


Abstract
Microcin C (McC) is a peptide–nucleotide antibiotic produced by Escherichia coli cells harboring a plasmid-borne operon mccABCDE. The heptapeptide MccA is converted into McC by adenylation catalyzed by the MccB enzyme. Since MccA is a substrate for MccB, a mechanism that regulates the MccA/MccB ratio likely exists. Here, we show that transcription from a promoter located upstream of mccA directs the synthesis of two transcripts: a short highly abundant transcript containing the mccA ORF and a longer minor transcript containing mccA and downstream ORFs. The short transcript is generated when RNA polymerase terminates transcription at an intrinsic terminator located in the intergenic region between the mccA and mccB genes. The function of this terminator is strongly attenuated by upstream mcc sequences. Attenuation is relieved and transcription termination is induced when ribosome binds to the mccA ORF. Ribosome binding also makes the mccA RNA exceptionally stable. Together, these two effects—ribosome-induced transcription termination and stabilization of the message—account for very high abundance of the mccA transcript that is essential for McC production. The general scheme appears to be evolutionary conserved as ribosome-induced transcription termination also occurs in a homologous operon from Helicobacter pylori.
INTRODUCTION
Microcins are a group of peptide antibiotics synthesized by enterobacteria (1). These compounds are produced under stress conditions and are effective against bacteria closely related to the producing strain. As a group, microcins are characterized by their small size (<10 kDa) and synthesis by ribosomes. The mechanisms of toxicity and targets vary for different microcins. While some microcins depolarize the cell membrane (2), others act on intracellular targets. For instance, microcin B targets DNA gyrase (3), microcin J targets the DNA-dependent RNA polymerase (4), while microcin C targets aspartyl-tRNA synthetase (5). Microcins acting on intracellular targets are extensively post-translationally modified and have unusual chemical structures (6).
Typically, microcin gene clusters are located on plasmids and contain genes coding for microcin precursor peptide, self-immunity proteins and modification enzymes (7). Microcin C (McC), the smallest microcin known, is produced by Escherichia coli cells harboring a plasmid-borne gene cluster mccABCDE (Figure 1A). Mature McC (Mw = 1178 Da) is a heptapeptide with covalently attached С-terminal adenosine monophosphate and a propylamine group attached to the phosphate (Figure 1B). McC enters susceptible E. coli cells through the YejABEF inner-membrane transporter (8). Inside the cell, the peptide part is processed by intracellular aminopeptidases (9) to release processed McC—a non-hydrolysable analog of aspartyl-adenylate, which specifically inhibits aspartyl-tRNA synthetase (Asp-RS), leading to cessation of translation (5).
Figure 1.
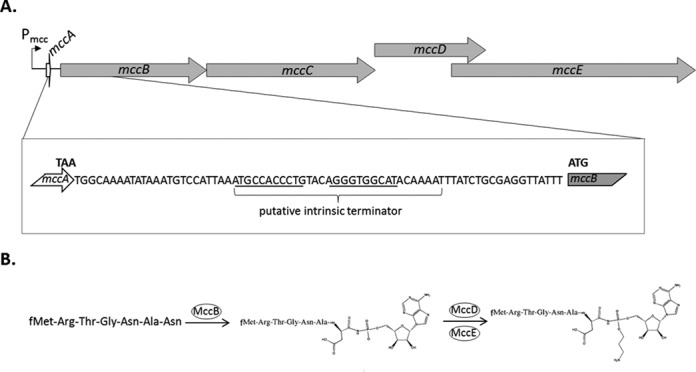
The mcc operon of E. coli and the biosynthesis of microcin C. (A) The mccABCDE operon is schematically presented at the top. Arrows indicate genes (drawn to scale). The position of the Pmcc promoter is indicated. The sequence of the intergenic region between mccA and mccB is expanded below. The last codon of the mccA ORF and the first codon of the mccB ORF are highlighted. The putative intrinsic transcription terminator is indicated and self-complementary regions capable of forming the terminator hairpin are underlined. (B) The pathway of McC biosynthesis is shown. The heptapeptide shown on the left, the product of mccA, is adenylated by MccB (10) and the resulting peptide-adenylate is decorated with an aminopropyl moiety in the reaction that requires MccD and MccE (11), resulting in mature McC.
The product of mccA, the first gene of the McC biosynthesis operon, is a heptapeptide MRTGNAN that is converted into biologically active McC maturation intermediate in an adenylation reaction catalyzed by the MccB enzyme (10–11). Aminopropylation of the product of MccB-catalyzed reaction requires MccD and MccE and increases the biological activity ca. 10-fold (11). MccE is also responsible for detoxification of processed McC that accumulates in the producing cell (12). The mccC gene encodes a pump that exports produced McC and its maturation intermediate and also renders the producing cells resistant to external McC (13).
The transcription of the mcc operon initiates from a CAP (catabolite activator protein)-dependent promoter located upstream of the mccA gene when the producer enters stationary phase or is nutrient deprived (14,15). Since both the mccA transcript, which codes for precursor peptide substrate, and the mccB transcript, which codes for the MccA peptide adenylation enzyme, are produced from the same promoter, there is likely a mechanism that regulates the substrate (MccA) to enzyme (MccB) ratio, so that a necessary level of McC production is achieved. In this work we uncover two mechanisms responsible for preferential synthesis of MccA compared to other products of the mcc operon. First, an intrinsic transcription terminator located between the mccA and mccB leads to the production of excess of monocistronic mccA transcripts. The function of this terminator requires ribosome binding to the mccA ORF. Second, ribosome binding leads to dramatic stabilization of the monocistronic mccA transcript. Together, these two effects lead to ca. 20-fold overproduction of mccA mRNA compared to mccB mRNA, so that a sufficiently high amount of substrate for McC synthesizing enzyme is achieved.
MATERIALS AND METHODS
Bacterial strains and DNA
E. coli strain BW25113 K-12 (16) was used if not indicated otherwise. E. coli BL21(DE3) was used as a sensitive strain for McC sensitivity tests. The pp70 plasmid is pBR322-based and bears a ∼6000 bp DNA fragment with the mcc gene cluster. To create pp70, a BstXI-АраLI fragment of plasmid pBM43 (17) was ligated in the EcoRI-AvaI fragment of the pBAD vector (Life Technologies). Both the vector and the insert were blunt ended before the ligation. The pp70 derivatives carrying mccA ATG1->TTA or Shine–Dalgarno ATAGGAGG→CCCTACTT (SD) mutations, as well as plasmid pp70Δ51 lacking nucleotides +59/+109 (the mcc transcription start is designated as +1) from the mccA–mccB intergenic region, were constructed by site-specific mutagenesis of pp70.
To create pFD_mcc_dir and pFD_mcc_inv plasmids, two pairs of oligonucleotides were annealed to yield two dsDNA fragments containing, in direct either inverted orientation, the mcc terminator hairpin along with 7 bp upstream and 13 bp downstream DNA flanked by AvaI (upstream) and EcoRV (downstream). The mcc part of resulting double-stranded DNA fragments corresponded to positions +71/+114 with respect to the mcc transcription start point. These fragments were ligated in the AvaI-EcoRV fragment of the pFD100 plasmid (18), between the lacUV5 promoter and the galK gene. To test galK gene expression, E. coli HB101 cells (ATCC33694) lacking functional galK gene were transformed with pFD-derivatives and streaked on McConkey indicator medium containing 1% galactose. The results were recorded after an overnight growth at 37°C.
RNA purification and differential RNA-seq
An aliquot of overnight culture was diluted hundred times with LB containing 100 μg/ml carbenicilline and grown for 4 h at 37°C until OD600 reached 2.0. Cells from 0.5 to 3 ml culture aliquots were collected by centrifugation, the supernatant was discarded, and cell pellets frozen in liquid nitrogen and stored at −70°C until further use. Cell pellets were resuspended in 30–50 μl of 20 mM Tris-HCl, pH 8.0, 1 mM EDTA, treated with 10 mg/ml lysozyme, mixed with 500 μl of TRIzol reagent (Invitrogen) and incubated for 5 min at room temperature. Hundred microliter of chloroform was added next and the samples were incubated for 15 min on ice until phase separation occurred. After 15-min centrifugation at 13,000×g the aqueous phase (∼300 μl) was transferred to a fresh tube. RNA was ethanol precipitated and then redissolved in 20–50 μl of DEPC-treated water. RNA concentration was determined using the NanoDrop spectrophotometer.
For whole transcriptome analysis by differential RNA-seq (dRNA-seq) (19) total RNA samples purified as described above were treated with DNase I (New England Biolabs) according to manufacturer's protocol to remove residual DNA from the samples. For depletion of processed transcripts, RNA was treated with Terminator™ 5′-phosphate-dependent exonuclease (TEX) (Epicentre) as previously described (19). cDNA libraries were constructed by vertis Biotechnology AG, Germany as described in (19). Sequencing of cDNA libraries was performed on an Illumina HiSeq2000 machine. In total, 1.8–2.4 million cDNA reads for each cDNA library were sequenced. The reads were mapped to the pp70 plasmid sequence. For data visualization, graphs representing the number of mapped reads per nucleotide were calculated and visualized using the Integrated Genome Browser (IGB) version 6.5.3 software from Affymetrix (http://genoviz.sourceforge.net/). The whole transcriptome dataset as well as further details of read mapping and sequencing statistics will be published elsewhere.
Northern blot hybridization
The RNA samples were diluted to a final concentration of 0.25 μg/μl with formamide loading buffer (95% formamide, 0.025% xylencyanol, 0.025% bromphenol blue), and 1 μg of RNA was loaded on a 10% (19:1) denaturing polyacrylamide gel. After separation at 10W for 15–20 min, RNA was electroblotted to Hybond XL membrane (Amersham) using wet transfer system (BioRad). Transfer was performed for 1 h at 250 mA in 0.5xTBE. The membrane was air-dried and baked using ‘optimal cross-link’ settings of UV crosslinker (UVP). The membrane was blocked with 5 ml of ExpressHyb buffer (Clontech) for 30 min at 37°C, 20 pmoles of 5′ 32P-labeled oligonucleotide probe was added, and hybridization was allowed to proceed for 1 h (or overnight) at 37°C. The membrane was washed twice for 10 min with 2xSSC, 0.1% SDS and once for 10 min with 0.1xSSC, 0.1% SDS. Results were visualized using Storm Phosphorimager (GE Healthcare).
In vivo transcript stability measurements
Cells were grown as described above. After 4 h of growth, 100 μg/μl rifampicin was added and growth was continued. Five hundred microliter culture aliquots were taken before and 2, 5, 10 and 20 min after rifampicin addition. Samples were immediately mixed with 100 μl stop solution (95% ethanol, 5% phenol) and snap-frozen in liquid nitrogen. After thawing on ice, samples were centrifuged for 5 min at 5000xg and RNA was purified as described above.
Estimation of in vivo termination efficiency
The monocistronic transcript (that we here denote A after mccA) and the polycistronic transcript (that we denote B after mccB) are generated from the same promoter, and their transcription rate is denoted as 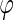 . Change of concentration for the two transcripts with time can be described by the following equations:
. Change of concentration for the two transcripts with time can be described by the following equations:
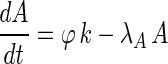 |
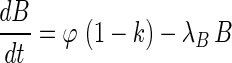 |
In the equations above, A and B are the concentration of the transcripts. k is the termination efficiency, which is defined as the ratio between the initiated transcripts that are terminated, and the total number of initiated transcripts (note that k < 1). 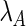 and
and 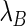 are, respectively, the decay rates of transcripts A and B. Consequently, the first terms on the right-hand sides of the equations describe the transcript generation, while the second terms describe the decay of the transcript concentration. In the steady state, we have
are, respectively, the decay rates of transcripts A and B. Consequently, the first terms on the right-hand sides of the equations describe the transcript generation, while the second terms describe the decay of the transcript concentration. In the steady state, we have 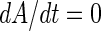 and
and 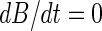 , and by solving the two equations above for
, and by solving the two equations above for 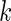 we obtain:
we obtain:
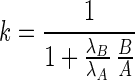 |
Furthermore, the decay rate 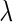 and the half-life are related by
and the half-life are related by 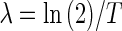 , where T is transcript half-life time, so that:
, where T is transcript half-life time, so that:
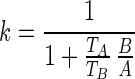 |
Primer extension reactions
1–6 μg RNA was reverse-transcribed using MuMLV reverse transcriptase (Invitrogen) according to manufacturer's instructions. Briefly, primers were annealed to RNA for 10 min at 65°C followed by transfer on ice. MuMLV reverse transcriptase was added to make up a total volume of 10 μl and a final MuMLV concentration of 10 u/μl in 1x reaction buffer (50 mM Tris-HCl, pH 8.3, 75 mM KCl, 3 mM MgCl2, 10 mM DTT); reactions were allowed to proceed at 37°C for 55 min. Reactions were terminated by the addition of an equal volume of formamide loading buffer and loaded on 8% denaturing polyacrylamide gel.
5′-RACE
Twenty microgram of total RNA was treated with DNAse I (New England Biolabs) according to manufacturer's instructions. To half of the sample (10 μg) five units of TAP (Tobacco Acid Pyrophosphatase, Epicentre) (+TAP reaction) was added. An equal amount of water was added instead of enzyme to another half, making control-TAP reaction. Reactions were incubated for 1 h at 37°C in 25 μl reaction volume in the presence of 20 units of RNaseOUT (Invitrogen). RNA was next phenol/chloroform extracted and ethanol precipitated. The pellet was resuspended in 50 μl ligation buffer (50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 1 mM DTT) with 120 units of T4 RNA ligase I (New England Biolabs) and 50 pmoles of RNA primer (5′ GGUAUUGCGGUACCCUUGUACGC 3′), and ligation was performed overnight at 16°C. Following phenol/chloroform extraction and ethanol precipitation, the RNA/primer pellet was resuspended in 10 μl of water, heated at 65°C for 10 min and immediately transferred on ice. The cDNA synthesis was carried out with 100 units of Super Script III from the First-Strand Synthesis System for RT-PCR (Invitrogen) according to manufacturer's instructions for 50 min at 55°C using a primer complementary to the beginning of mccB gene. Two microliter aliquots of cDNA synthesis reactions were used as templates for PCR (one primer was identical to RNA primer, another was complementary to the beginning of the mccB gene). PCR products were resolved on 2% agarose gels. Amplified fragments of expected length were excised from the gel and cloned into the pT7Blue cloning vector from the pT7Blue Perfectly Blunt Cloning Kit (Novagen). Inserts in several recombinant plasmids were sequenced using standard T7-promoter oligonucleotide primer. The 5′ ends of transcripts were determined by the position of the junction site between the sequence corresponding to the RNA primer and mcc sequences.
3′-RACE
Ten microgram of total RNA was mixed with 40 pmol of 5′ pre-adenylated DNA-adapter (New England Biolabs) and 200 units of truncated T4 RNA ligase 2 (New England Biolabs) in 50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 1 mM DTT, 10% PEG. Reactions proceeded at 25°C for 1 h and were terminated by incubation at 60°C for 25 min. Five microliter of reaction mixtures was used as templates for cDNA strand synthesis with 100 units SuperScript III reverse-transcriptase (Invitrogen) according to the manufacturer's protocol using 1 pmole of primer complementary to the DNA adapter. Two microliter of reverse-transcription reactions was used as a template for PCR amplification (one primer was complementary to the DNA adapter, another to the template strand of the mccA gene). The product of PCR amplification was cloned directly to the pGEM-T easy vector (Promega) using pGEM-T easy PCR ligation kit (Promega) according to the manufacturer's protocol. Plasmids from insert-positive clones were purified and insert sequences were determined.
In vitro transcription
Promoter complexes were allowed to form for 10 min at 37°C in 10 μl reactions containing 40 mM Tris-HCl, pH 8.0, 40 mM KCl, 10 mM MgCl2, 100 nM E. coli RNA polymerase σ70 holoenzyme (Epicentre) and 10 nM DNA templates. Reactions were supplemented with 10 μM CpApUpC, 25 μM ATP and GTP, and 0.33 μM α-32P-CTP (3000 Ci/mmol) and stalled elongation complexes were allowed to form for 5 min 37°C and reactions were diluted 10-fold with a chase mix that resulted in the final concentrations of 500 mM KCl, 2.5 μM of ATP and GTP, and 10 μM of UTP and CTP. Reactions were incubated for 15 min at 25°C and terminated by the addition of formamide loading buffer.
For chase reactions coupled with in vitro translation, stalled elongation complexes were formed as described above except that 50 mM NaCl was used instead of KCl. Stalled elongation complexes were diluted 10-fold with in vitro translation mix (In Vitro Protein Synthesis, ΔRibosome, or Δ(aa, tRNA) PURExpress kit, New England Biolabs) and reactions were allowed to proceed for 15 min at 25°C. Reaction volume was made up to 100 μl with ASE buffer (0.3 M NaAc, 0.5% SDS, 5 mM EDTA, pH5.3) and reactions were extracted twice with acid phenol/chloroform, precipitated with ethanol and dissolved in formamide loading buffer.
For single-round in vitro transcription promoter complexes were allowed to form for 10 min at 37°C in 10 μl reactions containing 40 mM Tris-HCl, pH 8.0, 40 mM KCl, 10 mM MgCl2, 100 nM E. coli RNA polymerase σ70 holoenzyme (Epicentre) and 10 nM DNA templates. Reactions were supplemented with 50 μg/ml heparin, 100 μM ATP, 100 μM GTP, 100 μM UTP, 10 μM CTP and 0.33 μM α-32P-CTP (3000 Ci/mmol). Reactions were incubated for 15 min at 37°C to allow single-round RNA synthesis and terminated by addition of 10 μl formamide loading buffer.
Isolation of ribosomal fractions
Cells were resuspended in 25 mM Tris, pH 8.0, 60 mM KCl, 10 mM MgCl2, 20% sucrose and lysed by five cycles of freezing in liquid nitrogen—thawing at 37°C. The suspension was next diluted 4-fold by the same buffer and supplemented with 0.5% Brij58, 10 u/ml DNAse I, 0.15% deoxycholic acid and 3 mM MgSO4. The suspension was incubated on ice for 30 min and cell debris was pelleted by 30-min centrifugation at 30,000xg. The lysate was loaded onto a 10–40% sucrose gradient (in 25 mM Tris, pH 7.8, 50 mM KCl, 10 mM MgCl2, 3 mM DTT) and centrifuged for 16 h at 18,000 rpm in SW38Ti rotor. Fractions were collected by dripping from percolated bottom of centrifuge tube.
Toeprinting
The mcc RNA (+1/+146 with respect to the natural transcription start site) was transcribed in vitro using T7 High Yield RNA Synthesis Kit (New England Biolabs) and an appropriate DNA template generated by PCR, and purified with RNA Clean&Concentrator 25 kit (ZymoResearch).
One pmol of purified in vitro transcribed mcc RNA was mixed, in a total volume of 5 μl, with 2 pmoles of 32P-labeled primer complementary to the 3′-end of RNA and in vitro translation mix (In Vitro Protein Synthesis, ΔRibosome or Δ(aa, tRNA) PURExpress kit, New England Biolabs) components. When added, thiostrepton was used at a final concentration of 50 μM and was preincubated with in vitro translation mix components for 3 min at 37°C. Ribosomes were allowed to bind for 15 min at 37°C and then reverse transcription was performed using Superscript III reverse transcriptase (Invitrogen) according to manufacturer's instructions. Reactions were extracted twice with acid phenol/chloroform, precipitated with ethanol, dissolved in formamide loading buffer and loaded on 5% denaturing polyacrylamide gel along with sequencing reactions prepared using Thermo Sequenase Cycle Sequencing Kit (Affymetrix).
Probing of RNA structure
In vitro synthesized mcc RNA (above) was dephosphorylated by calf intestinal phosphatase (Fermentas) and then 5′-end labeled with T4-polynucleotide kinase (Fermentas) according to manufacturer's protocols. 0.2 pmol of 5′-32P-labeled RNA was denatured for 5 min at 95°C in 10 μl of prefold buffer (40 mM HEPES pH 8.0, 100 mM NH4Cl, 4 mM DTT) and chilled on ice for 5 min. MgCl2 was added to final concentration 10 mM and RNA was allowed to fold by incubating the reactions for 10 min at 37°C followed by the addition of 1 μg of yeast RNA. Reactions were next supplied with 1 μl of RNase T1 (0.01 u/μl) or RNase T2 (0.01 u/μl) (Boehringer-Mannheim) and incubated for 5 min at 37°C. When probing the RNA structure in the presence of ribosomes, the in vitro translation mix (In Vitro Protein Synthesis PURExpress kit components, preincubated with thiostrepton) was added after RNA denaturation in prefold buffer.
RNase T1 marker ladders were obtained by incubating 0.2 pmol 5′-32P- labeled RNA in 1xSequencing buffer (20 mM sodium citrate pH 5, 1 mM EDTA, 7 M urea) for 5 min at 95°C. Reactions were chilled and 1.5 μl of RNase T1 (0.1 U/μl) was added followed by 5 min incubation at 37°C. All reactions were stopped by the addition of equal volume (10 μl) of formamide loading buffer, except for reactions contained PURExpress system, which were stopped by the addition of 100 μl ASE buffer (0.3 M NaAc, 0.5% SDS, 5 mM EDTA, pH 5.3), extracted twice with acid phenol/chloroform, precipitated with ethanol and then dissolved in formamide loading buffer.
Plate assay for McC production
Hundred microliter of fresh night culture of E. coli BL21 (DE3) cells was mixed with 5 ml 0.7% (soft) LB agar and poured over a 1.5% LB agar plate. After drying, 5 μl aliquots of tested cell cultures or McC solutions of known concentrations (1–20 μM) were applied on the plate surface. Plates were incubated at 37°C for 3 h to allow the lawn to grow, and the sizes of growth inhibition zones formed around antibiotic solution drops were measured.
RESULTS
An abundant short mccA transcript is required for McC production
To get an overview of the mccABCDE operon transcription, total RNA was prepared from E. coli cells harboring the McC-production plasmid. Cells were grown in LB broth to late exponential phase, a condition, when the culture was actively synthesizing McC ((14) and data not shown). Total RNA was purified from a culture aliquot and subjected to the differential RNA-seq (dRNA-seq) procedure that allows one to identify the 5′ ends of transcripts (19). During dRNA-seq, half of the RNA sample is subjected to TAP treatment prior to library construction, while another half is treated with 5′-phosphate-dependent exonuclease (TEX), an enzyme that destroys RNA molecules containing 5′-monophosphates, followed by TAP treatment and library construction. Sequencing reads present in libraries prepared from TEX treated RNA (+TEX) are enriched with sequences whose 5′ ends correspond to transcription initiation start points, while the TAP-only (−TEX) treated samples contain reads corresponding to both transcription initiation start points and exo- and endonucleolytic processing/degradation events. The results of dRNA-seq analysis for RNAs synthesized from the mcc operon are shown in Figure 2A. As can be seen, at conditions of McC production high coverage corresponding to the beginning of the mcc operon was observed in the −TEX library. Coverage was decreased ca. 30-fold in samples treated with TEX. The location of 5′ ends of these reads corresponds to the published location of the start point of Pmcc, a CAP-cAMP-dependent promoter located upstream of mccABCDE (14,15). Inspection of Figure 2A also reveals the apparent absence of other strong transcription initiation points within the mccABCDE operon, indicating that transcription from Pmcc is primarily responsible for operon expression at conditions tested. Several potential minor start points within the mccB and mccE gene are apparent. These start points are not preceded by recognizable promoter consensus elements and their significance is unknown. The mRNAs initiated from these hypothetical start points do not contain full-sized mccB ORF and so can have no influence on the MccA/MccB ratio in the producing cell, which is the subject of this study.
Figure 2.
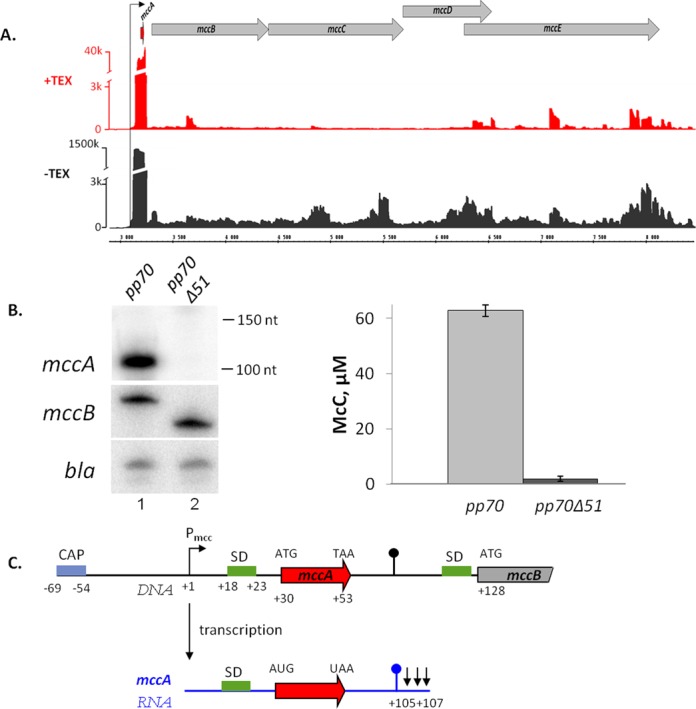
In vivo analysis of the mcc operon transcripts. (A) Analysis of the mccABCDE operon of E. coli using differential RNA-Seq. RNA was extracted from E. coli cells harboring the McC-production plasmid pp70 and grown to late exponential phase in LB medium. Half of RNA was treated with Terminator™ 5′-phosphate-dependent exonuclease (TEX) (+TEX), while another half was left untreated (−TEX) and then converted into a cDNA library, which was subjected to high-throughput Illumina sequencing. cDNA sequencing reads were aligned to the mcc operon and visualized as coverage plots representing the number of reads per nucleotide (‘k’ for thousands of reads) in the IGB (Affymetrix). In the IGB screenshot shown plasmid coordinates are indicated in the middle; gray arrow represent annotated ORFs, black arrow denotes Pmcc and the ORF of the small peptide mccA is shown as a red arrow. (B) At the left-hand side, results of detection of mcc transcripts in cells harboring the wild-type McC-production plasmid pp70 and its derivative pp70Δ51 harboring a deletion in the mccA–mccB region are shown: top—Northern blotting with an mccA-specific probe. Positions of Decade™ RNA Markers are indicated on the right. Middle—primer extension using a primer annealing at the beginning of the mccB gene. Bottom—primer extension using a primer annealing to plasmid-borne bla gene. At the right-hand side, the levels of McC production by cells harboring the wild-type or mutant McC-production plasmids and used for transcript analysis are shown. (C) Summary of mccA transcript mapping. A fragment of the mccABCDE operon DNA (in black) and the short mccA transcript (in blue) are schematically shown, arrows in the end of mccA transcript indicate results of 3′ end mapping by 3′-RACE. SD sequences of mccA and mccB ORFs are shown in green.
Northern-blotting analysis using an oligonucleotide complementary to the mccA ORF was also performed. A prominent ∼100 nt long RNA was present in McC-producing cells (Figure 2B, left, top panel, lane 1) but not in control cells lacking the mcc plasmid (data not shown). This RNA was also not detected in cells harboring an mcc plasmid with a deletion that removed the putative terminator in the intergenic region between the mccA and mccB genes (Figure 2B, left, top panel, lane 2). The amount of control transcript from plasmid-borne bla gene was unaltered by the deletion (Figure 2B, left, bottom panel, compare lanes 1 and 2).
The 5′ and 3′ ends of the mccA RNA were identified using RACE. A single 5′ end located 29 nt upstream of the mccA ATG codon was detected, coinciding with the previously reported Pmcc start point (15) and dRNA-seq data (above). The 3′ ends of the mccA transcripts mapped to three consecutive adenine residues downstream of the stem-loop formed by the putative intergenic transcription terminator hairpin. Thus, the size of the short mccA transcript is ∼105–107 nt. It contains the entire mccA ORF as well as a 52–54-nt 3′ untranslated region.
To monitor the polycistronic (mccA and downstream genes) mcc transcript, primer extension reactions with a primer annealing in the mccB gene were performed. A single primer extension product was observed (Figure 2B, left, middle panel, lane 1) and its 5′ end matched the transcription start point of the monocistronic mccA transcript (data not shown). The amount of polycistronic transcript appears to be unchanged in cells harboring a plasmid with deletion of the putative terminator (Figure 2B left, middle panel, compare lanes 1 and 2).
Semi-quantitative analysis using in vitro transcribed mono- and polycistronic mcc RNA as markers allowed us to estimate that there are ca. 20 molecules of monocistronic transcript for every polycistronic mcc transcript molecule present in McC-producing cells (data not shown). Cells harboring an mcc plasmid with disrupted putative terminator produced ca. 30 times less McC than cells harboring the wild-type plasmid (Figure 2B, right). The result thus suggests that the short mccA transcript is required for high McC production, presumably by allowing the synthesis of sufficient amounts of the MccA peptide to be converted into McC by MccB. The results of mccA transcript mapping are summarized in Figure 2C.
Short mccA transcript is highly stable and is ribosome-bound
The data presented above can be explained by either transcription termination or transcript processing between the mccA and the mccB ORFs. A processing event would be expected to generate a new 5′ end that should be detectable by primer extension with oligonucleotides annealing to mccB. No such primer extension product was detected (data not shown), suggesting that transcription termination is likely responsible for the appearance of the short mccA transcript. The high ratio of monocistronic to polycistronic mccA transcripts may result from very efficient transcription termination in the intergenic region or it may be caused by differential transcript stability (a combination of these two effects is also possible). To determine transcript stabilities, McC-producing cells were treated with rifampicin, an inhibitor of transcription initiation, and decay of mcc RNA abundance was followed over time by Northern blotting (for mccA transcript) and primer extension (for polycistronic transcript). The results are presented in Figure 3A. As can be seen, the half-life of polycistronic transcript was several minutes, i.e. comparable to half-lives of most E. coli mRNAs (20). In contrast, the short mccA transcript was exceedingly stable (almost no change in transcript abundance after a 20-min incubation with rifampicin). The exceptional stability of the mccA transcript may be responsible for the abnormally low (∼3%) ratio of TEX-resistant transcripts revealed by dRNA-seq (Figure 2A), since transcripts that persist for a long time could lose their 5′ triphosphate moiety (21).
Figure 3.
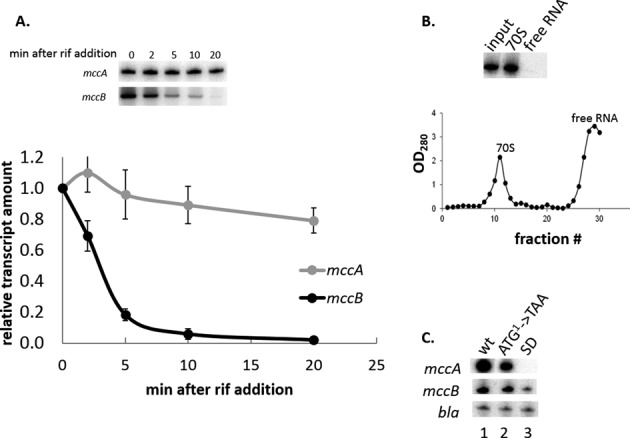
The mccA transcript is stable and ribosome-bound. (A) An McC-producing culture of E. coli harboring the pp70 plasmid was treated with rifampicin. At times indicated, aliquots of the culture were withdrawn, total RNA was purified and amounts of mccA and mccB transcripts were measured by Northern blotting and primer extension, respectively, as described in Figure 2B legend. The results were quantified and the mean values and standard deviations in mRNA abundance obtained in three independent experiments are presented. (B) Lysates of McC-producing cells were subjected to sucrose gradient centrifugation, fractions were collected and the amounts of mccA RNA were determined by Northern blotting. Gradient profile of sucrose centrifugation is shown below the gel. (C) Total RNA was purified from McC-producing E. coli culture harboring the pp70 plasmid or from equally grown cultures harboring pp70 derivatives with a mutated mccA gene initiating codon (ATG1->TAA) or multiple substitutions in the SD sequence of mccA and amounts of the mccA transcript (top), mccB transcript (middle) or bla transcript (bottom) were determined by Northern blot (mccA) or primer extension (mccB, bla) using appropriate primers.
We next wondered what could be the reason of exceptional stability of the mccA transcript, an obviously important part of the mechanism that contributes to preferential accumulation of the mccA message in the cell. The short transcript could be intrinsically stable (for example, there are known cases of 3′ end stem-loops stabilizing bacterial RNA (22–25)) or it could be stabilized by ribosome binding to and/or translation of the mccA ORF (26) (since the mccA ORF is only seven codons long, only one ribosome can bind to mccA RNA). Using sucrose gradient centrifugation, we showed that the mccA transcript is only present in the ribosome-containing fraction and is undetectable in the free RNA fraction (Figure 3B), consistent with idea that ribosome-binding and/or translation may have a stabilizing effect on short mccA mRNA. We next generated mutant variants of McC-producing plasmid that contained (i) a TAA instead of the initiating ATG codon of the mccA gene or (ii) down-mutations in the Shine-Dalgarno (SD) sequence of the mccA gene. As expected, cells harboring mutant plasmids did not produce McC (data not shown). Primer extension analysis using the mccB-specific primer indicated that compared to the wild-type, mutation in the initiating codon had no effect on transcript abundance, while mutation of the SD sequence decreased the abundance of this transcript ca. 3-fold (Figure 3C). The latter effect appears to have been caused by increased mRNA degradation, since introduction of substitutions in the mccA SD sequence led to the appearance of multiple additional primer extension products shorter than the primary product extending to the mcc transcript start point (data not shown). In contrast, Northern blot analysis indicated that the initiating codon mutation decreased, while SD mutation abolished the production of the short transcript. In the case of initiating codon mutant, the residual short transcript was found to be ribosome-bound and was stable (data not shown).
In vitro analysis of transcription termination in the mccA–mccB intergenic region
While our inability to detect the mccA transcript in cells where ribosome binding to mccA was disrupted made it impossible to determine stabilities of mutant transcripts, the result hinted that the free short transcript is unstable or, more interestingly, that ribosome binding may somehow stimulate short transcript production. To address this issue, in vitro transcription and transcription-translation experiments were performed. Since mcc promoter has very low basal transcription activity in vitro (data not shown), we used a chimeric transcription template containing a strong T7 A1 promoter with modified initial transcribed sequence (extending from positions −67 to +30, 5′-tccagatcccgaaaatttatcaaaaagagtattgacttaaagtctaacctataggatacttacagccAtcgagagggccacggcgaacagcaaccca-3′, the promoter consensus elements are underlined, the +1 position is capitalized) fused to a fragment of mcc DNA from position +1 to +154 with respect to the mcc promoter transcription start point. Transcription templates containing mutations at the mccA initiating codon and/or its SD sequence (above) were also created. Transcription templates were combined with E. coli RNA polymerase σ70 holoenzyme and transcription was initiated with a C−1pA+1pU+2pC+3 primer, ATP, GTP and radioactive CTP. Since the first adenine in the template strand is located in position +35 of the template, transcription is stalled before this position. The stalled transcription complexes contain a radioactive nascent 34-nt long RNA whose extension can be monitored upon the addition of UTP. As can be seen from Figure 4A, lane 2, very little, if any, transcription termination was detected with the wild-type template. However, when elongation of the nascent 34-mer RNA was performed in the presence of defined PURExpress in vitro translation system, a new transcript whose 3′ end matched the 3′ end of mccA RNA observed in vivo was produced on the wild-type template (Figure 4A, lane 5). The PURExpress system used in Figure 4A, lane 5, contains all components of the translation machinery and ribosomes. To determine whether transcription termination in the mccA–mccB intergenic region requires translation of the mccA gene, in vitro transcription of wild-type template was performed in the presence of PURExpress (Δaa, tRNA) system lacking amino acids and tRNA (Figure 4A, lane 4), or in the presence of PURExpress ΔRibosome system that lacks ribosomes but contains all other translation system components (Figure 4A, lane 3). As can be seen, transcription termination required the presence of ribosomes but was not dependent on the presence of amino acids and tRNA.
Figure 4.
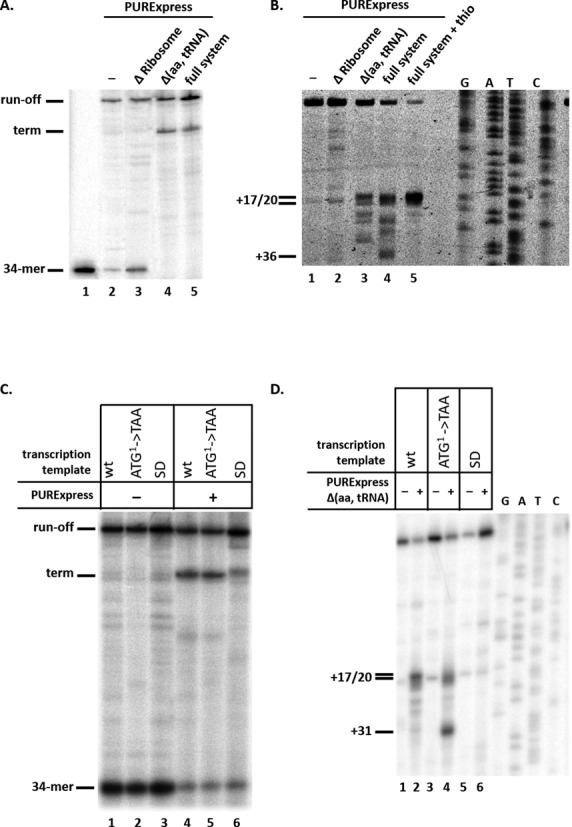
Ribosomes stimulate transcription termination in the mccA–mccB intergenic region in vitro. (A) Stalled E. coli RNA polymerase transcription elongation complexes containing a 34-nt long radioactively labeled RNA were formed on a DNA template containing the T7 A1 promoter fused to a fragment of mcc operon extending from position +1 to position +154. Transcription elongation was allowed to resume by the addition of NTPs (lane 1), or NTPs with PURExpress ΔRibosome (lane 2), Δ(aa, tRNA) (lane 3) or full system (lane 4). An autoradiograph of a denaturing gel showing reaction product separation is shown. (B) In vitro transcribed mccA RNA was combined with PURExpress ΔRibosome (lane 2), Δ(aa, tRNA) (lane 3) or full system (lanes 4 and 5) at conditions identical to those used in panel A experiment, and toeprinting was performed. In lane 5, reaction contained 50 μM thiostrepton. The products of toeprinting reactions were resolved by denaturing gel-electrophoresis and revealed by autoradiography. The 3′ end of the cDNA products synthesized in the toeprint experiment and labeled +17/20 correspond to bound ribosomes with the first (+17) or second (+20) codons of mccA located in the ribosomal P site (27). (C) Transcription complexes were formed as described in A on wild-type or mutant transcription templates with mutated first codon of the mccA gene (ATG1->TAA) or substitutions in the mccA SD sequence and transcription elongation was allowed to proceed. (D) In vitro transcribed wild-type mccA RNA or RNAs harboring mccA (ATG1->TAA) or SD sequence substitutions were subjected to toeprinting reaction in the presence or in the absence of PURExpress Δ(aa, tRNA) system. All designations are as in panel B.
Our data suggest that the presence of ribosomes, even in the absence of translation, stimulates transcription termination in the mccA–mccB intergenic region. To detect ribosome binding to the mccA transcript, a toeprinting experiment was conducted using purified monocistronic mccA mRNA prepared by in vitro transcription with T7 RNAP from an appropriate DNA template. In the absence of ribosomes reverse transcriptase stalled at several points along the transcript (Figure 4B, lane 1). The stalling apparently increased in the presence of PURExpress ΔRibosome system compared to what was observed with just pure RNA (Figure 4B, compare lanes 2 and 1). In the presence of PURExpress (Δaa, tRNA) system, a prominent toeprint was observed (Figure 4B, lane 3). The major toeprints were located 17/20 nt downstream of the mccA ORF start point, corresponding to the ribosome location at the first and second codons of mccA (27). Additional minor toeprints were located further downstream (up to position +31) and correspond to ribosome binding within the mccA ORF. A toeprinting experiment with PURExpress full system was also conducted (Figure 4B, lane 4). At these conditions, which support the synthesis of the MccA heptapeptide, a change in the toeprinting pattern was observed. In addition to toeprints corresponding to ribosome bound at the beginning and within the mccA ORF, another toeprint, at position +36, which corresponds to ribosome stalled at the last sense codon of mccA, was detected. The addition of thiostrepton, an antibiotic that inhibits ribosome translocation (28), to reactions containing full system led to accumulation of the toeprint at position +17 corresponding to ribosome bound at the initiating codon and eliminated the downstream toeprints (Figure 4B, lane 5).
Since the PURExpress (Δaa, tRNA) system causes efficient transcription termination (Figure 4A, lane 3) and appearance of a toeprint (Figure 4B, lane 3), we infer that ribosome binding to the mccA ORF in the absence of translation stimulates downstream transcription termination. The fact that efficient transcription termination is observed in the presence of the full system and thiostrepton (Supplementary Figure S1) is also consistent with this inference. Further support to the idea that ribosome binding is sufficient to induce transcription termination came from in vitro transcription termination analysis using mutant mccA templates. As can be seen (Figure 4C, compare lanes 4 and 5) transcription termination in the presence of PURExpress full system was unaffected by a mutation in the initiating codon of mccA but was strongly reduced by the SD sequence mutations (Figure 4C, lane 6). In the latter case, a diffuse band migrating just above a sharp band corresponding to the transcription termination event was observed. This band is caused by nucleolytic degradation of the full-sized transcript since it is also observed when phenol-extracted full-sized in vitro transcribed RNA is combined with transcription/translation reactions components (Supplementary Figure S2).
A toeprinting experiment with purified mccA mRNAs containing mutations in the initiating codon of mccA or in the SD sequence was next conducted in the presence of PURExpress (Δaa, tRNA) system (Figure 4D). A toeprint at positions corresponding to ribosome binding at the first and second mccA codons was seen with the wild-type template (Figure 4D, lane 2) while no toeprint was detected with mRNA containing substitutions in the SD sequence (Figure 4D, lane 6). The substitution of the initiating codon decreased the intensity of toeprint corresponding to ribosome bound at the initiating codon of mccA but led to a strong toeprint at position +31 (Figure 4D, lane 4), corresponding to ribosome bound at internal AUG codon located in the +1 reading frame within the mccA ORF.
We infer from these data that a ribosome bound upstream of or within mccA stimulates the formation of transcription terminator responsible for formation of the short mccA transcript that is required for McC synthesis. It therefore follows that the part of mccA RNA that is covered by the bound ribosome participates in interactions that inhibit the formation of the transcription terminator hairpin. Two types of experiments support this view. First, when transcription elongation was performed in the presence of an oligonucleotide annealing to mccA transcript positions +69/+91 (oligo 1) and thus covering the left arm of the putative stem-loop structure of the terminator (Figure 5D), transcription termination was low even in the presence of PURExpress system (Figure 5A, compare lanes 2 and 3). This result is consistent with an idea that for transcription termination to occur, a stem-loop structure at the terminator must form. On the other hand, when the transcription reaction was performed in the presence of DNA oligonucleotide annealing to mccA transcript positions +26/+48 (oligo 2) and thus overlapping with position of the bound ribosome, strong transcription termination was observed even in the absence of PURExpress system (Figure 5B, lane 1). Thus, oligo 2 mimicked ribosome binding, presumably by relieving inhibitory interactions in the nascent mccA RNA that suppressed termination. A DNA oligonucleotide annealing to mcc region +51 to +71 (oligo 3), immediately downstream of the bound ribosome also induced transcription termination (Figure 5B, lane 2), though the effect was weaker than with oligo 2. Thus, both the part of the mccA RNA that is covered by the ribosome and the segment located immediately downstream inhibit transcription termination.
Figure 5.
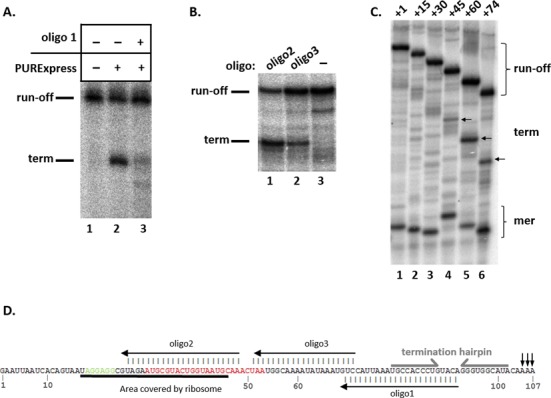
Mapping an element of the mccA transcript that suppresses transcription termination in the mccA–mccB intergenic region. (A) Stalled E. coli RNA polymerase transcription elongation complexes containing a 34-nt long radioactively labeled RNA were formed on a DNA template containing the T7 A1 promoter fused to a fragment of mcc operon extending from position +1 to position +154. Transcription elongation was allowed to resume by the addition of NTPs (lane 1), NTPs with PURExpress ΔRibosome (lane 2) or NTPs with PURExpress ΔRibosome in the presence of oligonucleotide oligo 1 (lane 3, see also panel D). An autoradiograph of a denaturing gel showing reaction product separation is shown. (B) Stalled E. coli RNA polymerase transcription elongation complexes containing a 34-nt long radioactively labeled RNA were formed on a DNA template containing the T7 A1 promoter fused to a fragment of mcc operon extending from position +1 to position +154. Elongation was resumed by the addition of NTPs in the presence or in the absence of DNA oligonucleotides oligo 2 or oligo3. (C) Single-round in vitro transcription reactions were performed on a DNA containing the T7 A1 promoter fused to a fragment of mcc operon extending from position +1 to position +154 (lane 1) and derivative templates that contained deletions extending from position +1 to a position, indicated in the template name (i.e. template labeled +15 lacked nucleotides +1/+14). The sites of termination are indicated by arrows. (D) The monocistronic mccA transcript. The SD sequence is shown in green, the mccA ORF in red. The area covered by a ribosome bound to the SD sequence, annealed oligos 1, 2 and 3, and the termination hairpin positions are indicated.
To better map the upstream boundary of the mccA RNA segment that affects the terminator formation, in vitro transcription reactions with a series of T7 A1-mcc templates containing deletions in the mcc part were performed in the absence of ribosomes (Figure 5C). As can be seen, removal of up to 29 nt of the initial transcribed mcc sequence had no effect on transcription termination. Removal of 44 nt caused weak termination, while removal of first 59 or 73 nt strongly stimulated transcription termination in the absence of ribosomes. Overall, the results of this analysis demonstrate that mccA RNA positions +30 to +60 inhibit the terminator function in nascent mccA RNA.
We next wished to probe the structure of the mccA RNA and determine whether this structure could be affected by ribosome binding. Both wild-type mccA RNA and mutant versions with substitutions in the initiating codon and the SD sequence were probed with RNases T1 and T2, which are specific to unpaired Gs (T1) and all unpaired nucleotides (T2). RNA digested with RNase T1 under denaturing conditions was used as a size marker. The results, presented in Figure 6A, are consistent with a structure presented in Figure 6B, showing the existence of two prominent stem-loop structural elements. The first element is located immediately downstream of bound ribosome, and contains an RNase T2 hypersensitive loop (loop 1), which is resistant to RNase T1 because it contains only A and U residues. Another stem-loop structure corresponds to the transcription terminator hairpin. Its loop, loop 2, is sensitive to both RNase T2 and RNase T1 (the sensitivity to the latter enzyme is due to the presence of G88). The two mutant RNA structures appear to be the same as that of the wild-type mccA RNA.
Figure 6.
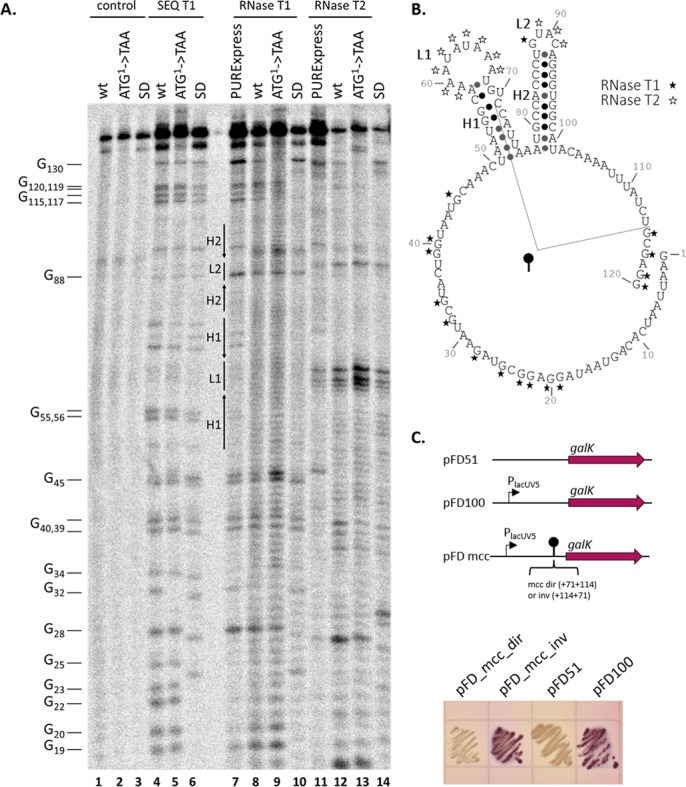
Structural and functional probing of mccA transcript. (A) 32P-5′-end labeled in vitro synthesized wild-type or mutant mccA RNA was digested with RNase T1 or RNase T2. Reaction products were resolved by denaturing PAGE and revealed by autoradiography. Lanes labeled ‘control’ show material that was not treated with RNAses. Lanes labeled ‘SEQ T1’ are marker lanes, showing digestion pattern obtained with RNase T1 under denaturing conditions (cleavages at every G). In lanes labeled ‘RNase T1’ and ‘RNase T2’ transcripts were re-folded in the presence of 10 mM MgCl2 and then digested with RNases. In lanes labeled ‘PURExpress’ wild-type mccA RNA was folded in the presence of PURExpress full system in the presence of thiostrepton. (B) A secondary structure of mccA RNA produced by RNAfold software (http://rna.tbi.univie.ac.at/) and consistent with RNase probing results. Cleavage positions by RNase T1 and RNase T2 are shown, respectively, with black and white asterisks. Elements of the structure (loops, L, and hairpins, H) are also marked on the gel shown in panel A. A fragment of mccA tested for in vivo termination activity is indicated. (C) A DNA fragment coding for a hairpin structure and adjacent sequences shown in panel C was cloned in direct (pFD_mcc_dir) and inverted (pFD_mcc_inv) orientations between the galK reporter and the lacUV5 promoter of plasmid pFD100. The resulting plasmids, as well as control pFD51 plasmid lacking the promoter, were transformed into galK−E. coli cells and transformants were then tested for GalK activity on McConkey indicator agar plates. Overnight growth of cells is shown.
We next performed the probing experiment in the presence of PURExpress full system. While ribosomes seem to have generally protected mccA RNA from RNase T2 digestion, the hypersensitivity in both loops persisted (Figure 6A, compare lanes 7 and 8, and lanes 10 and 11). The result either suggests that changes caused by bound ribosome are not sensed by the probing method used (for example, they may involve a tertiary, rather than secondary structure change) or, that such changes only occur in the context of shorter nascent RNAs, which fold co-transcriptionally.
Since results presented above failed to demonstrate a conformational change in mccA transcript caused by ribosome binding, we wished to prove that mccA terminator alone, in the absence of interfering upstream mccA sequences, can cause transcription termination in vivo. Accordingly, we cloned a fragment of mcc DNA from position +71 to +114 with respect to the transcription start point in between the lacUV5 promoter and the galK gene of plasmid pFD100 (18). This fragment contains the mccA terminator hairpin, the A/T-rich downstream region and a fragment of the upstream mcc hairpin revealed by footprinting experiments (Figure 6B). Plasmids containing mcc inserts in both orientations were constructed. E. coli galK− cells transformed with pFD100 plasmid formed purple-colored streaks on McConkey agar plates (Figure 6C) due to transcription from the lacUV5 promoter. Control cells transformed with pFD51 plasmid lacking this promoter formed white-color streaks. Cells transformed with a plasmid containing the mcc insert in ‘direct’ orientation also formed white-color streaks, demonstrating that the cloned segment of mcc DNA efficiently prevented galK transcription from upstream promoter. In contrast, cells transformed with a plasmid containing the mcc insert in ‘inverted’ orientation formed purple-color streaks. We infer from this experiment that a fragment of mcc DNA carrying the inverted repeat from the mccA–mccB intergenic region and 7 upstream and 13 downstream base pairs functions as a unidirectional transcription terminator in vivo.
The ribosome-dependent mechanism of mccA RNA production is evolutionarily conserved
The mcc-like operons appear to be widespread in phylogenetically diverse bacteria (6). While no production of McC-like compounds by these operons has been shown yet, some of these compounds were recently validated by enzymatic synthesis of in vitro using cognate pairs of MccB-like enzymes/predicted MccA-like substrates (29). It is reasonable to expect that the main features controlling the synthesis of these compounds should be conserved. Indeed, for all mcc-like operons identified to date, a long intergenic region separating the mccA and mccB homologs and containing terminator-like sequences can be identified (6). In Helicobacter pylori strain G27 (30), the mcc-like operon is encoded on a 10,032-bp plasmid pHPG27 (NC_011334) and contains three genes, mccA, mccB and mccC (Figure 7A). The intergenic region between pHPG27 mccA and mccB is 84 bp long (the corresponding region in E. coli mcc is 74 bp long, Figure 7A). To determine whether a short H. pylori mccA transcript is produced and, if so, whether its production can be stimulated by ribosome binding, an in vitro transcription experiment was performed using as a template a DNA fragment containing the T7 A1 promoter fused to the initial transcribed region of H. pylori mcc operon and in the presence of PURExpress system. In the absence of ribosomes, E. coli RNAP produced a single full-sized transcript, which extended into mccB (Figure 7B, lane 1). In contrast, in the presence of ribosomes an additional radioactive band was observed (Figure 7B, lanes 2 and 3). Electrophoretic mobility of this band was consistent with a transcription product whose 3′ end is located shortly after the hairpin of predicted terminator. Since production of the terminated product was observed both in the presence and in the absence of amino acids and tRNA (Figure 7B, compare lanes 2 and 3), the result indicates that similarly to the situation observed in E. coli, ribosome binding causes transcription termination in the mccA–mccB intergenic region of H. pylori.
Figure 7.
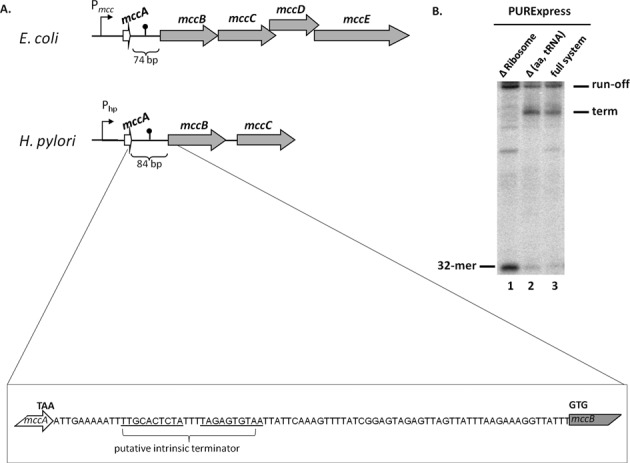
H. pylori mcc operon analysis. (A) Schematic representations of E. coli and H. pylori mcc operons (drawn not to scale). All designations are as in Figure 1C. The mccA–mccB intergenic region of H. pylori is expanded below with the stop codon of mccA, the start codon of mccB and the location of inverted repeat that may form a transcription terminator indicated. (B) Stalled E. coli RNA polymerase transcription elongation complexes containing a 32-nt long radioactively labeled RNA were formed on a DNA template containing the T7 A1 promoter fused to a fragment of H. pylori mcc operon extending from position +1 to position +137. Transcription elongation was allowed to resume by the addition of NTPs in the presence of PURExpress ΔRibosome (lane 1), Δ(aa, tRNA) (lane 2) or full (lane 3) translation systems. An autoradiograph of a denaturing gel showing reaction product separation is shown.
DISCUSSION
In this work, we studied transcriptional control mechanisms that ensure high levels of production of peptide–nucleotide antibiotic microcin C. The transcription of the entire mcc operon occurs from a single upstream promoter. The activity of this promoter requires CAP-cAMP and thus McC production only occurs during nutrient deprivation (14,15). Once transcription of mcc genes is initiated, a proper ratio of the MccA peptide, a product of promoter-proximal gene and a substrate of promoter distal biosynthetic genes products MccB, MccD and MccE, needs to be maintained to assure continuous and high levels of McC production. We here show that the intergenic region between the mccA and mccB genes plays an important role in controlling the overall level McC synthesis. A deletion in this region dramatically reduces McC production while leaving all the coding sequences of mcc genes intact. The deletion also abolishes the production of a highly abundant 105–107-nt transcript that contains the mccA ORF. Consequently, most of the MccA propeptide is synthesized from this short mccA RNA and the observed high abundance of this transcript evidently contributes to efficient McC production.
The monocistronic mccA RNA is produced at transcription terminator located in the intergenic region between mccA and mccB. However, the function of this terminator remains masked by secondary or tertiary interactions within the mccA RNA, such that in the absence of ribosome binding/mccA translation the RNA polymerase proceeds to transcribe downstream mcc genes. If a ribosome binds to the SD sequence upstream of the mccA, transcription termination becomes possible. While the ribosome binding is essential for transcription termination in the mccA–mccB intergenic region, translation of the mccA ORF is not. This effect appears to be due to both the very efficient interaction of ribosome with the mccA SD sequence and the presence of additional sites, including internal out-of-frame ATG codons within mccA where a ribosome can reside causing transcription termination. Presumably, a ribosome bound upstream of or within mccA causes a change in the nascent transcript structure that allows transcription terminator structure to form, although experiments with in vitro transcribed mccA RNA failed to reveal a structural change at least at the level of RNA secondary structure. The mechanism of short mccA transcript generation through ribosome-induced transcription termination may be a conserved feature of predicted mcc-like operons in diverse bacteria. This possibility is clearly implied by our in vitro analysis of transcription of H. pylori mcc operon. The result is particularly striking since sequence conservation between the mccA genes and untranslated regions of E. coli and H. pylori is lacking.
High (∼40% and more) termination efficiencies at the mccA–mccB intergenic terminator can be obtained in vitro in the presence of ribosomes. The large ratio in monocistronic and polycistronic mcc transcripts abundance observed in vivo suggests similarly high termination efficiencies in vivo. The steady-state ratio of monocistronic to polycistronic transcripts (ca. 20:1) is determined by their respective synthesis and degradation rates. Since both transcripts are synthesized from the same promoter, the ratio of their synthesis rates is determined by the termination efficiency. At steady state, the transcript synthesis has to be matched by the transcript decay; from this, it is straightforward to obtain a relationship that connects the termination efficiency with the ratio of the transcript concentrations, and the ratio of their half-lives (see Materials and Methods for derivation). The half-life of the polycistronic transcript can be estimated at ∼3 min, while the monocistronic transcript is stable on the timescale of the cell division, so that its half-life is determined by the cell doubling time (∼30 min). From the above it can then be estimated that the in vivo termination efficiency should be around 65%.
It is clear that exceptional stability of mccA mRNA is responsible for the large ratio of monocistronic to polycistronic transcripts observed. This stability is also due to ribosome binding, so that a ribosome bound to mccA ORF dramatically stabilizes the transcript. Since in the absence of translation the mccA transcript decays rapidly, it is very tempting to speculate that once bound to the mccA message, a ribosome does not dissociate from it anymore, shifting back to the beginning of the seven-codon message once translation of the MccA heptapeptide is completed. Such mechanism would provide an additional boost in the amount of MccA substrate synthesized and made available for adenylation by the MccB. Experiments aimed at testing this scenario are currently in progress in our laboratory.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Russian Academy of Sciences Presidium programs in Molecular and Cellular Biology and Nanotechnology grants; Ministry of Education and Science of Russian Federation [project 14.B25.31.0004 to K.S.]; European Community [PIRG08-GA-2010-276996 to M.D.]; Ministry of Education and Science of the Republic of Serbia under project No ON173052 and SNSF SCOPES (IZ73Z0_152297) to M.D. Funding for open access charge: Russian Academy of Sciences.
Conflict of interest statement. None declared.
REFERENCES
- 1.Asensio C., Pérez-Díaz J.C., Martínez M.C., Baquero F. A new family of low molecular weight antibiotics from enterobacteria. Biochem. Biophys. Res. Commun. 1976;69:7–14. doi: 10.1016/s0006-291x(76)80264-1. [DOI] [PubMed] [Google Scholar]
- 2.Riley M.A., Wertz J.E. Bacteriocins: evolution, ecology, and application. Annu. Rev. Microbiol. 2002;56:117–137. doi: 10.1146/annurev.micro.56.012302.161024. [DOI] [PubMed] [Google Scholar]
- 3.Hernandez-Chico C., Castillo I., Moreno F. The peptide antibiotic microcin B17 induces double-strand cleavage of DNA mediated by E. coli DNA gyrase. EMBO. 1991;10:467–76. doi: 10.1002/j.1460-2075.1991.tb07969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delgado M.A., Rintoul M.R., Farías R., Farías R.N., Salomón R.A. Escherichia coli RNA polymerase is the target of the cyclopeptide antibiotic microcin J25. J. Bacteriol. 2001;183:4543–4550. doi: 10.1128/JB.183.15.4543-4550.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Metlitskaya A., Kazakov T., Kommer A., Pavlova O., Praetorius-Ibba M., Ibba M., Krasheninnikov I., Kolb V., Khmel I., Severinov K. Aspartyl-tRNA synthetase is the target of peptide nucleotide antibiotic microcin C. J. Biol. Chem. 2006;281:18033–18042. doi: 10.1074/jbc.M513174200. [DOI] [PubMed] [Google Scholar]
- 6.Severinov K., Semenova E., Kazakov A., Kazakov T., Gelfand M.S. Low-molecular-weight post-translationally modified microcins. Mol. Microbiol. 2007;65:1380–1394. doi: 10.1111/j.1365-2958.2007.05874.x. [DOI] [PubMed] [Google Scholar]
- 7.Duquesne S., Destoumieux-Garzon D., Peduzzi J., Rebuffat S. Microcins, gene-encoded antibacterial peptides from enterobacteria. Nat. Prod. Rep. 2007;24:708–734. doi: 10.1039/b516237h. [DOI] [PubMed] [Google Scholar]
- 8.Novikova M., Metlitskaya A., Datsenko K., Kazakov T., Kazakov A., Wanner B., Severinov K. The Escherichia coli Yej transporter is required for the uptake of translation inhibitor microcin C. J. Bacteriol. 2007;189:8361–8365. doi: 10.1128/JB.01028-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kazakov T., Vondenhoff G.H., Datsenko K.A., Novikova M., Metlitskaya A., Wanner B.L., Severinov K. Escherichia coli peptidase A, B, or N can process translation inhibitor microcin C. J. Bacteriol. 2008;190:2607–2610. doi: 10.1128/JB.01956-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roush R.F., Nolan E.M., Löhr F., Walsh C.T. Maturation of an Escherichia coli ribosomal peptide antibiotic by ATP-consuming N−P bond formation in microcin C7. J. Am. Chem. Soc. 2013;130:3603–3609. doi: 10.1021/ja7101949. [DOI] [PubMed] [Google Scholar]
- 11.Metlitskaya A., Kazakov T., Vondenhoff G.H., Novikova M., Shashkov A., Zatsepin T., Semenova E., Zaitseva N., Ramensky V., Aerschot A., et al. Maturation of the translation inhibitor microcin C. J. Bacteriol. 2009;191:2380–2387. doi: 10.1128/JB.00999-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Novikova M., Kazakov T., Vondenhoff G.H., Semenova E., Rozenski J., Metlytskaya A., Zukher I., Tikhonov A., Van Aerschot A., Severinov K. MccE provides resistance to protein synthesis inhibitor microcin C by acetylating the processed form of the antibiotic. J. Biol. Chem. 2010;285:12662–12669. doi: 10.1074/jbc.M109.080192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonzalez-Pastor J.E., Millan J.L., Castilla M.A., Moreno F. Structure and organization of plasmid genes required to produce the translation inhibitor microcin C7. J. Bacteriol. 1995;177:7131–7140. doi: 10.1128/jb.177.24.7131-7140.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fomenko D., Veselovskii A., Khmel I. Regulation of microcin C51 operon expression: the role of global regulators of transcription. Res. Microbiol. 2001;152:469–479. doi: 10.1016/s0923-2508(01)01220-7. [DOI] [PubMed] [Google Scholar]
- 15.Moreno F., Gonzalez-Pastor J.E., Baquero M.R., Bravo D. The regulation of microcin B, C and J operons. Biochimie. 2002;84:521–529. doi: 10.1016/s0300-9084(02)01452-9. [DOI] [PubMed] [Google Scholar]
- 16.Datsenko K.A., Wanner B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurepina N.E., Basyuk E.I., Metlitskaya A.Z., Zaitsev D.A., Khmel I.A. Cloning and mapping of the genetic determinants for microcin C51 production and immunity. Mol. Gen. Genet. 1993;241:700–706. doi: 10.1007/BF00279914. [DOI] [PubMed] [Google Scholar]
- 18.Beletskaya I. V., Zakharova M. V., Shlyapnikov M.G., Semenova L.M., Solonin A.S. DNA methylation at the CfrBI site is involved in expression control in the CfrBI restriction-modification system. Nucleic Acids Res. 2000;28:3817–3822. doi: 10.1093/nar/28.19.3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dugar G., Herbig A., Förstner K.U., Heidrich N., Reinhardt R., Nieselt K., Sharma C.M. High-resolution transcriptome maps reveal strain-specific regulatory features of multiple Campylobacter jejuni isolates. PLoS Genet. 2013;9:e1003495. doi: 10.1371/journal.pgen.1003495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bernstein J.A., Khodursky A.B., Lin P., Lin-Chao S., Cohen S.N. Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays. Proc. Natl. Acad. Sci. 2002;99:9697–9702. doi: 10.1073/pnas.112318199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Celesnik H., Deana A., Belasco J.G. Initiation of RNA decay in Escherichia coli by 5′ pyrophosphate removal. Mol. Cell. 2007;27:79–90. doi: 10.1016/j.molcel.2007.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carrier T.A., Keasling J.D. Controlling messenger RNA stability in bacteria: strategies for engineering gene expression. Biotechnol. Prog. 1997;13:699–708. doi: 10.1021/bp970095h. [DOI] [PubMed] [Google Scholar]
- 23.Causton H., Py B., McLaren R.S., Higgins C.F. mRNA degradation in Escherichia coli: a novel factor which impedes the exoribonucleolytic activity of PNPase at stem-loop structures. Mol. Microbiol. 1994;14:731–741. doi: 10.1111/j.1365-2958.1994.tb01310.x. [DOI] [PubMed] [Google Scholar]
- 24.Stern D.B., Radwanski E.R., Kindle K.L. A 3′ stem/loop structure of the Chlamydomonas chloroplast atpB gene regulates mRNA accumulation in vivo. Plant Cell Online. 1991;3:285–297. doi: 10.1105/tpc.3.3.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen C.A., Beatty J.T., Cohen S.N., Belasco J.G. An intercistronic stem-loop structure functions as an mRNA decay terminator necessary but insufficient for puf mRNA stability. Cell. 1988;52:609–619. doi: 10.1016/0092-8674(88)90473-4. [DOI] [PubMed] [Google Scholar]
- 26.Deana A., Belasco J.G. Lost in translation: the influence of ribosomes on bacterial mRNA decay. Genes Dev. 2005;19:2526–2533. doi: 10.1101/gad.1348805. [DOI] [PubMed] [Google Scholar]
- 27.Orelle C., Szal T., Klepacki D., Shaw K.J., Vázquez-Laslop N., Mankin A.S. Identifying the targets of aminoacyl-tRNA synthetase inhibitors by primer extension inhibition. Nucleic Acids Res. 2013;41:e144. doi: 10.1093/nar/gkt526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orelle C., Carlson S., Kaushal B., Almutairi M.M., Liu H., Ochabowicz A., Quan S., Pham V.C., Squires C.L., Murphy B.T., et al. Tools for characterizing bacterial protein synthesis inhibitors. Antimicrob. Agents Chemother. 2013;57:5994–6004. doi: 10.1128/AAC.01673-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bantysh O., Serebryakova M., Makarova K., Dubiley S., Datsenko K. A., Severinov K. Enzymatic synthesis of bioinformatically predicted microcin C-like compounds encoded by diverse bacteria. MBio. 2014;5 doi: 10.1128/mBio.01059-14. doi:10.1128/mBio.01059-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baltrus D.A., Amieva M.R., Covacci A., Lowe T.M., Merrell D.S., Ottemann K.M., Stein M., Salama N.R., Guillemin K. The complete genome sequence of helicobacter pylori strain G27. J. Bacteriol. 2009;191:447–448. doi: 10.1128/JB.01416-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.