

Abstract
Abstract
Considerable data have accumulated over the past 20 years, indicating that the human kidney is involved in the regulation of glucose via gluconeogenesis, taking up glucose from the circulation, and by reabsorbing glucose from the glomerular filtrate. In light of the development of glucose-lowering drugs involving inhibition of renal glucose reabsorption, this review summarizes these data. Medline was searched from 1989 to present using the terms ‘renal gluconeogenesis’, ‘renal glucose utilization’, ‘diabetes mellitus’ and ‘glucose transporters’. The human liver and kidneys release approximately equal amounts of glucose via gluconeogenesis in the post-absorptive state. In the postprandial state, although overall endogenous glucose release decreases substantially, renal gluconeogenesis increases by approximately twofold. Glucose utilization by the kidneys after an overnight fast accounts for ∼10% of glucose utilized by the body. Following a meal, glucose utilization by the kidney increases. Normally each day, ∼180 g of glucose is filtered by the kidneys; almost all of this is reabsorbed by means of sodium–glucose co-transporter 2 (SGLT2), expressed in the proximal tubules. However, the capacity of SGLT2 to reabsorb glucose from the renal tubules is finite and, when plasma glucose concentrations exceed a threshold, glucose appears in the urine. Handling of glucose by the kidney is altered in Type 2 diabetes mellitus (T2DM): renal gluconeogenesis and renal glucose uptake are increased in both the post-absorptive and postprandial states, and renal glucose reabsorption is increased. Specific SGLT2 inhibitors are being developed as a novel means of controlling hyperglycaemia in T2DM.
Diabet. Med. 27, 136–142 (2010)
Keywords: gluconeogenesis, kidney, sodium glucose co-transporter 2, Type 2 diabetes mellitus
Introduction
The kidney’s involvement in glucose homeostasis was first described in the 1930s 1. Despite the large body of evidence amassed over the ensuing years, the kidney is still often overlooked as an important player in glucose metabolism. However, awareness of renal mechanisms of glucose homeostasis is likely to increase in the near future because novel glucose-lowering drugs are being developed that target one aspect of renal glucose handling, namely reabsorption of glucose from the glomerular filtrate [the sodium–glucose co-transporter 2 (SGLT2) inhibitors]. This article reviews our current understanding of the role of the kidney in normal glucose homeostasis and abnormalities in patients with Type 2 diabetes mellitus (T2DM). Medline was searched from 1989 to present using the terms ‘renal gluconeogenesis’, ‘renal glucose utilization’, ‘diabetes mellitus’ and ‘glucose transporters’.
Overview of renal glucose homeostasis
The human kidney is involved in the regulation of glucose homeostasis and in abnormalities found in diabetes mellitus via three different mechanisms: (i) release of glucose into the circulation via gluconeogenesis; (ii) uptake of glucose from the circulation to satisfy its energy needs; and (iii) reabsorption into the circulation of glucose from glomerular filtrate to conserve glucose carbon.
Plasma glucose concentrations are determined by the relative rates of glucose entry into, and removal from, the circulation. Normally, despite wide daily fluctuations in the rate of delivery of glucose into the circulation (e.g. meal ingestion) and in the demands of tissues for glucose (e.g. during exercise), plasma levels are maintained within a relatively narrow range throughout the day. Maximal plasma concentrations following meal ingestion are usually < 9.0 mmol/l 2 and minimal concentrations, after moderate fast or exercise, are usually > 3.0 mmol/l 3,4. This is in contrast to other substrates such as glycerol, lactate, free fatty acids (FFAs) and ketone bodies, for which daily fluctuation is much greater 5. Teleologically, this can be explained by the fact that, on the one hand, the body must defend itself from hyperglycaemia, which is associated with both chronic effects (including retinopathy, neuropathy, nephropathy and premature atherosclerosis 6–9) and acute effects (including diabetic ketoacidosis and hyperosmolar hyperglycaemic state, which have significant associated morbidity and mortality); on the other hand, the body must also defend itself against hypoglycaemia, which can cause cardiac arrhythmias, neurological dysfunction, coma, seizures and death 10. Brain function is particularly dependent on having adequate levels of plasma glucose because the brain is unable to either store or produce glucose and alternative sources of energy are either in short supply (e.g. ketone bodies) or are unable to pass the blood–brain barrier (e.g. FFAs) 10.
The precise regulation of plasma glucose concentrations is mainly determined by hormonal and neural factors, which regulate endogenous production of glucose 10. Acute glucoregulatory mechanisms involve insulin, glucagon and catecholamines, which can effect changes in plasma glucose levels over a matter of minutes. Insulin suppresses glucose release in both the liver and kidney by direct enzyme activation/deactivation, as well as by reducing the availability of gluconeogenic substrates and actions on gluconeogenic activators 11. Glucagon has no effect on the kidney, but increases both gluconeogenesis and glycogenolysis in the liver 12. Catecholamines have multiple acute actions, including stimulation of renal glucose release, inhibition of insulin secretion, stimulation of glucagon secretion, and increases in gluconeogenic substrate supply, stimulation of lipolysis and reduced tissue glucose uptake.
Growth hormone, thyroid hormone and cortisol influence glucose levels over a period of hours by altering the sensitivity of the liver, kidney, adipose tissue and muscle to insulin, glucagon and catecholamines, and by altering the activity of key enzymes, which effect glycogen stores and availability of gluconeogenic precursors (lactate, glycogen and amino acids) 10. In the post-absorptive state, glucose uptake by tissues is largely dependent on tissue needs and the mass-action effects of the ambient plasma glucose concentration and, to a lesser extent, on the permissive actions of insulin and counter-regulatory hormones (e.g. thyroid hormones, growth hormone, catecholamines and cortisol). In these circumstances, most uptake of glucose occurs in tissues that do not require insulin (e.g. brain, gastrointestinal tract, renal medulla). However, in the postprandial state, although insulin and other hormones exert greater influence on tissue uptake of glucose, changes in hepatic and renal glucose release into the circulation are still quite important (Table 1) 10.
Table 1.
Proportion of glucose utilization as a result of specific tissues in the fasting and postprandial state 5,57
| Tissue/organ | Post-absorptive state, ∼11.1 μmol/(kg min) (mainly insulin-independent) | Postprandial state, ∼55 μmol/(kg min) (mainly insulin-stimulated) |
|---|---|---|
| % of total | % of total | |
| Brain | 40–45 | ∼10 |
| Muscle | 15–20 | 30–35 |
| Liver | 10–15 | 25–30 |
| Gastrointestinal (GI) tract | 5–10 | 10–15 |
| Kidney | 5–10 | 10–15 |
| Other (e.g. skin, blood cells) | 5–10 | 5–10 |
Renal gluconeogenesis
The post-absorptive state
After a 14- to 16-h overnight fast, glucose is released into the circulation at a rate of approximately 10 μmol/(kg min) 10,13,14. Approximately 50% of this is the result of the breakdown of glycogen (glycogenolysis) stored in the liver and the other half is because of the production of new glucose molecules from precursors such as lactate, glycerol, alanine and other amino acids (gluconeogenesis) by liver and kidneys (Table 2) 10,13,14. The kidney is unable to release glucose through glycogenolysis because it contains very little glycogen and those renal cells that are able to synthesize glycogen lack the enzyme glucose-6-phosphatase and therefore cannot release glucose 14. In humans, only the liver and kidney contain significant amounts of the enzyme glucose-6-phosphatase and therefore are the only organs that are able to perform gluconeogenesis. Research over the last 15–20 years has established that the human liver and kidneys provide about equal amounts of glucose via gluconeogenesis in the post-absorptive state (Table 2). Consequently, after an overnight fast, 75–80% of glucose released into the circulation derives from the liver and the remaining 20–25% derives from the kidneys. As the duration of fasting increases, glycogen stores in the liver become further depleted until, after 48 h, virtually all the glucose released into the circulation is derived from gluconeogenesis 4,13. Consequently, as the length of fast increases, the proportion of overall glucose release accounted for by renal gluconeogenesis increases 15.
Table 2.
Mechanisms and sources of glucose release into the circulation in the post-absorptive state 10,13,14
| Overall rate [∼μmol/(kg min)] | 10 |
|---|---|
| Hepatic contribution [∼μmol/(kg min)] | 7.5–8.0 (75–80%) |
| Glycogenolysis [∼μmol/(kg min)] | 4.5–5.0 (45–50%) |
| Gluconeogenesis [∼μmol/(kg min)] | 2.5–3.0 (25–30%) |
| Renal contribution [∼μmol/(kg min)] | 2.0–2.5 (20–25%) |
| Glycogenolysis [∼μmol/(kg min)] | 0 |
| Gluconeogenesis [∼μmol/(kg min)] | 2.0–2.5 (20–25%) |
It is important to note that kidney and liver differ in their use of gluconeogenic precursors and the effect of hormones on their release of glucose. As shown in Table 3, lactate is the predominant gluconeogenic precursor in both organs, but otherwise the kidney preferentially uses glutamine 16, whereas the liver preferentially uses alanine 17.
Table 3.
Utilization of substrates for gluconeogenesis 17
| Lactate (n = 16) | Glycerol (n = 9) | Glutamine (n = 37) | Alanine (n = 9) | |
|---|---|---|---|---|
| Overall gluconeogenesis [∼μmol/(kg min)] | 1.88 ± 0.15 | 0.53 ± 0.09 | 0.58 ± 0.04 | 0.68 ± 0.07 |
| Renal gluconeogenesis [∼μmol/(kg min)] (% of overall gluconeogenesis) | 0.89 ± 0.09 (47 ± 8) | 0.17 ± 0.03 (32 ± 4) | 0.36 ± 0.02 (62 ± 3) | 0.02 ± 0.01 (3 ± 1) |
| Hepatic gluconeogenesis [∼μmol/(kg min)] (% of overall gluconeogenesis) | 0.97 ± 0.18 (53 ± 8) | 0.39 ± 0.8 (68 ± 4) | 0.23 ± 0.02 (38 ± 3) | 0.67 ± 0.08 (97 ± 1) |
With respect to hormonal influences, insulin suppresses glucose release by both organs with roughly comparable efficacy 18, whereas glucagon normally stimulates hepatic glucose release only, mainly via an early action on glycogenolysis 12. Catecholamines normally exert a direct effect on renal glucose release only 19,20, although they may indirectly affect both hepatic and renal glucose release by increasing availability of gluconeogenic substrates and by suppressing insulin secretion. Cortisol, growth hormone and thyroid hormones have long-term stimulatory influences on hepatic glucose release (over a period of days) 10. Their effects on renal glucose release in humans have yet to be determined.
The postprandial state
Classically, metabolic studies have usually been undertaken in the post-absorptive state (i.e. 12–16 h after the last meal). However, most of the day people are in the postprandial state as this includes 4–6 h on three occasions during the day.
Postprandial plasma glucose levels are critically influenced by insulin and glucagon levels. Following ingestion of glucose, plasma glucose levels peak in 60–90 min and slowly return to post-absorptive levels after 3–4 h. This profile is mirrored by a fourfold increase in plasma insulin and a reciprocal suppression of plasma glucagon levels of ∼50% 10. Meyer et al. (2002) demonstrated that, after meal ingestion, overall endogenous glucose release decreases by ∼61%, with hepatic glycogenolysis virtually ceasing in the 4- to 6-h period 21. Teleologically, this is understandable because this period is responsible for replenishment of hepatic glycogen stores. Furthermore, suppression of endogenous glucose release limits postprandial hyperglycaemia. Hepatic gluconeogenesis also decreases by ∼82% and glucose molecules generated through this pathway are not generally released in the circulation, but are largely directed into hepatic glycogen. Perhaps surprisingly, renal gluconeogenesis actually increases by approximately twofold and accounts for ∼60% of endogenous glucose release in the postprandial period 21. This has been hypothesized to facilitate efficient repletion of glycogen stores in the liver 21.
These differences in regulation and reciprocal change in renal and hepatic glucose release have led to the concept of hepatorenal glucose reciprocity 22. This concept refers to the situations in which a physiological or pathological decrease in glucose release by kidney or liver is associated with a compensatory increase in glucose release by liver or kidney so as to prevent hypoglycaemia or to optimize homeostasis. Examples of this include the anhepatic phase after liver transplantation, prolonged fasting, acidosis, meal ingestion and insulin overdoses in diabetes mellitus 22–24.
Renal glucose utilization
In the post-absorptive setting after an overnight fast, the kidneys utilize approximately 10% of all glucose utilized by the body. After meal ingestion their glucose utilization increases in an absolute sense. In terms of whole-body glucose economy, normally approximately 45% of ingested glucose is thought to be converted to glycogen in the liver, ∼30% is taken up by skeletal muscle and later converted to glycogen, ∼15% is taken up by the brain, ∼5% is taken up by the adipose tissue and ∼10% is taken up by the kidneys 10,21. The metabolic fate of glucose is different in different regions of the kidney. Because of its low oxygen tension, and low levels of oxidative enzymes, the renal medulla is an obligate user of glucose for its energy requirement and does so anaerobically. Consequently, lactate is the main metabolic end product of glucose taken up in the renal medulla, not carbon dioxide (CO2) and water. In contrast, the renal cortex has little glucose phosphorylating capacity but a high level of oxidative enzymes. Consequently, this part of the kidney does not take up and use very much glucose, with oxidation of FFAs acting as the main source of energy. A major energy-requiring process in the kidney is the reabsorption of glucose from glomerular filtrate in the proximal convoluted tubule 25.
Renal glucose reabsorption
In addition to releasing glucose into the circulation by synthesizing new glucose molecules via gluconeogenesis and its utilization of glucose, the kidney can also influence glucose homeostasis by returning glucose to the circulation via the reabsorption of glucose from glomerular filtrate. Normally, approximately 180 l of plasma are filtered by the kidneys each day. As the average plasma glucose concentration throughout a 24-h period is ∼5.5 mmol/l (100 mg/dl), ∼180 g of glucose is filtered by the kidneys each day. In healthy individuals, virtually all of this is reabsorbed into the circulation and the urine is essentially free from glucose. To put this into perspective, in a given day, the kidneys produce 15–55 g glucose via gluconeogenesis and metabolize 25–35 g glucose. Therefore, in terms of glucose economy, it is clear that renal reabsorption is the primary mechanism by which the kidney influences glucose homeostasis. Alterations in renal tubular glucose reabsorption may therefore be expected to have a considerable impact on glucose homeostasis.
Reabsorption of glucose from glomerular filtrate occurs by means of sodium–glucose co-transporters (SGLTs) in the proximal convoluted tubulae. There are six members of this family (Table 4) 26. In animal models, approximately 90% of glucose is reabsorbed by SGLT2, a high-capacity low-affinity glucose transporter (Km ∼10 mmol/l; Vmax ∼10 nmol/(min mg) protein 27). SGLT2 is thought to be located exclusively on the luminal surface of the epithelial cells lining the S1 and S2 segments of the proximal tubule 28,29. Transport of sodium and glucose by SGLT2 occurs in a 1:1 ratio 27,30. The remaining ∼10% of glucose reabsorption is mediated by SGLT1, a high-affinity, low-capacity glucose/galactose transporter (Km ∼0.2 mmol/l; Vmax ∼10 nmol/(min mg) protein; sodium:glucose coupling ratio = 2:1) located on the luminal surface of epithelial cells lining the S3 segment of the proximal tubule 27,30. SGLT1 is also extensively expressed in the small intestine and in other tissues 30. Glucose reabsorbed from the proximal tubules by SGLTs is then released into the circulation through the action of facilitative glucose transporters (GLUTs) at the basolateral membrane of the epithelial cells lining the proximal tubules (GLUT2 in the S1/2 segments and GLUT1 in the S3 segment) 31. SGLT-mediated glucose transport is an active process, moving glucose against a concentration gradient, utilizing energy derived from the sodium electrochemical potential gradient across the brush border membrane and maintained by the transport of intracellular sodium into the blood via sodium:potassium adenosine triphosphatase (ATPase) pumps at the basolateral membrane 26. In contrast, GLUTs facilitate passive transport (equilibration) of glucose across membranes and do not require an energy source 26.
Table 4.
The sodium glucose co-transporter family 26
| Co-transporter | Gene | Substrate | Tissue distribution |
|---|---|---|---|
| SGLT1 | SLC5A1 | Glucose, galactose | Intestine, trachea, kidney, heart, brain, testis, prostate |
| SGLT2 | SLC5A2 | Glucose | Kidney, brain, liver, thyroid, muscle and heart |
| SGLT4 | SLC5A9 | Glucose, mannose | Intestine, kidney, liver, brain, lung, trachea, uterus, pancreas |
| SGLT5 | SLC5A10 | Not known | Kidney |
| SGLT6 | SMIT2/SLC5A11 | Glucose, myo-inositol | Brain, kidney, intestine |
| SMIT1 | SLC5A3 | Glucose, myo-inositol | Brain, heart, kidney, lung |
Figure 1 describes the renal handling of filtered glucose 32. Glucose is freely filtered in the glomerulus, so that, as plasma glucose levels increase, the amount of glucose in the glomerular filtrate increases linearly. Reabsorption of filtered glucose also increases linearly until the maximal reabsorptive capacity is exceeded. This is often referred to as the renal threshold and equates to a filtration rate of 260–350 mg/min per 1.73 m2 33, which occurs at plasma glucose concentrations of 11.0 mmol/l in healthy adults 34. Above this plasma glucose concentration, the percentage of filtered glucose that is reabsorbed decreases and the percentage of the filtered load of glucose that is excreted in the urine increases, resulting in glucosuria. The ‘rounding’ of the titration curve seen around the transition from complete reabsorption to urinary excretion of excess glucose (shown in Fig. 1 as ‘splay’) can be accounted for by heterogeneity in the glomerular filtration rate and glucose reabsorptive capacity of different individual nephrons 32.
Fig 1.
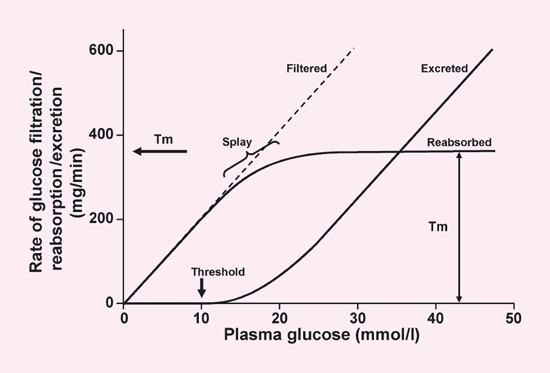
Renal glucose handling. Tm, transport maximum. Adapted with permission from Silverman & Turner (1992) 32. Copyright © 1992 by the American Physiological Society. By permission of Oxford University Press Inc.
The renal threshold for glucose is decreased in individuals with a rare condition known as familial renal glucosuria (FRG), caused by a range of mutations to the SLC5A2 gene, which encodes SGLT2 35. Depending on the nature of the mutations, these individuals have varying degrees of glucosuria, but in the most severe form (so-called ‘Type 0’ disease) they can lose > 100 g glucose per day to the urine 35. Interestingly, the large majority of patients exhibit no symptoms and their condition is only identified incidentally. Typically, they do not become hypoglycaemic or dehydrated and have no electrolyte imbalance or increased risk of urinary tract infections 35. Even the most severe form of the condition appears to carry a favourable prognosis 36 (although it should be noted that only small numbers of patients have been described in the literature). In contrast, patients with SGLT1 gene mutations have low levels of glucosuria but suffer from glucose–galactose malabsorption in the gut, which can be associated with life-threatening severe diarrhoea and dehydration unless a glucose- and galactose-free diet is carefully followed 37.
The kidney in diabetes mellitus
All of the ways in which the kidney normally affects glucose homeostasis are altered in patients with diabetes mellitus.
Renal gluconeogenesis in the post-absorptive state
Consistent with numerous studies in diabetic animal models 38–44, patients with T2DM have an increased release of glucose into the circulation by the kidney in the fasting state 45. Although the liver is commonly viewed as being largely responsible for increased release of glucose into the circulation in T2DM, the absolute increase in renal glucose release is comparable in magnitude (2.60 and 2.21 μmol/(kg min) for liver and kidneys, respectively; P = 0.26) 45. In fact, the relative increase in renal gluconeogenesis is substantially greater than the increase in hepatic gluconeogenesis (300 vs. 30%). Similar to the liver, the increased glucose release by the kidney in the fasting state is solely, if not exclusively, a result of gluconeogenesis 45.
Postprandial renal glucose release
After meal ingestion, renal glucose release increases to a greater extent in people with T2DM than in people with normal glucose tolerance 46. Meyer et al. (2004) found that over a 4.5-h period following ingestion of 75 g glucose, systemic glucose appearance was significantly greater in patients with T2DM than in normal individuals (100.0 ± 6.3 vs. 70.0 ± 3.3 g; P < 0.001). Much of this difference was as a result of an increase in endogenous glucose release because the systemic appearance of ingested glucose was only 7 g greater in the diabetic patients. Forty per cent of the increased endogenous glucose release was because of increased renal glucose release. This was primarily a result of impaired suppression of endogenous glucose release and to a lesser extent of reduced initial splanchnic sequestration of ingested glucose. Considering renal glucose release is regulated by insulin 11, this effect is not unexpected in diabetic patients for whom postprandial insulin release is reduced and insulin resistance is present. Other possible explanations could include a ‘hangover’ of the increased renal gluconeogenesis seen in the post-absorptive state 45, high FFA levels seen in patients with T2DM (because FFAs are known to stimulate renal and hepatic gluconeogenesis in animal models) 47,48 and an increased availability of gluconeogenic precursors seen in patients with T2DM 21.
Renal glucose uptake
In addition to increased glucose production, renal glucose uptake is increased in both the post-absorptive and postprandial states in patients with T2DM 45,46. Meyer et al. (1998) showed that, in the post-absorptive state, renal glucose uptake is significantly greater in patients with T2DM than in normal individuals (353 ± 48 vs. 103 ± 10 μmol/min; P < 0.001), actually exceeding increased glucose production to result in a net glucose uptake of 92 μmol/min. This contrasts with a net output of 21 μmol/min in non-diabetic individuals 45. In the postprandial state, uptake of glucose by tissues is increased in patients with T2DM and its distribution and fate are altered 46. Glucose uptake by the kidneys is raised by more than twofold [21.0 ± 3.5 vs. 9.8 ± 2.3 g in diabetic vs. non-diabetic individuals (P < 0.03) during a 4.5-h period following ingestion of 75 g of glucose 46], whereas glucose uptake in muscle is not significantly altered 46. Moreover, less glucose is oxidized 46.
Renal glucose reabsorption
It is well recognized that glucosuria in diabetic patients does not occur at plasma glucose levels that would normally produce glucosuria in non-diabetic individuals 49. This is the result of increased glucose reabsorbtion from glomerular filtrate in people with diabetes mellitus. The transport maximum (Tm) for glucose is increased and glucosuria only begins to occur at higher than normal plasma glucose levels. In one study, the Tm increased from approximately 350 mg/min in normal individuals to approximately 420 mg/min in those with diabetes mellitus 49. Studies of renal cells isolated from the urine of people with diabetes as well as cells from several animal models have demonstrated enhanced expression of SGLT2 transporters 50,51. Hyperglycaemia, albumin and angiotensin II have all been reported to up-regulate expression of SGLT2 in T2DM 50. The role of altered renal glucose reabsorption in the pathogenesis of diabetic nephropathy is unclear.
Therapeutic implications
Inhibitors of SGLT2 are currently undergoing clinical trials in patients with T2DM as a novel means of reducing hyperglycaemia. Phlorizin, a non-specific SGLT1/2 inhibitor first isolated from the root bark of the apple tree in 1835 52, was found to increase glucosuria and reduce hyperglycaemia and normalize insulin sensitivity in a partial pancreatectomized animal model of T2DM 53. However, it has not been developed as a treatment for diabetes because of a number of practical shortcomings. It is non-selective and inhibits SLGT1 at the intestinal brush border, which is responsible for absorption of dietary glucose 26. Inhibition of SGLT1, therefore, has the potential to result in glucose–galactose malabsorption and thus diarrhoea, as occurs in naturally occurring SGLT1 deficiency 54. Furthermore, phlorizin is poorly absorbed in the intestine and is readily hydrolysed to phloretin, a compound that blocks the facilitative glucose transporter, GLUT1. This might lead to interference with glucose uptake in a number of tissues 55. Highly-specific inhibitors of SGLT2 would overcome some of these shortcomings. However, this approach to lowering hyperglycaemia in T2DM has a number of attractions. For example, unlike the insulin secretagogues [e.g. sulphonylureas, glinides, glucagon-like peptide 1 (GLP-1) analogues and dipeptidyl peptidase-4 (DPP-4) inhibitors] and insulin sensitizers (metformin and thiazolidinediones), the action of SGLT2 inhibitors should not be dependent on pancreatic B-cell function, which deteriorates over time. The insulin-independence of their action may also mean that hypoglycaemic episodes are less likely. Furthermore, the glucosuric effects of these drugs mean that they may not cause weight gain and may even cause weight loss. Finally, SGLT2 inhibitors have an osmotic diuretic effect that may be beneficial for patients with elevated blood pressure. Theoretical safety and tolerability concerns include polyuria, electrolyte imbalance, urinary tract infection, genital fungal infection and impairment in renal function, although it is interesting to note that patients with FRG do not suffer from these adverse effects 35,36. Clinical trials are underway to assess clinical efficacy and safety of several SGLT2 inhibitors 56.
Competing interests
JEG discloses the following: Advisory Boards/Consultant for Amylin, AstraZeneca, Bristol-Myers Squibb, Centocor, Eisai, Elixir, Forest Laboratories, GlaxoSmithKline, Johnson & Johnson, Kowa, Lifescan, MannKind Corporation, Merck, Novartis, Novo Nordisk, Pfizer, Sankyo, Sanofi-Aventis, and Sitris; Speaker’s Bureau for GlaxoSmithKline, Lifescan, Merck, Novartis, Novo Nordisk, Pfizer, Sanofi-Aventis; Grants/Clinical Trials from/for Boehringer Ingelheim, Bristol-Myers Squibb, GlaxoSmithKline, Kowa, Novartis, Novo Nordisk, Pfizer, Sankyo, Sanofi-Aventis, Takeda.
Acknowledgments
Editorial support was provided by Mark Davies of Wolters Kluwer Health and was funded by Bristol-Myers Squibb and AstraZeneca. The author is solely responsible for the content of this manuscript.
Glossary
- FFA
free fatty acid
- FRG
familial renal glucosuria
- GLUT
glucose transporter
- SGLT1
sodium–glucose co-transporter 1
- SGLT2
sodium glucose co-transporter 2
- SGLTs
sodium–glucose co-transporters
- T2DM
Type 2 diabetes mellitus
- Tm
transport maximum
References
- 1.Bergman H, Drury DR. The relationship of kidney function to the glucose utilization of the extra abdominal tissues. Am J Physiol. 1938;124:279–284. [Google Scholar]
- 2.Rizza RA, Gerich JE, Haymond MW, Westland RE, Hall LD, Clemens AH, et al. Control of blood sugar in insulin-dependent diabetes: comparison of an artificial endocrine pancreas, continuous subcutaneous insulin infusion, and intensified conventional insulin therapy. N Engl J Med. 1980;303:1313–1318. doi: 10.1056/NEJM198012043032301. [DOI] [PubMed] [Google Scholar]
- 3.Wahren J, Felig P, Hagenfeldt L. Physical exercise and fuel homeostasis in diabetes mellitus. Diabetologia. 1978;14:213–222. doi: 10.1007/BF01219419. [DOI] [PubMed] [Google Scholar]
- 4.Consoli A, Kennedy F, Miles J, Gerich J. Determination of Krebs cycle metabolic carbon exchange in vivo and its use to estimate the individual contributions of gluconeogenesis and glycogenolysis to overall glucose output in man. J Clin Invest. 1987;80:1303–1310. doi: 10.1172/JCI113206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerich JE. Control of glycaemia. Baillieres Clin Endocrinol Metab. 1993;7:551–586. doi: 10.1016/s0950-351x(05)80207-1. [DOI] [PubMed] [Google Scholar]
- 6.UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33) Lancet. 1998;352:837–853. [PubMed] [Google Scholar]
- 7.Holman RR, Paul SK, Bethel MA, Neil HA, Matthews DR. Long-term follow-up after tight control of blood pressure in type 2 diabetes. N Engl J Med. 2008;359:1565–1576. doi: 10.1056/NEJMoa0806359. [DOI] [PubMed] [Google Scholar]
- 8.The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 9.Nathan DM, Cleary PA, Backlund JY, Genuth SM, Lachin JM, Orchard TJ, et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005;353:2643–2653. doi: 10.1056/NEJMoa052187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerich JE. Physiology of glucose homeostasis. Diabetes Obes Metab. 2000;2:345–350. doi: 10.1046/j.1463-1326.2000.00085.x. [DOI] [PubMed] [Google Scholar]
- 11.Meyer C, Dostou J, Nadkarni V, Gerich J. Effects of physiological hyperinsulinemia on systemic, renal, and hepatic substrate metabolism. Am J Physiol. 1998;275:F915–F921. doi: 10.1152/ajprenal.1998.275.6.F915. [DOI] [PubMed] [Google Scholar]
- 12.Stumvoll M, Meyer C, Kreider M, Perriello G, Gerich J. Effects of glucagon on renal and hepatic glutamine gluconeogenesis in normal postabsorptive humans. Metabolism. 1998;47:1227–1232. doi: 10.1016/s0026-0495(98)90328-6. [DOI] [PubMed] [Google Scholar]
- 13.Landau BR, Wahren J, Chandramouli V, Schumann WC, Ekberg K, Kalhan SC. Contributions of gluconeogenesis to glucose production in the fasted state. J Clin Invest. 1996;98:378–385. doi: 10.1172/JCI118803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stumvoll M, Meyer C, Mitrakou A, Nadkarni V, Gerich JE. Renal glucose production and utilization: new aspects in humans. Diabetologia. 1997;40:749–757. doi: 10.1007/s001250050745. [DOI] [PubMed] [Google Scholar]
- 15.Davidson MB, Peters AL. An overview of metformin in the treatment of type 2 diabetes mellitus. Am J Med. 1997;102:99–110. doi: 10.1016/s0002-9343(96)00353-1. [DOI] [PubMed] [Google Scholar]
- 16.Meyer C, Stumvoll M, Dostou J, Welle S, Haymond M, Gerich J. Renal substrate exchange and gluconeogenesis in normal postabsorptive humans. Am J Physiol Endocrinol Metab. 2002;282:E428–E434. doi: 10.1152/ajpendo.00116.2001. [DOI] [PubMed] [Google Scholar]
- 17.Stumvoll M, Meyer C, Perriello G, Kreider M, Welle S, Gerich J. Human kidney and liver gluconeogenesis: evidence for organ substrate selectivity. Am J Physiol. 1998;274:E817–E826. doi: 10.1152/ajpendo.1998.274.5.E817. [DOI] [PubMed] [Google Scholar]
- 18.Cersosimo E, Garlick P, Ferretti J. Insulin regulation of renal glucose metabolism in humans. Am J Physiol. 1999;276:E78–E84. doi: 10.1152/ajpendo.1999.276.1.E78. [DOI] [PubMed] [Google Scholar]
- 19.Meyer C, Stumvoll M, Welle S, Woerle HJ, Haymond M, Gerich J. Relative importance of liver, kidney, and substrates in epinephrine-induced increased gluconeogenesis in humans. Am J Physiol Endocrinol Metab. 2003;285:E819–E826. doi: 10.1152/ajpendo.00145.2003. [DOI] [PubMed] [Google Scholar]
- 20.Stumvoll M, Chintalapudi U, Perriello G, Welle S, Gutierrez O, Gerich J. Uptake and release of glucose by the human kidney. Postabsorptive rates and responses to epinephrine. J Clin Invest. 1995;96:2528–2533. doi: 10.1172/JCI118314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meyer C, Dostou JM, Welle SL, Gerich JE. Role of human liver, kidney, and skeletal muscle in postprandial glucose homeostasis. Am J Physiol Endocrinol Metab. 2002;282:E419–E427. doi: 10.1152/ajpendo.00032.2001. [DOI] [PubMed] [Google Scholar]
- 22.Meyer C, Dostou JM, Gerich JE. Role of the human kidney in glucose counterregulation. Diabetes. 1999;48:943–948. doi: 10.2337/diabetes.48.5.943. [DOI] [PubMed] [Google Scholar]
- 23.Joseph SE, Heaton N, Potter D, Pernet A, Umpleby MA, Amiel SA. Renal glucose production compensates for the liver during the anhepatic phase of liver transplantation. Diabetes. 2000;49:450–456. doi: 10.2337/diabetes.49.3.450. [DOI] [PubMed] [Google Scholar]
- 24.Gerich JE. Hepatorenal glucose reciprocity in physiologic and pathologic conditions. Diabetes Nutr Metab. 2002;15:298–302. [PubMed] [Google Scholar]
- 25.Brenner BM. Brenner & Rector’s The Kidney. 7th edn. Philadelphia: W.B. Saunders Company; 2004. [Google Scholar]
- 26.Wright EM, Hirayama BA, Loo DF. Active sugar transport in health and disease. J Intern Med. 2007;261:32–43. doi: 10.1111/j.1365-2796.2006.01746.x. [DOI] [PubMed] [Google Scholar]
- 27.Brown GK. Glucose transporters: structure, function and consequences of deficiency. J Inherit Metab Dis. 2000;23:237–246. doi: 10.1023/a:1005632012591. [DOI] [PubMed] [Google Scholar]
- 28.Lee YJ, Han HJ. Regulatory mechanisms of Na(+)/glucose cotransporters in renal proximal tubule cells. Kidney Int Suppl. 2007;106:S27–S35. doi: 10.1038/sj.ki.5002383. [DOI] [PubMed] [Google Scholar]
- 29.Chen J, Feder J, Neuhaus I, Whaley JM. Tissue expression profiling of the sodium–glucose co-transporter (SGLT) family: implication for targeting SGLT2 in type 2 diabetes patients (2493-PO) Diabetes. 2008;57:A682. [Google Scholar]
- 30.Wright EM. Renal Na(+)-glucose cotransporters. Am J Physiol Renal Physiol. 2001;280:F10–F18. doi: 10.1152/ajprenal.2001.280.1.F10. [DOI] [PubMed] [Google Scholar]
- 31.Hediger MA, Rhoads DB. Molecular physiology of sodium-glucose cotransporters. Physiol Rev. 1994;74:993–1026. doi: 10.1152/physrev.1994.74.4.993. [DOI] [PubMed] [Google Scholar]
- 32.Silverman M, Turner JR. Glucose transport in the renal proximal tubule. In: Windhager EE, editor. Handbook of Physiology. New York: Oxford University Press; 1992. pp. 2017–2038. [Google Scholar]
- 33.Zelikovic I. Aminoaciduria and glycosuria. In: Avner ED, Harmon WE, Niaudet P, editors. Pediatric Nephrology. 5th edn. Lippincott Williams & Wilkins; 2004. pp. 701–728. . In:, eds.. Philadelphia: [Google Scholar]
- 34.Moe OW, Wright SH, Palacin M. Renal handling of organic solutes. In: Brenner BM, editor. Brenner & Rector’s The Kidney. 8th edn. Saunders Elsevier; 2008. pp. 214–247. . In:, ed.. Philadelphia: [Google Scholar]
- 35.Santer R, Kinner M, Lassen CL, Schneppenheim R, Eggert P, Bald M, et al. Molecular analysis of the SGLT2 gene in patients with renal glucosuria. J Am Soc Nephrol. 2003;14:2873–2882. doi: 10.1097/01.asn.0000092790.89332.d2. [DOI] [PubMed] [Google Scholar]
- 36.Scholl-Burgi S, Santer R, Ehrich JH. Long-term outcome of renal glucosuria type 0: the original patient and his natural history. Nephrol Dial Transplant. 2004;19:2394–2396. doi: 10.1093/ndt/gfh366. [DOI] [PubMed] [Google Scholar]
- 37.Turk E, Zabel B, Mundlos S, Dyer J, Wright EM. Glucose/galactose malabsorption caused by a defect in the Na+/glucose cotransporter. Nature. 1991;350:354–356. doi: 10.1038/350354a0. [DOI] [PubMed] [Google Scholar]
- 38.Teng CT. Studies on carbohydrate metabolism in rat kidney slices. II. Effect of alloxan diabetes and insulin administration on glucose uptake and glucose formation. Arch Biochem Biophys. 1954;48:415–423. doi: 10.1016/0003-9861(54)90358-6. [DOI] [PubMed] [Google Scholar]
- 39.Landau BR. Gluconeogenesis and pyruvate metabolism in rat kidney, in vitro. Endocrinology. 1960;67:744–751. doi: 10.1210/endo-67-6-744. [DOI] [PubMed] [Google Scholar]
- 40.Flinn RB, Leboeuf B, Cahill GF., Jr Metabolism of C14-labeled substrates in kidney cortical slices from normal and alloxan-diabetic rats. Am J Physiol. 1961;200:508–510. doi: 10.1152/ajplegacy.1961.200.3.508. [DOI] [PubMed] [Google Scholar]
- 41.Joseph PK, Subrahmanyam K. Effect of growth hormone, insulin, thyroxine and cortisone on renal gluconeogenesis. Arch Biochem Biophys. 1968;127:288–291. doi: 10.1016/0003-9861(68)90228-2. [DOI] [PubMed] [Google Scholar]
- 42.Kamm DE, Cahill GF., Jr Effect of acid-base status on renal and hepatic gluconeogenesis in diabetes and fasting. Am J Physiol. 1969;216:1207–1212. doi: 10.1152/ajplegacy.1969.216.5.1207. [DOI] [PubMed] [Google Scholar]
- 43.Chang AY, Schneider DI. Rate of gluconeogenesis and levels of gluconeogenic enzymes in liver and kidney of diabetic and normal Chinese hamsters. Biochim Biophys Acta. 1970;222:587–592. doi: 10.1016/0304-4165(70)90184-4. [DOI] [PubMed] [Google Scholar]
- 44.Triscari J, Stern JS, Johnson PR, Sullivan AC. Carbohydrate metabolism in lean and obese Zucker rats. Metabolism. 1979;28:183–189. doi: 10.1016/0026-0495(79)90084-2. [DOI] [PubMed] [Google Scholar]
- 45.Meyer C, Stumvoll M, Nadkarni V, Dostou J, Mitrakou A, Gerich J. Abnormal renal and hepatic glucose metabolism in type 2 diabetes mellitus. J Clin Invest. 1998;102:619–624. doi: 10.1172/JCI2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meyer C, Woerle HJ, Dostou JM, Welle SL, Gerich JE. Abnormal renal, hepatic, and muscle glucose metabolism following glucose ingestion in type 2 diabetes. Am J Physiol Endocrinol Metab. 2004;287:E1049–E1056. doi: 10.1152/ajpendo.00041.2004. [DOI] [PubMed] [Google Scholar]
- 47.Krebs HA, Speake RN, Hems R. Acceleration of renal gluconeogenesis by ketone bodies and fatty acids. Biochem J. 1965;94:712–720. doi: 10.1042/bj0940712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Williamson JR. Mechanism for the stimulation in vivo of hepatic gluconeogenesis by glucagon. Biochem J. 1966;101:11C–14C. doi: 10.1042/bj1010011c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mogensen CE. Maximum tubular reabsorption capacity for glucose and renal hemodynamcis during rapid hypertonic glucose infusion in normal and diabetic subjects. Scand J Clin Lab Invest. 1971;28:101–109. doi: 10.3109/00365517109090668. [DOI] [PubMed] [Google Scholar]
- 50.Rahmoune H, Thompson PW, Ward JM, Smith CD, Hong G, Brown J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes. 2005;54:3427–3434. doi: 10.2337/diabetes.54.12.3427. [DOI] [PubMed] [Google Scholar]
- 51.Vestri S, Okamoto MM, De Freitas HS, Aparecida Dos Santos R, Nunes MT, Morimatsu M, et al. Changes in sodium or glucose filtration rate modulate expression of glucose transporters in renal proximal tubular cells of rat. J Membr Biol. 2001;182:105–112. doi: 10.1007/s00232-001-0036-y. [DOI] [PubMed] [Google Scholar]
- 52.Ehrenkranz JR, Lewis NG, Kahn CR, Roth J. Phlorizin: a review. Diabetes Metab Res Rev. 2005;21:31–38. doi: 10.1002/dmrr.532. [DOI] [PubMed] [Google Scholar]
- 53.Rossetti L, Smith D, Shulman GI, Papachristou D, DeFronzo RA. Correction of hyperglycemia with phlorizin normalizes tissue sensitivity to insulin in diabetic rats. J Clin Invest. 1987;79:1510–1515. doi: 10.1172/JCI112981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wright EM. I. Glucose galactose malabsorption. Am J Physiol. 1998;275:G879–G882. doi: 10.1152/ajpgi.1998.275.5.G879. [DOI] [PubMed] [Google Scholar]
- 55.Isaji M. Sodium–glucose cotransporter inhibitors for diabetes. Curr Opin Investig Drugs. 2007;8:285–292. [PubMed] [Google Scholar]
- 56.List JF, Woo V, Morales E, Tang W, Fiedorek FT. Sodium–glucose co-transport inhibition with dapagliflozin in Type 2 diabetes. Diabetes Care. 2009;32:650–657. doi: 10.2337/dc08-1863. [Epub ahead of print 29 December 2008] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Woerle HJ, Meyer C, Dostou JM, Gosmanov NR, Islam N, Popa E, et al. Pathways for glucose disposal after meal ingestion in humans. Am J Physiol Endocrinol Metab. 2003;284:E716–E725. doi: 10.1152/ajpendo.00365.2002. [DOI] [PubMed] [Google Scholar]