


Abstract
The RAS/ERK pathway is commonly activated in carcinomas and promotes oncogenesis by altering transcriptional programs. However, the array of cis-regulatory elements and trans-acting factors that mediate these transcriptional changes is still unclear. Our genome-wide analysis determined that a sequence consisting of neighboring ETS and AP-1 transcription factor binding sites is enriched near cell migration genes activated by RAS/ERK signaling in epithelial cells. In vivo screening of candidate ETS proteins revealed that ETS1 is specifically required for migration of RAS/ERK activated cells. Furthermore, both migration and transcriptional activation through ETS/AP-1 required ERK phosphorylation of ETS1. Genome-wide mapping of multiple ETS proteins demonstrated that ETS1 binds specifically to enhancer ETS/AP-1 sequences. ETS1 occupancy, and its role in cell migration, was conserved in epithelial cells derived from multiple tissues, consistent with a chromatin organization common to epithelial cell lines. Genome-wide expression analysis showed that ETS1 was required for activation of RAS-regulated cell migration genes, but also identified a surprising role for ETS1 in the repression of genes such as DUSP4, DUSP6 and SPRY4 that provide negative feedback to the RAS/ERK pathway. Consistently, ETS1 was required for robust RAS/ERK pathway activation. Therefore, ETS1 has dual roles in mediating epithelial-specific RAS/ERK transcriptional functions.
INTRODUCTION
The RAS/RAF/MEK/ERK (RAS/ERK) pathway is activated by many growth factors and regulates cellular proliferation, survival and motility. Mutations that constitutively activate the RAS/ERK pathway occur in one quarter of all tumors, including 95% of pancreatic cancers, 35% of lung cancers and 30% of melanomas (1). Activation of this pathway modulates the function of transcription factors and results in altered gene expression programs (2). Despite the clinical importance of this signaling pathway, we lack a thorough understanding of both the cis-regulatory sequences and trans-activating factors that orchestrate RAS/ERK-responsive gene expression in vivo.
Early studies focusing on the polyoma virus enhancer discovered a sequence motif that, when present in the promoter of a reporter gene, would activate expression in response to RAS/ERK signaling (3). This sequence consisted of neighboring binding sites for ETS and AP-1 family transcription factors (ETS/AP-1) (4). There are 28 ETS transcription factors in humans, which can be divided into 13 subfamilies of one to three members. All ETS proteins share a conserved DNA binding domain that mediates monomeric binding of the ETS binding sequence, consisting of an invariant GGA core and, most commonly, the extended sequence CCGGAAGT (5). AP-1 is a dimer of bZIP proteins that binds the sequence TGA(C/G)TCA and consists of either homodimers from the JUN family, or heterodimers of a JUN protein bound with a FOS, ATF or MAF family member (6). After discovery within the polyoma enhancer, ETS/AP-1 RAS-responsive elements were found in the promoters of a handful of endogenous genes, in particular, three matrix metalloproteases involved in cellular invasion and migration (MMP1, MMP9 and PLAU) (7–9). Despite the identification of ETS-AP1 sequences as RAS-response elements 25 years ago, the role these regulatory sequences play in regulating specific gene expression programs is largely unknown.
There are a number of candidates for a RAS-responsive ETS that might function through genomic ETS/AP-1 sequences. When overexpressed, the ETS proteins ETS1, ETS2 and ETV1 can activate ETS/AP-1 regulated reporter genes (10–12), but this function has not been tested for most ETS factors. At least nine ETS proteins can be phosphorylated by ERK, and in eight of these cases this modification increases transcriptional activation (13). A deletion of ETS2 in mice results in a phenotype similar to a deletion of the RAS/ERK activator EGFR (14), and a deletion of both ETS1 and ETS2 in mouse embryonic fibroblasts inhibits HrasG12V transformation (15), but it is not known what cis-regulatory sequences mediate these effects. Previous genome-wide mapping of ETS1 did not reveal occupancy of ETS/AP-1 sequences (16,17) and ETS2 mapping has not been reported due to a lack of specific antibodies. The ETS proteins ERG, FLI1, ETV1 and ETV4 can bind genomic ETS/AP-1 sequences when overexpressed (18,19). However, normal adult epithelial cells and many RAS-transformed cancer cell lines do not express any of these ETS factors (20,21). Furthermore, when introduced into epithelial cells with an active RAS/ERK pathway, these ETS factors do not activate an ETS/AP-1 transcriptional program (21). Thus, although ETS/AP-1 was one of the first described RAS-response elements, it is still unclear which endogenous ETS protein binds ETS/AP-1 sequences across the genome to mediate the RAS/ERK response.
Here, we report that ETS/AP-1 sequence motifs across the human genome occur specifically at RAS/ERK-responsive genes involved in cell migration. A comparison of multiple ETS proteins indicated that only ETS1 was required for RAS/ERK-mediated cell migration, and ERK phosphorylation of ETS1 was required for this function. Chromatin immunoprecipitation coupled with next generation sequencing (ChIP-seq) identified the ETS/AP-1 sequence as the most enriched motif in enhancers bound by ETS1, but not other ETS proteins. These ETS1-bound enhancers were conserved in RAS/ERK active prostate, lung and pancreatic cancer cells. Transcriptome analysis revealed that ETS1 and RAS/ERK signaling regulates a largely overlapping set of genes that neighbor ETS/AP-1 sequences. Further RNA-sequencing analysis showed that ETS1 activates the RAS/ERK-responsive genes involved in cell migration. However, we also discovered a surprising group of genes that are activated by RAS/ERK signaling and repressed by ETS1. These were largely genes that provide negative feedback to the RAS/ERK pathway. Therefore, ETS1 mediates a positive feedback loop that activates the RAS/ERK pathway by disrupting the negative feedback loop that normally attenuates RAS/ERK signaling. Together these findings indicate that ETS1 is a major downstream effector of RAS/ERK signaling in epithelial cells.
MATERIALS AND METHODS
Genomic analysis of ETS/AP-1 sequences
Datasets from Caco2 cells (Ha-RAS and Ki-RAS) and from human mammary epithelial cell (HMEC) (epithelial and mesenchymal phenotypes), NCBI GEO accessions GSE26541 and GSE12203, are used in Figure 1 and Supplementary Figure S1. DNase sensitivity data is from ENCODE (22) with GEO accession GSM736500 Sequences for each factor are: ETS, MGGAWRH; AP-1, TGASTCA; CRE, TSACGTMA; SP1, KGGGVGGRRK; mutETS, NGCGWNNN; mutAP-1, TAGSCTCA (M = A or C; W = A or T; R = A or G; H = A, C or T; S = G or C; K = G or T; V = G, A or C; N = A, C, G or T). Sequences were analyzed in pairs with all combinations of orientation for each sequence and a spacing of between −1 and +20. Data in Table 1 and Supplementary Table S1 use the same sequences, but filter with RWPE1 DNase sensitivity data from ENCODE with UCSC accession wgEncodeEH0022553 (22).
Figure 1.
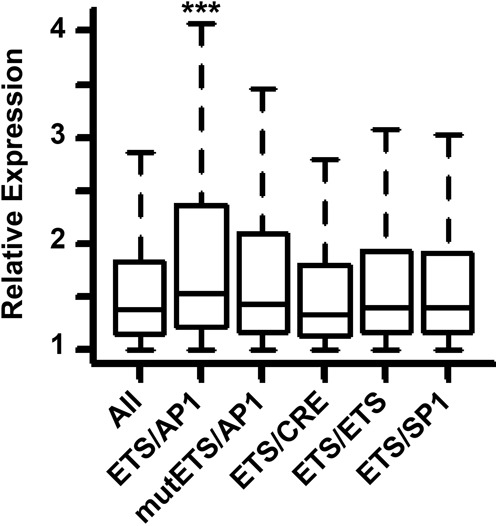
ETS/AP-1 sequences predict increased activation by RAS signaling. Box plots show all genes or genes with the indicated neighboring sequence, activated by expression of Ha-RASV12 or Ki-RASV12 in Caco2 or HMEC cells. Genes are only included if the indicated sequence is in open chromatin in the cell line where expression changed, and has the nearest transcription start site within 25 kb. Plotted gene numbers are All: 24826, ETS/AP-1: 3849, mutETS/AP-1: 1691, ETS/CRE: 334, ETS/ETS: 1037, ETS/SP1: 2331. P-values calculated by a Welch Two Sample t-test: ***<0.0001, unmarked are >0.05.
Table 1. Roles of genes near ETS/AP-1 sequences in the genome.
| Dataset | Most overrepresented ontologies | P-value |
|---|---|---|
| Unfiltered | Cellular component movement | 1.4×10−19 |
| Locomotion | 8.7×10−19 | |
| Cell development | 8.7×10−19 | |
| Response to endogenous stimulus | 1.5×10−16 | |
| Cell morphogenesis | 5.8×10−16 | |
| RWPE1 | Cellular component movement | 9.2×10−12 |
| Locomotion | 9.6×10−11 | |
| Cell morphogenesis | 9.4×10−10 | |
| Cell motility | 2.5×10−9 | |
| Localization of cell | 2.5×10−9 |
The unfiltered dataset includes all ETS/AP-1 motifs (MGGAARHN(0–20)TGASTCA or MGGAWRTGASTCA) on either strand, located within 25 kb of a RefSeq transcription start site (TSS). The RWPE1 dataset is the subset of sites within RWPE1 cell DNase sensitive regions.
The most overrepresented ontologies of the genes with the nearest TSS to sites in each category from GProfiler. Only biological process ontologies with <2000 genes are listed.
Cell culture and viral transduction
Cell lines were cultured according to ATCC recommendations as follows: EBNA293, HEK-293T, DU145 and PANC-1 were grown in Dulbecco's modified Eagle's medium (DMEM) [Sigma] with 10% fetal bovine serum (FBS) [Sigma]. RWPE (RWPE-1) and RWPE-KRAS (RWPE-2) were cultured in Keratinocyte SFM with supplemental epidermal growth factor, Bovine Pituitary Extract (Invitrogen). PC3 cells were cultured in F12K medium (Mediatech-Cellgro) with 10% FBS. All cell lines were cultured in media containing 1% Penicillin/Streptomycin 100X solution (Mediatech-Cellgro).
Lentivirus for ETS factor shRNA creation was produced by co-transfection of pLKO.1 (Addgene plasmid 8453) (23) containing specific shRNA sequences (Supplementary Table S4) in HEK293T cells with pMDLg/pRRE (Addgene plasmid 12251), pRSV-Rev (Addgene plasmid 12253) and pMD2.G (Addgene plasmid 12259) packaging plasmids (24) as previously described (21).
Transwell migration and soft agar assays
Transwell migration assays were carried out as described previously (18), with minor modifications. In brief, 1×105 cells were introduced to the transwell (8 μM pore size; BD Bioscience) and incubated for 12 h (PANC-1), 24 h (DU145), 48 h (PC3) and 60 h (RWPE-1, RWPE-KRAS). All biological replicates are the mean of two technical replicates.
Soft agar assays were carried out as described previously (25), with minor modifications. In brief, 2×105 cells were cultured in 0.3% agarose in DMEM on top of a lower 0.5% agarose layer. Cells were cultured at 37°C for four weeks and colonies counted with TotalLab Quant software (TotalLab).
MTT proliferation assays
Proliferation assays using 5 mg/ml MTT (Calbiochem) in phosphate buffered saline were completed as previously described (21) with minor changes. In brief, 800 cells of each cell line were plated in triplicate on a 96-well flat bottom culture dish and incubated at 37°C for up to 5 days. MTT reagent was added to each well daily and incubated for 4 h. After incubation, media was removed and 150 μl DMSO was added to each well. Absorbance readings at 600 nm used micro-plate reader ELx8200 (Biotek Instruments). Each biological replicate is the mean of three technical replicates.
Reporter assays
A Dual Luciferase Reporter Assay System (Promega) was used according to manufacturer's instructions with some modifications. Wild type and mutant ETS/AP-1 reporter plasmids were previously described (21). Cells were plated at ∼50% confluency in a 6-well plate (2.5×105 cells/well) 24 h before transfection. Cells were transfected with 1 μg of firefly and renilla plasmid using TransIT Prostate Transfection Kit (Mirus). After 24 h, media was removed, cells were resuspended in 250 μl 1xPLB and disrupted by three freeze/thaw cycles. Luciferase activity was measured in 20 μl of cell lysate using Appliskan Multimode Microplate reader (Thermo Scientific). Firefly values were normalized to renilla values.
Protein immunoblotting
Total protein extract from an equal number of cells was separated on 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis gels, transferred to nitrocellulose membrane (Bio-Rad), blocked in 5% milk in TBS (10 mM Tris, pH8.0, 150 mM NaCl), incubated with primary and secondary antibodies and visualized by ECL (Thermo Scientific) using standard procedures. Antibodies were the same as for ChIP plus ETV4 (Aviva), ETS2 (sc-351, Santa Cruz) and Tubulin (Sigma).
Chromatin immunoprecipitation, sequencing and analysis
ChIP was carried out as previously described (18). In short, cells were crosslinked using 1% v/v Formaldehyde (Fisher Scientific) for 15 min and quenched with 2M Glycine for 5 min. Isolated cells were lysed and sonicated for 4 cycles (20 s on, 40 off, 40 amplitude) [Misonix]. Nuclear lysate was rotated with specific antibody for 4 h at 4°C, washed and DNA isolated by phenol/chloroform. Antibodies used in ChIP were from Santa Cruz Biotechnology: GABPA (sc-22810, lot #B2009), ETS1, (sc-350, lot #F2811 [DU145], lot #F1312 [PANC-1]), ELF1 (sc-631, lot #L2702), ELK4 (sc-13030, lot #G2711, JUND (sc-74, lot #K1111). A pool of at least three biological ChIP replicates was sheared to ∼150 nucleotides using a Diagenode BioRuptor and size confirmed by gel electrophoresis. End repair used Klenow DNA polymerase (New England BioLabs), T4 DNA polymerase (New England BioLabs) and T4 DNA ligase (New England BioLabs) before QIAquick PCR purification (Qiagen). T4 DNA ligase (New England BioLabs) ligated adapters. Products were size selected between 200 and 300 bp by 2% agarose gel, then purified by Gel Extraction kit (Qiagen). Universal and indexing adapters were from the TruSeq sample preparation kit (Illumina). Libraries were multiplexed at a concentration of 20 nM and deep-sequenced with an Illumina HiSeq®2000 instrument by manufacturer's protocol.
Post sequencing, data was analyzed through a bioinformatic pipeline previously described (18). Raw .FASTQ files were obtained from Illumina and were mapped to the human genome (UCSC, v19). Peak calling was completed using the Useq8.4 platform (26), available: http://useq.sourceforge.net/. A549 ETS1 and JUND ChIP-seq data is from Richard Meyers group, accession numbers GSM1010829 and GSM1010723.
Centered enriched motif analysis of ChIP-seq datasets
Centered enriched motif analysis of each ChIP-seq dataset used the GEM algorithm, Genome wide event finding and motif discovery (http://www.psrg.csail.mit.edu/gem/) (27). Genomic locations were expanded 50 bp upstream and downstream and searched by RSA-tools-peak-motifs analysis http://rsat.ulb.ac.be/ (28) to determine enriched motifs. Motif size was 7 nucleotides and remaining parameters were as default. Motifs were only considered and presented if they occur in the top three of all enriched motifs.
RNA sequencing and analysis
Total RNA for three independent biological replicates was isolated from DU145 cells transduced with lentiviral shRNA knockdown vectors (see above), or U0126 inhibition at 50 μM for 6 h (Cell Signaling) using the RNeasy mini kit (Qiagen) according to manufacturer's instructions. Sequencing libraries for whole transcriptome analysis were generated using a modified Illumina TruSeq sample preparation protocol. Sequencing libraries for whole transcriptome analysis were generated using a modified Illumina TruSeq sample preparation protocol. Total RNA was DNase treated with TURBO DNase (Invitrogen). The DNase treated RNA was polyA selected with oligo(dT) beads (Invitrogen). A Superscript III Reverse Transcriptase First-Strand Synthesis (Invitrogen) system was used to generate cDNA from the polyA selected RNA with random hexamer primers (Invitrogen). After first strand synthesis a second strand was generated using Escherichia coli DNA ligase (New England BioLabs) and E. coli DNA polymerase I (New England BioLabs). The double-stranded cDNAs were sheared to ∼150 nucleotides using a Diagenode BioRuptor and the size was confirmed by DNA gel electrophoresis. Following sonication, library preparation was completed as described under ‘Chromatin Immunoprecipitation’ methods. Deep-sequencing was performed on an Illumina HiSeq®2000 instrument by the manufacturer's protocol. The Tuxedo Suite RNA sequencing pipeline was used to determine differential gene expression (29) with some modifications. Raw FASTQ files were obtained from Illumina and were mapped to the human genome (UCSC release, version 19) using TopHat2 utilizing Bowtie2. Differential expression of genes and transcripts used Cuffdiff.
RESULTS
ETS/AP-1 sequences define the cis-regulatory elements of RAS-inducible cell migration genes
Neighboring ETS/AP-1 sequences can mediate RAS/ERK-responsive gene expression in reporter assays, but the role of these sequences in RAS/ERK signaling genome-wide has not been characterized. To test if ETS/AP-1 sequences associate with RAS-regulated genes across the human genome, we searched for positions where an ETS binding sequence (A/C GGAA A/G A/C/T) was within 20 bp of an AP-1 binding sequence (TGA C/G TCA). This spacing was chosen because it was enriched in our previous genomic mapping of oncogenic ETS binding sites (18). Only sequences within 25 kb of a transcription start site (TSS) were considered, as these are the most likely to be regulatory. We then incorporated data from two epithelial cell lines with published datasets for both DNase sensitivity, to predict open chromatin, and microarray analysis to determine genes activated by RAS signaling. These were a colorectal adenocarcinoma cell line (Caco2; GEO accession GSE12203 (30)) and HMECs (GEO accession GSE26541). Genes with a neighboring ETS/AP-1 sequence, in open chromatin in that cell line, were significantly (P = 4.4 × 10−6) more activated by RAS than all activated genes (Figure 1). Individual Caco2 and HMEC datasets show the same result as the combined dataset (Supplementary Figure S1). Multiple control sequences were tested to verify significance, including a sequence with a point mutation in the ETS sequence (mutETS/AP-1), and three sequences that reflect other known ETS partnerships: ETS/ETS, ETS/CRE and ETS/SP1. Like ETS/AP-1, ETS/ETS and ETS/SP1 sites have also been identified as RAS-responsive in reporter assays (31). In contrast to the ETS/AP-1 sequence, none of the control sequences significantly predicted highly RAS-activated genes (P > 0.05). Therefore, the ETS/AP-1 sequence can define the cis-regulatory regions that control a RAS/ERK-regulated gene expression program in multiple epithelial cell types.
Neighboring ETS and AP-1 binding sequences can regulate the RAS/ERK response of a handful of genes involved in cellular migration and invasion. To determine whether migration and invasion genes are the main targets of ETS/AP-1 sequences across the genome, all genes with a neighboring ETS/AP-1 sequence (regardless of chromatin accessibility) were subjected to an ontology search. This gene list was most enriched in the functional categories of cellular component movement (a category that includes both cell migration and axonal projection) and locomotion (Table 1). These categories were not identified using ETS/ETS, ETS/CRE or ETS/SP1 sequences (Supplementary Table S1). To test if these ETS/AP-1 sequences are accessible in epithelial cells, genomic ETS/AP-1 sequences were filtered with a list of DNase sensitive regions in RWPE1 prostate epithelial cells. Genes nearest these accessible ETS/AP-1 sequences were enriched for functions including cellular component movement, locomotion and cell motility (Table 1). These results indicate that ETS/AP-1 sequences can predict a RAS-responsive, cell migration gene expression program.
ETS1, but not other ETS proteins, is required for migration of a RAS/ERK activated cell line
In a subset of prostate cancers, aberrantly high expression of an oncogenic ETS protein (ERG, ETV1 or ETV4) leads to ETS/AP-1 occupancy and activation of cell migration in backgrounds of low RAS/ERK signaling (18,21). However, normal epithelial cells, and many carcinomas, do not express high levels of these oncogenic ETS proteins, but instead can activate ETS/AP-1 reporter genes and migrate in response to high levels of RAS/ERK signaling (21). The ETS protein that binds ETS/AP-1 sequences across the genome and mediates this RAS/ERK response has not been identified. We used a shRNA knockdown approach to test cell migration roles of five candidate ETS proteins in DU145, a prostate cancer cell line that does not express high levels of oncogenic ETS proteins (21), but has an active RAS/ERK pathway due to a chromosomal rearrangement of KRAS (32). A lentiviral vector was used to make stable lines with shRNA-mediated depletion of ETS1, ETS2, ELF1 or GABPA (Figure 2A). Despite very low ETV4 protein levels in this cell line (21), we were also able to deplete and test ETV4. In each case, lowering the level of one ETS protein did not affect the levels of the others (Figure 2A). A transwell assay tested the migration of each knockdown cell line in comparison to a control (luciferase) knockdown. Loss of ETS1, and no other ETS protein, resulted in a dramatic decrease in cell migration (Figure 2B and Supplementary Figure S2A). A second shRNA targeting ETS1 had a similar effect (Supplementary Figure S2B). To verify that this was not due to cell death, or reduced cell growth, the proliferation rate of ETS1 knockdown cells was tested. ETS1-depleted cells proliferated at a similar rate to control knockdown cells (Figure 2C). While depletion of ELF1, GABPA and ETV4 had no effect on cell migration, knockdown of ETS2, a close homolog of ETS1, actually increased cell migration (Figure 2B), without affecting proliferation (Supplementary Figure S2C), indicating a possible attenuating function for this factor.
Figure 2.
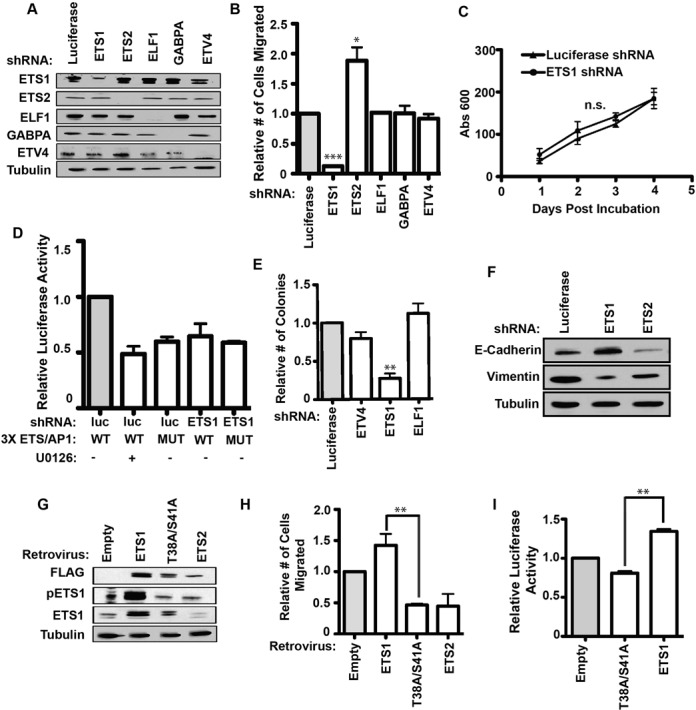
Phospho-ETS1 is required for the migration of the RAS-active prostate cancer line, DU145. (A) Immunoblot with antibodies shown (left) of DU145 cells with shRNA mediated knockdown of five ETS factors (top). An shRNA targeting luciferase is a negative control. Tubulin is a loading control. (B) A transwell assay measured relative cell migration of DU145 cells with indicated knockdown. Mean and SEM of ≥ 3 biological replicates shown. P-values (*<0.05, **<0.01, ***<0.001) are calculated by two-tailed T-test. (C) An MTT assay compared proliferation of DU145 cells with ETS1 and control shRNA knockdowns. Three biological replicates shown. Proliferation rates are not significantly different (P = 0.73). (D) A reporter assay compares relative luciferase units (firefly/renilla) from DU145 cells expressing control luciferase (luc) shRNAs or ETS1 shRNAs and treated with the MEK inhibitor, U0126 50 μM, as indicated. The firefly luciferase vector has three copies of the ETS/AP1 element (WT) 5′ to the minimal promoter or the same vector with point mutations in each ETS binding site (MUT). Values are shown as a ratio to the first column and are the mean and SEM of three biological replicates. P-values are as in (B). (E) Anchorage-independent growth by soft agar assay for selected ETS knockdown in DU145 cells. Number of colonies calculated relative to luciferase shRNA. Mean and SEM of three biological replicates reported. (F) Immunoblots show levels of E-cadherin and Vimentin in DU145 cells with the indicated shRNA knockdown. (G) Immunoblot with antibodies shown (left) for DU145 cells transduced with a retrovirus to stably overexpress empty vector, Flag-ETS1, Flag-T38A/S41A ETS1 and Flag-ETS2. (H) A transwell assay measured relative cell migration of DU145 cells with indicated ETS overexpression constructs as in (G). Mean and SEM of ≥3 biological replicates each containing two technical replicates are shown. P-value of ETS1 compared to T38A/S41A ETS1 by T-test. (I) Relative luciferase expression as in (D) for a 3xETS/AP-1 reporter in DU145 cells overexpressing indicated ETS construct.
To test if ETS1 was functioning through ETS/AP-1 sequences, DU145 cells were transfected with a firefly luciferase reporter with three copies of the ETS/AP-1 sequence cloned upstream of a minimal promoter. Repressing the RAS/ERK pathway by the MEK inhibitor U0126 decreased reporter expression about 50% (Figure 2D). Mutation of the ETS binding sequences (ETSmut/AP-1) in the reporter decreased expression to a similar extent, indicating that binding of an ETS factor is required for RAS/ERK mediated activation. Depletion of ETS1 by shRNA resulted in a decrease in reporter expression that approximated the decrease by MEK inhibition or ETS site mutation (Figure 2D). This indicates that ETS1 is required for RAS/ERK signaling to activate transcription through ETS/AP-1 sequences.
When overexpressed in prostate cells, oncogenic ETS proteins can promote anchorage-independent growth and epithelial-mesenchymal transition (EMT) in addition to cell migration (25,33). If ETS1 is mediating RAS/ERK signaling through the same cis-regulatory sequences bound by oncogenic ETS proteins, it might have the same phenotypes. In DU145 cells, knockdown of ETS1, but not ETV4 or ELF1 decreased anchorage independent growth (Figure 2E). ETS1 knockdown also increased E-Cadherin and decreased Vimentin expression consistent with a role for ETS1 in promoting EMT (Figure 2F). Interestingly, an ETS2 knockdown reduced E-Cadherin expression, again indicating an opposite or attenuating function. Therefore, in a cell line with an activated RAS/ERK pathway, ETS1 is required for transactivation through ETS/AP-1 sequences and promotes cellular phenotypes associated with ETS/AP-1 target genes, and ETS2 can have opposite roles.
The role of ETS1 in cell migration requires ERK phosphorylation
ERK can phosphorylate ETS1 at T38 and S41 residues and this promotes transactivation by increasing affinity for the co-activator CBP/p300 (12,34). To test the role of these residues in the ETS1 cell migration function, a retroviral vector system was used to stably overexpress FLAG-tagged versions of either wild-type ETS1 or ETS1 with two alanine substitutions (ETS1 T38A/S41A) in DU145 cells. As a control, FLAG-ETS2 was also overexpressed. Each tagged protein was expressed at similar levels, but only wild-type ETS1 is phosphorylated at T38 (Figure 2G). Overexpression of wild-type ETS1 increased migration of DU145 cells, but ETS1 T38A/S41A acted in a dominant negative fashion and decreased cell migration (Figure 2H). ETS2 had a similar effect as ETS1 T38A/S41A, again pointing to a role for ETS2 in attenuating migration. None of these overexpression constructs altered the proliferation rate of DU145 cells (Supplementary Figure S3). The ETS/AP-1 regulated luciferase reporter was then used to test the role of ETS1 phosphorylation in transactivation. Similar to the cell migration results, wild-type ETS1 increased reporter expression, and ETS1 T38A/S41A decreased reporter expression (Figure 2I). Therefore the ERK phosphorylation sites in ETS1 are required for activation of cell migration through ETS/AP-1 cis-regulatory sequences.
If RAS/ERK signaling functions through ETS1 to activate a cell migration gene expression program, then ETS1, despite being ubiquitously expressed, should only be necessary for cell migration in cell lines with high levels of RAS/ERK activity. To test this, we first examined PC3 prostate cancer cells, which have low ERK activation, but migrate due to high expression of the oncogenic ETS gene ETV4 (21,25). In PC3 cells, depletion of ETV4 decreased cell migration, but depletion of ETS1 had no effect (Figure 3A and B), indicating no ETS1 role when RAS/ERK activity is low. In addition, the RWPE1/2 cell system was used to directly compare ETS1 roles in cell lines where the only difference is in RAS/ERK activation. RWPE1 cells are derived from normal prostate epithelia and have low levels of ERK activation (21). Expression of Ki-RAS in RWPE1 cells activates ERK signaling, and these cells are called RWPE2. We have previously shown that overexpression of oncogenic ETS proteins such as ERG and ETV1 increases migration of RWPE1, but not RWPE2 cells (21). Here, in contrast, we show that ETS1 overexpression has no effect on RWPE1 migration (Figure 3C), but significantly increases the migration of RWPE2 cells (Figure 3D). Consistent with this observation, depleting endogenous ETS1 from RWPE2 cells results in a significant loss of cell migration (Figure 3E). In conclusion, the ability of ETS1 to transactivate through ETS/AP-1 sequences and drive cell migration requires RAS/ERK activation and the ERK phosphorylation residues T38 and S41.
Figure 3.
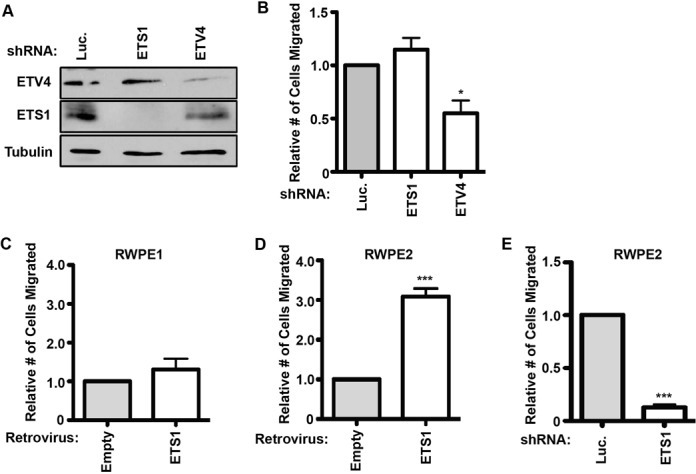
ETS1 only regulates cell migration in cells with an activated RAS/ERK pathway. (A) Immunoblot of PC3 cells stably expressing either luciferase, ETS1 or ETV4 shRNAs (top) demonstrates knockdown of each ETS factor. (B) Transwell assay of PC3 cells with indicated knockdowns. Replicates and significance as in Figure 1B. Transwell assays of RWPE1 (C) or KiRAS expressing RWPE2 (D) cells stably overexpressing retroviral ETS1. Replicates and significance as in Figure 1B. (E) Transwell assay of RWPE2 cells transfected with stable lentiviral expression of indicated shRNA. Replicates and significance as in Figure 1B.
ETS1 binds ETS/AP-1 sequences across the genome
These results indicate that ETS1 mediates RAS/ERK activation through ETS/AP-1 sequences. However, previous mapping of ETS1 binding sites in T cells by ChIP-seq did not identify the ETS/AP-1 sequence as a common ETS1 binding motif (16,17). To test if this result was due to the differences in cell type, we measured genomic occupancy of four ETS proteins in DU145 cells by ChIP-seq (Bound regions available in Supplementary Table S2). These included three ETS proteins previously mapped in Jurkat T cells (ETS1, GABPA and ELF1) and an additional RAS/ERK regulated ETS protein, ELK4. As a control, two biologically independent replicates of ETS1 ChIP were sequenced, and these showed significant convergence (Supplementary Figure S4). In total, ETS1 bound 9994 regions and GABPA bound 2489 regions in DU145 cells (Figure 4A). These are similar to the number of bound regions reported for these factors in T cells (17,35). ELF1 only occupied 247 regions in DU145 cells, possibly due to a low expression level in this cell type. ELK4 also bound a small number of regions (338). Although ELK4 occupancy has not been published, this number is very similar to the 529 bound regions reported for the close ELK4 homolog, ELK1, in breast epithelia (36). ChIP/qPCR was used to confirm enrichment of five randomly selected regions in each dataset (Supplementary Figure S5). An overlap of ETS1, GABPA, ELK4 and ELF1 binding sites (Figure 4A) revealed a pattern similar to that identified in T cells (17), with GABPA binding primarily in promoter sequences (<300 bp to TSS) that were co-occupied by other ETS factors (redundant sites), and the other ETS proteins having a mixture of redundant promoter binding and specific distal enhancer binding sites (>300 bp to TSS). The GEM algorithm (27) was used to identify centered binding sites for each of the ETS tested. Following GEM, windows were extended 50 bp in either direction of the centered motif. The PeakMotifs algorithm (28) was used to conduct an unbiased search for the most over-represented sequence motifs in either promoter or enhancer binding sites. Motifs identified in promoter regions (Supplementary Figure S6) were similar to those previously reported in other systems (16,17), and none included AP-1. In enhancers, GABPA and ELF1 sites were enriched for ETS consensus sequences (Figure 3B). ELK4 sites were enriched for both an ETS site and a site for serum response factor (SRF), the factor known to cooperatively bind with ELK4 (Figure 4B). Strikingly, at ETS1 enhancer sites, the most enriched sequence was a composite ETS/AP-1 sequence (CCGGAAGTGACTCA). These data indicate that ETS1, and not GABPA, ELF1 or ELK4, binds to ETS/AP-1 sequences in the enhancers of DU145 cells.
Figure 4.
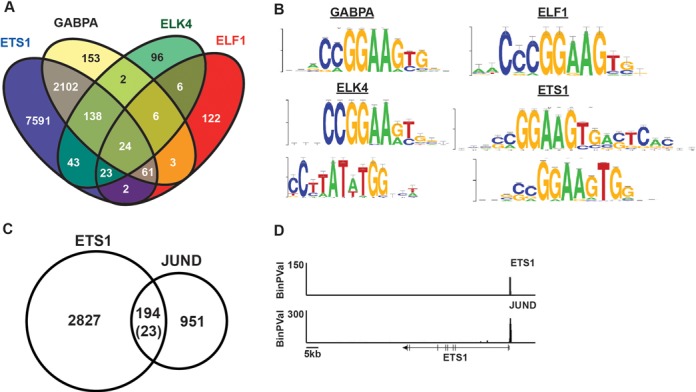
ETS1 binds ETS/AP1 sequences in vivo. (A) Venn diagram demonstrates overlap of all ChIP-seq bound regions in DU145 cells for ETS factors tested using Useq platform. (B) Most enriched motifs for bound enhancer regions (>300 bp from a TSS) as determined by RSAT peak-motifs algorithm after GEM analysis. For GABPA and ELF1, only one motif was identified. (C) Overlap of ETS1 bound enhancer regions with JUND bound regions in DU145 as determined by ChIP-seq analysis. Number in parenthesis indicates the number of expected overlaps using the assumption that any region of open chromatin is potential binding site (determined by RWPE1 DNase-seq). (D) ETS1 and JUND occupancy is plotted by -log binary P-value near the ETS1 gene.
To test if ETS1 co-occupies potential enhancers with AP-1, the AP-1 subunit JUND was mapped by ChIP-seq in DU145 cells. JUND occupancy was identified at 1145 enhancer regions (>300 bp to TSS). ETS1 and JUND showed significantly overlapping binding in enhancer binding sites with 8.5-fold more overlap than would be expected at random (Figure 4C). However, these data also indicate that the 194 ETS1 and JUND co-occupied sites represent a subset of the total binding sites for each protein, consistent with our previous observations that the ETS/AP-1 motif mediates a subset of both ETS and AP-1 regulatory functions (18). ETS1 and JUND co-occupy the promoter of ETS1 itself (Figure 4D), consistent with an autoregulatory function (37). In summary, consistent with ETS1 being required for the functions of ETS/AP-1 associated genes, ETS1 binds ETS/AP-1 sequences across the genome.
ETS/AP-1 sequences are accessible and bound by ETS1 in multiple carcinoma cell lines
The observation that ETS1 binds to ETS/AP-1 sequences in an epithelial-derived cell line contrasts with the previous finding that ETS1 binds to ETS/RUNX and not ETS/AP-1 sequences in Jurkat T cells (17). To test if chromatin accessibility explains the differences between epithelial cells and T cells, we compared previously published ENCODE DNase sensitivity and ChIP-seq data for Jurkat T cells with A549 lung cancer cells, PANC1 pancreatic cancer cells and DU145 prostate cancer cells (Table 2). The fraction of regions co-occupied by ETS1 and JUND in A549 and DU145 cells or co-occupied by ETS1 and RUNX in Jurkat cells that were in open chromatin were compared for two epithelial derived cell lines (A549 and PANC1) and Jurkat T cells. A strikingly higher percentage of ETS1/JUND co-occupied regions were in accessible chromatin in epithelial cells than in T-cells, even when comparing epithelial cells originating from different tissues (Table 2). In contrast ETS1/RUNX1 co-occupied regions were much more likely to be in open chromatin in Jurkat T cells. Therefore the difference in ETS1 binding site occupancy correlates with differences in chromatin accessibility in these cell types.
Table 2. Cell-type specificity of open chromatin at ETS/AP-1 and ETS/RUNX bound regions.
| Cell line | Bound factors | Regions | A549 DNase | Jurkat DNase | PANC1 DNase |
|---|---|---|---|---|---|
| A549 | ETS1/JUND | 4673 | 4570 (98%) | 1767 (39%) | 4121 (88%) |
| DU145 | ETS1/JUND | 383 | 368 (96%) | 229 (60%) | 323 (86%) |
| Jurkat | ETS1/RUNX | 1070 | 134 (13%) | 690 (65%) | 125 (12%) |
A549, DU145 and PANC1 epithelial lines are derived from lung, prostate and pancreatic carcinomas respectively. Jurkat cells are a lymphoid line derived from a T cell leukemia.
The number of genomic regions where both ETS1 and JUND (ETS/AP-1) or both ETS1 and RUNX bind, as indicated, based on ChIP-sequencing (17,38). Note that the difference in the number of ETS1/JUND sites in A549 and DU145 cells is due to an ∼10-fold difference in the number of reported JUND sites, likely due to differences in read number and peak calling in these two studies. The number of ETS1/JUND or ETS1/RUNX1 bound regions that are in open chromatin in the indicated cell line based on global mapping of DNase sensitivity.
RAS pathway activating mutations are more common in pancreatic cancer and lung cancer than in prostate cancer. To test if ETS1 also plays a role in the RAS/ERK response in these cell types, we mapped ETS1 binding sites in PANC1 cells by ChIP-seq. Like in DU145 cells, unbiased motif searching identified ETS and AP-1 sequences at ETS1-bound enhancers in PANC1 cells (Supplementary Figure S6). A comparison of ETS1-bound regions in PANC1, DU145 (Figure 5A) and ENCODE project data for A549 (38) revealed very high overlap across these cell lines, with 67% of DU145 sites, 60% of A549 sites and 89% of PANC1 sites also occupied in another cell type. For example, the promoter of the gene encoding Vimentin (VIM) is shown in Figure 5B, with co-occupancy of ETS1 and JUND in DU145, A549 and PANC1 cell lines. A potential enhancer is present at the 3′ end of VIM, with ETS1 occupancy in DU145 and A549, but not PANC1 cells. To test the role of ETS1 in PANC1 cells, ETS1 was depleted by shRNA (Figure 5C). Loss of ETS1 significantly decreased the migration of PANC1 cells (Figure 5D). Together, these data indicate that ETS1 has similar roles in regulating cell migration through ETS/AP-1 sequences in distinct epithelial derived cell lines.
Figure 5.
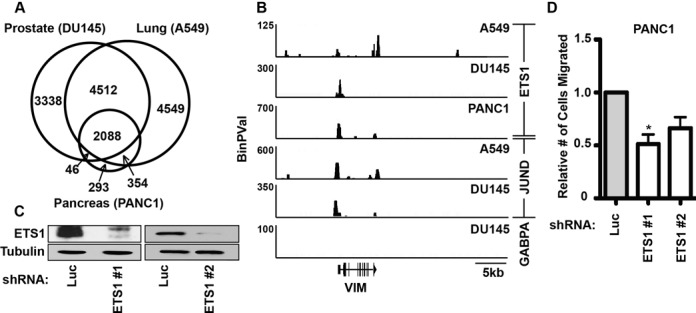
ETS1 is required for migration and demonstrates genome-wide enrichment for ETS/AP1 across multiple RAS-active cancer cell types. (A) Overlap of ETS1-bound regions in RAS-active prostate (DU145), lung (A549) and pancreatic (PANC1) cell lines. A549 data is from ENCODE (Richard Myers Group, GSM1010829). (B) ETS1, JUND and GABPA occupancy near the gene encoding vimentin (VIM) is plotted by -log binary P-value from ChIP-seq data in the indicated cell lines. (C) Immunoblots compare ETS1 and tubulin (loading control) levels in PANC1 cells after treatment with shRNAs targeting luciferase (negative control) or two independent shRNAs targeting ETS1. (D) A Boyden chamber transwell assay measures relative cell migration of PANC1 cells expressing each lentiviral shRNA of ETS1 or luciferase shRNA control. Mean and SEM of four biological replicates shown for ETS1 shRNA #1 and two replicates for shRNA #2. P-value (*<0.05) by two-tailed T-test.
ETS1 has dual roles in regulating RAS/ERK signaling
To test if ETS1 regulates expression of the same genes that are activated by RAS/ERK signaling, DU145 cells were treated with the MEK inhibitor U0126, and mRNA levels were measured in triplicate by RNA-seq (Gene expression changes are detailed in Supplementary Table S3). Inhibition of the RAS/ERK pathway decreased expression of 673 genes, and a significant fraction of these (40%), were direct ETS1 target genes based on neighboring ETS1-bound regions in the ChIP-seq dataset (Figure 6A). This represents a >3-fold increase over the random expectation (P < 0.001). Gene expression in DU145 cells depleted for ETS1 was then measured by three independent RNA-seq experiments. The majority (69%) of the RAS/ERK regulated, direct ETS1 target genes (as determined by ChIP-Seq, Figure 4A), changed expression when ETS1 was lost (Figure 6B). This represents a significant enrichment over the random expectation (P < 0.001). Surprisingly, an equal number of these direct targets were inhibited by ETS1 as were activated by ETS1. To interpret this finding, the ontologies of the genes either activated, or inhibited, by ETS1 were identified using GoMiner (Figure 6C). As expected, the genes activated by ETS1 were most enriched for categories associated with cell migration (Response to wounding; Positive regulation of epithelial cell migration). The genes repressed by ETS1 were enriched for other categories including ‘Negative regulation of MAPK cascade’. The genes in this category with the highest expression changes included DUSP4 and DUSP6 (Table 3). DUSPs are phosphatases that directly inactivate MAPKs, including ERK, and are known to be upregulated by RAS/ERK signaling as part of a negative feedback loop. Our data indicate that ETS1 might disrupt this negative feedback loop by directly inhibiting DUSP expression. To test this, we measured ETS1 and activated ERK (pERK) levels in DU145 cells by immunoblotting (Figure 6D). Treatment with the MEK inhibitor U0126 depleted pERK and caused a decrease in ETS1 expression consistent with ETS1 regulating its own promoter via an ETS/AP-1 binding site (Figure 4D). However, knockdown of ETS1 resulted in only a slight decrease in steady-state pERK levels (Figure 6D). Signaling events that activate the RAS/ERK pathway usually result in a peak in ERK phosphorylation followed by a decline, due to the function of negative regulators such as the DUSPs (39). Therefore, we hypothesized that ETS1 repression of DUSP expression might modulate this transient activation of ERK rather than steady-state pERK levels. DU145 cells were serum starved overnight, and then activated with phorbol ester (PMA) for 5 min. ETS1-depleted cells showed significantly less activation of RAS/ERK signaling than control shRNA treated cells (Figure 6E and F). Therefore, we concluded that ETS1 functions through two positive feedback loops (see the model in Figure 6G). First, ETS1 activates its own promoter in response to RAS/ERK signaling. Second, ETS1 represses DUSP expression, leading to a more robust RAS/ERK pathway activation in response to signaling.
Figure 6.
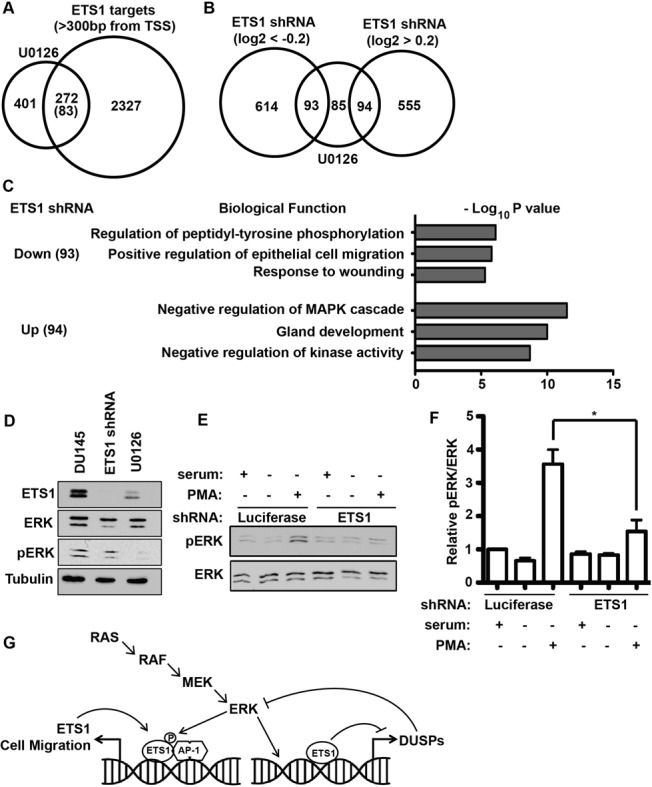
ETS1 regulates two positive feedback loops through two distinct mechanisms. (A) Venn diagram displaying overlap between genes downregulated by the MEK inhibitor U0126 and genes neighboring an ETS1-bound enhancer as determined by ChIP-seq. Downregulated genes are those with significant (P-value < 0.05) decreases after 6 h of 50 μM U0126 compared to mock treated cells as measured by RNA-seq of three biological replicates. Parenthesis indicates randomly predicted overlap. (B) The 272 genes overlapping in (A) were compared to genes with expression changes of greater than log2 = 0.2 or −0.2 in ETS1 shRNA compared to control shRNA by RNA-seq. (C) Ontology analysis of ETS1 enhancer targets from overlaps in (B) using g:Profiler. The top three informative, non-redundant, categories are listed along with their representative P-values. (D) Immunoblot of ETS1, ERK, pERK or tubulin in DU145 cells treated with ETS1 shRNA or U0126, as indicated. (E) Representative immunoblot of DU145 cells expressing luciferase (control) or ETS1 shRNAs and serum starved (12 h) and treated with 50 mg/ml PMA for 5 min as indicated. (F) Quantification of immunoblots as in (E), by ImageJ. P-value (*<0.05) by two-way T-test after three independent biological replicates. (G) Model demonstrating ETS1 regulating two distinct positive feedback loops perpetuating RAS/ERK signaling and cell migration.
Table 3. Genes repressed by MEK inhibition that are most activated by ETS1 knockdown.
| Gene | ETS1 shRNA | MEK inhibition |
|---|---|---|
| DUSP4 | 3.7 | –1.4 |
| SPRY4 | 3.1 | –2.4 |
| SPDEF | 3.0 | –2.0 |
| DUSP6 | 2.9 | –2.6 |
| DHRS3 | 2.6 | –0.77 |
| MLPH | 2.5 | –1.0 |
| FOXL1 | 2.4 | –1.4 |
| DKK1 | 2.3 | –0.57 |
| PRSS22 | 2.1 | –2.2 |
| SUSD2 | 2.1 | –0.70 |
The 10 genes with the largest increase in expression after ETS1 shRNA treatment among genes that are direct targets of ETS1 by ChIP-seq and are inhibited by a MEK inhibitor are shown. Column 2 is the Log (base 2) transformed fold change in expression after ETS1 knockdown in DU145 cells. Column 3 is the Log (base 2) transformed fold change in expression after treatment with the MEK inhibitor U0126 in DU145 cells.
DISCUSSION
Here we report that the presence of an ETS/AP-1 sequence in gene regulatory sequences can define a RAS-responsive cell migration gene expression program, and ETS1 executes this program. ETS1 was unique among ETS factors tested in that it was required for migration in cell lines with an active RAS/ERK signaling pathway. Consistent with our hypothesis, ETS1 had no ability to promote cell migration in cell lines lacking RAS/ERK activation, and the ERK phosphorylation sites T38 and S41 were absolutely required for this ETS1 function. Furthermore, ETS1 bound to ETS/AP-1 sequences genome-wide and was required for activation of target genes associated with cell migration. Surprisingly, we also discovered a novel repressive function of ETS1 at a subset of RAS-activated genes. This function repressed negative regulators of RAS/ERK signaling, allowing robust activation of the pathway. Therefore, ETS1 represents a critical effector of RAS/ERK signaling in epithelial cells.
ETS1 is ubiquitously expressed (20) and therefore has the potential to bind to ETS/AP-1 sites in any cell type where this sequence is accessible. Hence, ETS1 gives the RAS/ERK pathway the potential to activate cell migration in any cell type with the appropriate chromatin structure. We have previously reported that the ETS proteins that are aberrantly expressed in prostate cancer due to chromosome rearrangements, ERG, ETV1, ETV4 and ETV5, can also bind to ETS/AP-1 sequences across the genome and promote prostate cell migration (18). However, these four ETS proteins have restricted expression patterns (20). The PEA3 subfamily ETS proteins, ETV1, ETV4 and ETV5, are expressed during embryonic development, but in adults, high-level expression is restricted to certain tissue-specific stem cells, and their supporting cells (40,41). PEA3 proteins have two major roles, promoting self-renewal of stem cells (42,43) and promoting cellular movement, including both neuronal and epithelial branching (44–46), with this latter role possibly occurring through ETS/AP-1 cis-regulatory sequences. ERG has similar developmental and stem cell roles (47–49), but adult expression is restricted to the hematopoietic and endothelial compartments (20). The diversity of ETS proteins that can activate transcription upon binding of ETS/AP-1 sequences may allow cells to alter the specificity and sensitivity of ETS/AP-1 target genes to various signaling pathways by altering which ETS protein is bound. For example, we have recently shown that ERG binding of ETS/AP-1 sequences allows the PI3K/AKT pathway to regulate these gene targets in prostate epithelial cells (21). However, the ubiquitous presence of ETS1 provides a default setting of RAS/ERK regulation for ETS/AP-1 targets.
Despite the critical role described here for ETS1 in regulating the RAS/ERK response, mice can survive without ETS1. Homozygous disruption of ETS1 leads to an increase in perinatal mortality, but surviving mice only exhibit hematopoietic defects (50) that may be related to ETS/RUNX target genes. The lack of major developmental phenotypes might be attributed to embryonic-specific roles of ETV1, ETV4, ETV5 and ERG at ETS/AP-1 sequences. However, another critical player might be the close homolog of ETS1, ETS2. In fact, loss of ETS1 causes lethality due to vascular rupture when combined with a hypomorphic allele of ETS2 (51), indicating genetic redundancy in endothelial cells. Whether ETS1 and ETS2 ever have redundant roles in epithelial cells is not clear. ETS1 and ETS2 have been reported to have distinct roles in some systems (52), but the mechanisms mediating these differences are not understood. In this study we find that ETS1 and ETS2 have opposite roles in the migration of prostate cancer cells (Figure 2B). This is consistent with previous studies indicating that high ETS1 expression levels correlate with increased invasion and poor prognosis in breast and colon cancer (53,54), while ETS2 is tumor suppressive (55,56). Therefore, our data are consistent with a specific function of ETS1 mediating the RAS/ERK response in epithelial-derived tumor cells.
ETS1 is generally considered an activator of transcription, however, our data indicate that a similar number of direct ETS1 target genes are activated as are repressed when ETS1 is depleted. There have been a few reports of ETS1 acting to repress transcription, such as TGF-β dependent repression of collagens (57). The ability of ETS1 to switch to a repressing function is likely through context-dependent association with other factors. An example is the ability of ETS1 to repress transcription of BAX when the ETS binding sequence is directly adjacent to a GFI1 binding sequence (58). Strikingly, three of the top four genes repressed by ETS1 were negative regulators of the RAS/ERK pathway, DUSP4, SPRY4 and DUSP6 (Table 3). Other RAS/ERK inhibitors, including DUSP5 and SPRY2, were also in this category. Our data indicate that increased levels of these negative regulators in ETS1-depleted cells blunt the RAS/ERK response (Figure 6E and F). Interestingly, the third most repressed gene by ETS1 was the ETS factor SPDEF. SPDEF is an activator of E-cadherin (59,60), and thus could be mediating the changes in E-cadherin expression shown in Figure 1F.
Taken together, our findings indicate that ETS1 is a major effector of RAS/ERK signaling in epithelial cells through both gene activation and repression. Therefore, mechanisms of ETS1 function represent a new set of therapeutic targets for the RAS/ERK signaling nexus, which is often aberrantly activated in carcinomas.
ACCESSION NUMBERS
ChIP-seq and RNA-seq data from this study are available from NCBI's Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/), accession numbers GSE59021 and GSE59020.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank the ENCODE project consortium and the Richard Myers, Greg Crawford and John Stamatoyannopoulous labs for the use of datasets.
FUNDING
Research Scholar Award (RSG-13-215-01-DMC) from the American Cancer Society (to P.C.H.). Funding for open access charge: Indiana University School of Medicine Funds.
Conflict of interest statement. None declared.
REFERENCES
- 1.Fernandez-Medarde A., Santos E. Ras in cancer and developmental diseases. Genes Cancer. 2011;2:344–358. doi: 10.1177/1947601911411084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pratilas C.A., Taylor B.S., Ye Q., Viale A., Sander C., Solit D.B., Rosen N. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc. Natl Acad. Sci. U.S.A. 2009;106:4519–4524. doi: 10.1073/pnas.0900780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Imler J.L., Schatz C., Wasylyk C., Chatton B., Wasylyk B. A Harvey-ras responsive transcription element is also responsive to a tumour-promoter and to serum. Nature. 1988;332:275–278. doi: 10.1038/332275a0. [DOI] [PubMed] [Google Scholar]
- 4.Wasylyk B., Wasylyk C., Flores P., Begue A., Leprince D., Stehelin D. The c-ets proto-oncogenes encode transcription factors that cooperate with c-Fos and c-Jun for transcriptional activation. Nature. 1990;346:191–193. doi: 10.1038/346191a0. [DOI] [PubMed] [Google Scholar]
- 5.Hollenhorst P.C., McIntosh L.P., Graves B.J. Genomic and biochemical insights into the specificity of ETS transcription factors. Annu. Rev. Biochem. 2011;80:437–471. doi: 10.1146/annurev.biochem.79.081507.103945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eferl R., Wagner E.F. AP-1: a double-edged sword in tumorigenesis. Nat. Rev. Cancer. 2003;3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 7.Gavrilov D., Kenzior O., Evans M., Calaluce R., Folk W.R. Expression of urokinase plasminogen activator and receptor in conjunction with the ets family and AP-1 complex transcription factors in high grade prostate cancers. Eur. J. Cancer. 2001;37:1033–1040. doi: 10.1016/s0959-8049(01)00077-6. [DOI] [PubMed] [Google Scholar]
- 8.Gutman A., Wasylyk B. The collagenase gene promoter contains a TPA and oncogene-responsive unit encompassing the PEA3 and AP-1 binding sites. EMBO J. 1990;9:2241–2246. doi: 10.1002/j.1460-2075.1990.tb07394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D'Orazio D., Besser D., Marksitzer R., Kunz C., Hume D.A., Kiefer B., Nagamine Y. Cooperation of two PEA3/AP1 sites in uPA gene induction by TPA and FGF-2. Gene. 1997;201:179–187. doi: 10.1016/s0378-1119(97)00445-9. [DOI] [PubMed] [Google Scholar]
- 10.Yang B.S., Hauser C.A., Henkel G., Colman M.S., Van Beveren C., Stacey K.J., Hume D.A., Maki R.A., Ostrowski M.C. Ras-mediated phosphorylation of a conserved threonine residue enhances the transactivation activities of c-Ets1 and c-Ets2. Mol. Cell. Biol. 1996;16:538–547. doi: 10.1128/mcb.16.2.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bosc D.G., Goueli B.S., Janknecht R. HER2/Neu-mediated activation of the ETS transcription factor ER81 and its target gene MMP-1. Oncogene. 2001;20:6215–6224. doi: 10.1038/sj.onc.1204820. [DOI] [PubMed] [Google Scholar]
- 12.Foulds C.E., Nelson M.L., Blaszczak A.G., Graves B.J. Ras/mitogen-activated protein kinase signaling activates Ets-1 and Ets-2 by CBP/p300 recruitment. Mol. Cell. Biol. 2004;24:10954–10964. doi: 10.1128/MCB.24.24.10954-10964.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charlot C., Dubois-Pot H., Serchov T., Tourrette Y., Wasylyk B. A review of post-translational modifications and subcellular localization of Ets transcription factors: possible connection with cancer and involvement in the hypoxic response. Methods Mol. Biol. 2010;647:3–30. doi: 10.1007/978-1-60761-738-9_1. [DOI] [PubMed] [Google Scholar]
- 14.Yamamoto H., Flannery M.L., Kupriyanov S., Pearce J., McKercher S.R., Henkel G.W., Maki R.A., Werb Z., Oshima R.G. Defective trophoblast function in mice with a targeted mutation of Ets2. Genes Dev. 1998;12:1315–1326. doi: 10.1101/gad.12.9.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kabbout M., Dakhlallah D., Sharma S., Bronisz A., Srinivasan R., Piper M., Marsh C.B., Ostrowski M.C. MicroRNA 17–92 cluster mediates ETS1 and ETS2-dependent RAS-oncogenic transformation. PLoS One. 2014;9:e100693. doi: 10.1371/journal.pone.0100693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hollenhorst P.C., Shah A.A., Hopkins C., Graves B.J. Genome-wide analyses reveal properties of redundant and specific promoter occupancy within the ETS gene family. Genes Dev. 2007;21:1882–1894. doi: 10.1101/gad.1561707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hollenhorst P.C., Chandler K.J., Poulsen R.L., Johnson W.E., Speck N.A., Graves B.J. DNA specificity determinants associate with distinct transcription factor functions. PLoS Genet. 2009;5:e1000778. doi: 10.1371/journal.pgen.1000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hollenhorst P.C., Ferris M.W., Hull M.A., Chae H., Kim S., Graves B.J. Oncogenic ETS proteins mimic activated RAS/MAPK signaling in prostate cells. Genes Dev. 2011;25:2147–2157. doi: 10.1101/gad.17546311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patel M., Simon J.M., Iglesia M.D., Wu S.B., McFadden A.W., Lieb J.D., Davis I.J. Tumor-specific retargeting of an oncogenic transcription factor chimera results in dysregulation of chromatin and transcription. Genome Res. 2012;22:259–270. doi: 10.1101/gr.125666.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hollenhorst P.C., Jones D.A., Graves B.J. Expression profiles frame the promoter specificity dilemma of the ETS family of transcription factors. Nucleic Acids Res. 2004;32:5693–5702. doi: 10.1093/nar/gkh906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Selvaraj N., Budka J.A., Jerde T.J., Ferris M.W., Hollenhorst P.C. Prostate cancer ETS rearrangements switch a cell migration gene expression program from RAS/ERK to PI3K/AKT regulation. Mol. Cancer. 2014;13:61. doi: 10.1186/1476-4598-13-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thurman R.E., Rynes E., Humbert R., Vierstra J., Maurano M.T., Haugen E., Sheffield N.C., Stergachis A.B., Wang H., Vernot B., et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stewart S.A., Dykxhoorn D.M., Palliser D., Mizuno H., Yu E.Y., An D.S., Sabatini D.M., Chen I.S., Hahn W.C., Sharp P.A., et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA. 2003;9:493–501. doi: 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dull T., Zufferey R., Kelly M., Mandel R.J., Nguyen M., Trono D., Naldini L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hollenhorst P.C., Paul L., Ferris M.W., Graves B.J. The ETS gene ETV4 is required for anchorage-independent growth and a cell proliferation gene expression program in PC3 prostate cells. Genes Cancer. 2011;1:1044–1052. doi: 10.1177/1947601910395578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nix D.A., Courdy S.J., Boucher K.M. Empirical methods for controlling false positives and estimating confidence in ChIP-Seq peaks. BMC Bioinformatics. 2008;9:523. doi: 10.1186/1471-2105-9-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo Y., Mahony S., Gifford D.K. High resolution genome wide binding event finding and motif discovery reveals transcription factor spatial binding constraints. PLoS Comput. Biol. 2012;8:e1002638. doi: 10.1371/journal.pcbi.1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas-Chollier M., Darbo E., Herrmann C., Defrance M., Thieffry D., van Helden J. A complete workflow for the analysis of full-size ChIP-seq (and similar) data sets using peak-motifs. Nat. Protoc. 2012;7:1551–1568. doi: 10.1038/nprot.2012.088. [DOI] [PubMed] [Google Scholar]
- 29.Trapnell C., Hendrickson D.G., Sauvageau M., Goff L., Rinn J.L., Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joyce T., Cantarella D., Isella C., Medico E., Pintzas A. A molecular signature for Epithelial to Mesenchymal transition in a human colon cancer cell system is revealed by large-scale microarray analysis. Clin. Exp. Metastasis. 2009;26:569–587. doi: 10.1007/s10585-009-9256-9. [DOI] [PubMed] [Google Scholar]
- 31.Galang C.K., Der C.J., Hauser C.A. Oncogenic Ras can induce transcriptional activation through a variety of promoter elements, including tandem c-Ets-2 binding sites. Oncogene. 1994;9:2913–2921. [PubMed] [Google Scholar]
- 32.Wang X.S., Shankar S., Dhanasekaran S.M., Ateeq B., Sasaki A.T., Jing X., Robinson D., Cao Q., Prensner J.R., Yocum A.K., et al. Characterization of KRAS rearrangements in metastatic prostate cancer. Cancer Discov. 2011;1:35–43. doi: 10.1158/2159-8274.CD-10-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brase J.C., Johannes M., Mannsperger H., Falth M., Metzger J., Kacprzyk L.A., Andrasiuk T., Gade S., Meister M., Sirma H., et al. TMPRSS2-ERG -specific transcriptional modulation is associated with prostate cancer biomarkers and TGF-beta signaling. BMC Cancer. 2011;11:507. doi: 10.1186/1471-2407-11-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nelson M.L., Kang H.S., Lee G.M., Blaszczak A.G., Lau D.K., McIntosh L.P., Graves B.J. Ras signaling requires dynamic properties of Ets1 for phosphorylation-enhanced binding to coactivator CBP. Proc. Natl Acad. Sci. USA. 2010;107:10026–10031. doi: 10.1073/pnas.0915137107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Valouev A., Johnson D.S., Sundquist A., Medina C., Anton E., Batzoglou S., Myers R.M., Sidow A. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat. Methods. 2008;5:829–834. doi: 10.1038/nmeth.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Odrowaz Z., Sharrocks A.D. ELK1 uses different DNA binding modes to regulate functionally distinct classes of target genes. PLoS Genet. 2012;8:e1002694. doi: 10.1371/journal.pgen.1002694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seth A., Papas T.S. The c-ets-1 proto-oncogene has oncogenic activity and is positively autoregulated. Oncogene. 1990;5:1761–1767. [PubMed] [Google Scholar]
- 38.Consortium E.P., Bernstein B.E., Birney E., Dunham I., Green E.D., Gunter C., Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caunt C.J., Keyse S.M. Dual-specificity MAP kinase phosphatases (MKPs): shaping the outcome of MAP kinase signalling. FEBS J. 2013;280:489–504. doi: 10.1111/j.1742-4658.2012.08716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurpios N.A., MacNeil L., Shepherd T.G., Gludish D.W., Giacomelli A.O., Hassell J.A. The Pea3 Ets transcription factor regulates differentiation of multipotent progenitor cells during mammary gland development. Dev. Biol. 2009;325:106–121. doi: 10.1016/j.ydbio.2008.09.033. [DOI] [PubMed] [Google Scholar]
- 41.Eo J., Han K., K M.M., Song H., Lim H.J. Etv5, an ETS transcription factor, is expressed in granulosa and cumulus cells and serves as a transcriptional regulator of the cyclooxygenase-2. J. Endocrinol. 2008;198:281–290. doi: 10.1677/JOE-08-0142. [DOI] [PubMed] [Google Scholar]
- 42.Tyagi G., Carnes K., Morrow C., Kostereva N.V., Ekman G.C., Meling D.D., Hostetler C., Griswold M., Murphy K.M., Hess R.A., et al. Loss of Etv5 decreases proliferation and RET levels in neonatal mouse testicular germ cells and causes an abnormal first wave of spermatogenesis. Biol. Reprod. 2009;81:258–266. doi: 10.1095/biolreprod.108.075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen C., Ouyang W., Grigura V., Zhou Q., Carnes K., Lim H., Zhao G.Q., Arber S., Kurpios N., Murphy T.L., et al. ERM is required for transcriptional control of the spermatogonial stem cell niche. Nature. 2005;436:1030–1034. doi: 10.1038/nature03894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arber S., Ladle D.R., Lin J.H., Frank E., Jessell T.M. ETS gene Er81 controls the formation of functional connections between group Ia sensory afferents and motor neurons. Cell. 2000;101:485–498. doi: 10.1016/s0092-8674(00)80859-4. [DOI] [PubMed] [Google Scholar]
- 45.Livet J., Sigrist M., Stroebel S., De Paola V., Price S.R., Henderson C.E., Jessell T.M., Arber S. ETS gene Pea3 controls the central position and terminal arborization of specific motor neuron pools. Neuron. 2002;35:877–892. doi: 10.1016/s0896-6273(02)00863-2. [DOI] [PubMed] [Google Scholar]
- 46.Chotteau-Lelievre A., Montesano R., Soriano J., Soulie P., Desbiens X., de Launoit Y. PEA3 transcription factors are expressed in tissues undergoing branching morphogenesis and promote formation of duct-like structures by mammary epithelial cells in vitro. Dev. Biol. 2003;259:241–257. doi: 10.1016/s0012-1606(03)00182-9. [DOI] [PubMed] [Google Scholar]
- 47.Iwamoto M., Higuchi Y., Koyama E., Enomoto-Iwamoto M., Kurisu K., Yeh H., Abrams W.R., Rosenbloom J., Pacifici M. Transcription factor ERG variants and functional diversification of chondrocytes during limb long bone development. J. Cell. Biol. 2000;150:27–40. doi: 10.1083/jcb.150.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Loughran S.J., Kruse E.A., Hacking D.F., de Graaf C.A., Hyland C.D., Willson T.A., Henley K.J., Ellis S., Voss A.K., Metcalf D., et al. The transcription factor Erg is essential for definitive hematopoiesis and the function of adult hematopoietic stem cells. Nat. Immunol. 2008;9:810–819. doi: 10.1038/ni.1617. [DOI] [PubMed] [Google Scholar]
- 49.Taoudi S., Bee T., Hilton A., Knezevic K., Scott J., Willson T.A., Collin C., Thomas T., Voss A.K., Kile B.T., et al. ERG dependence distinguishes developmental control of hematopoietic stem cell maintenance from hematopoietic specification. Genes Dev. 2011;25:251–262. doi: 10.1101/gad.2009211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barton K., Muthusamy N., Fischer C., Ting C.N., Walunas T.L., Lanier L.L., Leiden J.M. The Ets-1 transcription factor is required for the development of natural killer cells in mice. Immunity. 1998;9:555–563. doi: 10.1016/s1074-7613(00)80638-x. [DOI] [PubMed] [Google Scholar]
- 51.Wei G., Srinivasan R., Cantemir-Stone C.Z., Sharma S.M., Santhanam R., Weinstein M., Muthusamy N., Man A.K., Oshima R.G., Leone G., et al. Ets1 and Ets2 are required for endothelial cell survival during embryonic angiogenesis. Blood. 2009;114:1123–1130. doi: 10.1182/blood-2009-03-211391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.John S., Russell L., Chin S.S., Luo W., Oshima R., Garrett-Sinha L.A. Transcription factor Ets1, but not the closely related factor Ets2, inhibits antibody-secreting cell differentiation. Mol. Cell. Biol. 2014;34:522–532. doi: 10.1128/MCB.00612-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Y., Yan L.X., Wu Q.N., Du Z.M., Chen J., Liao D.Z., Huang M.Y., Hou J.H., Wu Q.L., Zeng M.S., et al. miR-125b is methylated and functions as a tumor suppressor by regulating the ETS1 proto-oncogene in human invasive breast cancer. Cancer Res. 2011;71:3552–3562. doi: 10.1158/0008-5472.CAN-10-2435. [DOI] [PubMed] [Google Scholar]
- 54.Nakayama T., Ito M., Ohtsuru A., Naito S., Sekine I. Expression of the ets-1 proto-oncogene in human colorectal carcinoma. Mod. Path. 2001;14:415–422. doi: 10.1038/modpathol.3880328. [DOI] [PubMed] [Google Scholar]
- 55.Munera J., Cecena G., Jedlicka P., Wankell M., Oshima R.G. Ets2 regulates colonic stem cells and sensitivity to tumorigenesis. Stem Cells. 2011;29:430–439. doi: 10.1002/stem.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sussan T.E., Yang A., Li F., Ostrowski M.C., Reeves R.H. Trisomy represses Apc(Min)-mediated tumours in mouse models of Down's syndrome. Nature. 2008;451:73–75. doi: 10.1038/nature06446. [DOI] [PubMed] [Google Scholar]
- 57.Czuwara-Ladykowska J., Sementchenko V.I., Watson D.K., Trojanowska M. Ets1 is an effector of the transforming growth factor beta (TGF-beta) signaling pathway and an antagonist of the profibrotic effects of TGF-beta. J. Biol. Chem. 2002;277:20399–20408. doi: 10.1074/jbc.M200206200. [DOI] [PubMed] [Google Scholar]
- 58.Nakazawa Y., Suzuki M., Manabe N., Yamada T., Kihara-Negishi F., Sakurai T., Tenen D.G., Iwama A., Mochizuki M., Oikawa T. Cooperative interaction between ETS1 and GFI1 transcription factors in the repression of Bax gene expression. Oncogene. 2007;26:3541–3550. doi: 10.1038/sj.onc.1210140. [DOI] [PubMed] [Google Scholar]
- 59.Pal M., Koul S., Koul H.K. The transcription factor sterile alpha motif (SAM) pointed domain-containing ETS transcription factor (SPDEF) is required for E-cadherin expression in prostate cancer cells. J. Biol. Chem. 2013;288:12222–12231. doi: 10.1074/jbc.M112.434225. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 60.Findlay V.J., Turner D.P., Yordy J.S., McCarragher B., Shriver M.R., Szalai G., Watson P.M., Larue A.C., Moussa O., Watson D.K. Prostate-derived ETS factor regulates epithelial-to-mesenchymal transition through both SLUG-dependent and independent mechanisms. Genes Cancer. 2011;2:120–129. doi: 10.1177/1947601911410424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.