


Abstract
Alternative splicing (AS) modulates many physiological and pathological processes. For instance, AS of the BCL-X gene balances cell survival and apoptosis in development and cancer. Herein, we identified the polypyrimidine tract binding protein (PTBP1) as a direct regulator of BCL-X AS. Overexpression of PTBP1 promotes selection of the distal 5′ splice site in BCL-X exon 2, generating the pro-apoptotic BCL-Xs splice variant. Conversely, depletion of PTBP1 enhanced splicing of the anti-apoptotic BCL-XL variant. In vivo cross-linking experiments and site-directed mutagenesis restricted the PTBP1 binding site to a polypyrimidine tract located between the two alternative 5′ splice sites. Binding of PTBP1 to this site was required for its effect on splicing. Notably, a similar function of PTBP1 in the selection of alternative 5′ splice sites was confirmed using the USP5 gene as additional model. Mechanistically, PTBP1 displaces SRSF1 binding from the proximal 5′ splice site, thus repressing its selection. Our study provides a novel mechanism of alternative 5′ splice site selection by PTBP1 and indicates that the presence of a PTBP1 binding site between two alternative 5′ splice sites promotes selection of the distal one, while repressing the proximal site by competing for binding of a positive regulator.
INTRODUCTION
Regulation of splicing plays a pivotal role in controlling gene expression and in generating proteomic diversity (1,2). Splicing allows removal of the non-coding introns and ligation of the coding exons in pre-mRNAs and is operated by a ribonucleoprotein complex, named spliceosome, aided by multiple factors (3–5). Notably, although many exons are constitutively spliced, the large majority of human genes undergoes alternative splicing (AS) of a substantial number of variable exons (6,7). In this way, AS generates distinct mRNAs from a single pre-mRNA, yielding multiple protein isoforms that often display different functions in the cell (5,8). Indeed, although exons are defined by canonical signals (5′ splice site, branch point, polypyrimidine tract and 3′ splice site) that are recognized by the spliceosome, these sequences are short and degenerate and their precise recognition requires additional factors (3,4). Cis-regulatory RNA elements in exons and introns act as enhancer or silencer of exon recognition, often by recruiting specific RNA binding proteins (RBPs) that assist the spliceosome (3,5). Moreover, RNA structures, transcription-related processes and epigenetic modifications of the chromatin have all been shown to contribute to AS regulation (9,10).
The serine–arginine rich (SR) proteins and the heterogeneous nuclear ribonucleoproteins (hnRNPs) are the most typical families of RBPs involved in splicing regulation (2,3). With some exceptions, SR proteins play a positive role in exon recognition by binding exonic and intronic enhancers nearby the alternative splice sites. Conversely, hnRNPs often bind to silencer elements and interfere with the recruitment of spliceosome component(s) or with that of positive regulator(s) (3). For instance, hnRNP I, also known as polypyrimidine tract binding protein 1 (hereafter named PTBP1), can repress splicing by preventing recruitment of U2AF65 (11) or correct base pairing of U2 snRNP (12), or by affecting the interaction of U1 snRNP with other components of the spliceosome (13). Furthermore, the activity of cis-regulatory RNA elements is also dependent on its context. The presence of binding sites for NOVA (14), RBFOX2 (15) and PTBP1 (16,17) upstream or within the target exon correlates with exon skipping, whereas their location downstream of the exon promotes inclusion. Thus, AS results from a complex interplay between cis-regulatory elements, their position in the gene and the activity of trans-acting factors. Since modulation of AS strongly contributes to developmental processes (8), whereas its aberrant regulation leads to various diseases (18), deciphering the ‘splicing code’ is likely important to predict or even correct AS outcome in physiological and pathological conditions.
Apoptosis is a physiological process tightly regulated by AS (19). Many eukaryotic apoptotic genes are alternatively spliced to yield proteins with different or opposite functions. Hence, modulation of AS in response to internal and external stimuli can finely regulate the balance between cell survival and death (19,20). A typical example is offered by the BCL-X gene, which generates two splice variants with antagonistic roles in cell survival (21). Selection of the proximal 5′ splice site (L) in exon 2 promotes the anti-apoptotic long variant, BCL-XL, whereas selection of the distal 5′ splice site (S) promotes the pro-apoptotic short variant, BCL-Xs (19). Notably, regulation of BCL-X AS is strictly controlled and it is linked to cell-cycle progression (22). Moreover, modulation of this splicing event is of clinical relevance in cancer, as high expression levels of the anti-apoptotic BCL-XL variant contribute to chemotherapeutic resistance and poor prognosis (23–25). In line with its key role, BCL-X AS is regulated by numerous splicing factors (26–30), whose activities are controlled by kinases (27,31), transcriptional regulators (32,33) and components of the exon junction complex (34). Since deregulation of apoptosis plays a critical role in tumorigenesis (19,23–25), understanding the mechanisms underlying splicing of the pro-apoptotic isoform of BCL-X could pave the way for the development of new therapeutic approaches (35,36).
Here, we identified the PTBP1 as a regulator of the pro-apoptotic variant of BCL-X. PTBP1 directly binds to a specific polypyrimidine tract located between the two alternative 5′ splice sites in exon 2, which is required for PTBP1-dependent BCL-X AS. Binding of PTBP1 to this site represses the downstream 5′ splice site and favors the upstream one. A similar regulation was observed for alternative 5′ splice site selection in USP5 exon 15. Mechanistically, binding of PTBP1 displaces SRSF1 from the proximal 5′ splice site and antagonizes its activity in the regulation of BCL-X AS. Thus, our results identify BCL-X as a splicing target of PTBP1 and suggest a potentially novel mechanism by which this splicing factor modulates alternative 5′ splice site selection in target exons.
MATERIALS AND METHODS
Plasmid constructs
The BCL-X, BCL-X Δ1-500 and X2.13 minigenes have been previously described (27,33,37). The USP5 minigene was amplified using primers #(1-2) from HeLa cell genomic DNA and cloned into the XhoI/HindIII restriction sites of pCDNA3.1(−) vector. The E2m1- and E2m2- BCL-X Δ1-500 and E15m1-USP5 minigenes were constructed using the mega-primer strategy (38) using primers #(3-5-4), #(3-6-4) and #(1-7-2), respectively. Amplified bands were cloned into XhoI/HindIII restriction sites of pCDNA3.1(−) vector. The human hnRNP F cDNA was amplified from HeLa cells using primers #(8-9) and cloned into HindIII/BamHI restriction sites of p3XFLAG (Sigma-Aldrich). The PTBP1 cDNA was amplified from pCMV-His-PTBP1 using primers #(10-11) and cloned into EcoRI/SalI restriction sites of pEGFPC1 vector (Clontech). The human SRSF3 cDNA was amplified from HeLa cells using primers #(12-13) and cloned into PstI-BamHI restriction sites of p3XFLAG vector (Sigma-Aldrich). All oligonucleotide sequences are listed in Supplementary Table S1. Polymerase chain reactions (PCRs) were performed using Phusion Hot Start High-Fidelity DNA polymerase (Finnzymes) according to manufacturer's instruction. All plasmids were sequenced and validated.
Cell cultures, transfections and cell extract preparation
Cell cultures, transfections and sample preparation were carried out by standard methods as previously described (33). Briefly, HEK293T cells were transfected with various combinations of vectors as indicated using Lipofectamine 2000 (Invitrogen). For RNAi, cells were transfected twice with 60 nM siRNAs (Sigma-Aldrich) using Lipofectamine RNAi Max (Invitrogen) and Opti-MEM medium (Invitrogen) according to manufacturer's instruction. siRNA for PTBP1/PTBP2 were purchased from Dharmacon (On target plus human PTBP1 5725 siRNA and On target plus human PTBP2 58155 siRNA). Sequences for scramble (CTRL), hnRNP F and SRSF1 siRNA are listed in Supplementary Table S1.
Biotin-RNA pulldown
Biotin-RNA pulldown experiments were carried out using HeLa cell nuclear extracts as previously described (39). The BCL-X and USP5 RNA probes were in vitro synthesized from a PCR product amplified using primers (see Supplementary Table S1) #(16-17) and #(33-34), respectively, from wt or mutant minigenes in the presence of biotin labeled dNTP (Roche). Extracts were pre-cleared for 1 h on protein-A-Sepharose beads (Sigma-Aldrich) and 1 h on streptavidin-Sepharose beads (Sigma-Aldrich). Pre-cleared extracts were then incubated with streptavidin-Sepharose beads in the presence of 0.1% bovine serum albumin and biotinylated RNA probe. After 2 h of incubation at 4°C under rotation, beads were washed three times with washing buffer [10 mM Tris/HCl pH 7.4, 25 mM MgCl2, 100 mM NaCl, 1 mM DTT, 0.5% Triton, 20 mM β-glicerolphosphate, 1 mM Dithiothreitol (DTT), 0.5 mM Na3VO4 plus protease inhibitor cocktail (Sigma-Aldrich) and RNAse inhibitor (Promega)], and proteins were eluted in SDS sample buffer for western blot analysis.
UV-crosslinked and RNA immunoprecipitation (CLIP) assays
CLIP assays were performed as previously described (33,40). Briefly, cells were washed with phosphate buffered saline, UV-irradiated (400 mJ/cm2) and incubated 10 min on ice in the presence of lysis buffer [50 mM Tris pH 8, 100 mM NaCl, 1% NP40, 1 mM MgCl2, 0.1 mM CaCl2, 0.5 mM Na3VO4, 1 mM dithiothreitol, protease inhibitor cocktail (Sigma-Aldrich), RNase inhibitor (Promega)]. Samples were briefly sonicated and incubated with DNase-RNase free (Ambion) for 3 min at 37°C and then centrifuged at 15 000 × g for 3 min at 4°C. Half extract (1 mg) was treated with Proteinase K for 30 min at 37°C and RNA was purified by standard procedure (input). The remaining half of the extract (1 mg) was diluted to 1 ml with lysis buffer and immunoprecipitated by using anti-PTBP1 (Santa Cruz), anti-SRSF1 (Santa Cruz) antibodies or IgGs (negative control) in the presence of protein-G magnetic dynabeads (Novox, Life Technologies). When indicated, 10 μl/ml of RNaseI 1:1000 (Ambion) were added. Immunoprecipitates (IPs) were then incubated for 2 h at 4°C under rotation. After two washes with high salt buffer supplemented with RNase inhibitor and one wash with Proteinase K buffer, the IPs were resuspended in 100 ml of Proteinase K buffer. An aliquot (10%) was kept as control of immunoprecipitation while the rest was treated with 50 mg of Proteinase K and incubated for 1 h at 55°C. RNA was then isolated by standard procedures.
Splicing assays and RT-PCR analyses
BCL-X and USP5 minigene splicing assays and reverse transcriptase PCR (RT-PCR) analyses were performed as previously described (27,33). Quantitative real-time PCRs (qPCRs) were performed using LightCycler 480 SYBR Green I Master and the LightCycler 480 System (Roche) following manufacturer's instructions. For CLIP analyses, each sample was normalized with respect to its input. BCL-X or USP5 RNA associated with PTBP1 or SRSF1 is represented as fold enrichment relative to IgG samples. The expression level of BCL-Xs (relative to BCL-XL isoform), or PTBP1 (relative to GAPDH), was calculated by ΔΔCt method (31,33).
RESULTS
Identification of splicing factors involved in the modulation of BCL-X alternative splicing
To identify sequence elements and splicing factors involved in the alternative 5′ splice site selection in BCL-X (Figure 1A; 21), we employed minigene-based systems in HEK293T cells (Figure 1B, upper panel). Splicing assays showed that the largest minigene (named ‘BCL-X’), which contains part of exon 1, exon 2, intron 2 and exon 3 (37), displays a splicing pattern similar to that of the endogenous BCL-X and mainly yields the BCL-XL variant (Figure 1B; Supplementary Figure S1A). However, progressive deletions of the 5′ region in the Δ1-500 (33) and X2.13 minigenes (26) enhanced splicing of BCL-Xs (Figure 1B). This observation is in line with the presence of cis-regulatory elements upstream of the distal 5′ splice site required for BCL-XL splicing (41). Notably, further deletion of the B2 region between the two alternative 5′ splice sites (26) completely abrogated splicing of the short variant (Figure 1B), suggesting that this region contains cis-regulatory elements that are required for distal 5′ splice site selection in exon 2.
Figure 1.
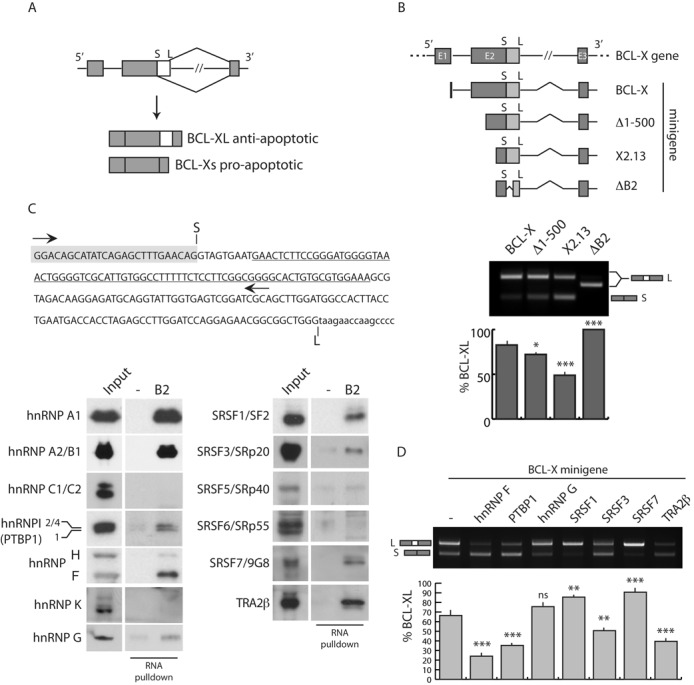
Identification of RNA-binding proteins involved in selection of distal 5′ splice site in BCL-X exon 2. (A) Schematic representation of BCL-X alternative splicing. Exons (boxes), introns (lines), and distal (S) and proximal (L) 5′ splice sites in exon 2 are indicated. (B) Scheme of BCL-X gene and mutant minigenes (upper panel). RT-PCR of in vivo splicing assay performed in HEK293T cells in the presence of indicated BCL-X minigenes (bottom panel). The bar graph shows the percentage of BCL-XL (mean ± SD, n = 3). (C) In the upper panel, sequence of BCL-X transcribed RNA using indicated primers (arrows) is shown. The sequences before the distal (S) 5′ splice sites (gray box) and deleted in ΔB2 minigene (underlined) are indicated. Western blot analysis of RNA-pulldown assay performed using biotin-labeled BCL-X B2 RNA or streptavidin (−), as negative control, and in the presence of commercial nuclear extracts from HeLa cells (bottom panel). (D) RT-PCR of in vivo splicing assay performed in HEK293T cells transfected with BCL-X minigene and the indicated splicing factors. The bar graph shows the percentage of BCL-XL (mean ± SD, n = 3). The P values of Student's t-test are reported. *P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., not significant.
To identify splicing factor(s) that bind the B2 element, we carried out RNA-pulldown assays using a biotin-labeled RNA overlapping this region (Figure 1C, upper panel) and nuclear extracts from HeLa cells. Western blot analyses showed that hnRNP A1, hnRNP A2/B1, hnRNP F/H, hnRNP G and PTBP1 (hnRNP I) bound the B2 element (Figure 1C, lower panel), whereas hnRNP K and hnRNP C1/C2 did not. Importantly, binding sites for hnRNP F/H were previously mapped in the B2 element (26), whereas hnRNP K was shown to bind upstream of the distal 5′ splice site (29), thus validating the fidelity of our analysis. In addition, we found that several SR proteins (SRSF1, SRSF3, SRSF7 and TRA2β) bind the B2 element.
Among the factors identified by our RNA-pulldown assay, SRSF1 (22,27), hnRNP A1 (27), hnRNP A2/B1 (42), hnRNP F/H (26) and hnRNP K (29) were already known to regulate BCL-X AS. To assess whether the other splicing factors were also functionally relevant, we performed splicing assays using the BCL-X minigene. We found that, with the exception of hnRNP G, all these RBPs modulated BCL-X AS. SRSF7 promoted splicing of the anti-apoptotic BCL-XL variant (Figure 1D) similarly to SRSF1 (22,27). By contrast, PTBP1, SRSF3 and TRA2β favored selection of the distal 5′ splice site (Figure 1D; Supplementary Figure S1B) like hnRNP F (26). These results suggest that the B2 element contains cis-regulatory element(s) that are bound by splicing factors involved in BCL-X AS.
PTBP1 binds exon 2 of BCL-X RNA in vivo
Given the relevance of BCL-X splicing regulation in cancer (19,35), we focused our attention on PTBP1 because this RBP plays oncogenic functions through splicing modulation of cancer-relevant genes (43–46).
PTBP1 was recently shown to play a role in glioblastoma (44–46). To evaluate the correlation between PTBP1 expression and BCL-X splicing, we selected three glioblastoma cell lines expressing different levels of PTBP1: A172, LN229 and LN18 (Supplementary Figure S2A and B). We observed that the expression of BCL-Xs directly correlates with that of PTBP1 in these cell lines (A172 > LN229 > LN18) (Supplementary Figure S2C). Next, we analyzed the response of glioblastoma cells to the chemotherapeutic agent temozolomide (the drug used in therapy) using the PTBP1-high A172 and the PTBP1-low LN18 cell lines. Treatment with the drug increased apoptosis (Supplementary Figure S2D) and the expression of BCL-Xs in the PTBP1-high A172 cells, but not in the PTBP1-low LN18 cells (Supplementary Figure S2E). These results suggest that high levels of PTBP1 are required for modulation of BCL-X splicing and apoptosis in live cells and support the involvement of PTBP1 in this regulation.
In line with this hypothesis, qPCR using specific exon junction primers (31,33) revealed that depletion of PTBP1 and 2 (PTBP1/2) in HEK293T cells significantly altered splicing of the endogenous BCL-X gene in favor of the anti-apoptotic BCL-XL variant (Figure 2A). A comparable effect was obtained by silencing hnRNP F, a previously validated regulator of BCL-X AS (24), whereas knockdown of SRSF1 exerted the opposite effect (31; Figure 2A).
Figure 2.
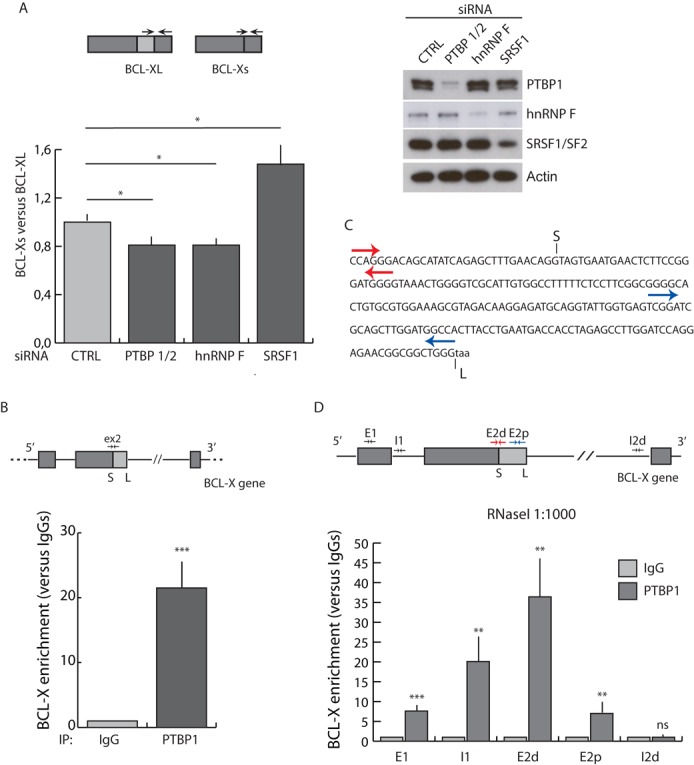
BCL-X is an RNA target of PTBP1 in live cells. (A) qPCR analysis of BCL-X isoforms in HEK293T cells transfected with scramble (CTRL), PTBP1 and PTBP2 (PTBP1/2), hnRNP F and SRSF1 siRNAs. The bar graph of fold variation of each sample was calculated by delta–delta Ct method as described in the Materials and Methods section (mean ± SD, n = 3). Scheme of exon junction primers used for qPCR (arrows) is also shown. Down-regulation of protein level was assessed by western blot analysis (right panel). (B) UV-crosslink and RNA immunoprecipitation (CLIP) of PTBP1 performed in HEK293T. Associated BCL-X RNA was quantified by qPCR using primers indicated in the upper BCL-X scheme and represented as fold enrichment relative to the IgG sample (mean ± SD, n = 3). (C) Schematic representation of distal (E2d; red arrows) and proximal (E2p; light blue arrows) exon 2 BCL-X primers used in (D). (D) CLIP of PTBP1 performed as in (B) in the presence of RNaseI (1:1000). (A, B and D) The P values of Student's t-test are reported. *P < 0.05, **, P < 0.01; ***, P < 0.001; n.s., not significant.
UV-crosslink and RNA immunoprecipitation (CLIP) assays indicated that PTBP1 binds the endogenous BCL-X mRNA in vivo in HEK293T cells (Figure 2B). To precisely map the binding site, we performed CLIP experiments in the presence of low concentrations of RNaseI (40; Supplementary Figure S3A and B). A progressive enrichment of PTBP1 was detected along the 5′ region of the BCL-X RNA, with a peak surrounding the B2 element in exon 2 (E2d in Figure 2C and D). PTBP1 binding was then decreased nearby the proximal 5′ splice site in exon 2 (E2p in Figure 2C and D) and was undetectable in the distal portion of the large intron 2 (I2d) near exon 3 (Figure 2D). As expected, no enrichment was observed if samples were treated with high concentration of RNaseI to completely degrade the RNA (Supplementary Figure S3C). These results confirmed that PTBP1 binds the B2 element of BCL-X RNA in vivo.
Binding of PTBP1 to the B2 element of BCL-X exon 2 favors selection of the distal 5′ splice site
Next, we tested whether the B2 region is involved in distal 5′ splice site selection by PTBP1 using the mutant BCL-X ΔB2 minigene (Figure 1B). As previously shown for the longer BCL-X minigene (Figure 1D), overexpression of PTBP1 promoted splicing of BCL-Xs in the wild-type (wt) minigene (X2.13; Figure 3A, lane 3). PTBP1 activity was similar to that of hnRNP F and opposite to that of SRSF1 (Figure 3A). By contrast, deletion of the B2 element completely abolished the activity of PTBP1 (Figure 3A), similarly to what was previously shown for hnRNP F that also binds this element (26; Figure 3A). These results suggest that PTBP1 binding to the B2 element is required for its effect on BCL-X AS.
Figure 3.
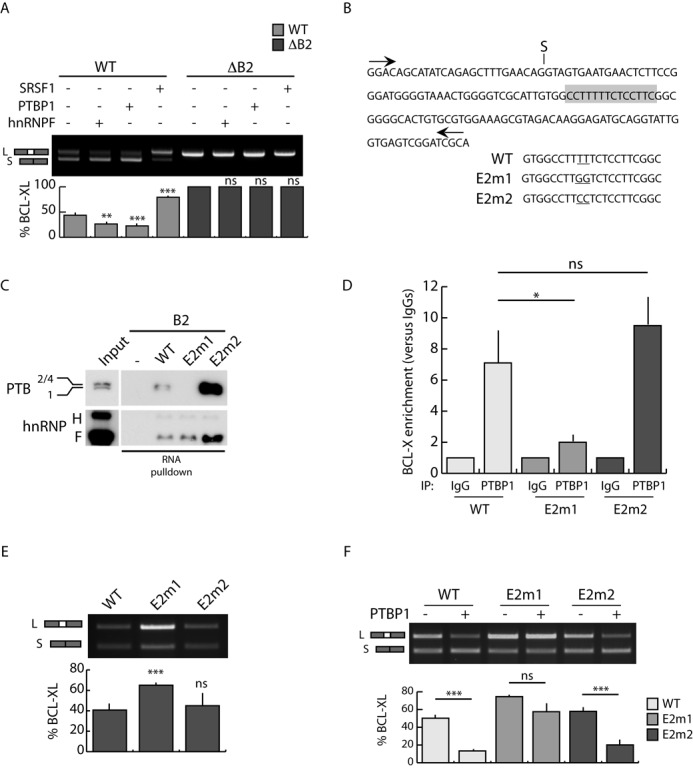
The B2 element is required for PTBP1-dependent BCL-Xs splicing. (A) RT-PCR analysis of the in vivo splicing assays performed in HEK293T cells transfected with WT or mutated (ΔB2) BCL-X minigenes and the indicated splicing factors. The bar graph of the percentage of BCL-XL is also shown (mean ± SD, n = 3). (B) Scheme of BCL-X exon 2 sequence. The putative PTBP1 binding site is highlighted (gray box). The mutated bases in E2m1 and E2m2 mutants are underlined. (C) Western blot analysis of RNA-pulldown assay using biotin-labeled BCL-X WT or mutant (E2m1 and E2m2) RNA. Streptavidin beads have been used as control (−). (D) CLIP experiments of PTBP1 performed in HEK293T transfected with the indicated minigenes. Associated BCL-X RNA was quantified by qPCR (primers used are indicated in Supplementary Table S1). Data are represented as fold enrichment relative to the IgG sample (mean ± SD, n = 3). (E and F) RT-PCR analysis of in vivo splicing assays performed in HEK293T cells transfected with wt and mutated (E2m1 and E2m2) minigenes in the presence (F) or absence (E) of PTBP1. The bar graph of the percentage of BCL-XL is shown (mean ± SD, n = 3). (A, D, E and F) The P values of Student's t-test are reported. *P < 0.05, **, P < 0.01; ***, P < 0.001; n.s., not significant.
Sequence analysis of the B2 element revealed the presence of a polypyrimidine tract that resembles a possible binding site for PTBP1 (gray box in Figure 3B). To test this possibility, we carried out RNA-pulldown assays using B2 biotin-labeled RNAs mutated in this polypyrimidine stretch. In E2m1, two pyrimidines (TT) were replaced with purines (GG) to disrupt the continuity of the polypyrimidine tract (Figure 3B). By contrast, in E2m2 we produced a conservative mutation in which the two thymidines were replaced with cytosines (CC), thus altering the nucleotide sequence but not the continuity of the pyrimidine stretch (Figure 3B). Binding of PTBP1 to the B2 element was strongly reduced when the polypyrimidine tract is disrupted (E2m1) (Figure 3C). This effect is specific as binding of hnRNP F was not altered by this mutation (Figure 3C). Remarkably, the conservative substitutions in E2m2 increased binding of PTBP1 to the B2 element (Figure 3C).
To test whether the integrity of this polypyrimidine tract was also required for the binding of PTBP1 in vivo, we inserted the E2m1 and E2m2 mutations in the BCL-X Δ1−500 minigene. CLIP assays in transfected HEK293T cells showed that recruitment of endogenous PTBP1 to the BCL-X RNA is strongly impaired when the polypyrimidine tract is disrupted (E2m1), whereas it is unaffected by the conservative E2m2 mutation (Figure 3D). Interestingly, splicing assays using the wt or mutated BCL-X minigenes showed that disruption of the PTBP1 binding site caused enhanced splicing of the E2m1 minigene toward the BCL-XL variant (Figure 3E), indicating that the endogenous PTBP1 participates in the splicing regulation of the minigene. Moreover, overexpression of PTBP1 significantly enhanced splicing of BCL-Xs in cells transfected with the wt and E2m2 minigenes, but its activity was strongly reduced in the presence of the E2m1 minigene (Figure 3F). The E2m1 and E2m2 mutations specifically affected the activity of PTBP1, as neither hnRNP F nor SRSF1 activities were influenced by them (Supplementary Figure S4A and B). Collectively, these results demonstrate that binding of PTBP1 to the polypyrimidine tract in the B2 element promotes selection of the distal 5′ splice site in BCL-X exon 2.
Binding of the PTBP1 between two 5′ alternative splice sites favors selection of the distal one
PTBP1 is a multifunctional RBP whose role in AS is well established (45). It can function both as repressor and activator, depending on the location where it binds relative to target exons (47). In particular, binding of PTBP1 upstream or within an exon represses its recognition by the spliceosome, whereas its binding downstream of the exon promotes inclusion (17,48). Since PTBP1 binds within BCL-X exon 2 between the two alternative 5′ splice sites, we reasoned that it may behave as a repressor for the proximal splice site (like binding within the exon in the proposed model) and as an activator for the distal one, with the binding site being regarded as part of the downstream intron. To test this hypothesis, we searched the literature for other alternative 5′ splicing events regulated by PTBP1. Similarly to BCL-X, selection of an alternative 5′ splice site in exon 15 of the USP5 gene generates a shorter isoform (Figure 4A), whose splicing was promoted by PTBP1 expression (49). We observed that the endogenous USP5 pre-mRNA was almost completely spliced to yield the shorter isoform 2 mRNA variant in HEK293T cells. However, knockdown of PTBP1/2 partially reverted this regulation and yielded equal amounts of isoform 1 splicing (Figure 4B).
Figure 4.
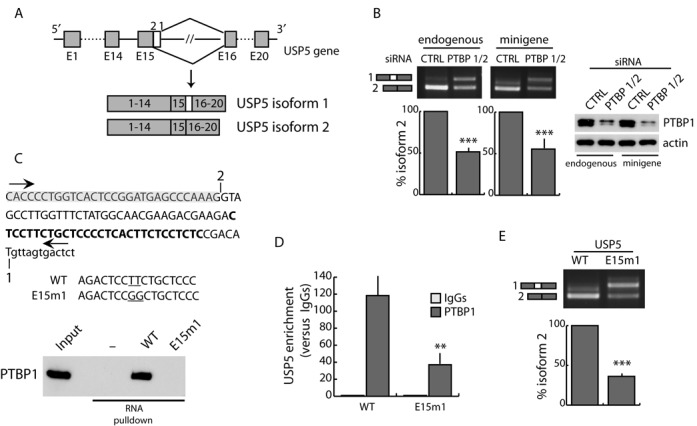
Binding of PTBP1 between two competing 5′ splice sites favors the selection of the distal one. (A) Schematic representation of USP5 alternative splicing. Exons (boxes), introns (lines), and distal (2) and proximal (1) 5′ splice sites in exon 15 are indicated. (B) RT-PCR of in vivo splicing assay performed in HEK293T cells transfected with control (CTRL) or PTBP1/2 siRNAs in the presence (right panel) or absence (left panel) of USP5 minigene. The bar graph shows the percentage of endogenous (left panel) or minigene-derived (right panel) USP52 variant (mean ± SD, n = 3). (C) Sequence of USP5 RNA transcribed using indicated primers (arrows). The sequence before the distal (2) 5′ splice site (gray box) and the putative PTBP1 binding site (bold) is indicated. The mutated bases in E15m1 mutant are underlined. Western blot analysis (bottom panel) of RNA-pulldown assay performed using biotin-labeled wt (WT) and mutated (E15m1) USP5 RNA, and streptavidin (−) as negative control. (D) CLIP experiments of PTBP1 performed in HEK293T transfected with wt (WT) or mutated (E15m1) minigenes. Associated USP5 RNA was quantified by qPCR (primers used are indicated in Supplementary Table S1). Data are represented as fold enrichment relative to the IgG sample (mean ± SD, n = 3). (E) RT-PCR of in vivo splicing assay performed in HEK293T cells transfected with WT or E15m1 mutant minigenes. The bar graph shows the percentage of USP52 isoform (mean ± SD, n = 3). (B, D and E) The P values of Student's t-test are reported. **, P < 0.01; ***, P < 0.001.
To investigate the mechanism of PTBP1-dependent USP5 AS, we generated a minigene encompassing the exon 14–exon 16 genomic region of USP5. The minigene recapitulated the regulation of the endogenous gene, as knockdown of PTBP1/2 enhanced splicing of the long isoform 1 variant (Figure 4B). Next, since the PTBP1 binding sites relevant for USP5 splicing regulation were not identified in the previous study (49), we searched the sequence for potential PTBP1-sensitive elements. As for BCL-X exon 2, analysis of the sequence located between the alternative 5′ splice sites in USP5 exon 15 revealed the presence of a polypyrimidine tract (in bold in Figure 4C), which resembles a putative PTBP1 binding site (16,17,47). RNA pulldown assays demonstrated that PTBP1 efficiently binds the sequence between the alternative 5′ splice sites and that mutation of the putative consensus abolished this interaction (Figure 4C). CLIP assays using wt and mutated USP5 minigenes confirmed that endogenous PTBP1 strongly binds the wt USP5 RNA in the exon 15 region, whereas this interaction was significantly impaired when the PTBP1 binding site was mutated (Figure 4D). Accordingly, splicing of the mutated minigene recapitulated the effect of PTBP1 knockdown on USP5 splicing (Figure 4E). These observations confirm that binding of PTBP1 between two alternative 5′ splice sites within an exon favors selection of the distal site while repressing the proximal one, suggesting that this is a general mechanism of action for PTBP1 in alternative 5′ splice site selection.
PTBP1 antagonizes SRSF1-dependent splicing of BCL-X
With the exception of a few examples, such as interference with U2 snRNP base pairing (13), the mechanism by which binding of PTBP1 within an exon represses its inclusion is still largely unknown. Due to the antagonistic role of hnRNPs and SR proteins (2,3,5), we reasoned that PTBP1 might either cooperate with hnRNP F or compete with SRSF1, two regulators of BCL-X splicing (22,26,27).
We found that concomitant knockdown of PTBP1/2 and hnRNP F did not exert additive effects (Supplementary Figure S4C). Moreover, co-overexpression of PTBP1 and hnRNP F elicited only a mild additive effect (Supplementary Figure S4D), indicating that the two splicing factors do not cooperate in the regulation of BCL-X AS. Next, we asked whether PTBP1 functions by antagonizing SRSF1 activity. First, we performed CLIP assays in HEK293T to evaluate whether SRSF1 directly binds BCL-X RNA in vivo. SRSF1 was enriched near the proximal 5′ splice site (E2p) (Figure 5A), indicating that it binds the BCL-X RNA just downstream of PTBP1 in vivo. Moreover, splicing assays using the wt minigene and suboptimal amounts of SRSF1 showed that increasing PTBP1 expression efficiently antagonizes SRSF1 activity in a dose-dependent manner (Figure 5B). By contrast, competition was lost when splicing assays were performed using the E2m1 minigene, in which the PTBP1 binding site is disrupted (Figure 5B). Likewise, in the presence of suboptimal amounts of PTBP1, SRSF1 antagonizes its activity in a dose-dependent manner (Supplementary Figure S5A). To test whether this competition was the result of a direct effect, we performed CLIP assays. In HEK293T cells transfected with the wt BCL-X minigene, PTBP1 was recruited efficiently to the BCL-X RNA whereas SRSF1 was barely detectable (Figure 5C). However, when cells were transfected with the E2m1 minigene, PTBP1 binding was significantly reduced whereas SRSF1 was recruited more efficiently (Figure 5C).
Figure 5.
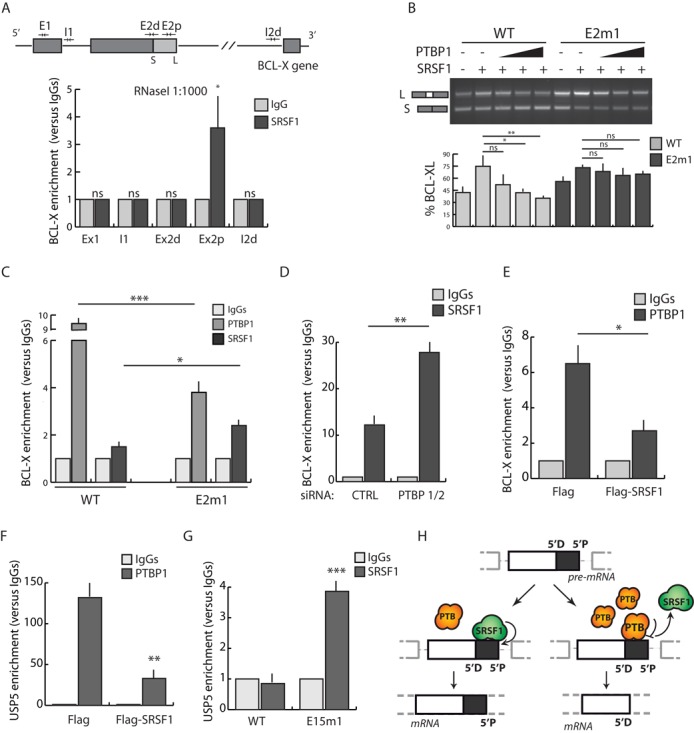
PTBP1 competes with SRSF1 for BCL-X and USP5 RNA binding. (A) CLIP experiment of SRSF1 performed in HEK293T in the presence of RNaseI (1:1000). Associated BCL-X RNA was quantified by qPCR using primers indicated in the upper BCL-X scheme (see also Supplementary Table S1). Data are represented as fold enrichment relative to the IgG sample (mean ± SD, n = 3). (B) RT-PCR of in vivo splicing assay performed in HEK293T transfected with the indicated minigenes, Flag-SRSF1 and increasing amounts of GFP-PTBP1. Bar graph (bottom panel) shows the percentage of BCL-XL (mean ± SD, n = 3). (C)–(G) CLIP experiment of SRSF1 (C, D and G) and PTBP1 (C, E and F) performed in HEK293T transfected with WT and E2m1 BCL-X minigenes (C), with WT (F and G) and E15m1 (G) USP5 minigenes, with the indicated siRNAs (D) or with Flag-SRSF1 (E and F). Associated BCL-X or USP5 RNAs were quantified by qPCR and represented as fold enrichment relative to the IgG sample (mean ± SD, n = 3). (A–G) The P values of Student's t-test are reported: *P < 0.05, **, P < 0.01; ***, P < 0.001; n.s., not significant. (H) Schematic model for the regulation of alternative 5′ splice site selection by PTBP1 as described in the text.
To confirm more directly the competition between PTBP1 and SRSF1 for binding to the BCL-X RNA, we modulated their expression in HEK293T cells. Notably, we observed a negative correlation between the binding of PTBP1 and SRSF1. Recruitment of the endogenous SRSF1 to BCL-X RNA was significantly increased after knockdown of PTBP1/2 (Figure 5D), whereas binding of the endogenous PTBP1 to BCL-X RNA was strongly reduced in cells overexpressing Flag-SRSF1 (Figure 5E).
To investigate whether a competition for binding between PTBP1 and an SR protein is a general mechanism for 5′ alternative splice site selection, the splicing activity of several SR proteins on USP5 minigene was tested. We found that SRSF1 was the most efficient in the regulation of USP5 isoform 1 (Supplementary Figure S5B). CLIP assays indicated that overexpression of SRSF1 displaced endogenous PTBP1 from USP5 RNA (Figure 5F). In addition, endogenous SRSF1 was not recruited to the USP5 RNA encoded by the wt minigene, but its binding was strongly increased in the presence of the mutated PTBP1 binding site (E15m1 minigene; Figure 5G). Collectively, these results suggest that USP5 AS is also regulated through an antagonistic activity of PTBP1 and SRSF1 and that binding of PTBP1 between the two alternative 5′ splice sites of target exons displaces SRSF1 from the proximal splice site and favors selection of the distal site (Figure 5H).
DISCUSSION
The flexibility of AS regulation is prone to errors and aberrant splicing contributes to many human diseases (18). In the last decade, antisense-based strategies that redirect splicing in favor of the advantageous splice variant have been developed (36). These methods represent promising therapeutic tools for cancer (35) and genetic disorders (50,51), as demonstrated by the clinical trial stage reached by some of these tools (36,50). Nevertheless, since these therapeutic approaches are based on affecting the recognition of splice sites and/or the recruitment of splicing factors, full elucidation of the molecular mechanisms involved in the regulation of specific splicing events is required to design effective tools.
Up-regulation of BCL-XL expression is a common feature in several tumors (23–25). For this reason, identifying strategies that switch BCL-X splicing in favor of the pro-apoptotic BCL-Xs variant may represent a valuable therapeutic tool for cancer therapy (35). Herein, by using a series of minigenes and in vitro pulldown assays, we identified several RBPs that affect BCL-X AS. Some of them, like SRSF1 (22,27), hnRNP F/H (26) and hnRNP A1 (27), were already known to be involved in BCL-X AS, confirming the fidelity of our analysis. Among the newly identified factors, we focused our attention on PTBP1 because this splicing factor is emerging as a key player in development and cancer (8,43–46). Thus, understanding the regulation of BCL-X splicing by PTBP1 may have strong relevance for various biological processes.
Deletion analysis using BCL-X minigenes identified a 77-bp-element region near the distal 5′ splice site that is strictly required for the effect of PTBP1 on BCL-Xs splicing. Previous studies showed that deletion of this region, named B2, had a strong impact on BCL-X AS (26), suggesting that this element is required for efficient selection of the distal 5′ splice site. We found that PTBP1 efficiently binds the B2 element in vitro and in vivo. Genome-wide analysis by CLIP-seq revealed that PTBP1 binds a large number of transcripts by recognizing single-strand RNA CU-rich motifs (16). Our CLIP analysis demonstrated that PTBP1 directly binds to the endogenous BCL-X RNA and allowed us to restrict the region of its binding in proximity of the B2 element. Furthermore, analysis of the B2 element sequence revealed the presence of a polypyrimidine stretch containing CU-rich motifs (CCUUUCU) that represent an optimal consensus for PTBP1. Disruption of the binding site by replacement of two pyrimidines with two purines (CCGGUCU; E2m1) strongly affected PTBP1 binding to BCL-X RNA and impaired its ability to modulate BCL-X AS. Notably, PTBP1 binding and splicing activity were not affected when these bases were substituted with two different pyrimidines (CCCCUCU; E2m2), confirming that this polypyrimidine stretch is a functional binding site for PTBP1.
Three observations indicate that PTBP1 is a physiologically relevant regulator of BCL-X AS. First, we found that glioblastoma cells expressing higher levels of PTBP1 display higher BCL-Xs levels and are more sensitive to drug-induced apoptosis. Next, knockdown of the endogenous PTBP1/2 in HEK293T cells enhanced usage of the proximal 5′ splice site of endogenous BCL-X pre-mRNA, thereby favoring the anti-apoptotic BCL-XL variant. The effect of PTBP1 was similar to that of hnRNP F and opposite to that of SRSF1, two previously validated regulators of this splicing event (22,26,27). Lastly, the splicing pattern of the E2m1 minigene, in which the binding site for PTBP1 is disrupted, was shifted in favor of BCL-XL. Thus, changes in the expression levels or in the activity of PTBP1 can influence BCL-X splicing in live cells. However, the apoptotic response of a given cell is likely to also depend on the relative abundance of other regulators of this splicing event. For instance, SRSF1, which competes with PTBP1 for binding to BCL-X exon 2 RNA (see below), is strongly up-regulated in various cancer cells (52) and might counteract PTBP1 activity. Thus, direct investigation of the function of PTBP1 in specific cancer cell models is required to define where and when regulation of BCL-X splicing participates in its oncogenic function.
PTBP1 is mostly known as a splicing repressor that binds silencer elements and represses exon cassettes. However, there are also examples in which PTBP1 activates exon inclusion (16,17). The outcome of PTBP1-mediated exon cassette splicing might be predicted on the basis of its position: binding upstream or within an exon typically causes repression, whereas binding downstream of an exon can favor inclusion (47). Although this model applies to exon cassettes and mutually exclusive exons, it cannot predict the outcome of PTBP1 binding in events characterized by alternative 5′ splice site selection within an exon. Since PTBP1 binds between the two alternative 5′ splice sites of BCL-X exon 2, we hypothesized that it may resemble its binding downstream of the exon for the distal site and within the exon for the proximal site. Thus, according to the proposed model, PTBP1 should act as an activator for the distal 5′ splice site. To test whether this could be applied to other alternative 5′ splice site events, we analyzed another PTBP1 target. Similarly to BCL-X, USP5 AS generates two isoforms, named 1 and 2, depending on the selection of two 5′ splice sites in exon 15 (49). PTBP1 was shown to favor selection of the distal splice site, but the functional binding site required for this effect was not mapped (49). We found that, in analogy with BCL-X exon 2, USP5 exon 15 contains a polypyrimidine stretch between the two alternative 5′ splice sites. Importantly, disruption of this polypyrimidine tract significantly reduced PTBP1 binding. Consistently, this mutation altered the splicing pattern of USP5 in favor of isoform 1 to the same extent as knockdown of endogenous PTBP1/2, suggesting that the presence of a PTBP1 binding site between two alternative 5′ splice sites generally promotes selection of the distal site while repressing the proximal one. Thus, our results suggest that regulation of alternative 5′ splice site selection by the binding of PTBP1 between two competing sites might represent a common mechanism of action of this splicing factor. Nevertheless, experiments to directly test whether PTBP1 activates the distal site and/or represses the proximal one will be required to fully elucidate the molecular mechanism of this regulation.
Several SR proteins are known to regulate BCL-X AS (22,27,28). Previous studies identified an enhancer region near the proximal 5′ splice site to which SRSF9 binds (28). However, depletion of this SR protein did not affect BCL-X AS, indicating that another factor mediates the enhancer activity of this cis-regulatory element (28). We observed an enrichment of SRSF1 binding in a region encompassing this element, suggesting that its enhancer activity might be mediated by SRSF1. Due to the antagonistic function of hnRNPs and SR proteins (2,3,5), we reasoned that PTBP1 might compete with SRSF1 in BCL-X AS regulation. Our results revealed that PTBP1 depletion, or disruptive mutation of its binding site, favors recruitment of SRSF1 to the BCL-X RNA. Similarly, overexpression of SRSF1 displaced PTBP1 from BCL-X RNA. Although we did not precisely map the binding site of SRSF1 on the BCL-X RNA, these results suggest that the presence of high levels of PTBP1 directly or indirectly interfere with its recruitment on the pre-mRNA. Since SR proteins are known to facilitate U1 snRNP recruitment (53), our results are compatible with a model in which SRSF1 recruits U1 snRNP to the proximal 5′ splice site, favoring its selection. When PTBP1 is up-regulated, it competes with SRSF1 for BCL-X binding, thus reducing U1 snRNP recruitment to proximal 5′ splice site and favoring the selection of the distal one. A similar competition was also observed in the regulation of USP5 exon 15 splicing, suggesting that this might represent a general mechanism for the regulation of alternative 5′ splice site selection in exons containing a binding site for PTBP1 between the competing sites (Figure 5H).
In conclusion, our results show that the BCL-X gene is a new target of PTBP1, which promotes splicing of the pro-apoptotic BCL-Xs variant by antagonizing SRSF1 activity. Moreover, our results reveal that the outcome of PTBP1-mediated 5′ AS events might be predicted on the basis of the presence of a PTBP1 binding site between the two competitive splice sites.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors wish to thank Dr Benoit Chabot for the gift of pX2.13 and ΔB2 (BCL-X minigenes); Dr Christopher W. J. Smith for the gift of pCMV-HisPTBP1; Dr James Stevenin for the gift of GFP-9G8pEGFPC1; and Dr Stefan Stamm for the gift of hnRNP F, hnRNP G and SRSF10 antibodies and pEGFPC2-hnRNP G.
Footnotes
Present addresses:
Matteo Bordi, European Molecular Biology Laboratory (EMBL) Developmental Biology Unit, Heidelberg, Germany.
Valentina Di Biasio, IRBM Science Park S.p.A., Pomezia, Rome, Italy.
FUNDING
Association for International Cancer Research (AICR) [12-0150]; Associazione Italiana Ricerca sul Cancro (AIRC) [14581]; Fondazione Santa Lucia Ricerca Corrente. Funding for open access charge: Association for International Cancer Research (AICR) [12-0150]; Associazione Italiana Ricerca sul Cancro (AIRC) [14581]; Fondazione Santa Lucia Ricerca Corrente.
Conflict of interest statement. None declared.
REFERENCES
- 1.Braunschweig U., Gueroussov S., Plocik A.M., Graveley B.R., Blencowe B.J. Dynamic integration of splicing within gene regulatory pathways. Cell. 2013;152:1252–1269. doi: 10.1016/j.cell.2013.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matera A.G., Wang Z. A day in the life of spliceosome. Nat. Rev. Mol. Cell Biol. 2014;15:108–121. doi: 10.1038/nrm3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Black D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 4.Wahl M.C., Will C.L., Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 5.Chen M., Manley J.L. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol. 2009;10:741–754. doi: 10.1038/nrm2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pan Q., Shai O., Lee L.J., Frey B.J., Blencowe B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 7.Wang E.T., Sandberg R., Luo S., Khrebtukova I., Zhang L., Mayr C., Kingsmore S.F., Schroth G.P., Burge C.B. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalsotra A., Cooper T.A. Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 2011;12:715–729. doi: 10.1038/nrg3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luco R.F., Allo M., Schor I.E., Kornblihtt A.R., Misteli T. Epigenetics in alternative pre-mRNA splicing. Cell. 2011;144:16–26. doi: 10.1016/j.cell.2010.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacobs E., Mills J.D., Janitz M. The role of RNA structure in posttranscriptional regulation of gene expression. J. Genet. Genomics. 2012;39:535–543. doi: 10.1016/j.jgg.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 11.Singh R., Valcárcel J., Green M.R. Distinct binding specificities and functions of higher eukaryotic polypyrimidine tract-binding proteins. Science. 1995;268:1173–1176. doi: 10.1126/science.7761834. [DOI] [PubMed] [Google Scholar]
- 12.Zheng X., Cho S., Moon H., Loh T.J., Oh H.K., Green M.R., Shen H. Polypyrimidine tract binding protein inhibits IgM pre-mRNA splicing by diverting U2 snRNA base-pairing away from the branch point. RNA. 2014;20:440–446. doi: 10.1261/rna.043737.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma S., Maris C., Allain F.H., Black D.L. U1 snRNA directly interacts with polypyrimidine tract-binding protein during splicing repression. Mol. Cell. 2011;41:579–588. doi: 10.1016/j.molcel.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ule J., Stefani G., Mele A., Ruggiu M., Wang X., Taneri B., Gaasterland T., Blencowe B.J., Darnell R.B. An RNA map predicting Nova-dependent splicing regulation. Nature. 2006;444:580–586. doi: 10.1038/nature05304. [DOI] [PubMed] [Google Scholar]
- 15.Yeo G.W., Coufal N.G., Liang T.Y., Peng G.E., Fu X.D., Gage F.H. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat. Struct. Mol. Biol. 2009;16:130–137. doi: 10.1038/nsmb.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue Y., Zhou Y., Wu T., Zhu T., Ji X., Kwon Y.S., Zhang C., Yeo G., Black D.L., Sun H., et al. Genome-wide analysis of PTB-RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Mol. Cell. 2009;36:996–1006. doi: 10.1016/j.molcel.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Llorian M., Schwartz S., Clark T.A., Hollander D., Tan L.Y., Spellman R., Gordon A., Schweitzer A.C., de la Grange P., Ast G., et al. Position-dependent alternative splicing activity revealed by global profiling of alternative splicing events regulated by PTB. Nat. Struct. Mol. Biol. 2010;17:1114–1123. doi: 10.1038/nsmb.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cooper TA., Wan L., Dreyfuss G. RNA and desease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwerk C., Schulze-Osthoff K. Regulation of apoptosis by alternative pre-mRNA splicing. Mol. Cell. 2005;19:1–13. doi: 10.1016/j.molcel.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 20.Thomas M.P., Lieberman J. Live or let die: posttranscriptional gene regulation in cell stress and cell death. Immunol. Rev. 2013;253:237–252. doi: 10.1111/imr.12052. [DOI] [PubMed] [Google Scholar]
- 21.Boise L.H., Gonzáles-Garcia M., Postema C.E., Ding L., Lindsten T., Turka L.A., Mao X., Nuñez G., Thompson C.B. bcl-x, a bcl-2-related gene that function as a dominant regulator of apoptotic cell death. Cell. 1993;74:507–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 22.Moore M.J., Wang Q., Kennedy C.J., Silver P.A. An alternative splicing network links cell-cycle control to apoptosis. Cell. 2010;142:625–636. doi: 10.1016/j.cell.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olopade O.I., Adeyanju M.O., Safa A.R., Hagos F., Mick R., Thompson C.B., Recant W.M. Overexpression of BCL-x protein in primary breast cancer is associated with high tumor grade and nodal metastases. Cancer J. Sci. Am. 1997;3:230–237. [PubMed] [Google Scholar]
- 24.Takehara T., Liu X., Fujimoto J., Friedman S.L., Takahashi H. Expression and role of Bcl-xL in human hepatocellular carcinomas. Hepatology. 2001;34:55–61. doi: 10.1053/jhep.2001.25387. [DOI] [PubMed] [Google Scholar]
- 25.Mercatante D.R., Mohler J.L., Kole R. Cellular response to an antisense-mediated shift of Bcl-x pre-mRNA splicing and antineoplastic agents. J. Biol. Chem. 2002;277:49374–49782. doi: 10.1074/jbc.M209236200. [DOI] [PubMed] [Google Scholar]
- 26.Garneau D., Revil T., Fisette J.F., Chabot B. Heterogeneous nuclear ribonucleoprotein F/H proteins modulate the alternative splicing of the apoptotic mediator Bcl-x. J. Biol. Chem. 2005;280:22641–22650. doi: 10.1074/jbc.M501070200. [DOI] [PubMed] [Google Scholar]
- 27.Paronetto M.P., Achsel T., Massiello A., Chalfant C.E., Sette C. The RNA-binding protein SAM68 modulates the alternative splicing of Bcl-x. J. Cell Biol. 2007;176:929–939. doi: 10.1083/jcb.200701005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cloutier P., Toutant J., Shkreta L., Goekjian S., Revil T., Chabot B. Antagonistic effects of the SRp30c protein and cryptic 5′ splice sites on the alternative splicing of the apoptotic regulator Bcl-x. J. Biol. Chem. 2008;283:21315–21324. doi: 10.1074/jbc.M800353200. [DOI] [PubMed] [Google Scholar]
- 29.Revil T., Pelletier J., Toutant J., Cloutier A., Chabot B. Heterogeneous nuclear ribonucleoprotein K represses the production of pro-apoptotic Bcl-xS splice isoform. J. Biol. Chem. 2009;284:21458–21467. doi: 10.1074/jbc.M109.019711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pedrotti S., Busà R., Compagnucci C., Sette C. The RNA recognition motif protein RBM11 is a novel tissue-specific splicing regulator. Nucleic Acids Res. 2012;40:1021–1032. doi: 10.1093/nar/gkr819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Naro C., Barbagallo F., Chieffi P., Bourgeois C.F., Paronetto M.P., Sette C. The centrosomal kinase NEK2 is a novel splicing factor kinase involved in cell survival. Nucleic Acids Res. 2014;42:3218–3227. doi: 10.1093/nar/gkt1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Montes M., Cloutier A., Sánchez-Hernández N., Michelle L., Lemieux B., Blanchette M., Hernández-Munain C., Chabot B., Suñé C. TCERG1 regulates alternative splicing of the Bcl-x gene by modulating the rate of RNA polymerase II transcription. Mol. Cell. Biol. 2011;32:751–762. doi: 10.1128/MCB.06255-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bielli P., Busà R., Di Stasi S.M., Munoz M.J., Botti F., Kornblihtt A.R., Sette C. The transcription factor FBI-1 modulates SAM68-mediated BCL-X alternative splicing and apoptosis. EMBO Rep. 2014;15:419–427. doi: 10.1002/embr.201338241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Michelle L., Cloutier A., Toutant J., Shkreta L., Thibault P., Durand M., Garneau D., Gendron D., Lapointe E., Couture S., et al. Proteins associated with the exon junction complex also control the alternative splicing of apoptotic regulators. Mol. Cell. Biol. 2012;32:954–967. doi: 10.1128/MCB.06130-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bauman J.A., Li S.D., Yang A., Huang L., Kole R. Anti-tumor activity of splice-switching oligonucleotides. Nucleic Acids Res. 2010;38:8348–8356. doi: 10.1093/nar/gkq731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kole R., Krainer A.R., Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012;11:125–140. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Massiello A., Salas A., Pinkerman R.L., Roddy P., Roesser J.R., Chalfant C.E. Identification of two RNA cis-elements that function to regulate the 5′ SS selection of Bcl-x premRNA in response to ceramide. J. Biol. Chem. 2004;279:15799–15804. doi: 10.1074/jbc.M313950200. [DOI] [PubMed] [Google Scholar]
- 38.Barik S. Site-directed mutagenesis in vitro by megaprimer PCR. Methods Mol. Biol. 1996;57:203–215. doi: 10.1385/0-89603-332-5:203. [DOI] [PubMed] [Google Scholar]
- 39.Pedrotti S., Bielli P., Paronetto M.P., Ciccosanti F., Fimia G.M., Stamm S., Manley J.L., Sette C. The splicing regulator Sam68 binds to a novel exonic splicing silencer and functions in SMN2 alternative splicing in spinal muscular atrophy. EMBO J. 2010;29:1235–1247. doi: 10.1038/emboj.2010.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Z., Tollervey J., Briese M., Turner D., Ule J. CLIP: construction of cDNA libraries for high-throughput sequencing from RNAs cross-linked to proteins in vivo. Methods. 2009;48:287–293. doi: 10.1016/j.ymeth.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 41.Revil T., Toutant J., Shkreta L., Garneau D., Cloutier P., Chabot B. Protein kinase C-dependent control of Bcl-x alternative splicing. Mol. Cell. Biol. 2007;27:8431–8441. doi: 10.1128/MCB.00565-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Z.Y., Cai L., Zhu J., Chen M., Chen J., Li Z.H., Liu X.D., Wang S.G., Bie P., Jiang P. Fyn requires HnRNPA2B1 and Sam68 to synergistically regulate apoptosis in pancreatic cancer. Carcinogenesis. 2011;32:1419–1426. doi: 10.1093/carcin/bgr088. [DOI] [PubMed] [Google Scholar]
- 43.Jin W., McCutcheon I.E., Fuller G.N., Huang E.S., Cote G.J. Fibroblast growth factor receptor-1 alpha-exon exclusion and polypyrimidine tract-binding protein in glioblastoma multiforme tumors. Cancer Res. 2000;60:1221–1224. [PubMed] [Google Scholar]
- 44.Clower C.V., Chatterjee D., Wang Z., Cantley L.C., Vander-Heiden M.G., Krainer A.R. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc. Natl. Acad. Sci. U.S.A. 2010;107:1894–1899. doi: 10.1073/pnas.0914845107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.David C.J., Chen M., Assanah M., Canoll P., Manley J.L. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463:364–368. doi: 10.1038/nature08697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferrarese R., Harsh G.R., IV, Yadav A.K., Bug E., Maticzka D., Reichardt W., Dombrowski S.M., Miller T.E., Masilamani A.P., Dai F. Lineage-specific splicing of a brain-enriched alternative exon promotes glioblastoma progression. J. Clin. Invest. 2014;124:2861–2876. doi: 10.1172/JCI68836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kafasla P., Mickleburgh I., Llorian M., Coelho M., Gooding C., Cherny D., Joshi A., Kotik-Kogan O., Curry S., Eperon I.C., et al. Defining the roles and interactions of PTB. Biochem. Soc. Trans. 2012;40:815–820. doi: 10.1042/BST20120044. [DOI] [PubMed] [Google Scholar]
- 48.Erkelenz S., Mueller W.F., Evans M.S., Busch A., Schöneweis K., Hertel K.J., Schaal H. Position-dependent splicing activation and repression by SR and hnRNP proteins rely on common mechanisms. RNA. 2013;19:96–102. doi: 10.1261/rna.037044.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Izaguirre D.I., Zhu W., Hai T., Cheung H.C., Krahe R., Cote G.J. PTBP1-dependent regulation of USP5 alternative RNA splicing plays a role in glioblastoma tumorigenesis. Mol. Carcinog. 2012;51:895–906. doi: 10.1002/mc.20859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rigo F., Hua Y., Krainer A.R., Bennett C.F. Antisense-based therapy for the treatment of spinal muscular atrophy. J. Cell Biol. 2012;199:21–25. doi: 10.1083/jcb.201207087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koo T., Wood M.J. Clinical trials using antisense oligonucleotides in duchenne muscular dystrophy. Hum. Gene Ther. 2013;24:479–488. doi: 10.1089/hum.2012.234. [DOI] [PubMed] [Google Scholar]
- 52.Das S., Krainer A.R. Emerging functions of SRSF1, splicing factor and oncoprotein, in RNA metabolism and cancer. Mol. Cancer Res. 2014;12:1195–1204. doi: 10.1158/1541-7786.MCR-14-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kohtz J.D., Jamison S.F., Will C.L., Zuo P., Lührmann R., Garcia-Blanco M.A., Manley J.L. Protein-protein interactions and 5′-splice-site recognition in mammalian mRNA precursors. Nature. 1994;368:119–124. doi: 10.1038/368119a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.