

This article reports the structure of pectin methylesterase from rice weevil (Sitophilus oryzae) which is the first structure of an animal pectin methylesterase.
Keywords: Sitophilus oryzae, rice weevil, pectin methylesterase
Abstract
Rice weevils (Sitophilus oryzae) use a pectin methylesterase (EC 3.1.1.11), along with other enzymes, to digest cell walls in cereal grains. The enzyme is a right-handed β-helix protein, but is circularly permuted relative to plant and bacterial pectin methylesterases, as shown by the crystal structure determination reported here. This is the first structure of an animal pectin methylesterase. Diffraction data were collected to 1.8 Å resolution some time ago for this crystal form, but structure solution required the use of molecular-replacement techniques that have been developed and similar structures that have been deposited in the last 15 years. Comparison of the structure of the rice weevil pectin methylesterase with that from Dickeya dandantii (formerly Erwinia chrysanthemi) indicates that the reaction mechanisms are the same for the insect, plant and bacterial pectin methylesterases. The similarity of the structure of the rice weevil enzyme to the Escherichia coli lipoprotein YbhC suggests that the evolutionary origin of the rice weevil enzyme was a bacterial lipoprotein, the gene for which was transferred to a primitive ancestor of modern weevils and other Curculionidae. Structural comparison of the rice weevil pectin methylesterase with plant and bacterial enzymes demonstrates that the rice weevil protein is circularly permuted relative to the plant and bacterial molecules.
1. Introduction
Plant cell walls consist of an organized arrangement of complex polysaccharides and proteins which serve as cell structure and plant defense. Nutritionally, plants are a rich source of food if an organism can disrupt and digest these plant products. Following the reports of Shen and coworkers (Shen et al., 1996 ▶, 1999 ▶, 2003 ▶, 2005 ▶), it has become clear that the family of Curculionoidae beetles and weevils uses cellulolytic and pectolytic enzymes to aid the invasion and digestion of cereal grains, especially rice and wheat. Initially, it was thought that these enzymes may have been present in the endosymbiotic organisms in the gut of the rice weevils (Sitophilus oryzae), but the study by Shen et al. (2005 ▶) demonstrated that the pectin methylesterase (PME) was encoded by the rice weevil genome. Amino-acid sequence comparisons suggest that similar enzymatic functions operate in the interactions between other Curculionoidae beetles and plants (see Pauchet et al., 2010 ▶).
Galacturonase and pectate lyase cannot act enzymatically on pectin, but require de-esterification of pectin to pectate in order to digest the polymer. Therefore, pectin methylesterase is central to an insect’s ability to use pectin and pectate as a food source. Additionally, the hydrolysis of these compounds allows the insect to access the cell contents as a source of further nutrition. The rice weevil is an abundant source of PME, which allowed its isolation, purification and characterization (Shen et al., 1999 ▶, 2005 ▶).
We initiated our structural studies of rice weevil PME (RW PME) 15 years ago, but we were unable to solve the crystal structure with the software and template structures available at the time. Recently, we succeeded in solving the structure using the BALBES molecular-replacement server (Long et al., 2008 ▶) and the automatic electron-density fitting program ARP/wARP (Langer et al., 2008 ▶). Also contributing to the success of this approach are the similar structures that have been solved and deposited recently (Jenkins et al., 2001 ▶; Johansson et al., 2002 ▶; Di Matteo et al., 2005 ▶; Eklöf et al., 2009 ▶; Boraston & Abbott, 2012 ▶).
2. Materials and methods
2.1. Protein expression, crystallization and diffraction data collection
RW PME was purified as described previously (Shen et al., 1999 ▶, 2005 ▶). Crystals of RW PME were obtained from hanging-drop vapor-diffusion experiments, but the details of the conditions have been lost owing to a hacked computer. Needles appeared shortly after the crystallization trials were set up, but it took a year for the crystals to assume a useable form. Heavy-atom soaks were performed, but the resulting crystals were non-isomorphous and diffracted poorly. Since only 4 mg of protein was available, all trial conditions were severely limited in scope. The space group of the crystals is C2221, with unit-cell parameters a = 37.43, b = 73.94, c = 204.90 Å and one RW PME molecule in the asymmetric unit. Diffraction data for RW PME were collected at the Structural Biology Center at the Advanced Photon Source at 100 K. Data sets were processed using DENZO and SCALEPACK (Otwinowski & Minor, 1997 ▶). A summary of the statistics is presented in Table 1 ▶.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the inner/outer shell.
| Diffraction data statistics | |
| Unit-cell parameters () | a = 37.43, b = 73.94, c = 204.90 |
| Space group | C2221 |
| Low-resolution limit () | 99.0 (99.0/1.85) |
| High-resolution limit () | 1.79 (3.85/1.79) |
| R merge | 0.085 (0.066/0.296) |
| No. of unique reflections | 23540 (2896/1357) |
| I/(I) | 12.7 (21.0/2.2) |
| Completeness (%) | 85.6 (98.1/50.7) |
| Multiplicity | 3.5 (6.0/1.3) |
| Refinement statistics | |
| Resolution () | 102.51.79 |
| No. of reflections, working set | 21081 |
| No. of reflections, test set | 1203 |
| R cryst, all data | 0.176 |
| R cryst, working set | 0.173 |
| R free, test set | 0.226 |
| Bond r.m.s. (restrained) () | 0.010 |
| No. of protein atoms | 2753 |
| No. of solvent atoms | 299 |
| Ramachandran outliers as determined by MolProbity (%) | 0.0 |
| Ramachandran favored (%) | 96.2 |
| PDB code | 4pmh |
2.2. Structure solution and refinement
The structure of RW PME was solved with BALBES (Long et al., 2008 ▶) and ARP/wARP (Langer et al., 2008 ▶). The amino-acid sequence used in the first structure determination had been truncated, but the initial model produced by BALBES and ARP/wARP and refined using REFMAC5 (Murshudov et al., 2011 ▶) yielded an R value of 0.20, indicating a correct solution. The resulting electron-density maps could be easily fitted with most of the residues missing in the input sequence file. A second run of BALBES and ARP/wARP with a corrected sequence produced a model lacking only seven residues. These were located in subsequent electron-density maps. The complete model was then refined using REFMAC5 in the CCP4 program suite (Winn et al., 2011 ▶). 5% of the reflections were reserved for calculation of R free (Brünger, 1992 ▶).
XtalView (McRee, 1999 ▶) and Coot (Emsley et al., 2010 ▶) were used to view σA-weighted |F o| − |F c| and 2|F o| − |F c| electron-density maps (Read, 1986 ▶) and to manipulate the molecular models. The structural models were evaluated during and after refinement using MolProbity (Chen et al., 2010 ▶) and ADIT (Berman et al., 2000 ▶). Figures were generated using MolScript (Kraulis, 1991 ▶) and Raster3D (Merritt & Bacon, 1997 ▶).
The final model for RW PME contains residues 1–366 and 299 water molecules. Refinement statistics for the structure are given in Table 1 ▶. Coordinates and structure factors for RW PME have been deposited in the Protein Data Bank (with code 4pmh).
Sequence alignments were constructed with the Clustal Omega web server (http://www.ebi.ac.uk/Tools/msa/clustalo/) using default parameters (Sievers et al., 2011 ▶).
The sequences were gathered using the sequence published by Shen et al. (2005 ▶) (AAW28928.1) and those for RW PME deposited by Pauchet et al. (2010 ▶). The sequence ADU33259.1 of Pauchet et al. (2010 ▶) is 98% identical to that of Shen et al. (2005 ▶). There were eight differences between these sequences. For seven of the eight differences the ADU33259.1 sequence fitted the electron density better than the AAW28928.1 sequence and therefore the former was used; the exception was position 61, where we have used the Ala from AAW28928.1. For our numbering of the sequence, we have used the start at DQTA as suggested by Shen et al. (2005 ▶) and this matches the electron density.
For the structural alignment of the carbohydrate esterase family (CE8), we used a locally written program which adaptively fits the translation/rotation matrix of a query to target proteins with sequence numbers increasing and within a given window and a specified tolerance based on a preliminary sequence alignment. Using this program, all of the PMEs in the Protein Data Bank were superposed on the RW PME coordinates, as was the YbhC lipoprotein of Escherichia coli (Eklöf et al., 2009 ▶).
Locally written programs were also used to demonstrate the structural similarity of the N-terminal region of RW PME and the C-terminal regions of plant and bacterial PMEs.
3. Results
The structures of all of the proteins downloaded from the Protein Data Bank under the name pectin methylesterase are similar right-handed β-helices (Fig. 1 ▶). A structural characteristic of β-helix proteins is the stacking of hydrophobic and aromatic side chains on the inside and outside of the β-helix (Jenkins & Pickersgill, 2001 ▶). Similar packings are seen in RM PME, where the arrangement of β-strands in the overall helix ensures that the side chains will be stacked in a slight twist along the direction of the helical axis.
Figure 1.

Stereo diagram of rice weevil pectin methylesterase. The β-strands of the right-handed β-helix are denoted by arrows. The coils show the few α-helices in the structure. Coloring is from blue at the N-terminus to red at the C-terminus. Note the close proximity of the N- and C-termini.
A sequence alignment of the PMEs (and the YbhC lipoprotein from E. coli; Eklöf et al., 2009 ▶; Supplementary Fig. S11) and a three-dimensional structure alignment generated using our adaptive superposition program (Table 2 ▶) provide a basis for comparing the proteins. From the results tabulated in the table, it is unclear whether RW PME is closer to the bacterial or plant PMEs in structure. It is clear that the two plant enzymes are similar to each other, as are the two bacterial PMEs.
Table 2. Distances () among PMEs measured via r.m.s. fit of C atoms using our alignment program.
The lower number is the number of distinct residues which were found to fit within 3 in the best-fit procedure. Our program adaptively fits the query and target proteins with the sequence numbers increasing within the given window (90 residues in this case) and within the specified tolerance (3 in this case). 4pmh is RW PME, 3grh is the YbhC lipoprotein from E. coli (Eklf et al., 2009 ▶), 1gq8 is the PME from carrot (Daucus carota; Johansson et al., 2002 ▶), 1xg2 is the PME from tomato (Solanum lycopersicum; Di Matteo et al., 2005 ▶), 1qjv is the PME from D. dadantii (Jenkins et al., 2001 ▶) and 3uw0 is the PME from Yersinia enterocolitica (Boraston Abbott, 2012 ▶).
A common structural core for the six proteins is made up of runs of consecutive residues that superpose to within 3 Å (see Fig. 2 ▶). Despite the remarkable superposition of the structures, the N-terminal region of RW PME is significantly longer than in the other enzymes and the C-terminal region is shorter. Noteworthy is the fact that the plant and bacterial enzymes have the N- and C-terminal residues rather close together. This is even more extreme in the case of RW PME as these two residues are only 5.8 Å apart at the Cα atoms. When all six proteins are superposed, one sees that the N-terminal region of RW PME aligns with the C-terminal regions of the plant and bacterial enzymes. This is shown in Fig. 3 ▶ for the Dickeya dadantii (formerly Erwinia chrysanthemi) enzyme compared with the RW PME. This circular permutation is discussed more fully below.
Figure 2.

Stereo diagram of RW PME with the structural core found in six superposed structures (Table 2 ▶) shown in magenta. The residues making up the core are consecutive runs of residues with Cα atoms that superpose to within 3 Å.
Figure 3.
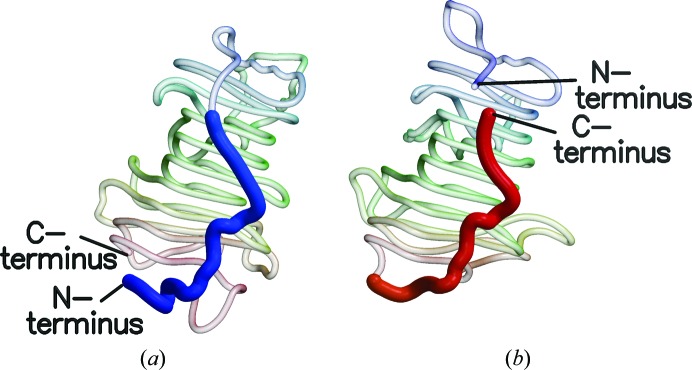
These diagrams of (a) RW PME and (b) D. dadantii PME demonstrate the circular permutation of the protein structures relative to one another. The N-terminal region of RW PME is extended, but when the structures are superposed it conforms to the C-terminal region of the bacterial and plant PMEs.
For the most part, the protein chain of RW PME tracks the other PMEs fairly closely. Between residues 113 and 142 there is an extra loop of two helices which is not present in the other enzymes but is present in the lipoprotein YbhC. This loop points away from the active-site groove, so it should not interfere with the enzyme activity. In the region of RW PME residues 171–176 the bacterial enzymes have a longer loop than the plant or RW PME enzymes. At the RW PME loop from residues 206 to 212 the lipoprotein YbhC has a longer loop (loop 2 in Fig. 2 of Eklöf et al., 2009 ▶), which is said to prevent pectin methylesterase activity. In the RW PME this seven-residue loop is longer than the plant and bacterial enzymes but protrudes into the active-site groove less than the 15-residue loop of YbhC. Indeed, the 15-residue loop of YbhC collides with the docked hexagalacturonic acid substrate (Fries et al., 2007 ▶), but the loop of RW PME does not. Unlike the YbhC protein, the RW PME is an active enzyme (Shen et al., 1999 ▶).
The shape of the loop of RW PME from residues 295 to 307 differs from that in the bacterial enzymes. The structure of the loop from residues 339 to 353 is also rather different from the rest of the plant and bacterial enzymes, but from residue 353 to the C-terminus at 366 the structures of all of the enzymes superpose well.
4. Discussion
There are two more observations to make about the comparison of the structures of RW PME and the E. coli lipoprotein YbhC. Firstly, the structure of YbhC is more similar to RW PME than are the plant or bacterial enzymes. In the structural superposition of YbhC on RW PME, the root-mean-square fit of the Cα atoms is 1.033 Å for 343 residues, which is almost the entire length of the RW PME enzyme (94% of the 366 residues; see Table 2 ▶).
Secondly, the enzymatic function of the YbhC lipoprotein remains unknown (Eklöf et al., 2009 ▶). It is not likely to be a PME, because an Asn residue in the YbhC lipoprotein superposes on a catalytic Asp in the PMEs (Table 3 ▶). The rice weevil has a putative protein, ADU33263.1, which similarly has Asn rather than Asp at the active center. The Asp at this position acts as a general acid–base in the reaction mechanism (Fries et al., 2007 ▶). Sequence alignment of the YbhC protein compared with the RW ADU33263.1 protein indicates that the loop that blocks pectin binding in YbhC (residues 229–243) is very much truncated in the RW homolog.
Table 3. Active-site residues in superposed CE8 proteins.
Note: residue numbers are those in the PDB files. 3grh is a lipoprotein with no PME activity.
Fries et al. (2007 ▶) determined the structures of seven PMEs, each of which had a hexasaccharide of varying methylation or pH of crystallization. We aligned these proteins with RW PME and then asked which residues of RW PME are in contact with the hexasaccharide ligand (Figs. 4 ▶ and 5 ▶, Table 3 ▶). The residues of RW PME in contact with these hexasaccharide ligands are completely compatible with those found for D. dadantii PME, and the enzyme mechanism proposed by Fries et al. (2007 ▶) can be assumed to apply directly to RW PME.
Figure 4.

Model of the binding of the hexasaccharide V of Fries et al. (2007 ▶) in the active site of RW PME. The hexasaccharide is shown in ball-and-stick representation on a ribbon background of the protein. The loop from a superposed YbcH lipoprotein that would interfere with binding of the hexasaccharide is shown as a blue coil.
Figure 5.
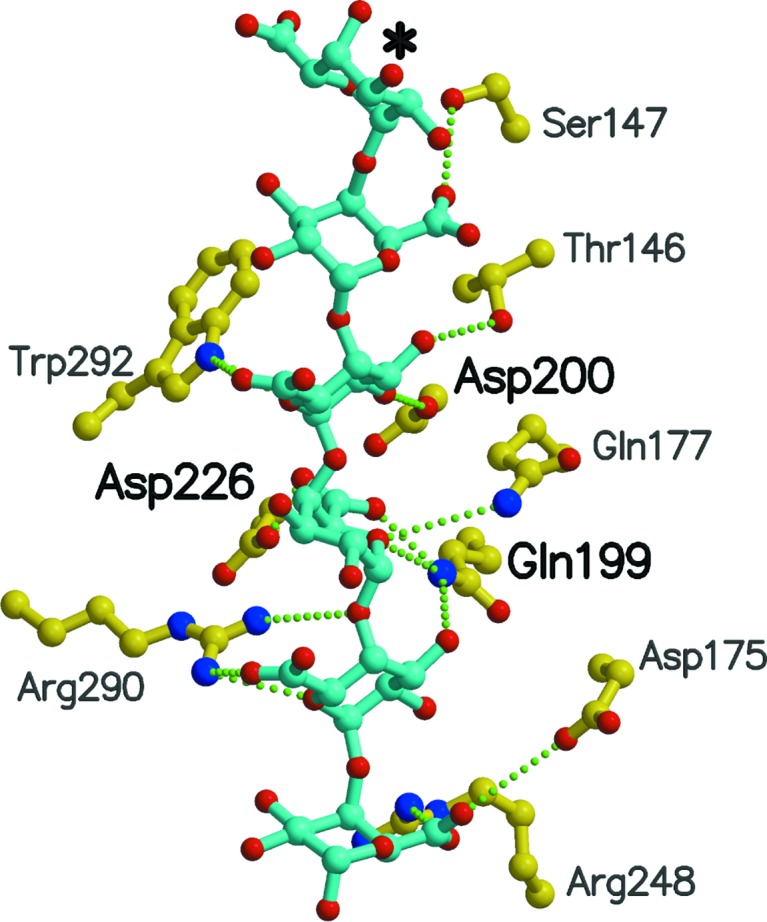
RW PME residues in contact with hexasaccharide V when its complex with YbhC (Fries et al., 2007 ▶) is superposed onto RW PME. This hexasaccharide V simulates the product of the reaction since it consists of galacturonic acid residues. Bold labels denote residues involved in catalysis and transition-state stabilization. The asterisk denotes the reducing end of the hexasaccharide.
The final issue to address in the comparison of these enzymes is the circular permutation of the sequences. Circular permutation of protein sequences is uncommon but does occur naturally (Lindqvist & Schneider, 1997 ▶). The permutations originate by either of two mechanisms: genetic or post-translational. The sequence of the lectin concanavalin A from jack bean is circularly permuted from that of favin (Cunningham et al., 1979 ▶) and occurs post-translationally. In the case of RW PME, the circular permutation is genetic in origin as seen from the encoding RNA sequences of all of the RW PME proteins.
As a final note, we have aligned the available sequences of the other Curculionidae PMEs (from the pine beetle Dendroctonus ponderosae and the sugarcane weevil Sphenophorus levis) with the rice weevil PME sequences. All of these proteins have the extended N-terminal region and truncated C-terminal region, indicating that the circular permutation relative to the plant and bacterial enzymes is characteristic of this insect family.
5. Conclusions
The structure of RW PME has been determined using techniques of molecular replacement which were unavailable 15 years ago. The structures of the members of the CE8 protein family, while sharing a core structure, differ considerably in the loops and turns between the strands in the β-helix motif. Clearly, current molecular-replacement and model-building methods can deal with the differences between the bacterial and plant PMEs and the final structure of this enzyme.
Eklöf et al. (2009 ▶) investigated the phylogeny of the YbhC lipoprotein from E. coli and concluded that it occurred in a separate branch of the CE8 family. This is the protein which is most similar to RW PME and is similarly circularly permuted from the remainder of bacterial and plant enzymes.
Our efforts have determined the structure of the first PME from an animal source. Carbohydrate esterase family 8 (CE8) enzymes have to date only been found in the family Curculionideae, the family of true weevils and bark beetles (Pauchet et al., 2010 ▶). Pauchet et al. (2010 ▶) identified sequences for five CE8 family pectolytic enzymes from rice weevil. Shen et al. (2005 ▶) determined the sequence of tissue-based PME, also from rice weevil. Their sequence is 98% identical to one of those deposited by Pauchet et al. (2010 ▶). The sequences differ in several positions, and based on our electron-density map the structural model has a sequence that is a combination of those two.
Sequence alignments of all of the known beetle PMEs indicate they are circularly permuted relative to the bacterial and plant PMEs. The YbcH lipoprotein, while found in a bacterium, is not a PME. We speculate that all of the isozymes of PME in rice weevil have evolved from a bacterial lipoprotein similar to the E. coli YbhC protein after horizontal transfer of the bacterial gene to the genome of a predecessor to curculionids.
6. Related literature
The following reference is cited in the Supporting Information for this article: Kabsch & Sander (1983 ▶).
Supplementary Material
PDB reference: pectin methylesterase, 4pmh
Supporting Information. . DOI: 10.1107/S2053230X14020433/dw5113sup1.pdf
Acknowledgments
We thank the Structural Biology Center, Argonne National Laboratory for beamtime for data collection. Use of the Advanced Photon Source, an Office of Science User Facility operated for the US Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the US DOE under Contract No. DE-AC02-06CH11357. The research reported in this publication was partially supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award No. T32GM008268. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Portions of this work were also supported by the Kansas Agricultural Experiment Station (contribution No. 15-027-J) and by grants from the National Science Foundation (MCB-9974805) and the National Research Initiative of the USDA Cooperative State Reseach, Education and Extension Service (grant No. 2001-35302-09978).
Footnotes
Supporting information has been deposited in the IUCr electronic archive (Reference: DW5113).
References
- Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N. & Bourne, P. E. (2000). Nucleic Acids Res. 28, 235–242. [DOI] [PMC free article] [PubMed]
- Boraston, A. B. & Abbott, D. W. (2012). Acta Cryst. F68, 129–133. [DOI] [PMC free article] [PubMed]
- Brünger, A. T. (1992). Nature (London), 355, 472–475. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Cunningham, B. A., Hemperly, J. J., Hopp, T. P. & Edelman, G. M. (1979). Proc. Natl Acad. Sci. USA, 76, 3218–3222. [DOI] [PMC free article] [PubMed]
- Di Matteo, A., Giovane, A., Raiola, A., Camardella, L., Bonivento, D., De Lorenzo, G., Cervone, F., Bellincampi, D. & Tsernoglou, D. (2005). Plant Cell, 17, 849–858. [DOI] [PMC free article] [PubMed]
- Eklöf, J. M., Tan, T.-C., Divne, C. & Brumer, H. (2009). Proteins, 76, 1029–1036. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Fries, M., Ihrig, J., Brocklehurst, K., Shevchik, V. E. & Pickersgill, R. W. (2007). EMBO J. 26, 3879–3887. [DOI] [PMC free article] [PubMed]
- Jenkins, J., Mayans, O., Smith, D., Worboys, K. & Pickersgill, R. W. (2001). J. Mol. Biol. 305, 951–960. [DOI] [PubMed]
- Jenkins, J. & Pickersgill, R. (2001). Prog. Biophys. Mol. Biol. 77, 111–175. [DOI] [PubMed]
- Johansson, K., El-Ahmad, M., Friemann, R., Jörnvall, H., Markovic, O. & Eklund, H. (2002). FEBS Lett. 514, 243–249. [DOI] [PubMed]
- Kabsch, W. & Sander, C. (1983). Biopolymers, 22, 2577–2637. [DOI] [PubMed]
- Kraulis, P. J. (1991). J. Appl. Cryst. 24, 946–950.
- Langer, G., Cohen, S. X., Lamzin, V. S. & Perrakis, A. (2008). Nature Protoc. 3, 1171–1179. [DOI] [PMC free article] [PubMed]
- Lindqvist, Y. & Schneider, G. (1997). Curr. Opin. Struct. Biol. 7, 422–427. [DOI] [PubMed]
- Long, F., Vagin, A. A., Young, P. & Murshudov, G. N. (2008). Acta Cryst. D64, 125–132. [DOI] [PMC free article] [PubMed]
- McRee, D. E. (1999). J. Struct. Biol. 125, 156–165. [DOI] [PubMed]
- Merritt, E. A. & Bacon, D. J. (1997). Methods Enzymol. 277, 505–524. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Pauchet, Y., Wilkinson, P., Chauhan, R. & ffrench-Constant, R. H. (2010). PLoS One, 5, e15635. [DOI] [PMC free article] [PubMed]
- Read, R. J. (1986). Acta Cryst. A42, 140–149.
- Shen, Z. C., Denton, M., Mutti, N., Pappan, K., Kanost, M. R., Reese, J. C. & Reeck, G. R. (2003). J. Insect Sci. 3, 1–9. [DOI] [PMC free article] [PubMed]
- Shen, Z., Manning, G., Reese, J. C. & Reeck, G. R. (1999). Insect Biochem. Mol. Biol. 29, 209–214.
- Shen, Z., Pappan, K., Mutti, N. S., He, Q.-J., Denton, M., Zhang, Y., Kanost, M. R., Reese, J. C. & Reeck, G. R. (2005). J. Insect Sci. 5, 21. [DOI] [PMC free article] [PubMed]
- Shen, Z., Reese, J. C. & Reeck, G. R. (1996). Insect Biochem. Mol. Biol. 26, 427–433.
- Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., Lopez, R., McWilliam, H., Remmert, M., Söding, J., Thompson, J. D. & Higgins, D. G. (2011). Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: pectin methylesterase, 4pmh
Supporting Information. . DOI: 10.1107/S2053230X14020433/dw5113sup1.pdf