


Abstract
The multifunctional RNA-dependent RNA polymerase L protein of vesicular stomatitis virus catalyzes unconventional pre-mRNA capping via the covalent enzyme-pRNA intermediate formation, which requires the histidine–arginine (HR) motif in the polyribonucleotidyltransferase domain. Here, the effects of cap-defective mutations in the HR motif on transcription were analyzed using an in vitro reconstituted transcription system. The wild-type L protein synthesized the leader RNA from the 3′-end of the genome followed by 5′-capped and 3′-polyadenylated mRNAs from internal genes by a stop–start transcription mechanism. Cap-defective mutants efficiently produced the leader RNA, but displayed aberrant stop–start transcription using cryptic termination and initiation signals within the first gene, resulting in sequential generation of ∼40-nucleotide transcripts with 5′-ATP from a correct mRNA-start site followed by a 28-nucleotide transcript and long 3′-polyadenylated transcript initiated with non-canonical GTP from atypical start sites. Frequent transcription termination and re-initiation within the first gene significantly attenuated the production of downstream mRNAs. Consistent with the inability of these mutants in in vitro mRNA synthesis and capping, these mutations were lethal to virus replication in cultured cells. These findings indicate that viral mRNA capping is required for accurate stop–start transcription as well as mRNA stability and translation and, therefore, for virus replication in host cells.
INTRODUCTION
Non-segmented negative strand (NNS) RNA viruses (e.g. rabies, measles, Ebola) belonging to the Mononegavirales order possess their multifunctional RNA-dependent RNA polymerase (RdRp) large (L) proteins, which catalyze all enzymatic reactions required for viral mRNA synthesis and processing (1–12). Virion-associated RdRp holoenzymes that are composed of the catalytic L protein and co-factor phosphoprotein (P or its counterpart) initiate primary transcription using their negative strand genomic RNAs encapsidated with nucleoproteins (N or NP) as templates in infected cells. Historically, vesicular stomatitis virus (VSV), an animal virus belonging to the Rhabdoviridae family, has been used as a prototypic model virus to investigate the fundamental molecular mechanisms of NNS RNA viral mRNA synthesis and processing.
The VSV genome of ∼11 kilo nucleotides (knt) is preceded and followed by the short 3′-leader and 5′-trailer sequences, respectively, and includes internal N, P, M, G and L genes arranged in tandem from the 3′- to the 5′-end. Each gene contains the conserved gene-start (3′-UUGUCDNUAG; D: A, U or G; N: any nucleotide) and gene-end (3′-MUACUUUUUUU; M: A or C) sequences, which serve as transcription initiation and termination signals, respectively. Earlier studies using in vitro transcription systems have shown that the VSV RdRp complex initiates transcription at the 3′-end of the genomic RNA to synthesize the 47-nt uncapped leader RNA (13–15) and then sequentially produces five 5′-capped and 3′-polyadenylated mRNAs from the internal genes by a stop–start mechanism (16–21). Reduction in the efficiency of transcription re-initiation at each gene-start sequence results in the generation of a gradient in transcript abundance in the following order: leader > N > P > M > G > L (18–21).
All VSV mRNAs are transcribed from each gene-start sequence, and 5′-capped and methylated (when S-adenosylmethionine is present) during in vitro transcription, generating a common 5′-terminal sequence with a cap 1 structure, m7GpppAmACAG-, in which 7-methylguanosine (m7G) is linked to 2′-O-methyladenosine (Am) via a 5′-5′ triphosphate bridge (16,22). In the absence of S-adenosylmethionine, VSV mRNAs with a 5′-cap core structure (GpppA) are synthesized in a methylation-independent manner (17). These capped mRNAs are co-transcriptionally polyadenylated at their 3′-ends by polymerase slippage on the poly(U) tract in the gene-end sequence (21,23–26).
In eukaryotic cells, the 5′-terminal mRNA cap structure (m7GpppN-) is formed by nuclear mRNA capping enzyme having the RNA 5′-triphosphatase (RTPase) and guanosine 5′-triphosphate (GTP):RNA guanylyltransferase (GTase) activities followed by mRNA cap (guanine-N7)-methyltransferase and plays essential roles in various steps of gene expression including mRNA splicing, translation and degradation (27–29). The mechanisms of VSV mRNA capping are distinctly different from those of eukaryotic mRNA capping (30,31). Using an in vitro capping assay with an oligo-RNA substrate, it has been demonstrated that the VSV L protein (241 kDa, 2109 amino acids) catalyzes a unique RNA capping reaction by the sequential action of the guanosine 5′-triphosphatase (GTPase) and RNA:guanosine 5′-diphosphate (GDP) polyribonucleotidyltransferase (PRNTase) activities (4,6,32).
In the first step of the VSV cap formation, the GTPase activity removes the γ-phosphate from GTP to produce GDP (4,32). Then, the PRNTase domain in the L protein specifically recognizes the 5′-triphosphorylated conserved mRNA-start sequence (5′-pppAACAG-), but not leader RNA-start sequence (5′-pppACGAA-), to form a covalent enzyme (L)-pRNA intermediate (4,6). In the L-pRNA intermediate, the 5′-phosphate group of the RNA is attached to the Nϵ2 position of a histidine residue at position 1227 (H1227) in the histidine–arginine (HR) motif of the VSV L protein with a phosphoamide bond (6). The L-pRNA intermediate subsequently transfers pRNA to GDP to liberate a 5′-capped RNA (GpppRNA) (6).
The HR motif is essential for the L-pRNA intermediate formation with the VSV and Chandipura virus L proteins and strikingly conserved in the L proteins of NNS RNA viruses belonging to different families (5,6,30,33), suggesting that it plays a fundamental role in NNS RNA viral mRNA capping. The R1221 residue of the VSV L protein as well as its counterpart of the Chandipura virus L protein is also required for the mRNA capping activity in the step of the L-pRNA intermediate formation, and its counterpart is conserved in the L proteins of vesiculoviruses (e.g. VSV, Chandipura virus), lyssaviruses (e.g. rabies virus) and ephemeroviruses (e.g. bovine ephemeral fever virus) belonging to the Rhabdoviridae family, but not in other NNS RNA viral L proteins (5,6,30,31).
Several studies suggested that pre-mRNA capping occurs at an early stage of mRNA chain elongation (20,34–36) and is required for production of full-length mRNAs (37–39). Li et al. (38,39) showed that cap-defective VSV L mutants terminate mRNA synthesis at various sites to produce heterogeneous 3′-truncated transcripts with 100–500 nt and are not able to polyadenylate mRNAs at 3′-ends, suggesting that pre-mRNA capping is required for processive elongation and polyadenylation of mRNA. However, how pre-mRNA capping with a transcribing modular L protein regulates each step of mRNA production still remains largely unknown.
To elucidate the molecular mechanisms of VSV mRNA synthesis and processing, an in vitro reconstituted transcription system with highly purified viral components (the N–RNA complex and the recombinant L and P proteins) has been developed (4,40). In this system, the wild-type L protein together with the P protein efficiently synthesizes the 47-nt uncapped leader RNA and full-length mRNAs (N, P, M and G) with a cap structure and a poly(A) tail (40). Here, using this in vitro system, the effects of cap-defective mutations in the HR motif (6) on transcription were re-evaluated. The cap-defective mutants synthesized the leader RNA, but exhibited unexpected aberrant stop–start transcription using cryptic transcription signals within the 3′-proximal N gene, resulting in sequential and discontinuous production of non-overlapping 5′-terminal (38 and 40 nt), internal (28 nt) and 3′-terminal (1.2 knt, 3′-polyadenylated) fragments of N mRNA with a 5′-triphosphate group. The latter two RNAs were found to be initiated with GTP, instead of canonical adenosine 5′-triphosphate (ATP), at different unusual start sites. These results indicate that the pre-mRNA capping activity of the L protein is required for accurate selection of transcription termination and re-initiation signals to synthesize full-length mRNAs by the stop–start mechanism, rather than increases in processibility of mRNA chain elongation and polyadenylation. Higher rates of incorrect transcription termination and re-initiation within the N gene resulted in a dramatic attenuation of synthesis of downstream mRNAs. Furthermore, this study, for the first time, shows that these cap-defective mutations are lethal to VSV replication in host cells, suggesting that the unique mRNA capping enzyme domain of NNS RNA viral L proteins is a potential target for developing anti-viral agents.
MATERIALS AND METHODS
In vitro transcription and capping with the VSV L protein
In vitro transcription was performed with the recombinant L protein, recombinant P protein and N–RNA complex as described previously (4,40), with some minor modifications (see the Supplementary Methods). Mutant L proteins including the HR-RH mutant were prepared as described previously (6). 32P-labeled transcripts were analyzed by electrophoresis in polyacrylamide gels containing 7-M urea [urea-polyacrylamide gel electrophoresis (PAGE)] followed by autoradiography (4,40). Unlabeled transcripts were analyzed by northern blotting with 5′-end-32P-labeled oligo-DNA probes as described (40,41) (see the Supplementary Methods). Unlabeled transcripts, synthesized with the HR-RH mutant L protein, were capped with the transcription-defective D714A L mutant (3,6) in the presence of [α-32P]GDP as described previously (40) (see the Supplementary Methods). Cap-labeled RNAs were digested with nuclease P1 (Sigma), and the resulting digests were analyzed along with cap analogs (GpppA and GpppG; New England Biolabs) by thin layer chromatography (TLC) on a polyethyleneimine (PEI) cellulose plate as described (40).
Enzymatic sequencing of transcripts
Unlabeled transcripts were synthesized with the wild-type or the HR-RH mutant L protein (6) and capped with vaccinia virus mRNA capping enzyme (Epicenter) in the presence of [α-32P]GTP as detailed in the Supplementary Methods. Major cap-labeled short RNAs were purified by urea-PAGE and subjected to partial digestions with RNase T1 or RNase A, essentially as described by Kuchino and Nishimura (42) (see the Supplementary Methods). The resulting digests were resolved on 20% polyacrylamide sequencing gels containing 7-M urea.
5′-RACE (rapid amplification of cDNA ends) analysis
Unlabeled transcripts, synthesized with the wild-type or HR-RH mutant L protein (see the Supplementary Methods), were incubated with or without 1 unit of calf intestine alkaline phosphatase (CIAP; Roche Applied Science) at 37°C for 30 min. After inactivation of CIAP, the RNAs were further incubated with or without 2.5 units of tobacco acid pyrophosphatase (TAP, Epicenter) at 37°C for 1 h. Then, 5′-monophosphate ends of the RNAs were ligated to a partially double-stranded adapter oligo-RNA with T4 RNA ligase 2 (New England Biolabs) according to the protocol described by Clepet (43). The resulting ligated RNAs were reverse-transcribed with AccuScript Hi-Fi reverse transcriptase (Agilent Technologies) and an oligo(dT) primer in 20-μl reaction mixtures. One-half-microliter aliquots of the resulting first-strand cDNAs were subjected to 28 cycles of polymerase chain reaction (PCR) amplification with KOD Hot Start DNA polymerase (Toyobo, Japan) using an adapter-specific forward primer with a 5′ EcoRI site and a reverse primer, complementary to nucleotide positions +479 to +500 of N mRNA, with a 5′ HindIII site. The resulting PCR products were separated by electrophoresis on a 2% agarose gel containing ethidium bromide and purified from the gel using QIAquick Gel Extraction Kit (Qiagen). The purified PCR products were digested with EcoRI and HindIII, inserted into the EcoRI and HindIII sites of the pUC18 vector and sequenced with the M13 RV-M primer.
Primer extension analysis
Unlabeled transcripts, synthesized with the wild-type or HR-RH mutant L protein (see the Supplementary Methods), were subjected to primer extension reactions at 50°C for 10 min with 0.03 units of Maxima H Minus reverse transcriptase (Thermo Scientific) and a 5′-end-32P-labeled oligo-DNA primer complementary to positions +168 to +188 of N mRNA. Primer-extended DNA products were analyzed along with sequencing ladders (see the Supplementary Methods) by electrophoresis in an 8% polyacrylamide sequencing gel containing 7-M urea followed by autoradiography.
Measurement of poly(A) tail length
Poly(A) tail lengths were measured essentially as described (44). Unlabeled transcripts were annealed with an oligo-DNA complementary to positions +1107 to +1126 of N mRNA in the presence or absence of oligo(dT) and digested with 0.3 units of RNase H at 37°C for 3 min. The resulting 3′-end fragments (200 nt) with or without a poly(A) tail were analyzed by northern blotting with a 5′-end-32P-labeled oligo-DNA probe complementary to positions +1241 to +1268 of N mRNA.
Generation of recombinant VSVs
Recombinant VSVs with the wild-type or mutant L gene were generated using the reverse genetics system as described by Lawson et al. (45) and Schnell et al. (46) with some modifications (see the Supplementary Methods). Viral titers were determined by a standard plaque assay.
RESULTS
Cap-defective mutant L proteins frequently use cryptic transcription termination and initiation signals within the N gene to produce aberrant short transcripts
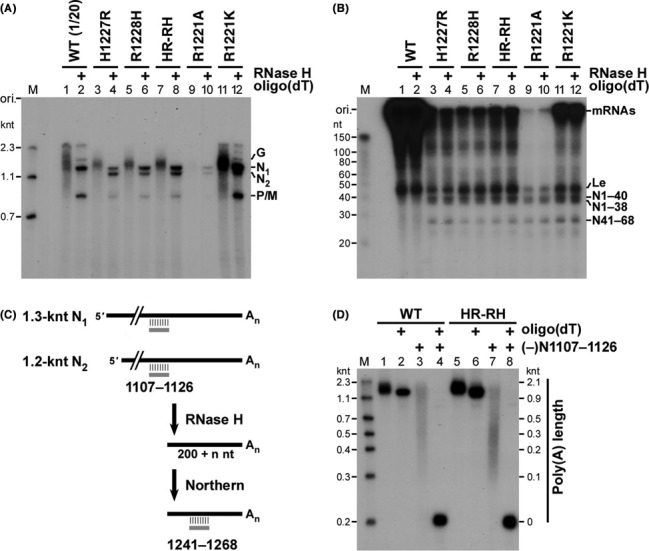
It has been previously shown that the capping activity of the VSV L protein is abolished by mutations in the conserved HR motif (H1227 and R1228) in the step of the L-pRNA intermediate formation (6). Here, to explore the effects of these cap-defective mutations in the VSV L protein on RNA synthesis, transcription reactions were reconstituted with the N–RNA complex, the recombinant P protein and the wild-type or mutant L protein. Internally 32P-labeled transcripts were deadenylated and analyzed by 5% (for mRNAs) or 20% (for the leader RNA) urea-PAGE. Complete results with the previously prepared recombinant L proteins with mutations in the HR motif (called HR mutants) or other residues are available in Supplementary Figure S1. Figure 1 shows a typical result using the HR-RH mutant, which possesses an RH sequence instead of the HR motif (6) and exhibits the highest RNA synthesis activity among the HR mutants, as a representative. As reported (40), the wild-type L protein synthesized 1.7-knt G, 1.3-knt N and 0.8-knt P/M mRNAs (Figure 1A, lane 2) and the leader RNA with ∼50 nt (Figure 1B, lane 2). In contrast, the HR-RH mutant showed an extremely weak mRNA synthesis activity (5% of the wild-type activity) (Figure 1A, lane 3), although its leader RNA synthesis activity was comparable to that of the wild-type L protein (Figure 1B, lane 3). Other HR mutants also showed similar transcription phenotypes to that of the HR-RH mutant (see Supplementary Figure S1B and C). These results indicate that the HR motif is not essential for leader RNA synthesis, but it appears to be required for efficient mRNA synthesis. Interestingly, all these cap-defective HR mutants were found to produce characteristic short RNAs (bands I and II, ∼40 nt; band III, ∼30 nt) and 1.2-knt RNA beside 1.3-knt N mRNA-like RNA (Figure 1 and Supplementary Figure S1).
Figure 1.
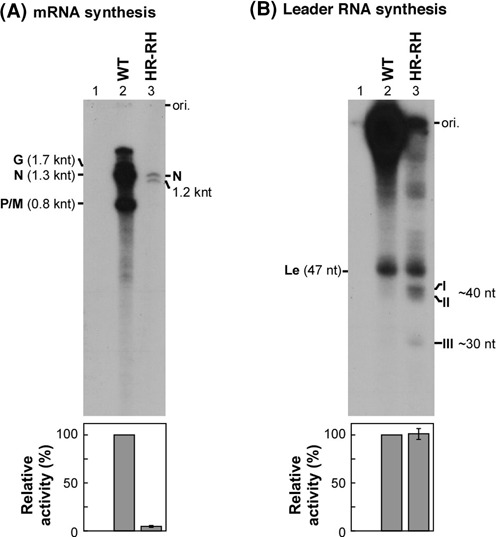
Cap-defective mutations in the HR motif of the L protein impair mRNA synthesis, but not leader RNA synthesis. The wild-type (WT) and HR-RH mutant L proteins were subjected to transcription reactions reconstituted with the N-RNA template and P protein. The HR-RH mutant possesses H1227R and R1228H mutations. 32P-labeled transcripts were deadenylated with RNase H in the presence of oligo(dT) and analyzed by 5% [(A) for the mRNAs] or 20% urea-PAGE [(B) for the leader RNA] followed by autoradiography. Lane 1 indicates no L protein. The positions of the viral mRNAs (G, N and P/M), unknown 1.2-knt RNA, leader RNA (Le), unknown short RNAs (I, II and III) and gel origins (ori.) are indicated. The lower panels show relative RNA synthesis activities of the WT (defined as 100%) and mutant L proteins. Columns and error bars represent the means and standard deviations, respectively, from three independent experiments.
To define a defective step(s) in mRNA synthesis by the HR mutants, short transcripts produced by these mutants (see Figure 1B, bands I, II and III) were further analyzed. First, in order to investigate whether these short transcripts have a 5′-di- or tri-phosphate end, transcripts were synthesized with the HR-RH mutant in the presence of unlabeled NTPs and then capped in the presence of [α-32P]GTP with vaccinia virus capping enzyme. This conventional capping enzyme with the GTase and RTPase domains is known to cap 5′-di- or tri-phosphorylated RNAs without strict RNA sequence specificity (47). As shown in Figure 2A (lane 2), the leader RNA in transcripts synthesized by the wild-type L protein was mainly capped with vaccinia virus capping enzyme, indicating that the leader RNA is a major RNA product with a 5′-di- or tri-phosphate end. In the case of transcripts synthesized by the HR-RH mutant (lane 3), three prominent RNA products with ∼40 nt (bands I and II) and ∼30 nt (band III) were found to be capped with vaccinia virus capping enzyme in addition to the leader RNA. As expected, the transcription-defective D714A mutant (3) did not produce any guanylyl acceptor substrates for vaccinia virus capping enzyme (lane 4).
Figure 2.
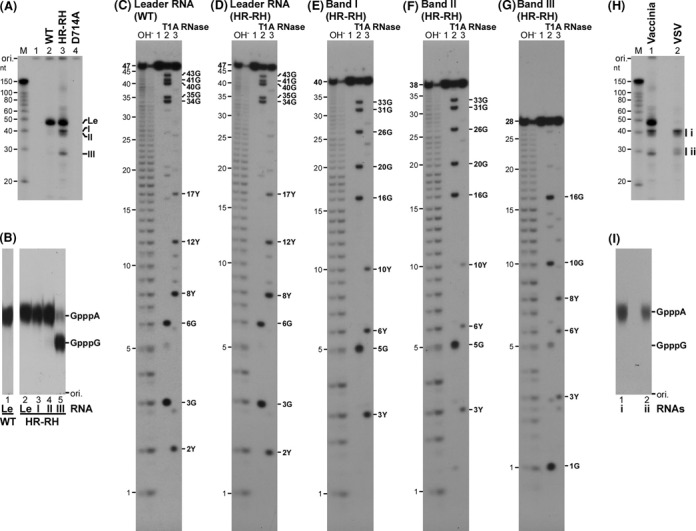
The cap-defective HR-HR mutant L protein produces aberrant short transcripts using cryptic signals within the N gene. (A) Transcripts, synthesized by the wild-type (WT), cap-defective (HR-RH) or transcription-defective (D714A) L protein, were post-labeled by vaccinia virus capping enzyme in the presence of [α-32P]GTP and analyzed by 20% urea-PAGE. Lane 1 indicates no L protein. (B) Cap-labeled short RNAs [Le, leader RNA; bands I–III, see panel (A)] were purified and digested with nuclease P1. The resulting digests were analyzed by PEI-cellulose TLC. The positions of standard cap analogs are shown on the right. (C–G) Purified cap-labeled short RNAs (lane 1) were partially digested with RNase T1 (lane 2) or RNase A (lane 3). The resulting digests were analyzed by 20% urea-PAGE followed by autoradiography. The OH− lanes indicate alkaline RNA ladders. The positions of digestion products with 3′-terminal Gp or Yp (Up or Cp) are indicated on the right. (H) Transcripts synthesized by the HR-RH mutant L protein were capped with vaccinia virus capping enzyme in the presence of [α-32P]GTP (lane 1, Vaccinia) or with the transcription-defective D714A mutant L protein in the presence of [α-32P]GDP (lane 2, VSV), and analyzed by 20% urea-PAGE. Short RNAs capped with the D714A mutant were grouped into i (∼40 nt) and ii (∼30 nt). (I) Capped short RNAs [groups i and ii; see panel (H)] were purified and digested with nuclease P1. The digests were analyzed as in panel (B).
To analyze the first bases of these short RNAs capped with vaccinia virus capping enzyme (see Figure 2A), they were subjected to PAGE purification and then digested with nuclease P1 (Figure 2B). The resulting 32P-labeled cap structures were separated with internal standards (GpppA and GpppG) by PEI-cellulose TLC. The cap structure formed on the leader RNA, synthesized with the HR-RH mutant (lane 2) as well as the wild-type L protein (lane 1), co-migrated with GpppA, indicating that the first base of the leader RNA is adenine as reported (13). The first base of RNAs, synthesized by the HR-RH mutant, in bands I and II was also identified as adenine (lanes 3 and 4). However, ∼80% and ∼20% of RNAs in band III were found to have guanine and adenine, respectively, as their first bases (lane 5).
In order to measure the exact lengths of these cap-labeled transcripts (see Figure 2A) and to obtain their partial RNA sequences, they were subjected to limited digestion (42,48) with alkali (Figure 2C–G, OH− lanes), RNase T1 (lane 2), which specifically cleaves RNA after G residues, or RNase A (lane 3), which cleaves RNA after pyrimidine residues. As expected, an RNase T1-digestion pattern of the leader RNA with 47 nt synthesized by the HR-RH mutant (Figure 2D, lane 2) as well as the wild-type L protein (Figure 2C, lane 2) matched a theoretical digestion pattern of the leader RNA (13). RNase A-digestion of the leader RNA produced some of the expected fragments (Figure 2C and D, lanes 3), but RNase A did not cleave all pyrimidine residues in the leader RNA as reported for other RNAs (48). It was found that RNase-digestion patterns of 40-nt (band I, Figure 2E) and 38-nt (band II, Figure 2F) RNAs, synthesized by the HR-RH mutant, match theoretical digestion patterns of the N mRNA-5′-terminal sequence, but not with those of the leader RNA- or other mRNA-5′-terminal sequences. These results indicate that these 5′-(p)ppA-initiated RNAs correspond to 5′-terminal fragments of N mRNA with residues +1 to +40 and +1 to +38 (designated as N1–40 and N1–38, respectively). Interestingly, the major 28-nt RNA in band III, synthesized by the HR-RH mutant, was suggested to be a 5′-(p)ppG-initiated RNA with residues +41 to +68 of N mRNA (referred to as N41–68) on the basis of its RNase-digestion patterns (Figure 2G, lanes 2 and 3).
As shown in Figure 2H (lane 1) as well as in Figure 2A (lane 3), vaccinia virus capping enzyme capped (p)ppRNAs (e.g. leader RNA, N1–40, N1–38 and N41–68), synthesized by the HR-RH mutant, in a sequence-independent manner. In contrast, the VSV L protein (PRNTase) preferentially caps 5′-triphosphorylated, but not 5′-diphosphorylated, RNAs with a 5′-terminal ARCNG sequence (R: A or G), in which the first A and third pyrimidine (C > U) residues are essential (4,32). To highlight RNAs with the 5′-pppARCNG sequence (e.g. pppAACAG-) in transcripts synthesized by the HR-RH mutant, they were capped in the presence of [α-32P]GDP with the transcription-defective D714A mutant L protein, which retains the PRNTase activity (6). As shown in Figure 2H (lane 2), N1–40 and N1–38 RNAs (group i) and minor RNAs with 28–30 nt (group ii) were capped with GDP, indicating that these RNAs are initiated with pppARCNG. To confirm their first bases are adenine, capped RNAs in groups i and ii were digested with nuclease P1 and CIAP, and the resulting digests were analyzed by PEI-cellulose TLC (Figure 2I). As expected, GpppA, but not GpppG, was detected in both digests (lanes 1 and 2). Further analyses of N1–40 and N41–68 RNAs revealed that these RNAs have exclusively a triphosphate group at their 5′-termini (Supplementary Figure S2). These results indicate that the unique short transcripts (bands I, II and III) produced by the HR-RH mutant are 5′-triphosphorylated N1–40, N1–38 and N41–68 RNAs.
In order to investigate whether other cap-defective mutants produce N1–40, N1–38 and N41–68 RNAs in addition to the leader RNA, short transcripts synthesized by selected mutants (H1227R, R1228H, HR-RH, R1221A and R1221K) were analyzed by northern blotting with 32P-labeled oligo-DNA probes complementary to the 5′-terminal 28 nt of the leader RNA, N1–40 and N41–68 [designated as (−)Le1–28, (−)N1–28 and (−)N41–68, respectively] (Figure 3A). Although these cap-defective mutants exhibited leader RNA synthesis activities comparable to or lower than that of the wild-type L protein (Figure 3B; see Supplementary Figure S1C), they produced significantly higher amounts of N1–40, N1–38 (Figure 3C, lanes 3–7) and N41–68 (Figure 3D, lanes 3–7) than the wild-type L protein (Figure 3C and D, lane 2). Note that these mutants also yielded unidentified minor products including 28–30-nt RNAs and ∼70-nt RNAs, which were detected with the (−)N1–28 (Figure 3C, marked by arrowheads) and (−)N41–68 (Figure 3D, marked by asterisks) probes, respectively. These results indicate that the cap-defective mutations cause aberrant stop–start transcription to produce short RNAs using cryptic transcription signals in the N gene.
Figure 3.
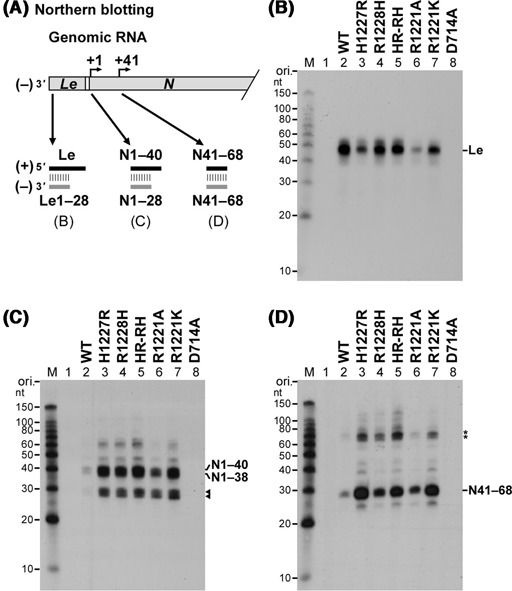
Cap-defective mutant L proteins produce large amounts of short N mRNA fragments including N1–40 and N41–68 RNAs. As illustrated in panel (A), short transcripts, synthesized by the wild-type (WT) or mutant L protein, were analyzed by northern blotting to detect the leader RNA (B), N1–40 RNA (C) and N41–68 RNA (D) with 32P-labeled oligo-DNA probes complementary to their 5′-terminal 28-nt sequences. Lane 1 indicates no L protein. Putative N1–28 and N1–30 RNAs are marked by arrowheads (C). Asterisks represent unidentified transcripts (∼70 nt) detected with the (−)N41–68 probe (D).
Cap-defective mutant L proteins synthesize an unusual 3′-polyadenylated long RNA using another cryptic transcription initiation signal within the N gene
To further examine the effects of the cap-defective mutations on synthesis of full-length mRNAs, long transcripts, synthesized by the wild-type or mutant L protein and deadenylated, were subjected to northern blotting with 32P-labeled oligo-DNA probes complementary to positions +401 to +428 of N, P, M and G mRNAs [referred to as (−)N401–428, (−)P401–428, (−)M401–428 and (−)G401–428, respectively] (Figure 4A). Since the VSV mRNAs are synthesized by the stop–start mechanism in a sequential and polar manner (18,19), an upstream gene, closer to the 3′-end of the genomic RNA, is transcribed much more efficiently than a downstream gene. As expected, the detection efficiency for mRNAs synthesized by the wild-type L protein followed the order of the genes in the genomic RNA (N > P > M > G) (Figure 4B–E, lane 2). To reduce signal intensities from hybridized wild-type mRNAs (lanes 2), its 20-fold dilution was also analyzed on the same blots [lanes 9, WT (1/20)].
Figure 4.
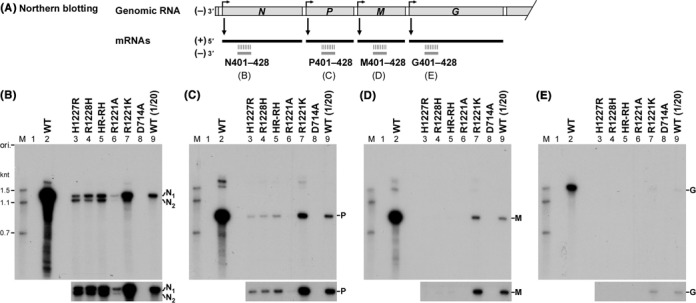
Cap-defective mutant L proteins are not able to synthesize mRNAs from downstream genes efficiently. mRNAs were synthesized with the wild-type (WT) or mutant L protein and deadenylated with RNase H in the presence of oligo(dT). Northern blotting [see panel (A)] was carried out to detect N (B), P (C), M (D) and G (E) mRNAs with 32P-labeled oligo-DNA probes complementary to positions +401 to +428 of respective mRNAs. Lane 1 indicates no L protein. In lane 9, one-twelfth of the RNA amount used in lane 2 was analyzed. The lower panels show portions of autoradiograms after a longer exposure. N1 and N2 indicate full-length N mRNA (1.3 knt) and its truncated form (1.2 knt), respectively.
As shown in Figure 4B (lane 9), the size of N mRNA lacking poly(A) tail, hybridized with (−)N401–428, was 1.3 knt, which is in agreement with its reported size (1326 nt). Interestingly, using the (−)N401–428 probe, two discrete RNAs with 1.3 and 1.2 knt (referred to as N1 and N2, respectively) were detected in transcripts synthesized by the cap-defective HR mutants (lanes 3–5). In contrast, the wild-type L protein produced no detectable N2 RNA (lane 9). Although the R1221A mutant showed a very low N1 RNA synthesis activity, it produced a trace amount of N2 RNA (lane 6, lower panel). The R1221K exhibited ∼10% of the wild-type N1 mRNA synthesis activity, but it also synthesized a small amount of N2 mRNA (lane 7). It is interesting to note that the N1-to-N2 ratios were different among these cap-defective mutants (H1227R, 4 to 6; R1228H, 8 to 2; HR-RH, 6 to 4; R1221A, 9 to 1; R1221K, 9 to 1).
The cap-defective HR mutants retained 1–2% of the wild-type P mRNA synthesis activities (Figure 4C, lanes 3–5), but did not produce detectable amounts of M and G mRNAs (Figure 4D and E, lanes 3–5). While the R1221A mutant did not synthesize detectable amounts of P, M and G mRNAs (Figure 4C–E, lanes 6), the R1221K mutant with a weak co-transcriptional capping activity (see Supplementary Figure S1D) retained the activity to synthesize all these mRNAs (Figure 4C–E, lanes 7), although to significantly lesser extents than the wild-type L protein.
In order to locate the 5′ and 3′-ends of N1 and N2 RNAs synthesized by the cap-defective HR-RH mutant, northern blotting was carried out with 32P-labeled oligo-DNA probes, such as (−)N1–28, (−)N81–108, (−)N161–188 and (−)N1291–1326, complementary to different regions of N mRNA (1326 nt) (Figure 5A). N1 RNA synthesized by the HR-RH mutant (lanes 2, 4, 6 and 8) as well as the wild-type L protein (lanes 1, 3, 5 and 7) was detected with all these probes, whereas N2 RNA was detected only in the HR-RH transcripts with (−)N161–188 (lane 6) or (−)N1291–1326 (lane 8), but not with (−)N1–28 (lane 2) or (−)N81–108 (lane 4). These results suggest that N1 and N2 RNAs are full-length N mRNA and its 5′-truncated form, respectively.
Figure 5.
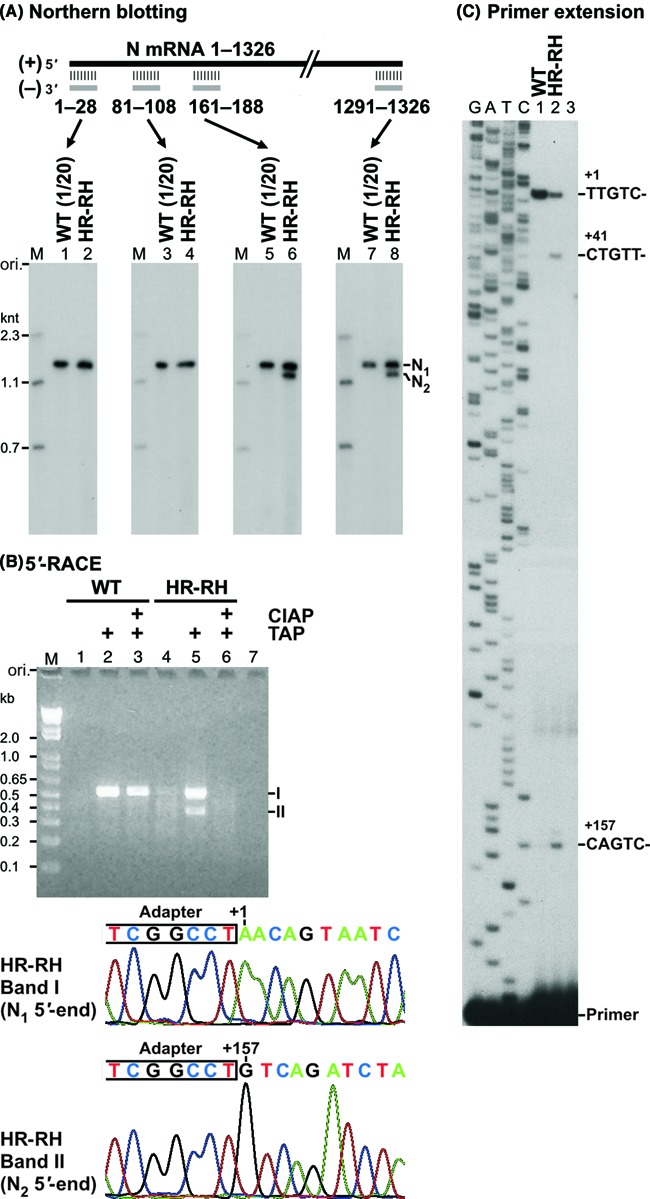
The cap-defective HR-RH mutant L protein uses another cryptic transcription initiation site to synthesize 1.2-knt N2 RNA. (A) Deadenylated RNAs, synthesized by either the wild-type (WT) or HR-RH mutant L protein (see Figure 4B, lanes 9 and 5), were analyzed by northern blotting with 32P-labeled oligo-DNA probes complementary to different regions of N mRNA. (B) Transcripts, synthesized by either the WT or HR-RH mutant L protein, were pretreated with or without calf intestine alkaline phosphatase (CIAP) followed by tobacco acid pyrophosphatase (TAP). After ligation of an adapter RNA to 5′-monophosphate ends of RNAs, first-strand cDNAs were synthesized with reverse transcriptase using oligo(dT) as a primer. The 5′-cDNA ends were amplified by PCR using an adapter-specific primer and an N gene-specific primer. The resulting PCR products were analyzed by 2% agarose gel electrophoresis. Lane 7 indicates no template. The amplified cDNA ends for N1 (band I) and N2 (band II) RNAs were purified from an agarose gel and cloned into a plasmid. Sequences of the adapter-ligated 5′-cDNA ends for N1 and N2 RNAs, synthesized by the HR-RH mutant, are shown with positions of their 5′-end residues in the N gene above electropherograms. (C) In vitro transcription was performed with the WT or HR-RH mutant L protein. Lane 3 indicates no L protein. Transcripts were subjected to reverse transcription with a 5′-end-32P-labeled primer complementary to positions +168 to +188 of N mRNA. Primer-extended products were analyzed along with sequencing ladders (G, A, T and C) by 8% urea-PAGE. DNA sequences complementary to 5′-terminal sequences of transcripts are shown at the positions of primer-extended products.
To identify 5′-terminal sequences of N1 and N2 RNAs, 5′-RACE analyses were performed using transcripts produced by the wild-type or HR-RH mutant L protein (Figure 5B). These transcripts were treated with TAP to generate a 5′-monophosphate end from a GpppN-cap, triphosphate or diphosphate end (lanes 2 and 5). In lanes 3 and 6, in order to determine the presence or absence of the cap structure on transcripts, they were pre-treated with CIAP to remove unblocked phosphate groups, and then their CIAP-resistant cap structure was converted into a 5′-monophosphate group with TAP. Five prime-monophosphorylated RNAs in untreated (lanes 1 and 4), TAP-treated (lanes 2 and 5) or CIAP- and TAP-treated (lanes 3 and 6) samples were ligated to a partially double-stranded oligo-RNA adapter with T4 RNA ligase 2, and 5′-adapter-ligated transcripts with a poly(A) tail were reverse-transcribed using an oligo(dT) primer to generate first-strand cDNAs. Then, 5′-cDNA ends were amplified using an adapter-specific forward primer and an N gene-specific reverse primer (complementary to positions +479 to +500 of N mRNA) and analyzed by agarose gel electrophoresis.
As expected, an ∼0.5-kb N1 cDNA end (band I) was amplified from the TAP-treated (Figure 5B, lane 2) or CIAP- and TAP-treated (lane 3) transcripts, but not from the untreated transcripts (lane 1), synthesized by the wild-type L protein, suggesting that N1 RNA is full-length N mRNA with a CIAP-resistant cap structure. In contrast, an ∼0.5-kb N1 (band I) and ∼0.35-kb N2 (band II) cDNA ends were efficiently amplified from the TAP-treated transcripts (lane 5), but not from the untreated (lane 4) or CIAP- and TAP-treated transcripts (lane 6), synthesized by the HR-RH mutant, suggesting that the majority of these N1 and N2 RNAs have a 5′-di- or tri-phosphate end. Thus, N2 RNA, synthesized by the HR-RH mutant, is not a degradation product of N mRNA, which should have a 5′-hydroxyl or monophosphate end. Consistent with these observations, vaccinia virus capping enzyme efficiently capped N1, N2 and P RNAs synthesized by the HR-RH mutant, but not any mRNAs synthesized by the wild-type L protein (Supplementary Figure S3). Taken together, these results indicate that mRNAs synthesized by the wild-type L protein are capped, whereas N1, N2 and P RNAs synthesized by the HR-RH mutant possess a 5′-di- or tri-phosphate end.
Sequencing analyses of cloned 5′-cDNA ends revealed that the transcription initiation site for N2 RNA is located 157 nt downstream of that for N mRNA (N1 RNA) and its initiator nucleotide is non-canonical GTP (Figure 5B, lower panels; see Supplementary Figure S4). Primer extension analyses also demonstrated that the HR-RH (Figure 5C) and other cap-defective mutants (Supplementary Figure S5) initiate transcription at position +157 as well as +41 at higher frequencies than the wild-type L protein. Although the 5′-RACE analyses suggested that the HR-RH mutant uses other minor transcription initiation sites within the N gene (Supplementary Figure S4), the primer extension analyses could not detect any of these putative minor transcripts (Figures 5C and Supplementary Figure S5). The 5′-RACE analysis of 3′-polyadenylated N RNAs did not detect cDNAs starting from position +41, suggesting that transcripts initiated at position +41 are not elongated to the N mRNA 3′-end or not polyadenylated.
In order to determine the presence or absence of the 3′-poly(A) tail on long transcripts synthesized by the cap-defective mutants, sizes of their transcripts were compared with those after deadenylation with RNase H in the presence of oligo(dT) (Figure 6A and B). Untreated mRNAs synthesized by the wild-type L protein migrated as a broad smear (Figure 6A, lanes 1), while deadenylated mRNAs were separated into three bands with 1.3 (N), 0.8 (P/M) and 1.7 (G) knt (Figure 6A, lanes 2), indicating that these wild-type mRNAs were co-transcriptionally polyadenylated as reported (40). Similarly, as shown in lanes 3–12, long transcripts with heterogeneous lengths (>0.8 knt) synthesized by these HR and R1221 mutants were found to be digested with RNase H in the presence of oligo(dT) into discrete RNA species (e.g. N1, N2, P). In contrast, sizes of short transcripts including the leader RNA, N1–40, N1–38 and N41–68 synthesized by the mutants were not different before and after RNase H treatment in the presence of oligo(dT) (Figure 6B). Thus, it was shown that mRNAs synthesized by the cap-defective mutants are efficiently polyadenylated during transcription.
Figure 6.
The cap-defective mutant L proteins polyadenylate N1 and N2 RNAs at their 3′-ends. (A, B) 32P-labeled transcripts, synthesized by the wild-type (WT) or mutant L protein, were treated with or without RNase H and oligo(dT) and analyzed by 5% (A) or 15% (B) urea-PAGE followed by autoradiography. In panel (A), lanes 1 and 2, the sample volume of transcripts synthesized by the WT L protein for the urea-PAGE analysis was 20-fold smaller than those of transcripts synthesized by the other mutants. (C, D) N1 (N mRNA) and/or N2 RNAs, synthesized by either the WT or HR-RH mutant L protein, were cleaved 200 nt upstream of the polyadenylation site with RNase H in the presence of an oligo-DNA complementary to positions +1107 to +1126 of N mRNA [(−)N1107–1126]. The resulting 3′-end fragments with poly(A) tails were detected by northern blotting with a 32P-labeled antisense oligo-DNA [(−)N1241–1268] [(D) lanes 3 and 7]. To estimate lengths of poly(A) tails on the 3′-end fragments (0.2 knt) of N1 and N2 RNAs, the positions of the marker RNAs (see the M lane) are shown on the right with lengths calculated by subtracting 0.2 knt from their actual lengths (D). Poly(A) tails were digested with RNase H in the presence of oligo(dT) with (lanes 4 and 8) or without (lanes 2 and 6) the (−)N1107–1126 oligo-DNA.
To measure the length of poly(A) tails generated on N transcripts by the wild-type or HR-RH mutant L protein, they were digested with RNase H in the presence of an oligo-DNA complementary to positions +1107 to +1126 of N mRNA [(−)N1107–1126], and the resulting 3′-terminal N mRNA fragments (200 nt) with different poly(A) tail lengths were analyzed by northern blotting with a 32P-labeled antisense probe [(−)N1241–1268] (see Figure 6C). As shown in Figure 6D (lanes 3 and 7), poly(A) tails generated by the wild-type and HR-RH mutant L proteins showed very broad size distributions ranging from 0.1 to 2 knt. The presence of long poly(A) tails on N1 (N mRNA) and N2 RNAs or on their 3′-fragments was confirmed by RNase H digestion of poly(A) tails in the presence of oligo(dT) (Figure 6D, lanes 2, 4, 6 and 8). These results indicate that the cap-defective mutants efficiently polyadenylate N1 and N2 RNAs at their 3′-termini.
Cap-defective mutations are lethal to VSV replication in host cells
Finally, the effects of these cap-defective mutations in the L gene on recombinant VSV generation were studied using the reverse genetics system (45,46). Three independent experiments were carried out to generate recombinant VSVs from pVSV plasmids (45,46) encoding a full-length positive strand VSV genome with the wild-type or mutant L gene in BHK-21 cells at 30°C (Table 1). In addition to the wild-type pVSV plasmid, a pVSV plasmid to generate an attenuated temperature-sensitive VSV with the cap-methylation-defective D1671V mutation (8) was used as a positive control. On the other hand, a pVSV plasmid with the transcription-defective D714A mutation (3) was used as a negative control. At 30°C (Table 1) as well as at 37°C (data not shown), wild-type VSV was efficiently generated from the plasmid, whereas viruses with the cap-defective mutations in the HR motif (H1227R, R1228H and HR-RH) were not, suggesting that the HR motif is essential for virus growth in cultured cells. While the R1221A mutation was also lethal to VSV, highly attenuated viruses were generated from a plasmid with the R1221K mutation at 30°C. The majority of recombinant R1221K viruses in the passage 1 stock formed pinpoint plaques similar to those of the D1671 mutant viruses (Table 1). In contrast to the wild-type virus, the R1221K virus as well as the D1671V virus was not generated at 37°C (data not shown), suggesting that the mutation may confer a temperature sensitivity to the virus.
Table 1. The cap-defective mutations in the L gene are lethal to VSV.
| L gene | Virus titer (pfu/ml)a | Plaque size (mm)b | ||
|---|---|---|---|---|
| Experiment | ||||
| 1 | 2 | 3 | ||
| WT | 1.8 × 109 | 1.7 × 109 | 3.2 × 109 | 3.1 ± 0.6 |
| H1227R | <10 | <10 | <10 | |
| R1228H | <10 | <10 | <10 | |
| HR-RH | <10 | <10 | <10 | |
| R1221A | <10 | <10 | <10 | |
| R1221K | 2.2 × 104 | 5.4 × 105 | 1.1 × 105 | 0.5 ± 0.2 |
| D714A | <10 | <10 | <10 | |
| D1671V | 1.4 × 107 | 1.3 × 107 | 2.0 × 107 | 0.7 ± 0.2 |
aRecombinant VSVs were generated from cDNAs encoding an anti-genome with the wild-type (WT) or mutant L gene using a reverse genetics system. Recovery of viruses was examined by a plaque assay. Three independent experiments were performed.
bThirty plaques of viruses, generated in Experiment 1, were randomly chosen to measure their sizes (the mean ± standard deviation).
RT-PCR and sequence analyses of viral genomic RNAs showed that the majority of the R1221K viruses generated at 30°C carry the R1221K mutation in their genomes (Supplementary Figure S6B). However, after further passaging of the R1221K viruses, revertant viruses with the wild-type phenotype and genotype were generated (Supplementary Figure S6C and D). Taken together, these results indicate that the H1227, R1228 and R1221 residues are essential for VSV replication in host cells as well as in vitro mRNA synthesis and capping. Consistent with the reduced levels of mRNA synthesis and RNA capping activities of the R1221K mutant (see Supplementary Figure S1), recombinant VSV with this mutation is highly attenuated in cell culture, but this mutation appears to be unstable during virus replication (Supplementary Figure S6).
DISCUSSION
Despite the diversity of NNS RNA viruses, their RdRp L proteins share a general organization of putative domains with highly conserved regions (30,49,50), suggesting that they catalyze common enzymatic reactions required for viral mRNA biogenesis. Here, using VSV as a model, it was demonstrated, for the first time, that the active site HR motif of the mRNA capping enzyme (PRNTase) domain of the L protein is required for accurate stop–start transcription to produce full-length mRNAs and virus growth in cultured cells. Since the HR motif is highly conserved in the L proteins of NNS RNA viruses (except for fish novirhabdoviruses) including many human pathogens, such as rabies, measles, respiratory syncytial and Ebola viruses (6,30,33), its functions in viral mRNA biogenesis appear to be fundamental to replication of these NNS RNA viruses in eukaryotic cells.
The cap-defective mutations in the HR motif or its adjacent R1221 residue of the VSV L protein significantly decreased the accuracy of selection of transcription termination and initiation signals for mRNA synthesis during in vitro transcription together with the P protein, leading to sequential production of non-overlapping 5′-triphosphorylated N mRNA fragments, such as N1–40, N41–68 and N2 (3′-polyadenylated N157–1326) RNAs, as depicted in Figure 7. In the presence of the P protein, the wild-type L protein produced the leader RNA and N mRNA at a molar ratio of 1:0.4 under the standard transcription conditions, whereas the HR-RH mutant synthesized the leader, N1–38/N1–40, N41–68, N1 (N mRNA) and N2 RNAs at a molar ratio of 1:0.7:0.3:0.02:0.01. Erroneous transcription termination and re-initiation using the cryptic signals within the N gene at higher frequencies resulted in a marked decrease in full-length N mRNA synthesis and attenuation of downstream mRNA production.
Figure 7.
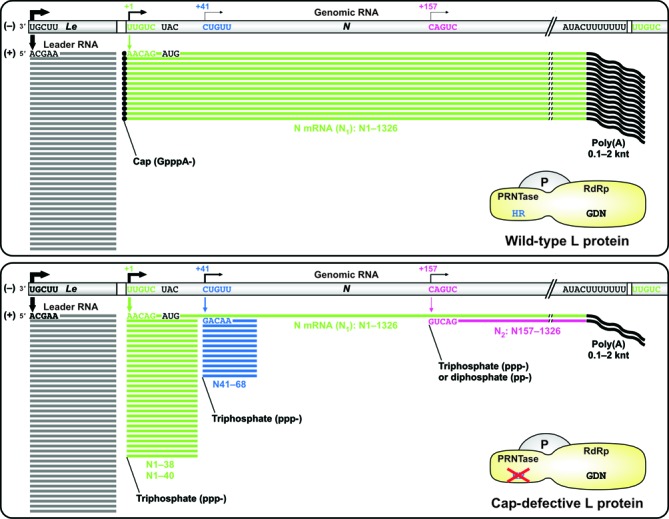
The cap-defective mutations in the L protein cause aberrant stop–start transcription. The cap-defective mutant L protein (lower) as well as the wild-type L protein (upper) together with the P protein synthesizes the leader RNA from the 3′-end of the genomic RNA. After synthesis of the leader RNA, the wild-type L protein productively synthesizes 5′-capped and 3′-polyadenylated N mRNA using the gene-start and gene-end sequences as transcription initiation and termination/polyadenylation signals, respectively. In contrast, the cap-defective mutant L proteins (e.g. HR-RH, H1227R, R1228H) frequently use cryptic transcription signals within the N gene, resulting in the generation of large amounts of 5′-uncapped short transcripts (e.g. N1–40, N41–68) and a small amount of 5′-uncapped and 3′-polyadenylated N2 RNA. GDN and HR indicate active site motifs for the RNA-dependent RNA polymerase (RdRp) and polyribonucleotidyltransferase (PRNTase) domains, respectively.
The P protein plays essential roles in the formation of the active RdRp complex, association of the complex with the N-RNA template and RNA chain elongation (1,2,51,52). In the reconstituted transcription system used in this study, synthesis of the leader RNA and mRNAs with the wild-type L protein completely depends on the presence of the P protein (40). Since the cap-defective HR-RH mutant L protein produced the leader RNA to a similar level as the wild-type L protein, it appears to form an RdRp complex with the P protein as the wild-type L protein does. Although the P protein is not required for the RNA capping activity of the L protein (4), the role of the P protein in the recognition of transcription stop–start sites is currently not known.
It is suggested that the failure in pre-mRNA capping at an early stage of N mRNA synthesis induces premature termination of N mRNA synthesis at positions +38 and +40. The minimum lengths of capped pre-mRNAs synthesized during in vitro transcription were reported to be 23–37 nt (34–36). Thus, the transcribing RdRp complex might be paused at a checkpoint for pre-mRNA capping soon after early mRNA chain elongation by recognizing the 5′-end capping signal (pppAACAG) on a nascent transcript with the PRNTase domain. It has been shown that the HR motif is essential for the specific recognition of the capping signal to form the L-pRNA intermediate (6,40). If pre-mRNA is not properly capped at this checkpoint by failing to form the L-pRNA intermediate, the RdRp complex might stop transcription mainly at position +40 to release uncapped transcripts before the transition into processive elongation of mRNA chain. If the RdRp complexes accidently terminate transcription within a gene, a part of them might abnormally seek proximal cryptic transcription initiation sites resembling the gene-start sequence without dissociating from the template. Termination of transcription at position +40 within the N gene results in the release of the uncapped 40-nt RNA and triggers erroneous re-initiation at position +41 using GTP, a non-canonical initiator nucleotide. It is noteworthy that similar stop–start transcription occurs naturally at the leader-N gene junction to release the uncapped 47-nt leader RNA and re-initiate transcription at the N-gene-start sequence.
Pre-mRNA capping appears to be a critical event for the transition of the RdRp into the continuing elongation mode for long mRNA synthesis, which enables the RdRp to ignore cryptic termination and re-initiation signals. However, externally added vaccinia virus mRNA capping enzyme or the transcription-defective D714A L protein with a full capping activity was not able to rescue the defect of the HR-RH mutant in mRNA synthesis (data not shown). Furthermore, the addition of a GpppA cap analog or capped 10-nt oligo-RNA with the 5′-terminal sequence of N mRNA in trans did not stimulate the production of full-length mRNAs with the cap-defective HR-RH mutant (data not shown). These observations suggest that a transcribing L protein must have the active PRNTase domain in the same molecule to produce capped full-length mRNAs via a concerted mechanism of stop–start transcription and co-transcriptional pre-mRNA capping.
The wild-type L protein uses ATP as the initiator nucleotide to start mRNA synthesis from each gene-start sequence (3′-UUGUCDNUAG) (20), in which the first three nucleotides (3′-UYG sequence) are essential for efficient transcription initiation, mRNA 5′-end modifications and production of full-length mRNA (37,53). In this study, the cap-defective L proteins were found to initiate transcription with ATP at the N gene-start sequence, but also with non-canonical GTP using cryptic initiation signals at positions +41 and +157 at significantly higher frequencies when compared with the wild-type L protein. Interestingly, among RNA sequences surrounding these cryptic initiation sites and the gene-start sequences (correct mRNA initiation site), there is a consensus sequence composed of an upstream AU-rich sequence (3′-UYUUWBWVA; Y: C or U; W: U or A; B: G, C or U; V: A, G or C) and 3′-YWGUY initiation sequence (see Supplementary Figure S7). Upstream AU-rich sequences have been found to significantly enhance transcription initiation from the gene-start sequence (54).
The VSV L protein is known to initiate transcription with GTP from suboptimal transcription initiation signals although to much lesser extents than with ATP (32,34,37,54,55). Schubert et al. (34) have shown that the VSV-associated RdRp complexes also infrequently terminate N mRNA synthesis at position +40 and re-initiate transcription at position +41 to synthesize 28-nt N41–68 RNA with 5′-GTP. Similarly, very small amounts of N1–40- and N41–68-like RNAs were synthesized with the recombinant wild-type L protein in the reconstituted transcription reaction (see Figure 3). Primer extension analyses also confirmed that the recombinant wild-type L protein initiates transcription at position +41 although to a much lesser degree than the cap-defective mutants (see Supplementary Figure S5B). Thus, the cap-defective mutations in the L protein appear to relax the internal-promoter specificity of the RdRp domain, resulting in more efficient initiation with GTP from the suboptimal cryptic initiation sites that are similar to the gene-start sequence. Currently it is not clear how the cap-defective mutant L proteins terminate transcription at position +68 and reach the downstream cryptic initiation site at position +157.
Li et al. (38,39) have also investigated the effects of cap-defective mutations (G1154A, T1157A, H1227A and R1228A) in the recombinant VSV L protein on mRNA synthesis using a reconstituted transcription system in the presence of rabbit reticulocyte lysates. In their system, the mutant L proteins initiate mRNA synthesis at the N gene-start sequence and terminate chain elongation within positions +100 to +500 of the N gene at various sites with a weak preference for U-rich template sequences, yielding heterogeneous 3′-truncated transcripts (100–500 nt) (38,39). Furthermore, they (39) reported that the cap-defective mutant L proteins are not able to polyadenylate mRNAs efficiently, producing mRNAs with 0–24 adenosine residues at their 3′-ends. In contrast, such heterogeneous transcripts ranging from 100 to 500 nt and non-polyadenylated full-length mRNAs were not observed in transcripts synthesized with the HR mutants in this study, even if the same H1227A and R1228A mutants were used. The reason for these discrepancies is currently unclear. G1154A and T1157A were identified as mutations significantly reducing the in vitro RNA capping activity of the VSV L protein by an unknown mechanism(s) (38). It would be interesting to examine the effects of these mutations on transcription using our reconstituted transcription system.
Using a model VSV replicon system, Stillman and Whitt (37) showed that transcription from suboptimal initiation sequences (e.g. 3′-CUG and UGG) is prematurely terminated, generating 40–200-nt transcripts lacking the cis-acting RNA capping signal (5′-ARC) in their 5′-ends. Liuzzi et al. (56) have found that anti-viral compounds against respiratory syncytial virus, belonging to the Paramyxoviridae family, inhibit in vitro synthesis of viral capped mRNAs, resulting in the production of 5′-triphosphorylated abortive transcripts (<50 nt). Furthermore, respiratory syncytial virus mutants resistant to these inhibitors were found to carry an amino acid substitution (I1381S) in the vicinity of the HR motif (H1338-R1339) of the L protein, suggesting that these inhibitors interact with the wild-type PRNTase domain, but not with the mutant domain. Taken together, these findings indicate that co-transcriptional pre-mRNA capping with PRNTase is essential for productive and accurate synthesis of full-length mRNAs by the stop–start transcription mechanism and PRNTase is an attractive target for developing anti-viral agents. PRNTase inhibitors may have potential to reduce levels of translatable viral mRNAs and to increase levels of viral 5′-triphosphorylated short RNAs, which are known to trigger anti-viral innate immunity through the RIG-I and IFIT pathways in virus-infected cells (57).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The author thanks Dr Amiya K. Banerjee (The Cleveland Clinic Foundation) for the reagents, helpful discussions and critical comments on the manuscript. Material was provided by Dr Amiya K. Banerjee with support from the NIH grant R01AI093569 to T.O. and NIH grant R0lAI026585 to A.K.B. All rights, title and interest in the Material are owned by The Cleveland Clinic Foundation. The author also thanks Drs Ganes C. Sen and Donal S. Luse (The Cleveland Clinic Foundation) for helpful discussions and Dr Timothy W. Nilsen (Case Western Reserve University) for critical comments on the manuscript. The author acknowledges Dr John K. Rose (Yale University) and Dr Sue A. Moyer (University of Florida) for the generous gifts of the plasmids.
FUNDING
National Institutes of Health (NIH) [AI093569]. Funding for open access charge: NIH [AI093569].
Conflict of interest statement. None declared.
REFERENCES
- 1.Emerson S.U., Yu Y. Both NS and L proteins are required for in vitro RNA synthesis by vesicular stomatitis virus. J. Virol. 1975;15:1348–1356. doi: 10.1128/jvi.15.6.1348-1356.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De B.P., Banerjee A.K. Requirements and functions of vesicular stomatitis virus L and NS proteins in the transcription process in vitro. Biochem. Biophys. Res. Commun. 1985;126:40–49. doi: 10.1016/0006-291x(85)90568-6. [DOI] [PubMed] [Google Scholar]
- 3.Sleat D.E., Banerjee A.K. Transcriptional activity and mutational analysis of recombinant vesicular stomatitis virus RNA polymerase. J. Virol. 1993;67:1334–1339. doi: 10.1128/jvi.67.3.1334-1339.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ogino T., Banerjee A.K. Unconventional mechanism of mRNA capping by the RNA-dependent RNA polymerase of vesicular stomatitis virus. Mol. Cell. 2007;25:85–97. doi: 10.1016/j.molcel.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 5.Ogino T., Banerjee A.K. The HR motif in the RNA-dependent RNA polymerase L protein of Chandipura virus is required for unconventional mRNA-capping activity. J. Gen. Virol. 2010;91:1311–1314. doi: 10.1099/vir.0.019307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ogino T., Yadav S.P., Banerjee A.K. Histidine-mediated RNA transfer to GDP for unique mRNA capping by vesicular stomatitis virus RNA polymerase. Proc. Natl Acad. Sci. U.S.A. 2010;107:3463–3468. doi: 10.1073/pnas.0913083107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horikami S.M., Moyer S.A. Host range mutants of vesicular stomatitis virus defective in in vitro RNA methylation. Proc. Natl Acad. Sci. U.S.A. 1982;79:7694–7698. doi: 10.1073/pnas.79.24.7694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grdzelishvili V.Z., Smallwood S., Tower D., Hall R.L., Hunt D.M., Moyer S.A. A single amino acid change in the L-polymerase protein of vesicular stomatitis virus completely abolishes viral mRNA cap methylation. J. Virol. 2005;79:7327–7337. doi: 10.1128/JVI.79.12.7327-7337.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J., Fontaine-Rodriguez E.C., Whelan S.P. Amino acid residues within conserved domain VI of the vesicular stomatitis virus large polymerase protein essential for mRNA cap methyltransferase activity. J. Virol. 2005;79:13373–13384. doi: 10.1128/JVI.79.21.13373-13384.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ogino T., Kobayashi M., Iwama M., Mizumoto K. Sendai virus RNA-dependent RNA polymerase L protein catalyzes cap methylation of virus-specific mRNA. J. Biol. Chem. 2005;280:4429–4435. doi: 10.1074/jbc.M411167200. [DOI] [PubMed] [Google Scholar]
- 11.Hunt D.M., Hutchinson K.L. Amino acid changes in the L polymerase protein of vesicular stomatitis virus which confer aberrant polyadenylation and temperature-sensitive phenotypes. Virology. 1993;193:786–793. doi: 10.1006/viro.1993.1187. [DOI] [PubMed] [Google Scholar]
- 12.Galloway S.E., Wertz G.W. S-adenosyl homocysteine-induced hyperpolyadenylation of vesicular stomatitis virus mRNA requires the methyltransferase activity of L protein. J. Virol. 2008;82:12280–12290. doi: 10.1128/JVI.01225-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Colonno R.J., Banerjee A.K. A unique RNA species involved in initiation of vesicular stomatitis virus RNA transcription in vitro. Cell. 1976;8:197–204. doi: 10.1016/0092-8674(76)90003-9. [DOI] [PubMed] [Google Scholar]
- 14.Colonno R.J., Banerjee A.K. Complete nucleotide sequence of the leader RNA synthesized in vitro by vesicular stomatitis virus. Cell. 1978;15:93–101. doi: 10.1016/0092-8674(78)90085-5. [DOI] [PubMed] [Google Scholar]
- 15.Emerson S.U. Reconstitution studies detect a single polymerase entry site on the vesicular stomatitis virus genome. Cell. 1982;31:635–642. doi: 10.1016/0092-8674(82)90319-1. [DOI] [PubMed] [Google Scholar]
- 16.Abraham G., Rhodes D.P., Banerjee A.K. The 5′ terminal structure of the methylated mRNA synthesized in vitro by vesicular stomatitis virus. Cell. 1975;5:51–58. doi: 10.1016/0092-8674(75)90091-4. [DOI] [PubMed] [Google Scholar]
- 17.Abraham G., Rhodes D.P., Banerjee A.K. Novel initiation of RNA synthesis in vitro by vesicular stomatitis virus. Nature. 1975;255:37–40. doi: 10.1038/255037a0. [DOI] [PubMed] [Google Scholar]
- 18.Abraham G., Banerjee A.K. Sequential transcription of the genes of vesicular stomatitis virus. Proc. Natl Acad. Sci. U.S.A. 1976;73:1504–1508. doi: 10.1073/pnas.73.5.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ball L.A., White C.N. Order of transcription of genes of vesicular stomatitis virus. Proc. Natl Acad. Sci. U.S.A. 1976;73:442–446. doi: 10.1073/pnas.73.2.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Testa D., Chanda P.K., Banerjee A.K. Unique mode of transcription in vitro by vesicular stomatitis virus. Cell. 1980;21:267–275. doi: 10.1016/0092-8674(80)90134-8. [DOI] [PubMed] [Google Scholar]
- 21.Iverson L.E., Rose J.K. Localized attenuation and discontinuous synthesis during vesicular stomatitis virus transcription. Cell. 1981;23:477–484. doi: 10.1016/0092-8674(81)90143-4. [DOI] [PubMed] [Google Scholar]
- 22.Rhodes D.P., Banerjee A.K. 5′-terminal sequence of vesicular stomatitis virus mRNA's synthesized in vitro. J. Virol. 1976;17:33–42. doi: 10.1128/jvi.17.1.33-42.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Banerjee A.K., Rhodes D.P. In vitro synthesis of RNA that contains polyadenylate by virion-associated RNA polymerase of vesicular stomatitis virus. Proc. Natl Acad. Sci. U.S.A. 1973;70:3566–3570. doi: 10.1073/pnas.70.12.3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barr J.N., Whelan S.P., Wertz G.W. cis-Acting signals involved in termination of vesicular stomatitis virus mRNA synthesis include the conserved AUAC and the U7 signal for polyadenylation. J. Virol. 1997;71:8718–8725. doi: 10.1128/jvi.71.11.8718-8725.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hwang L.N., Englund N., Pattnaik A.K. Polyadenylation of vesicular stomatitis virus mRNA dictates efficient transcription termination at the intercistronic gene junctions. J. Virol. 1998;72:1805–1813. doi: 10.1128/jvi.72.3.1805-1813.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barr J.N., Wertz G.W. Polymerase slippage at vesicular stomatitis virus gene junctions to generate poly(A) is regulated by the upstream 3′-AUAC-5′ tetranucleotide: implications for the mechanism of transcription termination. J. Virol. 2001;75:6901–6913. doi: 10.1128/JVI.75.15.6901-6913.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Furuichi Y., Shatkin A.J. Viral and cellular mRNA capping: past and prospects. Adv. Virus Res. 2000;55:135–184. doi: 10.1016/S0065-3527(00)55003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shuman S. Structure, mechanism, and evolution of the mRNA capping apparatus. Prog. Nucleic Acid Res. Mol. Biol. 2001;66:1–40. doi: 10.1016/s0079-6603(00)66025-7. [DOI] [PubMed] [Google Scholar]
- 29.Cougot N., van Dijk E., Babajko S., Seraphin B. ‘Cap-tabolism’. Trends Biochem. Sci. 2004;29:436–444. doi: 10.1016/j.tibs.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 30.Ogino T., Banerjee A.K. An unconventional pathway of mRNA cap formation by vesiculoviruses. Virus Res. 2011;162:100–109. doi: 10.1016/j.virusres.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogino T., Banerjee A.K. mRNA capping by vesicular stomatitis virus and other related viruses. In: Luo M., editor. Negative Strand RNA Virus. Singapore: World Scientific; 2011. pp. 79–94. [Google Scholar]
- 32.Ogino T., Banerjee A.K. Formation of guanosine(5′)tetraphospho(5′)adenosine cap structure by an unconventional mRNA capping enzyme of vesicular stomatitis virus. J. Virol. 2008;82:7729–7734. doi: 10.1128/JVI.00326-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koonin E.V., Moss B. Viruses know more than one way to don a cap. Proc. Natl Acad. Sci. U.S.A. 2010;107:3283–3284. doi: 10.1073/pnas.0915061107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schubert M., Harmison G.G., Sprague J., Condra C.S., Lazzarini R.A. In vitro transcription of vesicular stomatitis virus: initiation with GTP at a specific site within the N cistron. J. Virol. 1982;43:166–173. doi: 10.1128/jvi.43.1.166-173.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Piwnica-Worms H., Keene J.D. Sequential synthesis of small capped RNA transcripts in vitro by vesicular stomatitis virus. Virology. 1983;125:206–218. doi: 10.1016/0042-6822(83)90074-0. [DOI] [PubMed] [Google Scholar]
- 36.Tekes G., Rahmeh A.A., Whelan S.P. A freeze frame view of vesicular stomatitis virus transcription defines a minimal length of RNA for 5′ processing. PLoS Pathog. 2011;7:e1002073. doi: 10.1371/journal.ppat.1002073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stillman E.A., Whitt M.A. Transcript initiation and 5′-end modifications are separable events during vesicular stomatitis virus transcription. J. Virol. 1999;73:7199–7209. doi: 10.1128/jvi.73.9.7199-7209.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J., Rahmeh A., Morelli M., Whelan S.P. A conserved motif in region v of the large polymerase proteins of nonsegmented negative-sense RNA viruses that is essential for mRNA capping. J. Virol. 2008;82:775–784. doi: 10.1128/JVI.02107-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J., Rahmeh A., Brusic V., Whelan S.P. Opposing effects of inhibiting cap addition and cap methylation on polyadenylation during vesicular stomatitis virus mRNA synthesis. J. Virol. 2009;83:1930–1940. doi: 10.1128/JVI.02162-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ogino T. In vitro capping and transcription of rhabdoviruses. Methods. 2013;59:188–198. doi: 10.1016/j.ymeth.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pall G.S., Hamilton A.J. Improved northern blot method for enhanced detection of small RNA. Nat. Protoc. 2008;3:1077–1084. doi: 10.1038/nprot.2008.67. [DOI] [PubMed] [Google Scholar]
- 42.Kuchino Y., Nishimura S. Enzymatic RNA sequencing. Methods Enzymol. 1989;180:154–163. doi: 10.1016/0076-6879(89)80099-0. [DOI] [PubMed] [Google Scholar]
- 43.Clepet C. RNA captor: a tool for RNA characterization. PLoS ONE. 2011;6:e18445. doi: 10.1371/journal.pone.0018445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rio D.C., Ares M., Jr, Hannon G.J., Nilsen T.W. RNA: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2011. Measuring the length of poly(A) tails; pp. 554–558. [Google Scholar]
- 45.Lawson N.D., Stillman E.A., Whitt M.A., Rose J.K. Recombinant vesicular stomatitis viruses from DNA. Proc. Natl Acad. Sci. U.S.A. 1995;92:4477–4481. doi: 10.1073/pnas.92.10.4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schnell M.J., Buonocore L., Whitt M.A., Rose J.K. The minimal conserved transcription stop-start signal promotes stable expression of a foreign gene in vesicular stomatitis virus. J. Virol. 1996;70:2318–2323. doi: 10.1128/jvi.70.4.2318-2323.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martin S.A., Moss B. mRNA guanylyltransferase and mRNA (guanine-7-)-methyltransferase from vaccinia virions. Donor and acceptor substrate specificites. J. Biol. Chem. 1976;251:7313–7321. [PubMed] [Google Scholar]
- 48.Knapp G. Enzymatic approaches to probing of RNA secondary and tertiary structure. Methods Enzymol. 1989;180:192–212. doi: 10.1016/0076-6879(89)80102-8. [DOI] [PubMed] [Google Scholar]
- 49.Poch O., Blumberg B.M., Bougueleret L., Tordo N. Sequence comparison of five polymerases (L proteins) of unsegmented negative-strand RNA viruses: theoretical assignment of functional domains. J. Gen. Virol. 1990;71:1153–1162. doi: 10.1099/0022-1317-71-5-1153. [DOI] [PubMed] [Google Scholar]
- 50.Bujnicki J.M., Rychlewski L. In silico identification, structure prediction and phylogenetic analysis of the 2′-O-ribose (cap 1) methyltransferase domain in the large structural protein of ssRNA negative-strand viruses. Protein Eng. 2002;15:101–108. doi: 10.1093/protein/15.2.101. [DOI] [PubMed] [Google Scholar]
- 51.Mellon M.G., Emerson S.U. Rebinding of transcriptase components (L and NS proteins) to the nucleocapsid template of vesicular stomatitis virus. J. Virol. 1978;27:560–567. doi: 10.1128/jvi.27.3.560-567.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.De B.P., Banerjee A.K. Specific interactions of vesicular stomatitis virus L and NS proteins with heterologous genome ribonucleoprotein template lead to mRNA synthesis in vitro. J. Virol. 1984;51:628–634. doi: 10.1128/jvi.51.3.628-634.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stillman E.A., Whitt M.A. Mutational analyses of the intergenic dinucleotide and the transcriptional start sequence of vesicular stomatitis virus (VSV) define sequences required for efficient termination and initiation of VSV transcripts. J. Virol. 1997;71:2127–2137. doi: 10.1128/jvi.71.3.2127-2137.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hinzman E.E., Barr J.N., Wertz G.W. Identification of an upstream sequence element required for vesicular stomatitis virus mRNA transcription. J. Virol. 2002;76:7632–7641. doi: 10.1128/JVI.76.15.7632-7641.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rose J.K. Heterogneeous 5′-terminal structures occur on vesicular stomatitis virus mRNAs. J. Biol. Chem. 1975;250:8098–8104. [PubMed] [Google Scholar]
- 56.Liuzzi M., Mason S.W., Cartier M., Lawetz C., McCollum R.S., Dansereau N., Bolger G., Lapeyre N., Gaudette Y., Lagace L., et al. Inhibitors of respiratory syncytial virus replication target cotranscriptional mRNA guanylylation by viral RNA-dependent RNA polymerase. J. Virol. 2005;79:13105–13115. doi: 10.1128/JVI.79.20.13105-13115.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rehwinkel J., Reis e Sousa C. Targeting the viral Achilles’ heel: recognition of 5′-triphosphate RNA in innate anti-viral defence. Curr. Opin. Microbiol. 2013;16:485–492. doi: 10.1016/j.mib.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.