

The recombinant production and structural analysis of catalytically active type II dehydroquinase from P. aeruginosa are presented.
Keywords: type II dehydroquinase, Pseudomonas aeruginosa, shikimate pathway
Abstract
Pseudomonas aeruginosa causes opportunistic infections and is resistant to most antibiotics. Ongoing efforts to generate much-needed new antibiotics include targeting enzymes that are vital for P. aeruginosa but are absent in mammals. One such enzyme, type II dehydroquinase (DHQase), catalyzes the interconversion of 3-dehydroquinate and 3-dehydroshikimate, a necessary step in the shikimate pathway. This step is vital for the proper synthesis of phenylalanine, tryptophan, tyrosine and other aromatic metabolites. The recombinant expression, purification and crystal structure of catalytically active DHQase from P. aeruginosa (PaDHQase) are presented. Cubic crystals belonging to space group F23, with unit-cell parameters a = b = c = 125.39 Å, were obtained by vapor diffusion in sitting drops and the structure was refined to an R factor of 16% at 1.74 Å resolution. PaDHQase is a prototypical type II DHQase with the classical flavodoxin-like α/β topology.
1. Introduction
Pseudomonas aeruginosa is one of the major causes of opportunistic infections (Chastre et al., 1988 ▶, 1998 ▶; Chastre & Fagon, 2002 ▶; Chastre & Trouillet, 1995 ▶). P. aeruginosa is resistant to many antibiotics and disinfectants and is capable of growing in a variety of habitats, including soil, marshes and coastal marine habitats as well as on plants and animal tissue, including the lungs of cystic fibrosis patients (Chastre & Fagon, 2002 ▶; Chastre et al., 1988 ▶, 1989 ▶, 1998 ▶; Chastre, 2001 ▶; Chastre, Fagon, Bornet-Lecso et al., 1995 ▶; Chastre, Fagon & Trouillet, 1995 ▶; Chastre & Trouillet, 1995 ▶). Long-term P. aeruginosa infection is the primary cause of mortality of cystic fibrosis patients. P. aeruginosa is also a primary source of nosocomial infection, causing bacteremia in burns victims and urinary-tract infections in catheterized patients as well as pneumonia in patients on ventilators and respirators (Chastre & Fagon, 2002 ▶; Fagon, Chastre, Hance, Montravers et al., 1993 ▶; Fagon, Chastre, Hance, Domart et al., 1993 ▶; Fagon, Chastre, Domart et al., 1996 ▶; Fagon, Trouillet et al., 1996 ▶; Fagon & Chastre, 2000 ▶; Fagon, Chastre, Vuagnat et al., 1996 ▶; Fagon et al., 2000 ▶). Thus, there is a need to develop new therapeutics to reduce the morbidity and mortality of P. aeruginosa infections because of its natural resistance to many conventional antibiotics.
Viable therapeutic targets include enzymes that are vital to bacteria but absent in mammals, such as enzymes from the shikimate pathway (Coggins et al., 2003 ▶). The shikimate pathway produces chorismate from erythrose 4-phosphate and phosphoenolpyruvate and leads to the biochemical synthesis of aromatic amino acids, folic acid, ubiquinone and other aromatic compounds (Mousdale & Coggins, 1991 ▶; Abell, 1999 ▶; Haslam, 1993 ▶). Dehydroquinases (DHQase) catalyze the interconversion of 3-dehydroquinate and 3-dehydroshikimate (Fig. 1 ▶ a), which is the third step in the shikimate pathway (Mousdale & Coggins, 1991 ▶; Abell, 1999 ▶). There are two classes of DHQase and both classes have different structural, sequence, physical and biochemical properties. Structures of representative members from both classes have been solved. Although they catalyze the same reaction, type I and type II enzymes are structurally distinct (Gourley et al., 1999 ▶). Type I DHQases typified by 3-dehydroquinate dehydratase from Salmonella typhimurium (Gourley et al., 1999 ▶) and S. enterica (Light et al., 2013 ▶) have the typical αβ-barrel topology, whereas type II DHQases typified by homologs from Mycobacterium tuberculosis (Gourley et al., 1999 ▶) and Streptomyces coelicolor (Roszak et al., 2002 ▶) have a prototypical flavodoxin-like α/β topology. The active sites of type I and type II enzymes are different and the difference is evident when the structures of representative enzymes in complex with 3-dehydroshikimate are compared (Figs. 1 ▶ b and 1 ▶ c). The active site of the representative type I structure from S. enterica (PDB entry 4gug; Light et al., 2013 ▶) cannot be superposed with that of the representative type II structure from M. tuberculosis (PDB entry 3n59; Dias et al., 2011 ▶).
Figure 1.

DHQase enzymes. (a) The reaction catalyzed by DHQase. LigPlot (Wallace et al., 1996 ▶) representation of interactions in the active site of a prototypical (b) type 1 enzyme and (c) type II enzyme in complex with 3-dehydroshikimate (DHS) reveals differences in the two classes of enzymes. The representative type I structure is PDB entry 4gug from S. enterica (Light et al., 2013 ▶) while the representative type II structure is PDB entry 3n59 from M. tuberculosis (Dias et al., 2011 ▶).
This paper focuses on type II DHQase from P. aeruginosa because no type I DHQase was identified in the Pseudomonas genome using the Pseudomonas database (http://www.pseudomonas.com; Winsor et al., 2011 ▶). Additionally, inhibitors of type II DHQase have been investigated as potential therapeutic targets for tuberculosis and have been shown to inhibit the growth of M. tuberculosis (Frederickson et al., 1999 ▶). Type II DHQases form stable trimers and dodecamers and the functionally active oligomer is believed to be the dodecamer (Dias et al., 2011 ▶; Robinson et al., 2006 ▶). The crystal structure of recombinant catalytically active type II DHQase from P. aeruginosa (PaDHQase) is reported here.
2. Materials and methods
2.1. Recombinant expression and production of PaDHQase
The amino-acid sequence of P. aeruginosa strain PAO1 (NCBI Reference Sequence NP_253533.1) was used as a template for the chemical synthesis of DNA expression constructs encoding the 147-amino-acid protein with an N-terminal hexahistidine tag followed by a TEV protease cleavage site (GenScript USA Inc., Piscataway, New Jersey, USA). The synthetic construct was codon-optimized for expression in Escherichia coli and cloned into the HindIII/NdeI sites of pET-28a vector (Novagen, Madison, Wisconsin, USA). The plasmid was transformed into E. coli BL21-CodonPlus-(DE3)-RIPL (Stratagene) by heat shock. Positive colonies were identified by colony PCR with T7 promoter and T7 terminator primers (EMD Millipore, Rockland, Massachusetts, USA). A single positive colony was selected, verified and used for large-scale expression. Each 1 l culture was grown from a 25 ml overnight starter culture using 50 µg l−1 kanamycin. Protein induction was initiated by the addition of IPTG to a final concentration of 0.5 mM for 16 h at 28°C using NZYM medium.
The cell pellet was harvested by centrifugation and stored at −80°C until used; a pellet from a 1 l culture was resuspended in lysis buffer [100 ml phosphate-buffered saline (PBS) pH 7.4, 10%(v/v) glycerol, 5%(v/v) Triton X-100, 0.05%(v/v) β-mercaptoethanol and one Roche cOmplete EDTA-free protease-inhibitor tablet] and lysed under high pressure in an Emulsiflex homogenizer (Avestin, Canada). The supernatant was separated from the cell debris by centrifugation at 8000g for 10 min. PaDHQase from the clarified supernatant was purified by Ni-affinity chromatography on a 5 ml HisTrap FF crude column (GE Healthcare, Piscataway, New Jersey, USA) using an ÄKTApurifier (GE Healthcare, Piscataway, New Jersey, USA). The binding buffer was PBS pH 7.4 with 20 mM imidazole. Protein elution was preceded with extensive washing with binding buffer to remove nonspecifically bound proteins. Upon elution with 250 mM imidazole, ∼99% pure protein was obtained as assessed by Coomassie-stained SDS–PAGE (Fig. 2 ▶ a) and the final yield was ∼40 mg from 1 l culture.
Figure 2.

Purification and crystallization of recombinant PaDHQase. (a) Coomassie Blue-stained reduced SDS–PAGE gel of recombinant PaDHQase; the monomeric, trimeric and dodecameric species are indicated by the 16, 48 and 292 kDa molecular-weight markers, respectively. The molecular-weight-marker lane is labeled M, while the protein lane is labeled 1. (b) Crystals of PaDHQase. (c) Sample diffraction image of PaDHQase.
2.2. Enzyme kinetics
Enzymatic activity was determined by monitoring the characteristic absorbance change at 234 nm resulting from the production of the product 3-dehydroshikimate. All kinetics runs were carried out at room temperature with reagents kept on ice until added to the well for a run. Reactions were incubated for approximately 10 s before starting a run. The substrate, 3-dehydroquinic acid (catalog No. 40216, molecular mass 228.24 g mol−1), was purchased from Sigma–Aldrich, St Louis, Missouri, USA. A 0.2 M stock solution of the substrate was prepared by dissolving 5 mg in 985 µl water just prior to performing each assay. Recombinant His-tagged PaDHQase was stored at −80°C in 50 µl aliquots for one-time thaw and use. Kinetic studies were performed using a μQuant spectrophotometer and KC Junior software (BioTek, Winooski, Vermont, USA). The receptacle for each reaction was one well of a 96-well plate, with an extrapolated path length of 1 cm (for purposes of software absorbance calculation). All reaction conditions were prepared in a final volume of 100 µl. Varying concentrations of enzyme and substrate were added to determine the linear range of enzymatic activity and it was determined that 40 nM enzyme was within this range. Catalytic constants were determined from triplicate assays using 40 nM recombinant His-tagged PaDHQase in 50 mM Tris–HCl pH 7.0 at 25°C and concentrations of 3-dehydroquinic acid ranging from 2 to 15 mM. The absorbance measurements for each reaction were taken every 4 s for 6 min. The velocity values were calculated from the slopes of the absorbance versus time curves. The coefficient of molar absorption for the enone-carboxylate chromophore is 1.2 × 104 (González-Bello & Castedo, 2007 ▶).
2.3. Crystallization and data collection
Crystals were grown at 20°C by vapor diffusion in sitting drops. Drops were prepared by mixing 2 µl protein solution at 4 mg ml−1 in 50 mM Tris–HCl pH 7.0 with 1 µl reservoir solution. The reservoir solution for the largest crystal consisted of a mixture of 0.8 M potassium sodium tartrate tetrahydrate, 0.1 M 2-[4-(2-hydroxyethyl)-1-piperazinyl]ethanesulfonic acid (HEPES) pH 7.5. Large tetrahedral crystals were obtained after two weeks (Fig. 2 ▶ b). A single crystal of approximately 0.3 × 0.4 × 0.6 mm in size was briefly soaked in cryoprotecting solution [0.52 M potassium sodium tartrate, 35%(v/v) glycerol, 0.065 M HEPES pH 7.5] prior to flash-cooling directly in the stream of liquid nitrogen. The reservoir and cryoprotecting solutions were prepared by mixing appropriate volumes of stock solutions from Hampton Research. The stock solutions were 1.0 M HEPES sodium pH 7.5 (CAS 75277-39-3, catalog No. HR2-733), 1.5 M potassium sodium tartrate tetrahydrate (CAS 6381-59-5, catalog No. HR2-539), 100% glycerol (CAS 56-81-5, catalog No. HR2-623) and no further pH adjustments were made.
X-ray diffraction data were collected at the Baylor College of Medicine core facility using a Rigaku HTC detector. The X-ray source was a Rigaku FR-E+ SuperBright microfocus rotating-anode generator with VariMax HF optics. A data set was collected from a single crystal with a crystal-to-detector distance of 115 mm and exposure times of 59 s for 0.5° oscillations, using the CrystalClear (d*TREK) package (Pflugrath, 1999 ▶). A sample diffraction image is shown in Fig. 2 ▶(c). Data were processed using iMosflm (Battye et al., 2011 ▶). The crystal belonged to the cubic space group F23 with unit-cell parameters a = b = c = 125.39 Å.
2.4. Structure determination
The structure was solved by molecular replacement with Phaser (McCoy et al., 2005 ▶; Storoni et al., 2004 ▶). The search model was obtained by stripping a monomer of DHQase from M. tuberculosis of its water molecules (Gourley et al., 1999 ▶). Molecular replacement was followed by iterative cycles of manual model building with Coot (Emsley et al., 2010 ▶) and structure refinement with REFMAC5 (Murshudov et al., 2011 ▶) within the CCP4 package (Winn et al., 2011 ▶). Unless otherwise noted, figures were generated using PyMOL (DeLano, 2002 ▶). The refined model statistics are shown in Table 1 ▶. The atomic coordinates have been deposited with the RCSB PDB with accession code 4l8l.
Table 1. Structure and refinement statistics for PaDHQase (PDB entry 4l8l).
Values in parentheses are for the highest resolution shell.
| Data | |
| Space group | F23 |
| Unit-cell parameters (, ) | a = b = c = 125.39, = = = 90 |
| Resolution () | 31.41.74 (1.81.74) |
| R merge † (%) | 10.0 (21.5) |
| Completeness (%) | 99 (100) |
| Multiplicity | 41.8 (6.96) |
| I/(I) | 31.2 (3.5) |
| Refinement | |
| R factor‡ (%) | 16.2 (35.4) |
| R free § (%) | 19.8 (43.9) |
| Correlation coefficients | |
| F o F c | 0.969 |
| F o F c, free | 0.959 |
| Components of model | |
| Amino-acid residues | 138 |
| Water | 96 |
| Mean B factor (2) | 23.59 |
| R.m.s. deviation from ideal | |
| Bond length () | 0.029 |
| Bond angles () | 2.114 |
| Chiral (3) | 0.159 |
| Ramanchandran plot, residues in (%) | |
| Favored regions | 97.0 |
| Allowed regions | 3.0 |
| Outlier regions | 0 |
| PROCHECK ¶ | |
| Side-chain outliers (%) | 1.8 |
| RSZR outliers (%) | 0.7 |
R
merge = 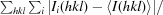
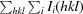 , where I
i(hkl) and I(hkl) are the intensity of the ith measurement of I(hkl) and the mean intensity of the reflection with indices hkl, respectively.
, where I
i(hkl) and I(hkl) are the intensity of the ith measurement of I(hkl) and the mean intensity of the reflection with indices hkl, respectively.
R
cryst = 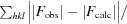
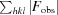 , where F
obs are observed and F
calc are calculated structure-factor amplitudes.
, where F
obs are observed and F
calc are calculated structure-factor amplitudes.
The R free set consisted of a randomly chosen 5% of reflections.
PROCHECK validation was used (Laskowski et al., 1993 ▶). RSZR is the root-mean-square of all Z scores.
3. Results and discussion
3.1. PaDHQase is catalytically active
Preliminary kinetic parameters for the PaDHQase catalytic conversion of dehydroquinic acid to dehydroshikimic acid at 40 nM enzyme, pH 7 and 25°C are shown as the Michaelis–Menten curve (Fig. 3 ▶ a). The derived V max from this curve is 848.76 nM s−1, with a K m of 2.94 mM and a k cat of 18.72 s−1. These results indicate that the recombinant enzyme is catalytically active and suggest that PaDHQase belongs to the slow class of type II DHQases such as the homolog from M. tuberculosis (5.2 s−1), which has significantly lower k cat values than fast type II DHQases such as S. coelicolor (124 s−1) with high k cat values from 100 to 1000 s−1 (Evans et al., 2002 ▶).
Figure 3.

Structure and activity of PaDHQase. (a) Enzymatic activity of 40 nM PaDHQase with increasing concentrations of the substrate 3-dehydroquinic acid. (b) Ribbon diagram of a monomer of PaDHQase rainbow colored from blue (N-terminus) to red (C-terminus); the missing lid loop is also indicated. (c) Dodecamer of PaDHQase with each monomer colored differently. (d) Fit of selected trimer interface residues in the final 2F o − F c electron-density map contoured at 1.6σ.
3.2. Overall structure
The structure of PaDHQase was solved in the cubic space group F23. The refined model of PaDHQase has one monomer in the asymmetric unit with the classic type II DHQase superfamily topology (Fig. 3 ▶ b). The classic dodecamer observed in other bacterial DHQases is generated from the lattice symmetry elements (Fig. 3 ▶ c). The dodecamer and trimer are stable enough to be observed on the reduced SDS–PAGE gels of purified protein after boiling for 10 min in the presence of reducing agent and SDS (Fig. 2 ▶ a). The trimer interface is mediated by strong electrostatic interactions: only one of these contacts involves binding-cavity residues, the intermonomer salt bridge between Asp88 and Arg112 (Fig. 3 ▶ d). This salt bridge is conserved in the structures from other species (Gourley et al., 1999 ▶; Roszak et al., 2002 ▶; González-Bello & Castedo, 2007 ▶; Blanco et al., 2013 ▶; Coderch et al., 2014 ▶). 138 of the 147 amino-acid residues are clearly visible in the 2F o − F c electron-density maps calculated from a molecular-replacement solution, and the quality of fit is evident from the final 2F o − F c electron-density maps (Fig. 3 ▶ d). The disordered regions in the structure are the N-terminal residues (hexahistidine tag as well as TEV cleavage site) and lid loop region. The quality of the final model is summarized in Table 1 ▶.
3.3. Active site of PaDHQase
The active site of PaDHQase is typical of type II DHQases. It is in a cleft surrounded by the C-terminal edge of β-sheets 1 and 3, which interacts with the α3 helix of a neighboring subunit at the trimer interface and is also held together by a salt bridge between Arg112 and Asp88 of these opposing subunits (Fig. 3 ▶ d). The active site typically includes a flexible lid, which closes over the active site upon substrate binding. The lid loop is disordered in our PaDHQase structure (Fig. 3 ▶ b), which is not unusual since the lid loop is disordered in other structures that lack a substrate or an inhibitor (Gourley et al., 1999 ▶). The active site includes a substrate-binding cavity and a proximal glycerol-binding cavity (Dias et al., 2011 ▶). Despite the use of glycerol as a cryoprotectant, there is no evidence of glycerol binding in the electron-density maps.
3.4. Comparison to other type II DHQases
The structures that were most similar to PaDHQase were identified using the Structure Similarity option of PDBeFold (http://www.ebi.ac.uk/msd-srv/ssm/). This method identifies similar structures using the following criteria: the r.m.s.d. for alignment of all main-chain atoms, alignment length and number of gaps. ClustalW (Larkin et al., 2007 ▶) alignment of the top hits reveals well conserved amino-acid sequences (Fig. 4 ▶). The structure of PaDHQase superposes quite well with these enzymes, with an r.m.s. deviation of all main-chain atoms ranging from 0.63 Å for the Actinobacillus pleuropneumoniae homolog to 1.2 Å for some of the M. tuberculosis homolog structures. The regions of highest structural variability for these structures tend to be in loop regions, most notably the flexible-lid regions, and are related to disorder in the absence of ligand binding and variable conformations dependent upon ligand type.
Figure 4.

Comparison of PaDHQase with other type II DHQases. The amino-acid sequence alignment showing structural elements of DHQase was generated with ESPript3.0 (Gouet et al., 2003 ▶). Secondary-structure elements are as follows: α-helices are shown as large squiggles labeled α, 310-helices are shown as small squiggles labeled η, β-strands are shown as arrows and are labeled β. Identical residues are shown on a red background, conserved residues are shown in red and conserved regions are shown in blue boxes. The DHQases used in alignment were PaDHQase, recombinant PaDHQase (rPaDHQase), and others identified using the Structure Similarity option of PDBeFold (http://www.ebi.ac.uk/msd-srv/ssm/) as most structurally similar to PaDHQase. ApHHQase is the crystal structure of DHQase from Actinobacillus pleuropneumoniae (PDB entry 1uqr), the most similar to PaDHQase, and has 66% sequence identity and an r.m.s.d. 0.63 Å (Maes et al., 2004 ▶). YpDHQase, the crystal structure of the homolog from Yersinia pestis (PDB entry 3lwz), is the next closest structural homolog with 68% sequence identity and an r.m.s.d. of 0.75 Å (Center for Structural Genomics of Infectious Diseases, unpublished work). CaDHQase is the homolog from Candida albicans (PDB entry 3kip), which has 56% sequence identity and an r.m.s.d. of 0.76 Å (Trapani et al., 2010 ▶). Other structurally similar type II DHQases include MtDHQase from M. tuberculosis (Gourley et al., 1999 ▶; Schmidt et al., 2013 ▶), BaDHQase from Bacillus subtilis (PDB entry 1gqo; D. A. Robinson, A. W. Roszak, J. R. Coggins & A. J. Lapthorn, unpublished work), TtDHQase from Thermus thermophilus (PDB entry 2uyg; H. Utsunomiya, Y. Agari, T. Imagawa & H. Tsuge, unpublished work) and ScDHQase from S. coelicolor (Roszak et al., 2002 ▶).
The enzymatic site of PaDHQase is accessible to small molecules and is quite similar to those of homologous enzymes, for example that from M. tuberculosis, and key residues involved in inhibitor and substrate binding are well conserved (Fig. 5 ▶). As was observed in other DHQases the active site of PaDHQase involves residues from two monomers (colored gray and aquamarine) and there are no amino-acid residues from either monomer obstructing access to the active site. The third monomer at the trimer interface (colored blue) is not in proximity to the active site (Fig. 5 ▶). There are no inter- or intra-dodecamer contacts obstructing the active site.
Figure 5.

Comparison of the active sites of PaDHQase and MtDHQase (M. tuberculosis DHQase). The superposed structure of complex of M. tuberculosis DHQase (gold) with compound 5 shows well conserved residues with PaDHQase (shown in stick representation). The location of the lid loop from the MtDHQase structure is also shown with the catalytic tyrosine labeled and shown in stick representation. Key binding-site residues from one monomer of PaDHQase (gray) are in proximity to a helix from another monomer (green). The cavity is large enough to bind compound 5 (in pink stick representation), an inhibitor of M. tuberculosis DHQase that extends into the glycerol cavity. The PDB entry corresponding to the complex of M. tuberculosis DHQase is 3n76 (Dias et al., 2011 ▶).
The similarity of the crystal form and accessibility of the active site suggest the possibility of soaking inhibitors or other small molecules into the preformed crystals. Additionally, the binding cavity of PaDHQase extends into the glycerol-binding cavity, revealing that inhibitors such as compound 5 from the Blundell group which extends from the active site to the glycerol site (Dias et al., 2011 ▶) are capable of fitting into the PaDHQase structure and providing good starting points for structure-based inhibitor design. Overall the extensive structural similarity of PaDHQase to other type II DHQases suggests the possibility of shared inhibitors.
4. Concluding remarks
Structural studies of catalytically active recombinant PaDHQase reveal a prototypical type II DHQase with extensive structural similarity to previously characterized enzymes. Kinetic activity suggests that PaDHQase belongs to the slow class of type II DHQases. Rational drug-development efforts will benefit from the wealth of existing knowledge of enzymes from other organisms, particularly other slow-class members.
Supplementary Material
PDB reference: type II dehydroquinase, 4l8l
Acknowledgments
SR was supported by a graduate student fellowship from the Toxicology Department, School of Public Health University at the University of Nebraska Medical Center, Omaha, Nebraska. MMM was an undergraduate intern from Rice University. GR is supported by an American Chemical Society Project SEED Fellowship. OAA and AK are supported by startup funds from the National School of Tropical Medicine at the Baylor College of Medicine.
References
- Abell, C. (1999). Comprehensive Natural Products Chemistry, edited by U. Sankawa, pp. 573–607. Amsterdam: Elsevier.
- Battye, T. G. G., Kontogiannis, L., Johnson, O., Powell, H. R. & Leslie, A. G. W. (2011). Acta Cryst. D67, 271–281. [DOI] [PMC free article] [PubMed]
- Blanco, B., Prado, V., Lence, E., Otero, J. M., Garcia-Doval, C., van Raaij, M. J., Llamas-Saiz, A. L., Lamb, H., Hawkins, A. R. & González-Bello, C. (2013). J. Am. Chem. Soc. 135, 12366–12376. [DOI] [PubMed]
- Chastre, J. (2001). Rev. Pneumol. Clin. 57, 113–123. [PubMed]
- Chastre, J. & Fagon, J.-Y. (2002). Am. J. Respir. Crit. Care Med. 165, 867–903. [DOI] [PubMed]
- Chastre, J., Fagon, J.-Y., Bornet-Lecso, M., Calvat, S., Dombret, M.-C., al Khani, R., Basset, F. & Gibert, C. (1995). Am. J. Respir. Crit. Care Med. 152, 231–240. [DOI] [PubMed]
- Chastre, J., Fagon, J.-Y., Domart, Y. & Gibert, C. (1989). Eur. J. Clin. Microbiol. Infect. Dis. 8, 35–39. [DOI] [PubMed]
- Chastre, J., Fagon, J.-Y., Soler, P., Bornet, M., Domart, Y., Trouillet, J.-L., Gibert, C. & Hance, A. J. (1988). Am. J. Med. 85, 499–506. [DOI] [PubMed]
- Chastre, J., Fagon, J.-Y. & Trouillet, J.-L. (1995). Clin. Infect. Dis. 21, S226–S237. [DOI] [PubMed]
- Chastre, J. & Trouillet, J.-L. (1995). Curr. Opin. Pulm. Med. 1, 194–201. [PubMed]
- Chastre, J., Trouillet, J.-L., Vuagnat, A., Joly Guillou, M.-L., Clavier, H., Dombret, M.-C. & Gibert, C. (1998). Am. J. Respir. Crit. Care Med. 157, 1165–1172. [DOI] [PubMed]
- Coderch, C., Lence, E., Peón, A., Lamb, H., Hawkins, A. R., Gago, F. & González-Bello, C. (2014). Biochem. J. 458, 547–557. [DOI] [PubMed]
- Coggins, J. R., Abell, C., Evans, L. B., Frederickson, M., Robinson, D. A., Roszak, A. W. & Lapthorn, A. P. (2003). Biochem. Soc. Trans. 31, 548–552. [DOI] [PubMed]
- Delano, W. L. (2002). PyMOL. http://www.pymol.org. [DOI] [PubMed]
- Dias, M. V., Snee, W. C., Bromfield, K. M., Payne, R. J., Palaninathan, S. K., Ciulli, A., Howard, N. I., Abell, C., Sacchettini, J. C. & Blundell, T. L. (2011). Biochem. J. 436, 729–739. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Evans, L. D., Roszak, A. W., Noble, L. J., Robinson, D. A., Chalk, P. A., Matthews, J. L., Coggins, J. R., Price, N. C. & Lapthorn, A. J. (2002). FEBS Lett. 530, 24–30. [DOI] [PubMed]
- Fagon, J.-Y. & Chastre, J. (2000). Ann. Intern. Med. 133, 1009. [DOI] [PubMed]
- Fagon, J.-Y., Chastre, J., Domart, Y., Trouillet, J.-L. & Gibert, C. (1996). Clin. Infect. Dis. 23, 538–542. [DOI] [PubMed]
- Fagon, J.-Y., Chastre, J., Hance, A. J., Domart, Y., Trouillet, J.-L. & Gibert, C. (1993). Chest, 103, 547–553. [DOI] [PubMed]
- Fagon, J.-Y., Chastre, J., Hance, A. J., Montravers, P., Novara, A. & Gibert, C. (1993). Am. J. Med. 94, 281–288. [DOI] [PubMed]
- Fagon, J.-Y., Chastre, J., Vuagnat, A., Trouillet, J.-L., Novara, A. & Gibert, C. (1996). JAMA, 275, 866–869. [PubMed]
- Fagon, J.-Y., Chastre, J., Wolff, M., Gervais, C., Parer-Aubas, S., Stéphan, F., Similowski, T., Mercat, A., Diehl, J. L., Sollet, J. P. & Tenaillon, A. (2000). Ann. Intern. Med. 132, 621–630. [DOI] [PubMed]
- Fagon, J.-Y., Trouillet, J.-L. & Chastre, J. (1996). Presse Med. 25, 1441–1446. [PubMed]
- Frederickson, M., Parker, E. J., Hawkins, A. R., Coggins, J. R. & Abell, C. (1999). J. Org. Chem. 64, 2612–2613. [DOI] [PubMed]
- González-Bello, C. & Castedo, L. (2007). Med. Res. Rev. 27, 177–208. [DOI] [PubMed]
- Gouet, P., Robert, X. & Courcelle, E. (2003). Nucleic Acids Res. 31, 3320–3323. [DOI] [PMC free article] [PubMed]
- Gourley, D. G., Shrive, A. K., Polikarpov, I., Krell, T., Coggins, J. R., Hawkins, A. R., Isaacs, N. W. & Sawyer, L. (1999). Nature Struct. Biol. 6, 521–525. [DOI] [PubMed]
- Haslam, E. (1993). Shikimic Acid: Metabolism and Metabolites. Chichester: John Wiley & Sons.
- Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J. & Higgins, D. G. (2007). Bioinformatics, 23, 2947–2948. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Light, S. H., Anderson, W. F. & Lavie, A. (2013). Protein Sci. 22, 418–424. [DOI] [PMC free article] [PubMed]
- Maes, D., Gonzalez-Ramirez, L. A., Lopez-Jaramillo, J., Yu, B., De Bondt, H., Zegers, I., Afonina, E., Garcia-Ruiz, J. M. & Gulnik, S. (2004). Acta Cryst. D60, 463–471. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Storoni, L. C. & Read, R. J. (2005). Acta Cryst. D61, 458–464. [DOI] [PubMed]
- Mousdale, D. M. & Coggins, J. R. (1991). Target Sites for Herbicide Action, edited by R. C. Kirkwood, pp. 29–56. New York: Plenum Press.
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Pflugrath, J. W. (1999). Acta Cryst. D55, 1718–1725. [DOI] [PubMed]
- Robinson, D. A., Stewart, K. A., Price, N. C., Chalk, P. A., Coggins, J. R. & Lapthorn, A. J. (2006). J. Med. Chem. 49, 1282–1290. [DOI] [PubMed]
- Roszak, A. W., Robinson, D. A., Krell, T., Hunter, I. S., Fredrickson, M., Abell, C., Coggins, J. R. & Lapthorn, A. J. (2002). Structure, 10, 493–503. [DOI] [PubMed]
- Schmidt, M. F., Korb, O., Howard, N. I., Dias, M. V., Blundell, T. L. & Abell, C. (2013). ChemMedChem, 8, 54–58. [DOI] [PubMed]
- Storoni, L. C., McCoy, A. J. & Read, R. J. (2004). Acta Cryst. D60, 432–438. [DOI] [PubMed]
- Trapani, S., Schoehn, G., Navaza, J. & Abergel, C. (2010). Acta Cryst. D66, 514–521. [DOI] [PubMed]
- Wallace, A. C., Laskowski, R. A. & Thornton, J. M. (1996). Protein Eng. 8, 127–134. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Winsor, G. L., Lam, D. K. W., Fleming, L., Lo, R., Whiteside, M. D., Yu, N. Y., Hancock, R. E. & Brinkman, F. S. L. (2011). Nucleic Acids Res. 39, D596–D600. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: type II dehydroquinase, 4l8l