of Mcm4-mEos3.1 proteins (Red). Linear fitting of the first four points of the curve leads to the determination of an apparent diffusion coefficient (D* = 0.7 μm2/s) that is underestimated because proteins diffuse in three dimension inside the nucleus. Simulations of 2D mean squared displacements from 3D diffusing molecules inside a sphere of radius 1 μm were performed for diffusion coefficients ranging from 1.5 to 2 μm2/s (n > 1000 tracks per simulation). The best correspondence was obtained for D = 1.7 ± 0.3 μm2/s (Black). (B) Representative FCS curve obtained in vivo in S. pombe cells with Mcm4 tagged with eGFP. FCS experiments were performed in the nuclei of four different cells, leading to an average diffusion coefficient of 1.6 ± 0.4 μm2/s. (C) Diffusing (diffusion coefficient = 1.7 μm2/s) and static molecules were simulated with different exposure times and processed using our super-resolution fitting routines. The percentage recall of simulated molecules was determined by comparing simulation and Gaussian-fitted data sets. Error bars represent standard deviation calculated from three repeats. Recall percentages from simulations were used to estimate the probability of detecting diffusing rather than static molecules at different exposure times (diffusing/(diffusing+static)). Error bars represent standard deviations from three experiments. (D) Average recall analysis of mixed populations of simulated molecules at the optimal exposure time. The percentage of static molecules was altered to observe any effect of a higher proportion of diffusing species on recall of static molecules at 350-ms exposure time. Error bars represent standard deviation calculated from three independent repeats.
Figure 3.
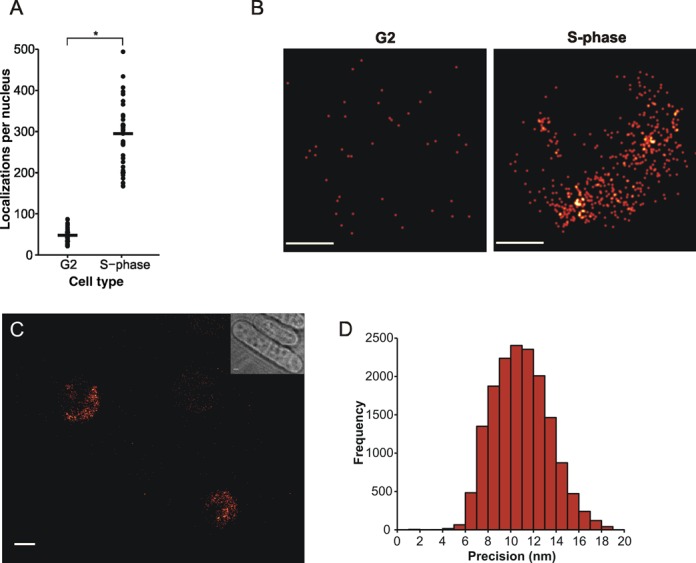
Visualization of DNA-bound Mcm4 in fission yeast. (A) Numbers of Mcm4-mEos3.1 fluorescent localizations per nucleus for both G2 and S-phase cells imaged at 350-ms exposure time using continuous wave (CW) activation. Raw data were processed with 2D-Gaussian fitting routines to score total localizations. G2 cells (n = 36) were harvested from lactose gradients and either directly imaged or cultured in YE media + 10-mM HU for 120 min to obtain S-phase cells (n = 39). Black solid line indicates median value (G2 = 47, S-phase = 295) *P = 1.5 × 10−21. (B) Typical Mcm4-mEos3.1 reconstructed nuclear localization patterns in both G2 and S-phase cells. Reconstructed PSFs were plotted with an experimental average width of 11 nm, the calculated average accuracy. Scale bar represents 0.5 μm. (C) Reconstructed localization image of G2 and S-phase fission yeast cells expressing Mcm4-mEos3.1. Cells were obtained from lactose gradient synchronization and the two cell types mixed and imaged on the same agarose pad. Similar localization patterning can be seen compared to when cell types were imaged separately. Scale bar represents 1 micron. (D) Representative histogram of mEos3.1 fluorescent localization precisions imaged at 350-ms exposure time (n = 16011, median = 10.83 nm).
Figure 4.
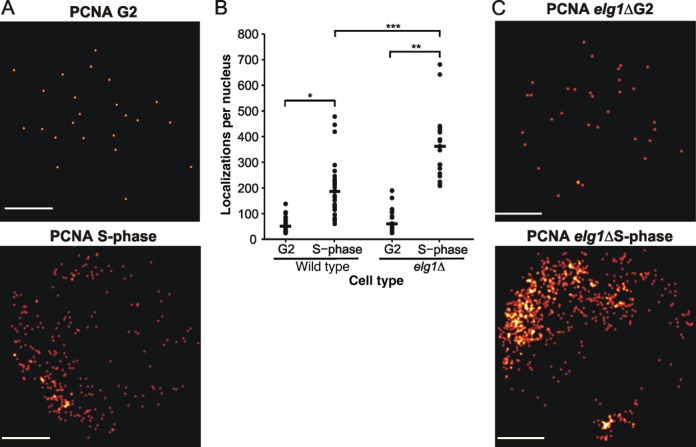
Visualization of DNA-bound PCNA in fission yeast. (A) Representative reconstructed images of mEos3.1-PCNA nuclear localization patterns in S-phase and G2 S. pombe cells. (B) Numbers of mEos3.1-PCNA fluorescent localizations per nucleus in elg1+ and elg1Δ cells imaged with an exposure time of 350 ms in either G2 or S-phase cell cycle stages (n = 19, 26, 21 and 20 for elg1+ G2, elg1+ S-phase, elg1Δ G2 and elg1Δ S-phase, respectively). Cells were treated with sodium azide to prevent ATP-dependent PCNA DNA unloading/loading during imaging. Horizontal bar represents median value (elg1+ G2 = 51, elg1+ S-phase = 187, elg1Δ G2 = 60, elg1Δ S-phase = 362). P-value determined by two-tailed students T-test, *P = 8.1 × 10−7, **P = 9.4 × 10−10 ***P = 5.9 × 10−5. (C) Representative reconstructed images of elg1+ and elg1Δ mEos3.1-PCNA nuclear localization patterns in S-phase cells.
Image analysis and reconstruction
Raw image data from experiments were processed using a custom ImageJ (http://rsb.info.nih.gov/ij/) 2D-Gaussian fitting routine (http://www.sussex.ac.uk/gdsc/intranet/microscopy/imagej/gdsc_plugins#install). Code available on GitHub: https://github.com/aherbert/GDSC-SMLM. Only single-molecule PSFs that were in focus and had a signal to background ratio >150 were retained. In order to quantify nuclear single-molecule localizations, binary images were produced where each localization was plotted as a single pixel and given a value of 1, pixels with no localizations had a value of 0. In order to identify nuclear regions maximum intensity projections of the raw data were produced and scaled to the same size as the binary images. Regions of interest (ROIs) were drawn around nuclear regions using ImageJ circular ROI selection tool. This ROI was then duplicated in exactly the same region on the binary image. The integrated density was then measured in the ROI, the value of which is the number of localizations detected in that area. Localizations were only counted in nuclei that were considered to be centred in the focal plane of the microscope, this was judged from maximum intensity projections. Localization images were reconstructed by plotting single-molecule positions with an average precision (20) of ∼11 nm.
Simulations
The simulation plugin uses an experimental 3D-PSF derived from imaging 20-nm Z-stacks of fluorescent beads (FluoSpheres® Polystyrene Microspheres, Life Technologies). The PSF stack was normalized using the total signal of the slice in the focal plane. The simulation creates an image by randomly positioning molecules and simulating fluorescent photon emissions and molecule diffusion over a specified time using configured intervals. Photons emitted per simulation step were rendered to pixels by selecting the appropriate slice from the PSF Z-stack and convolving the total photons with the PSF. Simulation steps were integrated to a specified output exposure time allowing diffusing molecules to move within one output frame. Each pixel was subjected to Poisson shot noise. Background noise, fluorophore intensity and blinking parameters were simulated to match experimental values observed under our optimized imaging conditions. All simulation software was written for the ImageJ program and is available from the link above.
Recall analysis
For each simulation a total of 500 molecules were simulated and randomly positioned in confined spherical regions with diameter of 2 microns in order to mimic the confinement of a fission yeast nucleus. Diffusing molecules were simulated in three dimensions with a depth of 2 microns, similar to the depth of a yeast cell. Static molecules were simulated in two dimensions within the confinement in order to mimic static molecules in a focal plane. Simulated data were fitted with our 2D Gaussian fitting routines and the results compared to the known simulation positions. Recall of single molecules was measured by calculating the percentage of molecules that had been correctly localized at least once within 50 nm of the true position. Analysis using the recall of all localizations showed similar results (data not shown).
Single-molecule tracking
S. pombe cells in G2 phase expressing Mcm4 labelled with mEos3.1 were harvested from lactose gradients and imaged with an acquisition time of 30 ms. Localization analysis was carried out on 8 nuclei using a custom-written plugin for ImageJ software. PSF candidates were identified by applying a threshold value of 40 nm on the precision of localization and 50 on the signal-to-noise estimate ratio, with a filter on the PSF width of two times the ‘in focus’ PSF width. Noise in the image was estimated by calculating the sum of the differences of each pixel with their four direct neighbours divided by 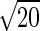 to form a pixel residual. The smallest half of the squared residuals (n) was then summed (sum) and used to estimate the noise as
to form a pixel residual. The smallest half of the squared residuals (n) was then summed (sum) and used to estimate the noise as 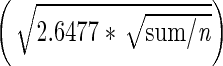 . This method provided a very stable noise estimate irrespective of the number of spots present in a given frame.
. This method provided a very stable noise estimate irrespective of the number of spots present in a given frame.
Peaks appearing in adjacent frames within a threshold distance of 800 nm were considered as belonging to the same molecular track. This choice of threshold ensured that the probability of detecting a molecule that has moved by a distance L during an acquisition time Δt of 0.03 s, given by (21): P(L, Δt) = 1−exp(−L2/(4DΔt)) is comprised between 93% and 99% for diffusion coefficients D ranging from 1 μm2/s to 2 μm2/s. Individual tracks of single diffusing proteins consisting in a minimum of four steps were retained for further diffusion analysis by calculating their mean square displacement (MSD). The MSD over all the trajectories collected at a lag time τ was computed relative to the first localization in the track ri(t), using the following expression:
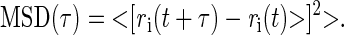 |
MSD analysis was used to determine an apparent diffusion coefficient (D* = 0.7 μm2/s) by linearly fitting the points associated with the first four lag times of the curve, following the relation 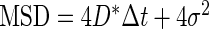 , where σ is the localization uncertainty (22). This diffusion coefficient is underestimated because of the confinement of the 3D motion of Mcm4 proteins inside the nucleus and motion blur (23). We therefore simulated a 3D Brownian motion inside a sphere of radius 1 μm, in order to retrieve a more accurate diffusion coefficient inside the nucleus. Individual tracks were generated by simulating random steps in 3D (100 steps for every frame simulated with an acquisition time of 30 ms) with a length taken from a Gaussian distribution, a mean of zero and a standard deviation of s, such as the step size in each dimension was
, where σ is the localization uncertainty (22). This diffusion coefficient is underestimated because of the confinement of the 3D motion of Mcm4 proteins inside the nucleus and motion blur (23). We therefore simulated a 3D Brownian motion inside a sphere of radius 1 μm, in order to retrieve a more accurate diffusion coefficient inside the nucleus. Individual tracks were generated by simulating random steps in 3D (100 steps for every frame simulated with an acquisition time of 30 ms) with a length taken from a Gaussian distribution, a mean of zero and a standard deviation of s, such as the step size in each dimension was 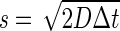 . The number of molecules in the field of view was adjusted to be suitable for single particle tracking analysis.
. The number of molecules in the field of view was adjusted to be suitable for single particle tracking analysis.
The range of diffusion coefficient simulated was comprised between 1.5 and 2 μm2/s, using an increment of 0.1 μm2/s. More than 2000 tracks were extracted for each simulated data set using exactly the same parameters as for experimental data. A diffusion coefficient of D = 1.7 ± 0.3 μm2/s for Mcm4-mEos3 proteins was found to match best the apparent diffusion coefficient, using a least square estimates method.
Fluorescence correlation spectroscopy
Mcm4-EGFP was preferred to mEos3.1 due to the better photostability of the former that was needed to obtain accurate autocorrelation curves (The Mcm4-EGFP strain was a gift from Meliti Skouteri). We assumed that there would be no significant changes in the diffusion coefficient of the fusion proteins because of the almost identical structures and molecular weights of the two fluorescent reporters. Fluorescence correlation spectroscopy (FCS) was performed on an inverted Nikon TE2000-E microscope. The excitation laser (473 nm, DHOM) was expanded using two matching lenses (Thorlabs LC1439 and LA1484) and an iris in order to slightly underfill the back aperture of the objective [1.40 NA, 100 × objective (Nikon, Plan-APO DICH)], which was demonstrated to provide the best Gaussian detection volume (24). The fluorescence emission was separated from the laser excitation using a dichroic mirror (Semrock FF495-Di02-25 × 36) and a bandpass filter (Semrock FF01-520/35-25) and was detected by an Avalance Photodiode detector (APD) (Perkin-Elmer SPCMAQR-14) linked to an autocorrelator (Flex2k, Autocorrelator.com).
For all experiments, microscope glass slides were carefully cleaned before use. No. 1 borosilicate coverslips were first ozonated for 30 min to remove any trace of autofluorescence. Cells were placed on a 1.5% (w/v) agarose pad placed between two ozonated coverslips sealed with paraffin wax. Experiments were carried out at 17.0 ± 0.5°C at a low excitation power of 0.45 μW at the sample, in order to reduce the photobleaching effect during the experiment.
A solution of 10 nM of commercial fluorescein (Aldrich) was used for the calibration of the detection volume. FCS experiments were performed at the power used in the cell experiment and the resulting curves were fitted by an equation of the type (25)
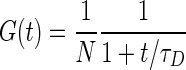 |
with N being the average number of molecule inside the detection volume and τD being the average diffusion time, linked to the lateral dimension of the detection volume wx,y and the diffusion coefficient D by the relation: 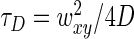 . The calibration experiment was repeated three times, leading to a determination of the lateral dimension of the detection volume wxy = 0.340 ± 0.004 μm, using a diffusion coefficient D = 4.25 ± 0.01 μm2/s for fluorescein (26) at 25°C, corrected according to the Stokes–Einstein relationship to D = 3.41 ± 0.01 μm2/s in order to account for the experimental temperature of 17.0 ± 0.5°C.
. The calibration experiment was repeated three times, leading to a determination of the lateral dimension of the detection volume wxy = 0.340 ± 0.004 μm, using a diffusion coefficient D = 4.25 ± 0.01 μm2/s for fluorescein (26) at 25°C, corrected according to the Stokes–Einstein relationship to D = 3.41 ± 0.01 μm2/s in order to account for the experimental temperature of 17.0 ± 0.5°C.
The laser beam was placed in the middle of four immobilized G2 cells and the signal intensity detected was recorded for 30 s. Resulting FCS curves were fitted using the model previously described. The average of the five associated diffusion coefficients determined leads to an estimate of the diffusion coefficient D = 1.6 ± 0.4 μm2/s.
RESULTS
Selective detection of DNA-associated proteins in unfixed fission yeast
Proteins involved in DNA metabolism are typically localized to the nucleus and many interact with DNA only transiently, e.g. at specific stages of the cell cycle or in response to DNA damage. Thus, in addition to DNA-associated molecules there is, at any given time, a considerable pool of diffusing molecules that are not specifically DNA associated. Fluorescence imaging protocols that involve chemical fixation immobilize all molecules, thus preventing the effective differentiation of DNA bound and freely diffusing species. For example, as shown in Figure 1A, the total population of Mcm4-mEos3 molecules are seen in both G2 and S-phase cells even though the G2 Mcm4-mEos3 is not chromatin associated. It is possible to extract the soluble (i.e. not chromatin associated) population of molecules from the cell, leaving only the chromatin-associated fraction (27). However, such methods are perturbative and, in our hands, time consuming, lack reproducibility and often lead to increased background fluorescence that is incompatible with SMLM. We thus chose to visualize the biologically active DNA-bound proteins in unfixed cells.
In order to distinguish DNA-bound proteins from unbound molecules, we targeted the difference in apparent diffusion speed: DNA-bound proteins will remain relatively static when compared to those that are diffusing. Our strategy was to utilize the effect of motion blurring (28) and perform PALM imaging using long exposure times (Figure 1B). Using extended exposure times allowed us to separate the fluorescent signal arising from diffusing and immobile populations: unbound proteins that are rapidly diffusing emit a fluorescent signal from multiple separated physical locations in the sample within the exposure time of each acquired frame. This results in a fluorescent signal detected that has a significantly different shape than the sharp PSF of a fixed molecule. Conversely, DNA-bound molecules are relatively static during the acquisition time of each frame, preventing motion blurring. Therefore, DNA-associated proteins can theoretically be selectively localized with a 2D Gaussian distribution to observed pixel intensities.
In order to test whether this approach could be used to selectively image DNA-bound proteins in S. pombe, we imaged unfixed cells expressing the replication helicase subunit, Mcm4, tagged with the photoconvertible fluorescent protein mEos3.1 at different exposure times and processed the raw data using a typical SMLM algorithm (the Materials and Methods section). The mini-chromosome-maintenance two–seven proteins form the heterohexameric replicative DNA helicase in eukaryotic cells. Replicative helicases associate with DNA just prior to S-phase and are tasked with unwinding the DNA duplex during replication [reviewed in (29)]. Upon completion of DNA replication (G2 phase) the replicative helicases disassociate from the DNA. We observed the average number of localizations in S-phase and G2 cells as a function of exposure time (Figure 1C). At short exposure times (∼30 ms) fluorescence from individual diffusing molecules is expected to appear as individual puncta and thus be indistinguishable from the static molecules. This would result in no discernable difference between cell cycle stage. Indeed, we observed no significant difference in the number of Mcm4-mEos3 molecules detected between S phase and G2 cells. As exposure time is increased however, the fluorescence from diffusing molecules is expected to become increasingly motion-blurred. In accordance with this expectation, a greater differentiation in the number of localizations between S phase and G2 cells was evident with increasing exposure times. This is consistent with there being more DNA-associated Mcm4 proteins in cells undergoing replication. The time for which single fluorophores were visualized formed an exponential distribution, with a median time of 40 ms and the 95th percentile of localizations falling at 97 ms. The decrease in the detection of bound molecules at higher exposure times is thus likely to be due to continued integration of background signal, limiting the localizations detected above background to a small population of long-lived fluorophores.
Modelling molecular diffusion for exposure time optimization
To computationally validate our observations and determine an optimum camera exposure time for visualizing DNA-bound Mcm4, we simulated a range of conditions. The starting point for simulations requires an accurate estimation of the Mcm4-mEos3.1 in vivo diffusion constant. Thus, we first estimated the diffusion coefficient inside the nucleus using single-particle tracking PALM (21) by following the motion of sparsely activated fluorescent proteins over time. Using a data set of 762 molecular tracks observed in eight S. pombe G2-phase nuclei (Figure 2A), we determined an apparent 2D diffusion coefficient of 0.7 μm2 s−1. To account for the systematic underestimation of the actual 3D diffusion coefficient (13,23), we simulated the diffusion of proteins in 3D (the Materials and Methods section) for a range of diffusion coefficients and empirically determined the best match with the experimental 2D data (D = 1.7 ± 0.3 μm2 s−1) (Figure 2A). This value was confirmed by an independent measurement using in vivo fluorescence correlation spectroscopy (D = 1.6 ± 0.4 μm2 s−1) (Figure 2B). D = 1.7 ± 0.3 μm2 s−1 was therefore used for subsequent simulations.
Observations of static and diffusing molecules that mimicked different exposure times were generated (the Materials and Methods section) and processed using our SMLM fitting algorithm. Subsequently, the recall percentage of molecules was determined for different exposure times (Figure 2C). The majority of 3D diffusing molecules were not recalled at exposure times >300 ms. The probability of detecting a diffusing molecule was typically <5%. Conversely, more than 95% of static molecules, simulated on a 2D plane and representing DNA-bound molecules in the focal depth of the objective lens, were recalled with exposure times >200 ms (Figure 2C). An optimal exposure time of 350 ms was chosen which enables the maximum discrimination between bound and unbound states with a robust P value (Figure 1C; inset) but without significant loss of the dynamic range that was experimentally observed at higher exposures times (Figure 1C). Finally, we modelled a variety of scenarios with different proportions of diffusing and static molecules in a mixed population to establish that mixed populations did not affect recall efficiency (Figure 2D).
Quantification of DNA-associated replication proteins in differing stages of the cell cycle and genetic backgrounds
At the optimal exposure time (350 ms) we were able to visualize a highly statistically significant difference (P = 1.5 × 10−21) in localizations of Mcm4-mEos3 between S-phase and G2 cells by comparing the total number of fluorescent localizations detected per nucleus (Figure 3A). This is fully consistent with the expectation that a proportion of the replicative helicase molecules are DNA associated in S-phase nuclei, but not in G2 nuclei. The spatial distribution of molecules can be visualized in reconstructed localization images (Figure 3B). We observed equivalent results when imaging G2 and S-phase cells simultaneously in the same field of view (Figure 3C). To ensure that our methodology was applicable to proteins other than Mcm4, we applied this approach to a second well-characterized DNA replication protein, PCNA. PCNA foci are often used as a marker of DNA synthesis in diffraction-limited microscopy experiments (17). Cells expressing an mEos3.1-tagged copy of Pcn1 (the PCNA orthologue in S. pombe) were imaged with the parameters previously applied to Mcm4. Comparing S-phase and G2 cells, we consistently observed significantly more fluorescent localizations in the S-phase nuclei (Figure 4A and B).
The advantage of yeasts as model eukaryotes is the ease with which sophisticated genetic experiments can be performed to elucidate important relationships between gene function and phenotype. Recent studies highlighted a role for Elg1 in unloading PCNA from the DNA during DNA replication (30,31). Deleting elg1 from cells caused an accumulation of PCNA on the DNA, as determined by biochemical fractionation analysis. To establish if our method was sufficiently sensitive to detect changes in DNA binding in vivo in single cells due to background genetic mutations, we introduced our mEos3.1-tagged PCNA construct into an elg1Δ genetic background and imaged both elg1+ and elg1Δ cells undergoing DNA synthesis with a 350-ms camera exposure time. As predicted from ensemble biochemical analysis, we observed a statistically significant increase in the number of single-molecule localizations in S-phase elg1Δ cells compared to wild type (Figure 4B and C). The reconstructed images of elg1Δ cells show increased mean localization numbers for the PCNA molecules, consistent with previously reported results and directly confirming that deletion of elg1 causes retention of PCNA on the DNA during replication.
DISCUSSION
In vitro and in some living (largely prokaryotic) cells, single-molecule localization microscopy has shown great promise in the study of complex processes relating to DNA metabolism. Extending these methodologies for use in eukaryotic model organisms is predicted to be of great benefit to the field (10). However, the future use of these technologies will rely on the development of a robust methodological toolkit that will enable direct characterization and visualization of specific phenomena. A detailed protocol for applying SMLM to the visualization of DNA-bound replication proteins in a eukaryotic model organism is described in this study and its efficacy in distinguishing the S phase-specific DNA-binding kinetics of two DNA replication proteins has been established. We have also demonstrate that, in a defined genetic background (elg1Δ) that causes an ∼2.5-fold increase in Pcn1PCNA association with chromatin when assayed by biochemical purification methods (data not shown), we can clearly distinguished increased Pcn1PCNA DNA association. Since the increase in apparent localizations between elg1Δ and elg1+ cells was ∼2-fold, there is reasonable concurrence between our method and biochemical purification.
The standard procedure of chemical fixation of cells is not compatible with SMLM experiments that are designed to visualize the DNA-bound species of a specific protein: the fixation processes immobilize individual proteins that would have otherwise been freely diffusing in a cell. The technique presented here demonstrates that simple and widely available wide-field PALM imaging can be exploited to characterize DNA-bound proteins in a eukaryotic system without the need for cell permeabilization or the extraction of unbound proteins. By stochastic activation of photo-activated fluorescence and the simple reduction of the camera exposure time, we demonstrate that motion blurring of individual mobile fluorophores selectively detects DNA-bound species with a sensitivity that is compatible with the exploration of protein kinetics in distinct cell cycle compartments and the exploitation of genetic backgrounds that subtly affect DNA-association dynamics. Our method thus facilitates relative quantification of DNA-binding behaviours inside cells with good dynamic sensitivity.
We have chosen to establish and validate our method using the fission yeast models system, where we routinely study DNA replication and repair protein dynamics. However, there is no a priori reason why the method cannot be extended to other eukaryotes. One limitation of our approach is that, because the chromatin moves during the time devoted to data acquisition (typically between 5 and 20 min, depending on protein abundance), the reconstructed pictures do not provide spatial information on protein localization within the cell at any one time. Indeed, the output is mainly limited to a quantitative measurement that represents the chromatin-associated fraction of protein that can be only interpreted between two or more specific conditions. Despite the fact that chromatin is mobile within the nucleus, this does not have a significant effect on our ability to measure chromatin association using motion blurring: the diffusion rate of a specific chromosomal locus has been shown in many cell types to lie between 10−4 and 10−3 μm2 s−1 (32). This would lead to a maximum movement of the locus of 45 nm during the 350-ms exposure time used, which is well within the size equating to a single image pixel (usually 100–110 nm) and does not affect the PSF Gaussian fitting. In situations where chromatin is more mobile [for example after DNA damage (32) or in certain cell types], we would recommend exploring the use of shorter exposure times and/or potentially using a more promiscuous Gaussian fitting procedure.
We envisage that, as SMLM becomes more common place in laboratories, our method can be used to complement and extend existing molecular biology techniques aimed at measuring the association of proteins with DNA, e.g. western blotting. Its ability to quantitatively measure protein behaviours at the single-cell level can be used to study crucial biological interactions, potentially revealing previously undetectable changes in DNA association.
Acknowledgments
We thank the members of the Carr, Murray and Osborne labs (University of Sussex) and the Klenerman and Laue labs (University of Cambridge). We also thank Alessandro Bianchi for the gift of plasmid DNA.
Authors Contributions: T.J.E. and S.F.L. conceived the approach. All authors contributed to the design of experiments. T.J.E. and R.L.B. performed microscope experiments. T.J.E. analysed localization numbers, reconstructed super-resolution images and performed simulations. R.L.B. performed single-particle tracking analysis. S.F.L., R.L.B. and S.G. designed and built the microscope. A.H. developed single-molecule fitting routines and simulation. T.J.E., A.W., Y.D. and S.G. created yeast strains. J.T., P.J. and M.A.O. designed the setup; R.L.B. and J.T. performed FCS experiments. T.J.E., R.L.B., S.F.L. and A.M.C. wrote the manuscript.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
FUNDING
European Research Council [268788-SMI-DDR to A.M.C.]. Funding for open access charge: European Research Council [268788-SMI-DDR].
Conflict of interest statement. None declared.
REFERENCES
- 1.Betzig E., Patterson G.H., Sougrat R., Lindwasser O.W., Olenych S., Bonifacino J.S., Davidson M.W., Lippincott-Schwartz J., Hess H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 2.Hess S.T., Girirajan T.P.K., Mason M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rust M.J., Bates M., Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat. Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Horrocks M.H., Palayret M., Klenerman D., Lee S.F. The changing point-spread function: single-molecule-based super-resolution imaging. Histochem. Cell Biol. 2014;141:577–585. doi: 10.1007/s00418-014-1186-1. [DOI] [PubMed] [Google Scholar]
- 5.Patterson G., Davidson M., Manley S., Lippincott-Schwartz J. Superresolution imaging using single-molecule localization. Annu. Rev. Phys. Chem. 2010;61:345–367. doi: 10.1146/annurev.physchem.012809.103444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heilemann M., van de Linde S., Schuttpelz M., Kasper R., Seefeldt B., Mukherjee A., Tinnefeld P., Sauer M. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew. Chem. Int. Ed. Engl. 2008;47:6172–6176. doi: 10.1002/anie.200802376. [DOI] [PubMed] [Google Scholar]
- 7.Szymborska A., de Marco A., Daigle N., Cordes V.C., Briggs J.A., Ellenberg J. Nuclear pore scaffold structure analyzed by super-resolution microscopy and particle averaging. Science. 2013;341:655–658. doi: 10.1126/science.1240672. [DOI] [PubMed] [Google Scholar]
- 8.Lando D., Endesfelder U., Berger H., Subramanian L., Dunne P.D., McColl J., Klenerman D., Carr A.M., Sauer M., Allshire R.C., et al. Quantitative single-molecule microscopy reveals that CENP-A(Cnp1) deposition occurs during G2 in fission yeast. Open Biol. 2012;2:120078. doi: 10.1098/rsob.120078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tokunaga M., Imamoto N., Sakata-Sogawa K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods. 2008;5:159–161. doi: 10.1038/nmeth1171. [DOI] [PubMed] [Google Scholar]
- 10.Stracy M., Uphoff S., Garza de Leon F., Kapanidis A.N. In vivo single-molecule imaging of bacterial DNA replication, transcription, and repair. FEBS Lett. 2014 doi: 10.1016/j.febslet.2014.05.026. doi:10.1016/j.febslet.2014.05.026. [DOI] [PubMed] [Google Scholar]
- 11.Reyes-Lamothe R., Sherratt D.J., Leake M.C. Stoichiometry and architecture of active DNA replication machinery in Escherichia coli. Science. 2010;328:498–501. doi: 10.1126/science.1185757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su'etsugu M., Errington J. The replicase sliding clamp dynamically accumulates behind progressing replication forks in Bacillus subtilis cells. Mol. Cell. 2011;41:720–732. doi: 10.1016/j.molcel.2011.02.024. [DOI] [PubMed] [Google Scholar]
- 13.Uphoff S., Reyes-Lamothe R., Garza de Leon F., Sherratt D.J., Kapanidis A.N. Single-molecule DNA repair in live bacteria. Proc. Natl Acad. Sci. 2013;110:8063–8068. doi: 10.1073/pnas.1301804110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watson A.T., Garcia V., Bone N., Carr A.M., Armstrong J. Gene tagging and gene replacement using recombinase-mediated cassette exchange in Schizosaccharomyces pombe. Gene. 2008;407:63–74. doi: 10.1016/j.gene.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 15.Bähler J., Wu J.-Q., Longtine M.S., Shah N.G., McKenzie Iii A., Steever A.B., Wach A., Philippsen P., Pringle J.R. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 1998;14:943–951. doi: 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 16.Forsburg S.L. Codon usage table for Schizosaccharomyces pombe. Yeast. 1994;10:1045–1047. doi: 10.1002/yea.320100806. [DOI] [PubMed] [Google Scholar]
- 17.Meister P., Taddei A., Ponti A., Baldacci G., Gasser S.M. Replication foci dynamics: replication patterns are modulated by S-phase checkpoint kinases in fission yeast. EMBO J. 2007;26:1315–1326. doi: 10.1038/sj.emboj.7601538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsuyama A., Shirai A., Yoshida M. A novel series of vectors for chromosomal integration in fission yeast. Biochem. Biophys. Res. Commun. 2008;374:315–319. doi: 10.1016/j.bbrc.2008.07.015. [DOI] [PubMed] [Google Scholar]
- 19.Henteges P., Van Driessche B., Tafforeau L., Vandenhaute J., Carr A.M. Three novel antibiotic marker cassettes for gene disruption and marker switching in Schizosaccharomyces pombe. Yeast. 2005;22:1013–1019. doi: 10.1002/yea.1291. [DOI] [PubMed] [Google Scholar]
- 20.Mortensen K.I., Churchman L.S., Spudich J.A., Flyvbjerg H. Optimized localization analysis for single-molecule tracking and super-resolution microscopy. Nat. Methods. 2010;7:377–381. doi: 10.1038/nmeth.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manley S., Gillette J.M., Patterson G.H., Shroff H., Hess H.F., Betzig E., Lippincott-Schwartz J. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat. Methods. 2008;5:155–157. doi: 10.1038/nmeth.1176. [DOI] [PubMed] [Google Scholar]
- 22.Spendier K., Lidke K.A., Lidke D.S., Thomas J.L. Single-particle tracking of immunoglobulin E receptors (FcϵRI) in micron-sized clusters and receptor patches. FEBS Lett. 2012;586:416–421. doi: 10.1016/j.febslet.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michalet X., Berglund A.J. Optimal diffusion coefficient estimation in single-particle tracking. Phys. Rev. E. 2012;85:061916. doi: 10.1103/PhysRevE.85.061916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim S.A., Heinze K.G., Schwille P. Fluorescence correlation spectroscopy in living cells. Nat. Methods. 2007;4:963–973. doi: 10.1038/nmeth1104. [DOI] [PubMed] [Google Scholar]
- 25.Krichevsky O., Bonnet G. Fluorescence correlation spectroscopy: the technique and its applications. Rep. Prog. Phys. 2002;65:251–297. [Google Scholar]
- 26.Culbertson C.T., Jacobson S.C., Michael Ramsey J. Diffusion coefficient measurements in microfluidic devices. Talanta. 2002;56:365–373. doi: 10.1016/s0039-9140(01)00602-6. [DOI] [PubMed] [Google Scholar]
- 27.Kearsey S.E., Montgomery S., Labib K., Lindner K. Chromatin binding of the fission yeast replication factor mcm4 occurs during anaphase and requires ORC and cdc18. EMBO J. 2000;19:1681–1690. doi: 10.1093/emboj/19.7.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elf J., Li G.W., Xie X.S. Probing transcription factor dynamics at the single-molecule level in a living cell. Science. 2007;316:1191–1194. doi: 10.1126/science.1141967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masai H., Matsumoto S., You Z., Yoshizawa-Sugata N., Oda M. Eukaryotic chromosome DNA replication: where, when, and how. Annu. Rev. Biochem. 2010;79:89–130. doi: 10.1146/annurev.biochem.052308.103205. [DOI] [PubMed] [Google Scholar]
- 30.Kubota T., Nishimura K., Kanemaki Masato T., Donaldson Anne D. The Elg1 replication factor C-like complex functions in PCNA unloading during DNA replication. Mol. Cell. 2013;50:273–280. doi: 10.1016/j.molcel.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 31.Shiomi Y., Nishitani H. Alternative replication factor C protein, Elg1, maintains chromosome stability by regulating PCNA levels on chromatin. Genes Cells. 2013;18:946–959. doi: 10.1111/gtc.12087. [DOI] [PubMed] [Google Scholar]
- 32.Dion V., Gasser S.M. Chromatin movement in the maintenance of genome stability. Cell. 2013;152:1355–1364. doi: 10.1016/j.cell.2013.02.010. [DOI] [PubMed] [Google Scholar]