


Abstract
The understanding of folding and function of RNA molecules depends on the identification and classification of interactions between ribonucleotide residues. We developed a new method named ClaRNA for computational classification of contacts in RNA 3D structures. Unique features of the program are the ability to identify imperfect contacts and to process coarse-grained models. Each doublet of spatially close ribonucleotide residues in a query structure is compared to clusters of reference doublets obtained by analysis of a large number of experimentally determined RNA structures, and assigned a score that describes its similarity to one or more known types of contacts, including pairing, stacking, base–phosphate and base–ribose interactions. The accuracy of ClaRNA is 0.997 for canonical base pairs, 0.983 for non-canonical pairs and 0.961 for stacking interactions. The generalized squared correlation coefficient (GC2) for ClaRNA is 0.969 for canonical base pairs, 0.638 for non-canonical pairs and 0.824 for stacking interactions. The classifier can be easily extended to include new types of spatial relationships between pairs or larger assemblies of nucleotide residues. ClaRNA is freely available via a web server that includes an extensive set of tools for processing and visualizing structural information about RNA molecules.
INTRODUCTION
Like proteins, RNA molecules fold hierarchically in time and space into complex 3D structures necessary for molecular function (1). When RNA molecules fold, ribonucleotide residues form various interactions, including canonical (cis Watson–Crick A-U and C-G) base pairs, ‘wobble’ G-U base pairs, other types of nucleotide pairs, different types of base stacking, as well as base–phosphate and base–ribose interactions. The rapidly increasing number of experimentally determined RNA structures revealed a wealth of local motifs that are formed by combinations of these interactions and play specific functional roles (2–4). Therefore, understanding RNA structure and function depends heavily on the identification and classification of interactions between residues in RNA structures.
A number of computational methods have been developed to perform automatic assignment of residue pairs from atomic coordinates of RNA 3D structures, based on different criteria, e.g. MC-Annotate (5), RNAView (6) and FR3D (7). In general, these methods exhibit a broad consensus as to the location of canonical base pairs and stacking interactions. However, they do not always agree about non-canonical pairs and they differ in the assessment of other types of interactions, e.g. those between the base and ribose or phosphate moieties. Further, these methods have been developed to analyze structures represented by full-atom models and they are not appropriate for analyzing models generated by coarse-grained methods that use reduced representations, e.g. for simulations of RNA folding.
In models of experimentally determined RNA structures available in databases such as Protein Data Bank (PDB) (8), not all interactions represent the ideal geometry in the active in vitro and especially the in vivo context. In fact, there is a ‘twilight zone’ of contacts, where the mutual orientation of interacting residues departs significantly from that of idealized structures. For such cases, one has to decide whether the observed deviation is genuine (e.g. due to intramolecular strain), and could be functionally and structurally important (9), or if it represents a modeling error or lack of resolution in the experimental structure due to motional averaging or multiple conformations. Hence, it is important to detect not only perfect interactions, but also ‘near matches’ for further analyses and possibly refinement. This is particularly important in modeling RNA structures with the use of low-resolution or sparse data, where details of the geometry are not always discernible, as well as in purely theoretical modeling that often produces models with globally correct topologies, but with flawed local geometries (10).
To address these issues, we have developed a new method called classification of contacts in RNA tertiary structures (ClaRNA). It is predictive in nature, and is robust to coordinate errors and can be used to define interactions even in poorly refined and low-resolution RNA structures, including coarse-grained representations that contain a reduced representation of the number of atoms per residue. We compared assignments made by ClaRNA with those given by RNAView, MC-Annotate and FR3D, and we found that our method agrees well with the consensus between the other methods and has a relatively small fraction of assignments that are not supported by other methods.
ClaRNA is also capable of identifying certain types of interactions that are common in RNA structures, but are not reported by other methods, and the method has been developed in such a way as to easily include additional types of interactions in the future.
Finally, our method provides valuable assignment of contacts in RNA structures that can aid in model analysis and refinement, and can be used for identification of recurrent structural motifs, alignment of RNA 3D structures and RNA model quality assessment.
MATERIALS AND METHODS
Classification of interactions using existing methods and preparation of training and testing data sets
The Leontis group has provided exemplars (centroids) of each type of base pair according to base combination (AA, GC, etc.) and base–pair type (WW-cis, HS-trans, etc.), which are available at http://rna.bgsu.edu/main/databases/#RNA_Basepair_Catalog. However, for some base combinations/pairs, single examples are insufficient to describe the diversity of geometries that fulfill the given interaction type. Classifiers developed to date often have different scope (e.g. some focus just on ribonucleotide pairs and pay less attention to stacking or base–phosphate interactions, etc.). They also sometimes disagree with each other even for the common classes such as different ribonucleotide pairs. Thus, we extracted experimentally determined high-resolution structures of RNA molecules from the PDB database, and carried out classification with several existing methods to identify interactions that can be clearly ascertained.
Atomic coordinates of 2432 macromolecular structures containing RNA molecules were downloaded from the PDB (8) (release date 16 November 2012). Structures solved by X-ray crystallography at resolution 3.0 Å or better that were released before 18 April 2012 (941 structures comprising 300 913 ribonucleotide residues) were used to establish the initial version of the training data set. The remaining structures, including those solved by nuclear magnetic resonance, at lower resolution than 3.0 Å or released after 18 April 2012 (1491 structures comprising 1 077 316 ribonucleotide residues) were used to establish the testing data set. The lists of structures included in either data set are provided in Supplementary File S1 and the classification of doublets according to the type of RNA structure is illustrated in Supplementary Figure S1.
For all structures we identified doublets of residues that were close in space according to the definition used by Sykes and Levitt (11), i.e. residues with at least one pair of non-hydrogen atoms within a distance of 4.0 Å. In the downloaded structures only canonical ribonucleotide residues (A, U, C, G) were retained and all modified residues and non-RNA residues were ignored. We identified 804 580 doublets in the training set and 2 756 404 doublets in the testing set. Each doublet is uniquely described using its PDB ID, chain ID, residue type and, additionally, the number of the first residue and chain, residue type and the number of second residue. For example 3B4B:A_G7/B_A35 denotes a doublet from a PDB file 3B4B, with the first residue in chain A, guanosine number 7, and the second residue in chain B, adenosine number 35.
For assignment of pairwise interactions between ribonucleotides in RNA structures, the following third-party classifiers were used: MC-Annotate (5), RNAView (6) and FR3D (7). We also used the ModeRNA stacking detection algorithm (12). The classifiers have been executed on the entire structures (on the complete Asymmetric Unit contents) from the above-mentioned data sets. We introduced a common dictionary to relate classes used by different classifiers with each other (available as Online Material O7 at http://iimcb.genesilico.pl/clarna/supp/).
The ribonucletide doublets are identified when they satisfy one of the following criteria:
The same or equivalent class of a residue pair was detected by at least two methods among RNAView, MC-Annotate and FR3D. For each combination of ribonucleotides (e.g. A-A, A-G, A-U, etc.), we considered 12 classes of interaction pairs according to the nomenclature proposed by Leontis and Westhof (13).
The same or equivalent type of stacking interaction was detected by at least two methods among MC-Annotate, FR3D and ModeRNA (RNAView results were not taken into account in identifying stacking interactions, because this program does not report sub-classes of these contacts). For each combination of ribonucleotides, we considered four types of stacking: 3′-5′ (>>), 5′-3′ (<<), 3′-3′ (><) and 5′-5′ (<>) defined by the RNA Ontology Consortium (14).
Base–phosphate and base–ribose interaction was detected by FR3D. For each combination of ribonucleotides, we considered 10 types of base–phosphate (BPh) interactions as defined by (15). We added W and H to the original cluster names to indicate interactions of the Watson and Hoogsteen edge, respectively, with the phosphate group. We also grouped together spatially overlapping clusters W_3BPh,W_4BPh and W_5BPh into a single class named W_345BPh, and in analogy we grouped H_7BPh, H_8BPh and H_9BPh clusters into a single class H_789BPh; see below. Base–ribose interactions were defined using the same method, according to FR3D (7).
It must be emphasized that one residue can be present in many doublets, and one doublet can be included in one or more classes of interactions, e.g. two residues can be involved in base–pairing and base–phosphate interactions at the same time.
In the course of the development of the training data set, doublets of the same chemical type (e.g. all G residues that interact with C residues) were clustered according to geometric similarity (based on location of C2, C4, C6 and C1′ atoms) using an in-house program (G. Chojnowski, unpublished data). Major groups of spatially similar doublets were then inspected by eye and analyzed in the context of the aforementioned consensus annotations. They were manually curated to remove clear outliers that were apparently misclassified (865 doublets total). A full list of removed pairs is available at the ClaRNA web page (Online material O6). After the manual curation, 430 809 doublets remained to be considered in the training data set.
Some theoretically possible nucleotide pairs are very rare or have not been found in structures deposited in the PDB. For such cases we prepared artificial reference doublets by comparative modeling, based on geometrically analogous doublets comprising residues of a different chemical type. Of course, it would be much better to have experimentally determined reference structures. With the growing size of RNA structure database, we intend to update ClaRNA periodically, especially with respect to those classes where the number of reference pairs is low. The artificial reference doublets were constructed by replacing the original bases with the desired ones, followed by energy minimization using the AMBER force field (16), with restraints on planarity of the purine and pyrimidine rings. These 100 additional artificial reference doublets were added to the final version of the training data set (PDB files with additional doublets are available as Online material O9). The final training data set is available on the ClaRNA web page; it contains the list of interaction classes, the respective number of doublets in each class and the corresponding PDB files (Online material O1, available for interactive browsing or for download as a compressed ZIP file, 12 MB). Additionally, the list of recognized classes accompanied with the exemplar doublets is available in the Online material O2.
For the testing data set, types of interactions within doublets were defined using the same consensus procedure as in the initial step of development of the training data set. However, no manual curation has been performed, to avoid biasing the assessment of our classifier. The overview of the ClaRNA preparation is presented in Figure 1.
Figure 1.
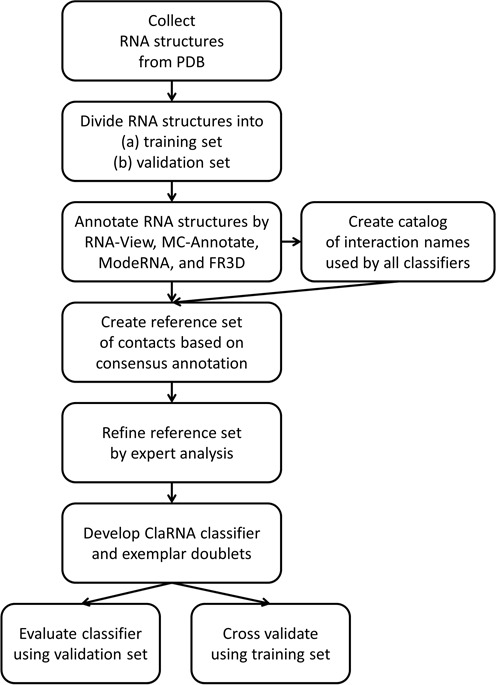
ClaRNA development workflow.
ClaRNA classifier algorithm
The ClaRNA method classifies ribonucleoside interactions in RNA 3D structures based on comparison to a reference/training data set. A distinguishing feature of this classifier, compared to the classifiers developed earlier (MC-Annotate (5), RNAView (6) and FR3D (7)), is that it relies on geometric matching rather than on detection of physical interactions, such as hydrogen bonds. In particular, except for the base–ribose and stacking interactions where methods similar to the ones used in MC-Annotate and FR3D are used, base doublets are classified based on a direct comparison with exemplary doublets from a manually curated reference database. Hence, ClaRNA can detect interactions in RNA structures represented in a coarse-grained fashion (e.g. it can detect tentative base pairing even in the structural models that lack explicit representation of bases) or can suggest ‘near matches’ in structural models that exhibit various distortions. In other words, for each doublet of residues that are spatially close, ClaRNA reports similarity to previously defined classes of interactions. This feature makes ClaRNA open to addition of new classes of interactions and spatial relations of residues that may be defined in the future (see below).
The classifier processes input files in four steps. First, the PDB-formatted file is parsed using the BioPython library (17). Geometry of each residue is analyzed, and in case of missing atoms or non-planarity of the atoms in the base, the residue is restored using idealized ribonucleotide bases. This approach and the idealized ribonucleotide base models were taken from FR3D (15). In the next step, the pairs of ribonucleotide residues (doublets) with the smallest distance between their non-hydrogen atoms below 4.0 Å are identified. The running time of this step is minimized by the use of the KD-Trees data structure (18). KD-Trees is a data structure used to organize points in k-dimensional space into a hierarchical system of subgroups that vastly simplifies the closest-neighbor searches. In the next step, doublets that are likely to form one of the types of contacts present in the training data set are identified based on the following parameters: (i) the angle between the base normal vectors (below 65°), (ii) the distance between base centers (between 4.0 and 8.5 Å), (iii) the smallest distance between non-hydrogen atoms of the bases (between 0.1 and 3.2 Å) and (iv) the angle between the base normal vector and the vector connecting the base centers (between 50° and 140°). Base doublets that fulfill all of the above-given criteria are compared with the entire set of representative doublets from each of the interaction classes (see Materials and Methods for the list of all types of interactions). Depending on the level of similarity of the query doublet to the closest reference doublet from the training data set, an interaction is assigned a score in the range from 0.0 to 1.0, where 1.0 corresponds to a perfect match and all scores below 1 indicate imperfect matches. A similar approach was introduced in the FR3D classifier, which uses a similar set of parameters for classifying doublets to particular interaction types. FR3D also reports near matches (e.g. ncWW, which stands for near WW-cis), but such matches are reported without a score, so nearly ideal near matches are not discriminated from poor near matches.
We use different parameters (mapped onto [0:1] range using linear functions) to score the similarity of different interaction types. The parameters, however, are always computed based on the relative position of the second moiety (base, ribose or sugar) after the optimal superposition of the first base from the query and reference doublet. Since we compare only bases of same type, the superposition is always unambiguous. We use the root mean square deviation (RMSD) of the query and the reference base, ribose and phosphate moieties to parameterize the base–pair, base–ribose and base–phosphate interactions, respectively. Stacking interactions are parameterized simply by the distance between query and the reference base centers (proper mutual orientation of the bases is guaranteed by the initial filtering). As a result, one query doublet can be matched to one or more classes of interactions. A doublet that exhibits a geometry that is intermediate between two well-separated classes may be reported as an imperfect match in both of these classes.
Base (residue) pair interactions
Base/residue pairs in the sense of the Leontis–Westhof classification (13) are detected based on the distance to the selected reference doublets (usually 10–16 doublets per interaction class). The distance between the query doublet and the reference doublet is calculated using two measures: (i) the RMSD between the doublet spatially superimposed with the use of the C1′, C2, C4 and C6 atoms (defined separately for A, C, G and U residues), and (ii) the deviation between the interatomic distance matrices that do not require superposition. Here, for a given interaction class, the query doublet is assigned a score that reflects the number of interatomic distances that fall into ranges derived from doublets in the training set (the details of this score are included in the Supplementary File S2). This measure of (dis)similarity is especially useful for the processing of reduced representations that do not contain all the atoms and/or for distorted structures that may not be easily superimposable onto reference structures. In the case of the reduced representations, only the common elements of the distance matrices are compared. In the case of missing atoms within the bases of the doublets, the classifier tries to rebuild the missing atoms, but this procedure can only succeed if there are at least three atoms available per base. If there is not enough information to identify the three atoms required for the superposition, then the RMSD-based score is not calculated and only the score based on the comparison of distance matrices is used. Hence, the classification is more precise for RNA representations that contain all or nearly all atoms or at least C1′, C2, C4 and C6 atoms, but in principle can be attempted for any coarse-grained representation that retains the planarity of the base moiety (e.g. at least three beads per base).
Stacking interactions
For detecting stacking interactions, we follow the conditions defined by Major and coworkers (5), that is, find the appropriate distance between bases and the angle between normal vectors. The ModeRNA stacking detection algorithm uses a precise version of this criterion; however, the current versions of MC-Annotate (5) and FR3D (7) appear to use a more relaxed criteria, as we found that they report stacking for some other residue pairs (e.g. pair 157D:A_C11/A_G12). Hence, we extended our classifier to detect stacking interactions recognized by both MC-Annotate and FR3D, and this has been done using additional parameterization to measure an overlap area of polygons spanned by the base atoms (including hydrogens) after projecting the analyzed base onto the plane defined by the reference base.
Base–phosphate and base–ribose interactions
Base–phosphate and base–ribose interactions are identified according to the conditions defined by the developers of FR3D (15). Base–phosphate interactions are detected based on the location of the phosphate and neighboring oxygen atoms (OP1, OP2, O5′ and O3′). Base–ribose interactions are detected based on the observed location of the oxygen atoms from the ribose moiety (O2′, O3′ and O4′). We found that the residue pairs involved in the base–phosphate interactions classified as W_3BPh, W_4BPh or W_5BPh by FR3D (7) formed geometrically overlapping clusters; hence, we grouped them into a single class named W_345BPh. Analogously, we found that residue pairs classified as H_7BPh, H_8BPh and H_9BPh were also indistinguishable from each other, and we grouped them into a single class H_789BPh. The merging of these classes can be additionally justified by comments made by the authors of FR3D, who noted that W_(3/4/5)BPh interactions as well as H_(7/8/9)BPh interactions may be interchangeable during conformational changes or thermal fluctuations of RNA (15). The superposition of spatially very close members of W_(3/4/5)BPh interaction classes and elements from merged base–phosphate classes (W_345BPh, H_789BPh) are presented in the Supplementary Figures S2 and S3.
Additional classes considered by classifier
Diagonal relations (consecutive and non-consecutive), sandwich (intercalation) and base–ribose stacking are detected analogously to the standard interaction types described above. In this case, we parameterized similarity with the RMSD of the second moiety atoms (base or ribose) after optimal superposition of the first bases from the query and the reference doublet.
Software
The new RNA contact classifier (ClaRNA) and additional scripts were developed based on the Python programming language, and Biopython (17) and SciPy (19) libraries. The web interface was developed using the Django framework (http://djangoproject.com).
Hardware
For calculations with third-party methods and for the training and benchmarking of the ClaRNA classifier, we used an in-house high performance computing cluster comprising 624 nodes (2.2 GHz processor and 2 GB of RAM each). The ClaRNA web server is hosted on a dual core virtual machine with 4 GB of RAM.
RESULTS
Comparison with other classifiers
ClaRNA is a multi-class and multi-label classifier, since each doublet can be annotated with multiple interactions. Therefore, its evaluation is much more complicated than that of classical binary classifiers.
To formalize the evaluation metric, for each interaction class (base–pair, stacking, base–phosphate, base–ribose) we introduce the following definitions.
True positive: the classifier returned some classification (unambiguous, with a maximum score) and the result agrees with at least two of the reference methods (or with FR3D as the only classifier in the case of base–ribose and base–phosphate interactions). Example: a doublet recognized by ClaRNA, FR3D and RNAView as WW_cis and unrecognized by MC-Annotate.
True negative: the classifier does not return any result, and there is no pair of reference methods that would give the same result (or FR3D alone does not return any result in the case of base–ribose and base–phosphate interactions). Example: a doublet unrecognized by ClaRNA, FR3D and MC-Annotate and recognized by RNAView as SS_trans.
False positive: the classifier returns a result that does not agree with the consensus result of the reference methods (or FR3D alone in case of the base–ribose and base–phosphate interactions). Example 1: a doublet recognized by ClaRNA and RNAView as SH_cis and unrecognized by FR3D and MC-Annotate. Example 2: a doublet recognized by ClaRNA SH_cis and recognized by FR3D, RNAView and MC-Annotate as HH_cis.
False negative: the classifier does not return any result, but the reference methods’ consensus exists. Example: a doublet unrecognized by ClaRNA and recognized by FR3D, RNAView and MC-Annotate as HH_cis.
It should be emphasized that the false positive definition includes two kinds of misclassifications: elements that were found to belong to some class while they should not be classified at all (e.g. SS_cis instead of the expected undetected) and elements with a wrong classification (e.g. WW_trans instead of the expected WW_cis). It must be also stressed that in most of the cases, the set of true negatives is the largest, which has a major influence on some of the evaluation parameters (e.g. the accuracy or false positive rate). Additionally, we provide a formal mathematical definition of the confusion table in Supplemenary File S2.
Using the above definitions, ClaRNA has been benchmarked using the testing set, and the following accuracies were obtained: 0.9974 for canonical base pairs (WW_cis for CG/GC or AU/UA residues), 0.9833 for all non-canonical residue–residue pairs including wobble WW_cis UG/GU pairs, 0.9841 for all non-canonical residue–residue pairs excluding wobble pairs, 0.9615 for stacking interactions, 1.000 for base–phosphate interactions and 0.9946 for base–ribose interactions. The Matthews correlation coefficients are as follows: 0.9842 for canonical base pairs (correlation for classifying ‘canonical base pairs’ against all remaining doublets), 0.8450 for non-canonical pairs including wobble pairs, 0.8166 for non-canonical pairs excluding wobble pairs, 0.9125 for stacking, 0.9988 for base–phosphate interactions and 0.9008 for base–ribose interactions. The Receiver Operating Characteristic (ROC) curve for ClaRNA is presented in Figure 2. Detailed lists of other confusion matrix parameters for ClaRNA and other classifiers are presented in Supplementary Table S1.
Figure 2.
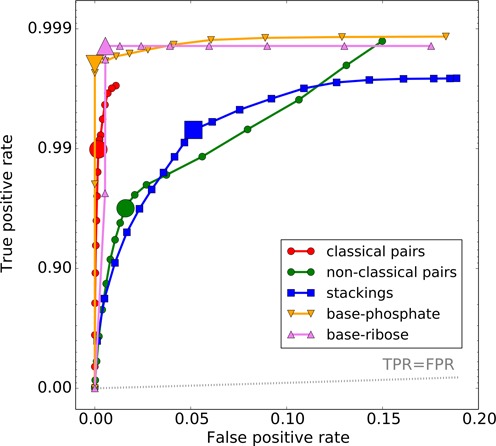
ROC curve for ClaRNA classifier for each contact class: classical pairs (red), non-classical pairs (green), stackings (blue), base–phosphate interactions (orange) and base–ribose (pink) interactions.
We also calculated simple similarity scores for results obtained from various classifiers including MC-Annotate, RNAView, FR3D and ClaRNA. Results presented in Supplementary Table S1 reveal that most classifiers generally agree on the classification of classical base pairs, but there are substantial differences between them with respect to the classification of non-classical base pairs. Also there is no universal agreement on the detection of stacking interactions between the classifiers tested in this work.
Many of the RNA structures from the PDB are redundant in the sense that they form groups of structures with globally similar tertiary folds. The geometries of equivalent residues among these structures are not completely independent, as they are subject to evolutionary constraints. Related structures are also often used as an aid in the process of structure determination. Besides, a good classifier of contacts should be able to perform well also on RNA structures that exhibit completely novel folds, without using information from structures of homologous molecules. To address these issues we performed an additional 13-cross-validation analysis based only on sets of structures that represent non-redundant classes defined in (20), and, in this case, ClaRNA obtained even better results (see Supplementary Table S1). During each cycle of the cross-validation procedure, the classifier parameters were automatically determined based on the training set doublets. The final parameters of the classifier released as a web server were determined using the complete training set.
We also investigated a different definition of the table of confusion, where doublets with uncertain classification (e.g. those reported by only one classifier) were completely omitted. The obtained results were marginally better (less than 1% difference) compared to the standard definition described earlier (data not shown).
We also evaluated the parameters using the methodology from (21) and obtained an overall percentage of correct predictions Q_total = 99.06%. The full evaluation and detailed parameters using this methodology have been presented in Supplementary Table S1.
Detailed presentation of ClaRNA results for particular interaction types (with visualization) is available in the Online materials O3–O5 at the program website (http://iimcb.genesilico.pl/clarna/supp/).
We also compared the similarity of the results obtained by ClaRNA and other classifiers (Figure 3). Each value corresponding to the intersection of sets CL, MC, RV and FR represents the percentage of doublets from the testing set recognized by ClaRNA, MC-Annotate, RNAView and FR3D, respectively, with the same interaction type. Here, 100% is arbitrarily defined as the number of doublets classified into the same interaction type by at least two methods. This analysis illustrates the tendency of individual methods to agree with each other as well as to propose solutions that are at odds with classifications made by other methods. For classical base pairs, all methods showed excellent agreement with each other, with over 94% of the pairs classified in the same way by all methods tested. RNAView reports the highest number of classical base pairs that are not identified by other methods (2.57%). In the case of non-classical pairs, the agreement between all methods is much lower, close to 67%, and each method reports a sizeable fraction of pairs that are not classified as pairs by any other method. FR3D is particularly generous in reporting non-canonical pairs, as the number of such pairs identified by this method alone is equal to as much as 30.70% of cases where at least two methods agree with each other. For stacking interactions, MC-Annotate and ModeRNA are quite conservative, and FR3D is the only method to report over 16% of doublets as stacked, compared to the number of doublets classified as stacked by at least two methods considered. For base–phosphate interactions, ClaRNA agrees with FR3D, and for base–ribose interactions, ClaRNA reports those identified by FR3D, as well as identifies 18.37% additional ones that are not identified by FR3D.
Figure 3.
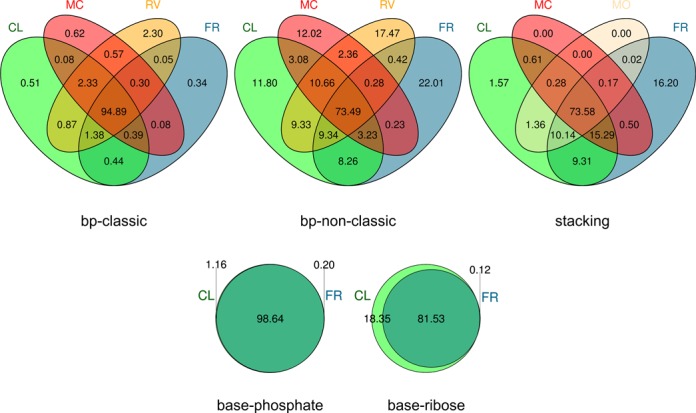
Comparison of the ClaRNA results for the testing set. Each value represents the percentage of doublets from the testing set detected by ClaRNA (CL), MC-Annotate (MC), RNAView (RV), FR3D (FR) and ModeRNA (MO). The number of canonical base pairs that were detected exclusively by ClaRNA was only 2516, as compared to the size of the test set of canonical base pairs (486 118). MC-Annotate, RNAView and FR3D returned 3060, 11 352 and 1684 assignments of canonical base pairs, respectively, that lacked support from other classifiers. The number of non-canonical base pairs (including wobble WW_cis UG/GU pairs) that were detected only by ClaRNA is 31 016 as compared to the size of the corresponding test set of non-canonical base pairs (246 290). MC-Annotate, RNAView and FR3D returned 31 592, 45 904 and 57 832 assignments of non-canonical base pairs, respectively, that lacked support from other classifiers. Furthermore, the number of stacking interactions detected by ClaRNA exclusively was 12 533 (the test set contains 721 851 doublets classified as ‘consensual’ by other methods). In comparison, MC-Annotate, ModeRNA and FR3D returned 0, 34 and 129 230 stacking assignments, respectively, that lacked support from other classifiers.
Many ‘families’ of interactions are geometrically similar to each other and may be difficult to separate. Therefore, we have compared the results obtained with different classifiers with a view that geometrically related pairs (IsoDiscrepancy Index (22) below 9) should be considered to be ‘the same’; i.e. a detection of a pairing type that was different, but geometrically similar to the one in the reference structure, was regarded as a true positive. In addition, we asked to what extent the results would be different if we regarded ‘all’ the non-canonical base–pairing families as equivalent (regardless of the IsoDiscrepancy Index value), and in this test we considered a detection of ‘any’ non-canonical base–pair type as a true positive in the case where the real structure contained a non-canonical base pair of the same or any other type. In both cases, the calculated accuracies of the classifiers were not significantly different from the ones obtained originally. This leads to the conclusion that the discrepancies among the classifiers are mostly due to two types of errors: (i) a classifier annotates a pairing that is not annotated by the reference methods (additional pairing that should not be reported at all) and (ii) a classifier reports ‘no pairing’ for a pair that has a consensus annotation according to the reference methods (missed true pairing).
Finally we have benchmarked ClaRNA using coarse-grained models. For this purpose, we reduced all the test set RNA structures to the representation of our in-house developed program SimRNA [the sugar–phosphate backbone represented by two pseudo-atoms (centered at P and C4′ atoms) and the bases represented by three points (centered at N9, C2, C6 or N1, C2, C4 atoms for purines and pyrimidines, respectively]. We obtained the following accuracies: 0.9974 for canonical base pairs (WW_cis for CG/GC or AU/UA residues), 0.9801 for all non-canonical residue–residue pairs (including wobble WW_cis UG/GU pairs), 0.9810 for all non-canonical residue–residue pairs excluding wobble pairs, 0.9614 for stacking interactions, 0.9566 for base–phosphate interactions and 0.8403 for base–ribose interactions. The Matthews correlation coefficients are as follows: 0.9842 for canonical base pairs, 0.8234 for non-canonical pairs (including wobble pairs), 0.7916 for non-canonical pairs (excluding wobble pairs), 0.9126 for stacking, 0.5325 for base–phosphate interactions and 0.2981 for base–ribose interactions. Detailed results of the benchmark are presented in the Supplementary Table S1.
Computing efficiency
We benchmarked the time required to process the PDB structures as a function of the number of the residues. The results are presented in Supplementary Figure S4. ClaRNA is slower than RNAView and MC-Annotate, but comparable in speed with FR3D. This is because thus far we spent most of the effort on optimizing the quality of results. The time efficiency will be improved in future versions of the classifier, in particular by implementation of the classifier in a programming language such as C++.
Additional classes considered by classifier
In addition to the annotation of contact types, for which we prepared reference data sets based on RNA structure annotation by other methods, we trained ClaRNA to recognize several other types of spatial relations between adjacent residues that have a substantial number of occurrences in the analyzed RNA structures and in our subjective opinion are important for a clear understanding of RNA 3D architectures. The current version of ClaRNA reports four additional classes of relations, including direct base–ribose stacking, and three types of indirect relations that involve stacking and base pairing with other residues (Figure 4). In our experience, the detection of these relations can greatly facilitate the inference of secondary structure from 3D coordinates.
Figure 4.
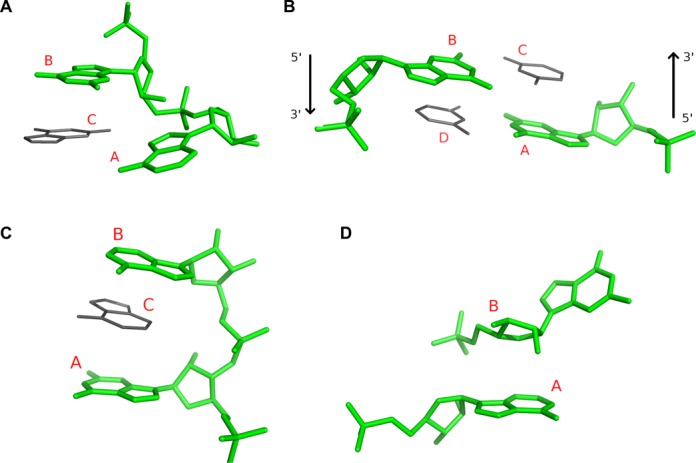
Additional classes of spatial relations between ribonucleotide residues detected and reported by ClaRNA: diagonal (consecutive) (A), diagonal (non-consecutive), arrows indicate main chain direction (B), sandwich (consecutive) (C) and base–ribose stacking (D). The pair of residues forming a contact is indicated by the green color, and possible neighboring residues are indicated by the gray color.
Diagonal (consecutive)
This class of spatial relations is composed of doublets of consecutive (covalently bound) residues denoted by A and B, oriented in a diagonal fashion. Residues A and B are located in a such way that there is enough space for another residue (denoted by C), with the additional condition that A and C could form WW_cis interaction and B and C could form a stacking interaction. ClaRNA detects this class of spatial relations between residues A and B without taking the presence or orientation of residue C into account. An example interaction of this type is presented in Figure 4A.
Diagonal (non-consecutive)
This class is composed of non-consecutive residues denoted by A and B, located in a diagonal fashion. In this case we require that there should be enough space for two neighboring residues C and D, such that there could be WW_cis interactions between A–D and B–C, and stacking interactions between A–C and B–D. ClaRNA detects this class of spatial relations between residues A and B without taking the presence or orientation of residues C and D into account. An example interaction of this type is presented in Figure 4B.
Sandwich (consecutive)
Doublets of consecutive residues A and B form this class of spatial relationship if they are placed in such a way that there is enough space for another residue C to be inserted (intercalated) between them and form stacking interactions with both A and B. Again, the detection of this relationship between A and B does not require the detection of the actual residue C. We used the RNA Bricks database (23) to test the specificity of ClaRNA in detecting this type of contact. Out of the 6705 residue pairs detected in the test set structures, all but two interact with a third intercalating base. The two exceptions are 1FCW:D_G18/D_G19 and 1S1H:A_C1501/A_A1502 from low-resolution structures modeled based on electron microscopy data. In both cases the interacting residues suffer from severe clashes that may suggest limited reliability of their geometry (data not shown). An example interaction of this type is presented in Figure 4C.
Base–ribose stacking
This class of doublets forms stacking interaction between the base moiety of one residue (denoted by A) and the ribose moiety of another residue (denoted by B). An example interaction of this type is presented in Figure 4D.
ClaRNA implementation and web server
ClaRNA classifier is available as a web server at http://iimcb.genesilico.pl/clarna/. The site provides a web interface for uploading input PDB files. A sample query is presented in Figure 5. Each query is queued and processed by the server. All queries have a unique job link that allows access to the results. With the currently available storage capacity, we store the results up to 1 month. Optionally, a user can provide an e-mail address for notification of the job completion.
Figure 5.
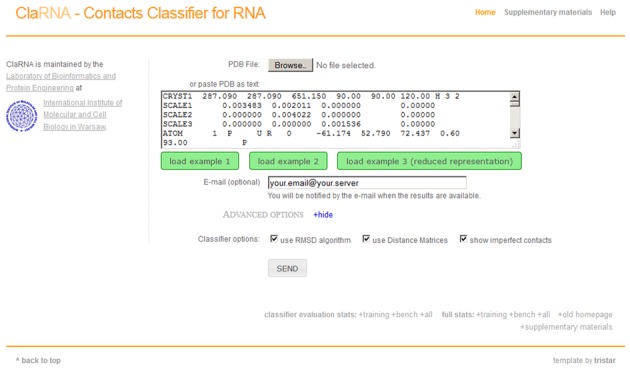
ClaRNA homepage, with a sample query.
Visualization of detected contacts
Query structures are annotated with the detected contacts. Since ClaRNA is not a binary classifier and was developed specifically to enable the detection of suboptimal matches, its results can be filtered by setting the threshold of the score. Each contact can be visualized in 3D using JSmol, an open source JavaScript-Based Molecular Viewer From Jmol (http://sourceforge.net/projects/jsmol/). Query structures are also processed by other classifiers including RNAView, MC-Annotate, ModeRNA and FR3D, and the results obtained are presented to the user for comparison. Secondary structure inferred from the contacts detected by both ClaRNA and other methods is also presented as a graph, using the Varna applet (24). We use an in-house modified version of Varna to visualize stackings, non-classical pairs, phosphate–base and ribose–base interactions. A user can also export results to a Comma-separated values file (CVS) or a JavaScript Object Notation contact graph file (JSON). A sample results’ page is presented in Figure 6.
Figure 6.
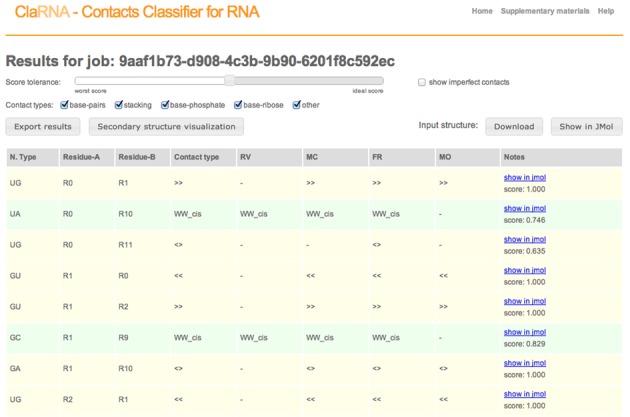
Sample classifier output.
DISCUSSION
Systematic differences with other classifiers
Problems with cis/trans pair discrimination
We found that algorithms for discrimination between cis and trans orientations of base pairs (13) implemented in MC-Annotate and RNAView in some cases produce inconsistent results for base pairs closely similar in geometry. For example, doublets such as 1N8R:A_U2889/A_C2868 and 1Q86:A_U2889/A_C2868 have almost the same structure, but MC-Annotate reports the first one as HH_trans and the second one as HH_cis (Figure 7). In order to avoid such problems, we decided to use the approach used by FR3D, i.e. to take into account the local RNA strand orientation for defining cis/trans orientations. Consequently, there are cases where ClaRNA generates results that generally agree with FR3D, but disagree with RNAView and MC-Annotate. A full list of differences is presented on the ClaRNA web page (Online material O4).
Figure 7.
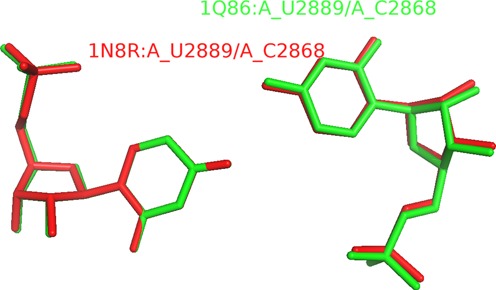
Example of two close doublets with different cis/trans orientations. The doublet 1N8R:A_U2889/A_C2868 (show in red color) is reported as HH_trans by MC-Annotate classifier and doublet 1Q86:A_U2889/A_C2868 (shown in green color) is reported as HH_cis by the same classifier.
Interactions with the sugar edge (SH_cis/CA)
In general our classifier mimics the consensus classification obtained from other classifiers. In some cases, however, the classifiers used as a reference returned predictions of pairs that could not be validated by the visual analysis. Such outliers were corrected in the ClaRNA reference set. For example, a small number of doublets annotated by MC-Annotate and RNAView as Watson–Hoogsteen cis pairs were found by visual inspection to be geometrically much closer to sugar–Hoogsteen cis pairs (as classified by FR3D) and, in fact, one of the residues in a doublet presented more of the sugar edge interacting with the other residue, compared to its Watson–Crick edge (Figure 8). We describe such cases on the ClaRNA web page (with interactive browser as Online material O4 and the full list of doublets as Online Material O8).
Figure 8.
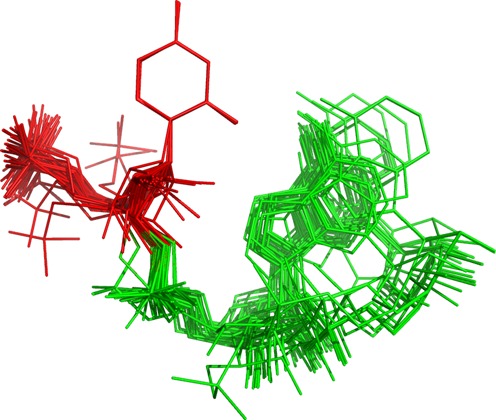
Example of doublets classified by MC-Annotate and RNAView as WH_cis, which FR3D and ClaRNA report as SH_cis. Each doublet is superimposed using base of first residue (show in red color), the other residue is shown in green color.
Base–ribose interactions
The version of FR3D tested at the time of writing this manuscript had a minor implementation issue that affected the detection of base–ribose contacts, as it also tested the position of a phosphate atom. Since the phosphate atom is irrelevant to base–ribose contacts, we removed that condition from our classifier. As a result, ClaRNA detects some pairs of residues as base–ribose interactions that are valid, but have been discarded by the FR3D implementation used at the time of ClaRNA development. We describe such cases on ClaRNA web page (with interactive browser as Online material O4 and the full list of doublets as Online Material O8). This issue has been communicated to the developers of FR3D.
CONCLUSIONS
ClaRNA is a new method for computational classification of contacts in RNA 3D structures. It uses a completely different approach than other methods developed so far, namely, it relies on a reference data set resulting from a consensus classification obtained from other methods, combined with expert assessment. The only exceptions are base–ribose and base–phosphate interactions which are currently detected exclusively by FR3D according to the classification proposed by the Leontis group. Therefore, for these types of contacts, FR3D is the only external classifier we could use to generate the reference set. Of course, this causes some bias, but to our knowledge, there is no other classifier that we could include. Unlike other methods, it was developed to detect suboptimal contacts, to facilitate model building based on limited experimental observations and to guide refinement of models obtained from homology modeling that may contain various distortions. The set of contact classes reported by ClaRNA can be easily extended to incorporate other types of structures or subtypes of spatial relations between ribonucleotide residues. In the future, ClaRNA will also be extended to enable detection and classification RNA–ligand and RNA–protein interactions. ClaRNA is freely available via a web server that includes an extensive set of tools for processing and visualizing structural information about RNA molecules, including comparison of results with those available from other methods.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We would like to thank Wayne Dawson, Michał Boniecki, Grzegorz Łach and Juliusz Stasiewicz for critical reading of the manuscript and useful comments. We also thank the developers of MC-Annotate, RNAView and FR3D for discussions and for making their software available. We would also like to thank Arkadiusz Chworoś for valuable comments on ClaRNA web sever.
FUNDING
European Research Council [ERC, StG RNA+P = 123D to J.M.B.]; National Science Center [2011/01/D/NZ1/00212 to G.Ch.]; Polish Ministry of Science and Higher Education [Iuventus 0586/IP3/2011/71 to T.W., POIG.02.03.00–00–003/09 to J.M.B.]. Funding for open access charge: ERC [StG RNA+P = 123D to J.M.B.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Thirumalai D., Hyeon C. RNA and protein folding: common themes and variations. Biochemistry. 2005;44:4957–4970. doi: 10.1021/bi047314+. [DOI] [PubMed] [Google Scholar]
- 2.Leontis N.B., Lescoute A., Westhof E. The building blocks and motifs of RNA architecture. Curr. Opin. Struct. Biol. 2006;16:279–287. doi: 10.1016/j.sbi.2006.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Halder S., Bhattacharyya D. RNA structure and dynamics: a base pairing perspective. Prog. Biophys. Mol. Biol. 2013;113:264–283. doi: 10.1016/j.pbiomolbio.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 4.Lee J.C., Gutell R.R. Diversity of base-pair conformations and their occurrence in rRNA structure and RNA structural motifs. J. Mol. Biol. 2004;344:1225–1249. doi: 10.1016/j.jmb.2004.09.072. [DOI] [PubMed] [Google Scholar]
- 5.Gendron P., Lemieux S., Major F. Quantitative analysis of nucleic acid three-dimensional structures. J. Mol. Biol. 2001;308:919–936. doi: 10.1006/jmbi.2001.4626. [DOI] [PubMed] [Google Scholar]
- 6.Yang H., Jossinet F., Leontis N., Chen L., Westbrook J., Berman H., Westhof E. Tools for the automatic identification and classification of RNA base pairs. Nucleic Acids Res. 2003;31:3450–3460. doi: 10.1093/nar/gkg529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarver M., Zirbel C.L., Stombaugh J., Mokdad A., Leontis N.B. FR3D: finding local and composite recurrent structural motifs in RNA 3D structures. J. Math. Biol. 2008;56:215–252. doi: 10.1007/s00285-007-0110-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perbandt M., Vallazza M., Lippmann C., Betzel C., Erdmann V.A. Structure of an RNA duplex with an unusual G.C pair in wobble-like conformation at 1.6 A resolution. Acta Crystallogr. D Biol. Crystallogr. 2001;57:219–224. doi: 10.1107/s0907444900017042. [DOI] [PubMed] [Google Scholar]
- 10.Cruz J.A., Blanchet M.F., Boniecki M., Bujnicki J.M., Chen S.J., Cao S., Das R., Ding F., Dokholyan N.V., Flores S.C., et al. RNA-Puzzles: A CASP-like evaluation of RNA three-dimensional structure prediction. RNA. 2012;14:610–625. doi: 10.1261/rna.031054.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sykes M.T., Levitt M. Describing RNA structure by libraries of clustered nucleotide doublets. J. Mol. Biol. 2005;351:26–38. doi: 10.1016/j.jmb.2005.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rother M., Rother K., Puton T., Bujnicki J.M. ModeRNA: a tool for comparative modeling of RNA 3D structure. Nucleic Acids Res. 2011;39:4007–4022. doi: 10.1093/nar/gkq1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leontis N.B., Westhof E. Geometric nomenclature and classification of RNA base pairs. RNA. 2001;7:499–512. doi: 10.1017/s1355838201002515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoehndorf R., Batchelor C., Bittner T., Dumontier M., Eilbeck K., Knight R., Mungall C.J., Richardson J.S., Stombaugh J., Westhof E., et al. The RNA Ontology (RNAO): an ontology for integrating RNA sequence and structure data. J. Appl. Ontol. 2011;6:53–89. [Google Scholar]
- 15.Zirbel C.L., Sponer J.E., Sponer J., Stombaugh J., Leontis N.B. Classification and energetics of the base-phosphate interactions in RNA. Nucleic Acids Res. 2009;37:4898–4918. doi: 10.1093/nar/gkp468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Case D.A., Cheatham T.E., 3rd, Darden T., Gohlke H., Luo R., Merz K.M., Jr, Onufriev A., Simmerling C., Wang B., Woods R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cock P.J., Antao T., Chang J.T., Chapman B.A., Cox C.J., Dalke A., Friedberg I., Hamelryck T., Kauff F., Wilczynski B., et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bentley J.L. Multidimensional binary search trees used for associative searching. Commun. ACM. 1975;18:509–517. [Google Scholar]
- 19.Oliphant T.E. Python for scientific computing. Comput. Sci. Eng. 2007;9:10–20. [Google Scholar]
- 20.Leontis N.B., Zirbel C.L. In: RNA 3D Structure Analysis and Prediction. Leontis N.B., Westhof E., editors. Berlin-Heidelberg: Springer; 2012. pp. 281–298. [Google Scholar]
- 21.Baldi P., Brunak S., Chauvin Y., Andersen C.A., Nielsen H. Assessing the accuracy of prediction algorithms for classification: an overview. Bioinformatics. 2000;16:412–424. doi: 10.1093/bioinformatics/16.5.412. [DOI] [PubMed] [Google Scholar]
- 22.Stombaugh J., Zirbel C.L., Westhof E., Leontis N.B. Frequency and isostericity of RNA base pairs. Nucleic Acids Res. 2009;37:2294–2312. doi: 10.1093/nar/gkp011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chojnowski G., Walen T., Bujnicki J.M. RNA Bricks—a database of RNA 3D motifs and their interactions. Nucleic Acids Res. 2014;42:D123–D131. doi: 10.1093/nar/gkt1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Darty K., Denise A., Ponty Y. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009;25:1974–1975. doi: 10.1093/bioinformatics/btp250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.