

Abstract
Improvements in childhood and adolescent cancer mortality and survival rates have continued in recent years, but the rate of progress has been greater in hematologic malignances than in solid tumors. A short reflection on these achievements is warranted before quickly turning our attention to the remaining challenges.
In this issue of Cancer, Smith et al report on mortality rates for children and adolescents with cancer from 1975 to 2010.1 They focused on the most recent period between 2002 and 2010 and demonstrated a rate of decline in mortality similar to that observed from 1975 to 1998 after an apparent plateau between 1998 and 2002. An annual percentage change (APC) in mortality rate of 2.4% was observed across both adolescents and children, with the most significant reductions reported in acute leukemias, lymphomas, neuroblastoma, gonadal cancers, and some central nervous system tumors. Increased survival rather than any change in incidence appears to explain the decreased mortality. The authors estimated that 45,000 cancer deaths have been avoided between 1975 and 2010 because of this sustained improvement.
The main strength of the article is the 35-year duration of mortality data from a significant proportion of the United States derived from the US Centers for Disease Control and Prevention. Survival estimates were derived from the Surveillance, Epidemiology, and End Results (SEER) 9 registries. Similar population-based analyses of mortality and survival for European children have been reported recently, allowing us to highlight comparisons that may aid in the interpretation of the findings.2,3 Bosetti et al used a similar joinpoint analysis method to assess trends in mortality rates for European childhood cancer over the period from 1970 to 2007. This demonstrated a continuous decline in mortality rates throughout the period of analysis, with an APC between 2% and 4% for all cancers combined and between 3% and 6% separately for leukemias. Although those authors did not comment on any obvious plateau in the rate of decline, there was considerable geographic variation in mortality and rates of change.
Two important messages emerge from the article by Smith et al. The first is that improvements in childhood cancer mortality and survival rates have continued in recent years, but the rate of progress has been greater in hematologic malignances than in solid tumors. This differential rate of improvement has been observed in both the United States and Europe, and the cancer types that have made the major contribution to increased survival rates are acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), and non-Hodgkin lymphoma (NHL). Here, the survival rates are comparable between the United States and the best of the European geographic regions (eg Northern and Central Europe). Unfortunately, there has been no significant improvement in survival for high-grade glioma and metastatic sarcomas on either continent. However, there are some notable differences, with the US data reporting significant improvements in 5-year relative survival in neuroblastoma and Hodgkin lymphoma that have yet to be observed across Europe.
The second important message is that these improvements in mortality and survival also have been observed in adolescents. Progress in adolescent cancer mortality has been impressive and indeed superior to that of children during the same time frame. This is despite data suggesting that adolescents and young adults did not have the same opportunity of being enrolled onto clinical trials with a possible disadvantage in outcome.4,5 This disparity may be caused in part by a different spectrum of malignancies, eg germ cell tumors, lymphoma, and leukemia, and because these cancers have benefited from survival increases as a result of previous successful clinical trial protocols that have now been adopted as standard therapies, with the result that adolescents and young adults are increasingly being treated either as part of a clinical trial or within units familiar with their administration, eg pediatric ALL protocols. However, it is vital that the opportunity to be enrolled onto suitable cooperative trials is extended to as many malignances as possible that affect teenagers and young adults by facilitating appropriate referral pathways.6
There are some limitations to the study of Smith and colleagues that should be considered when interpreting the data. The SEER 9 registry data cover only 10% of the United States, and it must be asked whether the data presented represent the whole US population. Furthermore, additional new data for the period from 2007 to 2010 has resulted in the emergence of new plateaus in the joinpoint analyses of earlier periods that were not observed in the initial publication, thus casting some doubt on the reproducibility of the findings. However, a similar deceleration in the rate of improvement has also been observed in some European mortality data, particularly in Northern and Western European countries, where the plateau appears to be continuing.2,7 By contrast, the presented US data demonstrate a further improvement in the most recent period studied from 2003 onward. Smith et al interpret these trends in the APC for mortality rates as a reflection of several years in which more effective treatments were not identified or broadly adopted for childhood cancers, but they offer no further explanation why the newly identified plateau spanning the period from 1998 through 2002 differs either from the preceding or subsequent US data or indeed in a comparison with European data. It would have been interesting to explore any correlation between clinical trial activity during the various periods studied and mortality and survival rates, as was published by the UK group.8 Details on cause of death would be valuable to determine whether improvements also may have been related to better supportive care and a reduction in the treatment-related mortality rate, a relevant factor in allowing treatment intensification.
Smith et al also report on improvements in relative survival from the SEER registries, demonstrating an estimated 5-year survival rate for all cancers combined of almost 84% for children and adolescents who were diagnosed between 2003 and 2007 and followed until 2010. This is slightly better than the equivalent rate of 79.1% for all childhood cancers diagnosed during 2005 to 2007 in Europe, as recently published by the EUROCARE-5 Childhood Cancer Survival Study. With participation from 29 countries and 74 population-based cancer registries, the EUROCARE-5 study was able to demonstrate the extent of geographic variation that may be observed, an aspect that has not been addressed in the current analysis of US data. Survival rates in the best performing European regions can match those reported in the United States. Such regional variations in outcomes highlight the importance of ensuring uniform population-level access to best practice. Indeed, efforts to introduce European standards of cancer care for children are in progress.9 However, making visible and then addressing inequalities in outcomes for children and young patients with cancer needs consideration on every continent.10
Over the last decade, concerns have been expressed in high-income countries that clinical trials have almost reached the limits for optimization of conventional surgical, radiotherapy, and cytotoxic-based therapies, with minimal further improvements possible.7 Therefore, it is reassuring that further reductions in mortality rates have been achieved in the United States. It is important to further explore the possible reasons behind these improvements for all ages. Taking ALL as an example, it seems unlikely that the most recent reductions in mortality and increases in survival have been caused by the introduction of novel targeted agents. It is more likely that the optimization of conventional therapies as a result of better patient risk stratification has allowed more accurate selection of high-risk groups for appropriate intensification of conventional therapies with a de-escalation of treatment for selected low-risk groups.
This paradigm of risk stratification using combined clinical, pathologic, and molecular biology data is being used increasingly in some solid tumors (eg neuroblastoma, nephroblastoma, and medulloblastoma), but its application is restricted to those solid tumors in which the clinical significance of the underlying tumor biology is well understood. Progress in this area has been slower and more recent in solid tumors than in hematologic malignancies, perhaps explaining the differential rate of improvement. Another possible cause for mortality reduction and increased overall survival may be the more systematic use at relapse of second-line “salvage” protocols, which, in some diseases, may have improved the 5-year overall survival rate among patients who had relapsed tumors that previously were considered incurable.11,12
In contrast to the continued improvements observed in hematologic malignancies, the lack of any significant reduction in mortality rates among patients with poor-prognostic solid tumors (ie, high-risk brain tumors, metastatic sarcomas, and neuroblastoma) remains a major concern. Deaths from leukemia continue to be the second largest cause of cancer deaths, and the identification of high-risk subgroups and improved salvage therapies remains a priority. Figure 1 illustrates the percentage contribution by category to all cancer deaths in children ages birth to 14 years in all 50 areas covered by population-based cancer registries that contributed data for the years 2000 through 2007 to the European Cancer Observatory. It is apparent from these data that the cause of cancer death in the group aged <15 years is very similar to that reported by Smith et al in the current study. This highlights the need for international collaboration and a refocusing of efforts on developing new treatment approaches to tackle the major causes of childhood cancer deaths if we are to witness further reductions in childhood cancer mortality in the future. The US data suggest that there have been modest improvements in survival among children with neuroblastoma, but it is not clear whether the survival gains demonstrated for high-risk neuroblastoma are a result of the introduction of immunotherapy using the disialoganglioside 2 (GD2)-targeted chimeric monoclonal antibody ch14.18.14 However, as an example of a new cancer therapeutic approach, it is hoped that immunotherapy may be a promising development not only in neuroblastoma but also in other childhood malignancies, such as treatment with chimeric antigen receptor-modified T cells in ALL.15 It is unlikely that the introduction of biologically targeted therapies or molecular-based risk stratification can explain the resumption of a decline in US mortality rates from 2002 onward, because their use is not yet widespread enough to affect mortality rates at the population level. However, it is hoped that the rapid expansion in knowledge of the underlying molecular etiology of pediatric solid and central nervous system tumors can translate into real survival benefits with the commencement of molecular-stratified clinical trials, eg for medulloblastoma.16
Figure 1.
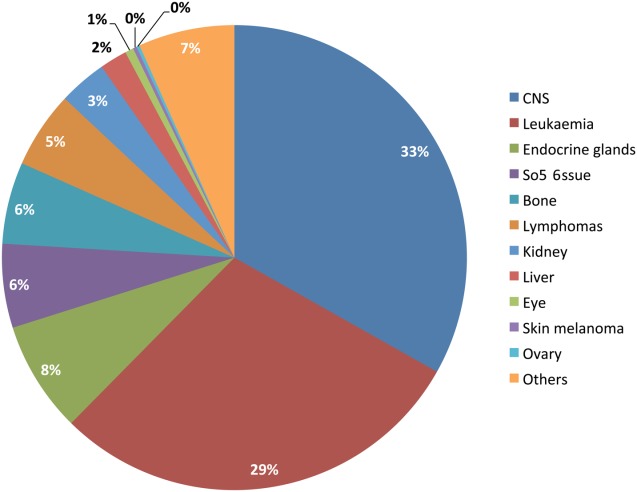
Percentages of known causes among all cancer deaths are illustrated in children (ages 0-14 years) for all 50 areas covered by population-based cancer registries that contributed data for the years 2000 through 2007 to the European Cancer Observatory (N = 6256).13 Causes of deaths are classified according to the International Classification of Diseases 10th edition. Note that endocrine gland tumor deaths are in the majority among patients with neuroblastoma. CNS indicates central nervous system.
In addition to better risk stratification, the objective of large-scale, consortia-based genomic studies is to identify and validate possible targetable mutations in pediatric malignancy. The results to date have identified relatively low frequencies of known actionable mutations, such as ALK or BRAF mutations in childhood cancers.17–19 To effectively study targeted agents against such low-frequency mutations will require close cooperation between academia, industry, and regulatory groups on a global basis because of the rarity of patients harboring these oncogenes.20 What is emerging from the interrogation of the genome, epigenome, and transcriptome of pediatric malignancies is the often distinctive and specific underlying molecular changes of childhood cancers, eg hedgehog pathway activation in some medulloblastomas and histone and chromatin remodeling genes in pediatric malignant and pontine gliomas.21–23 Some of these distinct targets already have novel agents developed against them, eg smoothened inhibitors, and are moving from early to later phase, biomarker-led clinical trials. However, for many pediatric known and emerging targets, there are no current targeted therapies, eg chromosomal translocations in sarcomas and MYCN in neuroblastoma, sarcomas, and medulloblastoma. The need to incentivize drug development for childhood cancer has been recognized; and, in both the United States and Europe, new regulations and legislation have been introduced to engage industry and encourage collaborations with the academic community.20 However, these have been only partially successful, and further adaptations and amendments will be needed to produce a sufficient investment in knowledge and funding to achieve the successful development of possible novel, pediatric-specific therapies.24,25 In addition, it appears that the global community needs to refocus on those tumors in which little progress has been made in the past 3 or 4 decades, such as malignant gliomas and metastatic sarcomas.
The data presented by Smith et al only describe mortality and do not indicate the cause of death or, importantly, the burden of cure on the patient and society. A major priority of pediatric oncology is not only to avoid as many deaths as possible in children with cancer but also to decrease the burden of treatment and to reduce both acute and long-term toxicity. Quality of survivorship is vitally important, and better risk stratification has led to a reduction in some toxic treatments for good-risk patients, eg those with Hodgkin lymphoma and nephroblastoma, by decreasing anthracycline and radiotherapy exposure. We hope that novel targeted agents may decrease long-term side effects by replacing conventional radiotherapy and chemotherapy; however, care needs to be taken, because many targeted pathways play important roles in normal development, and new toxicities and side effects may emerge in the acute and chronic settings. Therefore, it is important that investment is made into whole outcome research to estimate the burden of both existing and new therapies with appropriate long-term, validated outcome measures.
The report by Smith et al adds to data confirming the continued progress in reducing cancer mortality among both children and adolescents, and a short reflection on this achievement is warranted before we quickly turn our attention to the remaining challenges. Now, we need to ensure that every patient achieves the same outcomes wherever they may live, that we reinvigorate our efforts to improve survival in the cancers that kill the most patients, and that we continue to decrease the burden of treatment and improve the quality of survival for those we cure.
FUNDING SUPPORT
KP-J and DH are supported by the National Institute for Health Research Great Ormond Street Hospital UCL Biomedical Research Centre award.
CONFLICT OF INTEREST DISCLOSURES
Dr Hargrave has acted as an advisor for Astra-Zeneca, Boehringer-Ingelheim, GlaxoSmithKline, Merck, Pfizer and Roche-Genentech.
REFERENCES
- 1.Smith MA, Altekruse SF, Adamson PC, Reman GH, Seibel NL. Declining childhood and adolescent cancer mortality. Cancer. 2014;120:2497–2506. doi: 10.1002/cncr.28748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bosetti C, Bertuccio P, Chatenoud L, Negri E, Levi F, La Vecchia C. Childhood cancer mortality in Europe, 1970–2007. Eur J Cancer. 2010;46:384–394. doi: 10.1016/j.ejca.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 3.Gatta G, Botta L, Rolssi S, et al. EUROCARE Working Group. Childhood cancer survival in Europe 1999–2007: results of EUROCARE-5—a population-based study. Lancet Oncol. 2014;15:35–47. doi: 10.1016/S1470-2045(13)70548-5. [DOI] [PubMed] [Google Scholar]
- 4.Fern LA, Whelan JS. Recruitment of adolescents and young adults to cancer clinical trials—international comparisons, barriers, and implications [serial online] Semin Oncol. 2010;37:e1–e8. doi: 10.1053/j.seminoncol.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 5.Bleyer A, Siegel SE, Coccia PF, Stock W, Seibel NL. Children, adolescents, and young adults with leukemia: the empty half of the glass is growing. J Clin Oncol. 2012;30:4037–4038. doi: 10.1200/JCO.2012.44.7466. [DOI] [PubMed] [Google Scholar]
- 6.Kmietowicz Z. Teenagers and young adults with cancer need better access to clinical trials [serial online] BMJ. 2013;346:f1959. doi: 10.1136/bmj.f1959. [DOI] [PubMed] [Google Scholar]
- 7.Pritchard-Jones K, Pieters R, Reaman GH, et al. Sustaining innovation and improvement in the treatment of childhood cancer: lessons from high-income countries [serial online] Lancet Oncol. 2013;14:e95–e103. doi: 10.1016/S1470-2045(13)70010-X. [DOI] [PubMed] [Google Scholar]
- 8.Stiller CA, Kroll ME, Pritchard-Jones K. Population survival from childhood cancer in Britain during 1978–2005 by eras of entry to clinical trials. Ann Oncol. 2012;23:2464–2469. doi: 10.1093/annonc/mds183. [DOI] [PubMed] [Google Scholar]
- 9.Kowalczyk JR, Samardakiewicz M, Fitzgerald E, et al. Towards reducing inequalities: European Standards of Care for Children with Cancer. Eur J Cancer. 2014;50:481–485. doi: 10.1016/j.ejca.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 10.Sullivan R, Kowalczyk JR, Agarwal B, et al. New policies to address the global burden of childhood cancers [serial online] Lancet Oncol. 2013;14:e125–e135. doi: 10.1016/S1470-2045(13)70007-X. [DOI] [PubMed] [Google Scholar]
- 11.Ha TC, Spreafico F, Graf N, et al. An international strategy to determine the role of high dose therapy in recurrent Wilms' tumour. Eur J Cancer. 2013;49:194–210. doi: 10.1016/j.ejca.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 12.Parker C, Waters R, Leighton C, et al. Effect of mitoxantrone on outcome of children with first relapse of acute lymphoblastic leukaemia (ALL R3):an open-label randomised trial. Lancet. 2010;376:2009–2017. doi: 10.1016/S0140-6736(10)62002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.E Steliarova-Foucher, M O'Callaghan, J Ferlay, E Masuyer, D Forman, H Comber, F Bray. European Cancer Observatory: Cancer Incidence, Mortality, Prevalence and Survival in Europe. Version 1.0 (September 2012) European Network of Cancer Registries, International Agency for Research on Cancer. Available at http://eco.iarc.fr. Accessed January 9, 2014.
- 14.Yu AL, Gilman AL, Ozkaynak MF, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363:1324–1334. doi: 10.1056/NEJMoa0911123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ellison DW, Kocak M, Dalton J, et al. Definition of disease-risk stratification groups in childhood medulloblastoma using combined clinical, pathologic, and molecular variables. J Clin Oncol. 2011;29:1400–1407. doi: 10.1200/JCO.2010.30.2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pugh TJ, Morozova O, Attiyeh EF, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013;45:279–284. doi: 10.1038/ng.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J, Wu G, Miller CP, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet. 2013;45:602–612. doi: 10.1038/ng.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shern JF, Chen L, Chmielecki J, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014;4:216–231. doi: 10.1158/2159-8290.CD-13-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vassal G, Zwaan CM, Ashley D, et al. New drugs for children and adolescents with cancer: the need for novel development pathways [serial online] Lancet Oncol. 2013;14:e117–e124. doi: 10.1016/S1470-2045(13)70013-5. [DOI] [PubMed] [Google Scholar]
- 21.Jones DTW, Jager N, Kool M, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488:100–105. doi: 10.1038/nature11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwartzentruber J, Korshunov A, Liu X-Y, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 23.Wu G, Broniscer A, McEachron TA, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44:251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saint-Raymond A, Herold R. Medicines for pediatric oncology: can we overcome the failure to deliver? Expert Rev Clin Pharmacol. 2012;5:493–495. doi: 10.1586/ecp.12.51. [DOI] [PubMed] [Google Scholar]
- 25.Vassal G, Blanc P, Pearson A. Need for change in implementation of paediatric regulation. Lancet Oncol. 2013;14:1156–1157. doi: 10.1016/S1470-2045(13)70467-4. [DOI] [PubMed] [Google Scholar]