

Abstract
Background
A cDNA library made from 2 glioma cell lines, U87MG and T98G, was screened by serological identification of antigens by recombinant cDNA expression (SEREX) using serum from a glioblastoma patient. Elongation factor Tu GTP binding domain containing protein 1 (EFTUD1), which is required for ribosome biogenesis, was identified. A cancer microarray database showed overexpression of EFTUD1 in gliomas, suggesting that EFTUD1 is a candidate molecular target for gliomas.
Methods
EFTUD1 expression in glioma cell lines and glioma tissue was assessed by Western blot, quantitative PCR, and immunohistochemistry. The effect on ribosome biogenesis, cell growth, cell cycle, and induction of apoptosis and autophagy in glioma cells during the downregulation of EFTUD1 was investigated. To reveal the role of autophagy, the autophagy-blocker, chloroquine (CQ), was used in glioma cells downregulating EFTUD1. The effect of combining CQ with EFTUD1 inhibition in glioma cells was analyzed.
Results
EFTUD1 expression in glioma cell lines and tissue was higher than in normal brain tissue. Downregulating EFTUD1 induced G1 cell-cycle arrest and apoptosis, leading to reduced glioma cell proliferation. The mechanism underlying this antitumor effect was impaired ribosome biogenesis via EFTUD1 inhibition. Additionally, protective autophagy was induced by glioma cells as an adaptive response to EFTUD1 inhibition. The antitumor effect induced by the combined treatment was significantly higher than that of either EFTUD1 inhibition or CQ alone.
Conclusion
These results suggest that EFTUD1 represents a novel therapeutic target and that the combination of EFTUD1 inhibition with autophagy blockade may be effective in the treatment of gliomas.
Keywords: autophagy, EFTUD1, glioma, ribosome biogenesis, SEREX
Glioblastoma (GBM) is the most common and malignant type of primary brain tumor. Despite aggressive multimodal treatment with maximal surgical resection followed by temozolomide and radiation, the prognosis for patients with GBM remains poor, with a median survival of 14.6 months and a 3-year survival rate of only 10%.1 The development of new therapies for gliomas is therefore necessary.
We previously isolated several candidate glioma antigens using serological identification of antigens by recombinant cDNA expression (SEREX).2,3 Elongation factor Tu GTP-binding domain containing protein 1 (EFTUD1) was identified as one of the isolated antigens. Serological reactivity against recombinant EFTUD1 protein was not observed in 13 healthy individuals. Thus, EFTUD1 was recognized only by the serum from a GBM patient and not by normal donor serum. Following the primary screening by SEREX, in silico analysis of the Oncomine' cancer microarray database showed overexpression of EFTUD1 in glioma samples compared with matched normal samples (see Supplemental Fig. S1). Here, we focused on EFTUD1 as a candidate molecular target for gliomas.
EFTUD1 belongs to the GTP-binding elongation factor family and plays an important role in ribosome biogenesis and its translational activation (see Supplemental Fig. S2).4–7 In the nucleus/nucleolus, eukaryotic translation initiation factor 6 (eIF6) binds to 60S-ribosome subunits and exports them to the cytoplasm, where they undergo final maturation.4,7–11 In the cytoplasm, EFTUD1 triggers the GTP-dependent release of eIF6 from 60S-ribosome subunits together with Shwachman-Bodian-Diamond syndrome protein (SBDS).4,7,12,13 The release of eIF6 allows 60S-ribosome subunits to join 40S-ribosome subunits, resulting in the formation of actively translating 80S complexes.
Ribosome biogenesis represents a key metabolic requirement in proliferating cells, and its tight regulation is essential for normal cell growth and proliferation.14–21 Consequently, deregulation of this process is thought to contribute to tumor biology.18,21–23 Indeed, overexpression of ribosomal proteins and mutations in genes encoding for proteins that regulate ribosome biogenesis are associated with several cancers.19,20,24 However, the role of EFTUD1 in cancers, including glioma, remains unknown.
Here, we analyzed the expression and function of EFTUD1 in glioma to examine the efficacy as a candidate molecular target.
Materials and Methods
Tissue Samples and Cell Lines
All tumor tissue specimens were obtained from glioma patients who underwent surgery at the Department of Neurosurgery, Keio University School of Medicine. Written informed consent for the study was obtained from all participants, and the study was approved by the local ethical review board of Keio University (No. 12-21-2). Tumors obtained from surgical cases were classified according to the World Health Organization (WHO) criteria. Human adult brain-tissue samples were obtained from Biochain. Human glioblastoma cell lines (U87MG, U251MG, KNS81, T98G, SF126, and KALS-1) were maintained in Dulbecco's modified Eagle's medium (GIBCO) supplemented with 10% fetal bovine serum and antibiotics (50 IU/mL benzyl penicillin G potassium and 100 μg/mL streptomycin sulfate (Meiji) in a humidified incubator at 37°C and 5% CO2. U251MG cells were purchased from the RIKEN Cell Bank. KNS81, SF126, and KALS-1 cells were purchased from the Japanese Collection of Research Bioresources Cell Bank. Short tandem repeat DNA profiling of T98G and U87MG cells, a kind gift from collaborators (Keio University), was performed using the Cell ID System (Promega).
Immunoscreening of the cDNA Library With SEREX and Identification of Glioma Antigen
Total RNA was extracted from cultured glioma cell lines (U87MG and T98G) by the guanidinium thiocyanate method, and poly(A)+ RNA was purified. cDNA was ligated into a lambda ZAP expression vector (Stratagene), and a lambda-phage cDNA library was obtained through in vitro packaging. Immunoscreening for the detection of reactive clones was performed with serum from a glioma patient. E. coli transfected with recombinant lambda-ZAP phages were plated at a density of 104 plaque-forming units per 150-mm plate. After nitrocellulose membrane presoaked with 10 mM isopropyl β-D-thiogalactoside was placed onto plates, the membrane was incubated with the absorbed serum at a 1:100 dilution. The membrane was further incubated with alkaline phosphatase-conjugated goat antihuman IgG secondary antibodies, and reactive phage plaques were visualized with 5-bromo-4-chloro-3-indolyl-phosphate and nitroblue tetrazolium. Positive clones were subcloned twice and screened for purification. After amplification of cDNA inserts, sequencing of the cDNA fragments was performed using the BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems) and an automated DNA sequencer (ABI-PRISM 310 Genetic Analyzer; PerkinElmer). Comparison of DNA homology was performed with BLAST software on the GenBank database.
Plasmid Construction and Transfection
EFTUD1 full-length cDNA was subcloned into a pcDNA3myc vector to generate the pcDNA3myc/EFTUD1 construct. Monkey kidney fibroblast cell line COS-7 cells were transfected with a pcDNA3myc/EFTUD1 plasmid using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol.
Preparation of Polyclonal Antibody Against EFTUD1
The full cDNA of EFTUD1 was subcloned into a pMAL plasmid vector (New England Biolabs), and maltose-binding protein (MBP)-tagged protein was expressed in E. coli. The recombinant EFTUD1 protein was purified using an amylose resin column (New England Biolabs). The polyclonal antibody against EFTUD1 was made by immunizing rabbits against the recombinant EFTUD1 protein.
Western Blot Analysis
Cell lysates were prepared using a radio immunoprecipitation assay (RIPA) buffer (25 mM Tris–HCl, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, and 0.1% sodium dodecyl sulfate; pH 7.6) containing protease inhibitors (Cocktail Tablet; Roche Diagnostics). The protein concentration of each sample was determined using a Bio-Rad protein assay kit (Bio-Rad). Identical amounts of proteins were electrophoresed in 10% or Any kD Mini-PROTEAN TGX Precast Gel (Bio-Rad) and transferred to a nitrocellulose membrane. Blots were incubated with either a rabbit EFTUD1 antibody (1:500), a mouse anti-β-actin antibody (1:4,000; Sigma), a rabbit anti-p62 antibody (1:5,000; Sigma), a horseradish peroxidase (HRP)-conjugated anti-p53 (DO-1) antibody (1:5,000; Santa Cruz Biotechnology), a rabbit anti-p21(C-19) antibody (1:1,000; Santa Cruz Biotechnology), or a rabbit anti-LC3 antibody (1:2,000; PM036; MBL). The blots were incubated with HRP-conjugated secondary antibodies (1:20 000; anti-rabbit; MBL International; anti-mouse; GE Health Care Biosciences), and were then exposed to x-ray films.
Quantitative Polymerase Chain Reaction Analysis
cDNA was synthesized from 10 μg total RNA using reverse transcriptase XL (AMV; Takara Bio). The primers were designed as follows: for EFTUD1, forward primer, 5′-CTAGTATTCAGCCATTGGG-3′, and reverse primer, 5′-CTTGGTTCTCAGAGTCAG-3′; for glyceraldehyde 3-phosphate dehydrogenase (GAPDH), forward primer, 5′-CCCACTCCTCCACCTT TGAC-3′, and reverse primer, 5′-ATGAGGTCCACCACCCTGTT-3′. Quantitative reverse-transcriptase PCR analysis was performed using SYBR Green (PerkinElmer) and the ABI prism 7700 Sequence Detection System (PerkinElmer). The threshold-cycle value was defined as the value obtained in the PCR cycle when the fluorescence signal increased above the background threshold.
Immunohistochemical Staining
Paraffin-embedded tissue sections (5 μm) were deparaffinized in xylene and rehydrated. The sections were treated with a heat-based antigen retrieval method using a citrate solution (pH 7.0; 0.01 M). Nonspecific binding of antibodies was blocked by incubation in 2.5% normal horse serum (ImmPRESS reagent kit; Vector Laboratories) for 60 minutes. The slides were then incubated with a rabbit anti-human EFTUD1 polyclonal antibody (1:100) diluted with 0.02% bovine serum albumin in 0.01 M phosphate-buffered saline overnight at 4°C in a humidified box. The slides were then incubated with a secondary antibody conjugated with polymers of horseradish peroxidase (ImmPRESSTM reagent kit; anti-rabbit; Vector Laboratories) for 60 minutes at 37°C. The sections were developed with 3,3′-diaminobenzidene and then lightly counterstained with Mayer's hematoxylin.
siRNA/Lentivirus-mediated shRNA Gene Knockdown
Two short interference RNA (siRNA) oligonucleotide sequences for EFTUD1 were used: siRNA1 (sense strand, 5′-GCAGUCAUACACCAAAUGAAA-3′; antisense strand, 5′ -UCAUUUGGUGUAUGACUGCAA-3′) and siRNA2 (sense strand, 5′- CUAUGAAAUCCAGUGCCAUTT-3′; antisense strand, 5′- AUGGCACUGGAUUUCAUAGTT-3′). Predesigned RNAi (sense strand, 5′-GUACCGCACGUC AUUCGUAUC-30; antisense strand, 5′-UACGAAUGAC GUGCGGUACGU-3′) was used as a control siRNA. The final concentration of 20 nM siRNA was incubated with Lipofectamine RNAiMax (Invitrogen) according to the manufacturer's instructions.
Recombinant lentiviruses were produced by cotransfecting either control lentiviral vector (TRC2; Sigma) or validated EFTUD1 targeting shRNA vector (shEFTUD1-1; TRCN000023650, shEFTUD1-2; TRCN0000236501; Sigma) with 2 second-generation packaging plasmid psPAX2 (Addgene) and envelope plasmid pMD2G (Addgene), into 293T cells. The gene knockdown efficiency of the EFTUD1 targeting shRNA vector in U87MG cells was examined by quantitative RT-PCR.
Immunofluorescent Cell Staining
U87MG and U251MG cells were seeded on chamber slides (Nunc) at a density of 5 × 103/well. After siRNA transfection, cells were fixed, permeabilized, blocked, and incubated with either a rabbit anti-EFTUD1 antibody (1:500) or a rabbit anti-eIF6 antibody (1:100; Cell Signaling Technology). The cells were then incubated with Alexa-Fluor anti-rabbit IgG. Finally, they were treated with 4’,6-diamidino-2-phenylindole (DAPI) for 5 minutes for nuclear staining. The slides were washed, mounted, and imaged using fluorescence microscopy (Axioplan 2 imaging; Carl Zeiss).
Cell Viability Assay
Glioma cells were plated in 96-well plates at a seeding density of 3 × 104 cells/well. In the combined therapy (siRNA and chloroquine (CQ), CQ (50 μM; Sigma) was added 24 hours before analysis. Cell viability assay was analyzed 2, 4, and 6 days after transfection using the Cell Titer-Glo Luminescent Cell Viability assay kit (Promega) according to the manufacturer's protocol with a luminometer (Wallac ARVO 1420 multilabel counter; Wallac Oy). Results are presented as fold relative to the cell viability in control cells (mock-treated or control siRNA-treated cells) at day 0.
Flow Cytometry Assay
Cell-cycle analysis by DNA content was performed using flow cytometry. siRNA-transfected cells with or without nocodazole (final 0.1 μg/mL; Sigma) for 12 hours were stained with propidium iodide for 30 minutes at 37°C according to the manufacturer's protocol and then subjected to flow cytometry (Gallios; Beckman Coulter).
Clonogenicity Assay
U87MG cells were transfected with control siRNA and EFTUD1 siRNAs. Combined treatment (siRNA and CQ) included 3 hours of CQ (50 μM) administration beginning 24 hours after transfection. Subsequently, both CQ-treated and untreated cells (2 × 104) were mixed with 2 mL of culture medium containing 0.4% agar and 10% fetal calf serum (FCS) and then plated on 2 ml of the bottom layer containing 0.6% agar with 10% FCS in each well of a 6-well plate. Each experiment was performed in triplicate. After culturing for 4 weeks, colonies were counted after staining with MTT 3-(4,5-dimethyl-2-thia- zolyl)-2,5-diphenyl-2H-tetrazolium bromide.
Xenograft Tumor Models
For subcutaneous implantation, 12 six-week-old male BALB/c nude mice (Sippr-bk) were randomly divided into a control group (n = 6) or EFTDU1 knockdown group (n = 6). Mice received 106 U87 MG cells infected with lentivirus expressing either control shRNA or shRNA EFTDU1-1 (1 day after infection) injected into the right flank. Tumor length and width were measured with calipers, and measurements were performed at regular intervals. Animals were euthanized when tumors grew to nearly 2 cm in length. Tumor volume was calculated using the formula: volume = (length × width2)/0.5. Gene silencing of EFTUD1 in tumor samples obtained from euthanized animals was confirmed by quantitative real-time PCR analysis.
Apoptosis Assay
Caspase-3/7 activity in siRNA-transfected cells was measured using the Caspase-Glo 3/7 assay kit (Promega). U87MG and U251MG glioma cells were plated in 96-well plates at a seeding density of 5 × 103 cells/well and transfected by siRNAs after the 24-hour incubation. CQ (50 μM) was added 24 hours before analysis in the combined-treatment (siRNA and CQ) condition. Caspase-3/7 activity was measured 6 days after siRNA transfection. In parallel, signals of viable cells treated in a similar manner were measured by the Cell Viability Assay. The value of caspase-3/7 activity was normalized according to signals from viable cells. Relative caspase-3/7 activity was presented as fold relative to this normalized value found in control siRNA-transfected cells. Every experiment was performed in triplicate.
In Silico Analysis
Data mining and analysis of EFTUD1 mRNA expression were performed using some datasets from the Oncomine database. Details of the standardized normalization techniques and statistical calculations can be found on the Oncomine website (https://www.oncomine.com).
Statistical Analysis
Differences between groups in xenografted tumor models were analyzed using a Mann-Whitney U Test. Data are presented as the mean ± standard error of the mean (SEM). All other statistical analyses were performed with 1-way ANOVA or Student' t test. Data are presented as the mean ± standard deviation (SD). Differences were considered statistically significant at P < .05.
Results
Identification of EFTUD1 as a Glioma Antigen and It's Expression Analysis in Gliomas
To identify human glioma antigens, a cDNA library made from 2 human glioma cell lines, U87MG and T98G, was screened by SEREX with serum from a GBM patient (a 48-year-old male). Four different positive clones were identified by SEREX screening (not shown). Sequence analysis revealed that one of the isolated clones was EFTUD1. EFTUD1 was recognized exclusively only by the serum from the GBM patient but not by normal donor serum. Following the primary screening by SEREX, the expression analysis of EFTUD1 in gliomas was performed using in silico microarray data. EFTUD1-expression profiles from normal brain and malignant glioma (WHO grade III or IV) were analyzed in Oncomine. Significantly high expression of EFTUD1 in gliomas was confirmed in Oncomine analysis of 3 datasets (see Supplemental Fig. S1). These results indicated that EFTUD1 may be a candidate molecular target for gliomas.
We analyzed the expression of EFTUD1 in glioma cell lines and glioma tissue. Western blot analysis was performed against a positive control (lysate from pcDNA3myc/EFTUD1-transfected COS7), a negative control (lysate from COS7 cells), 6 glioma cell lines (U87MG, U251MG, T98G, KNS81, SF126, and KALS-1), 10 glioma tissues, and 2 normal brain tissues (Fig. 1A). EFTUD1 protein was highly expressed in all 6 glioma cell lines and 10 glioma tissue samples but was rarely expressed in the 2 normal brain tissues. Quantitative PCR analysis using a cDNA panel containing 5 glioma cell lines (U87MG, U251MG, T98G, KNS81, and SF126) and 10 glioma tissues also revealed that the expression of EFTUD1 was higher in gliomas than in normal brain tissue (Fig. 1B). Although we investigated the expression of EFTUD1 in a total of 31 glioma samples (Fig.1B and Supplementary Fig. S3), no apparent correlation was observed between EFTUD1 expression and the WHO grade of gliomas.
Fig. 1.

High expression of EFTUD1 in glioma cell lines and tissue. (A) Western blot analysis of EFTUD1 expression in a positive control, a negative control, 2 normal brain tissues, 6 glioma cell lines (U87MG, U251MG, T98G, KNS81, SF126, and KALS-1), and 10 glioma tissues. Cell lysate of COS-7 cells transfected with pcDNA3myc/EFTUD1 was prepared as a positive control, and COS-7 cells were prepared as a negative control. β-actin loading controls from the same blots are shown in the lower panels. (B) Quantitative PCR analysis of the EFTUD1 gene in glioma cell lines and tissue compared with normal brain tissue. The same glioma cells and tissue samples as Western blot analysis were investigated. Relative EFTUD1 expression level was normalized to GAPDH level in each sample and calculated as the threshold cycle (CT) value in each sample divided by the CT value in the normal brain. Mean ± S.D. (bars) for at least 3 independent experiments are shown.
We also immunohistochemically stained formalin-fixed paraffin-embedded sections of 8 tissues from different GBM patients and normal brain tissue with an EFTUD1 antibody (Fig. 2A). EFTUD1-immunopositive cells were detected in all 8 GBM tissues but not in the normal brain tissue. Expression of EFTUD1 in glioma cells was observed predominantly in the cytoplasm and slightly in the nucleus. The same result was confirmed by immunofluorescent cell staining of U87MG and U251MG glioma cells with an EFTUD1 antibody (Fig. 2B).
Fig. 2.
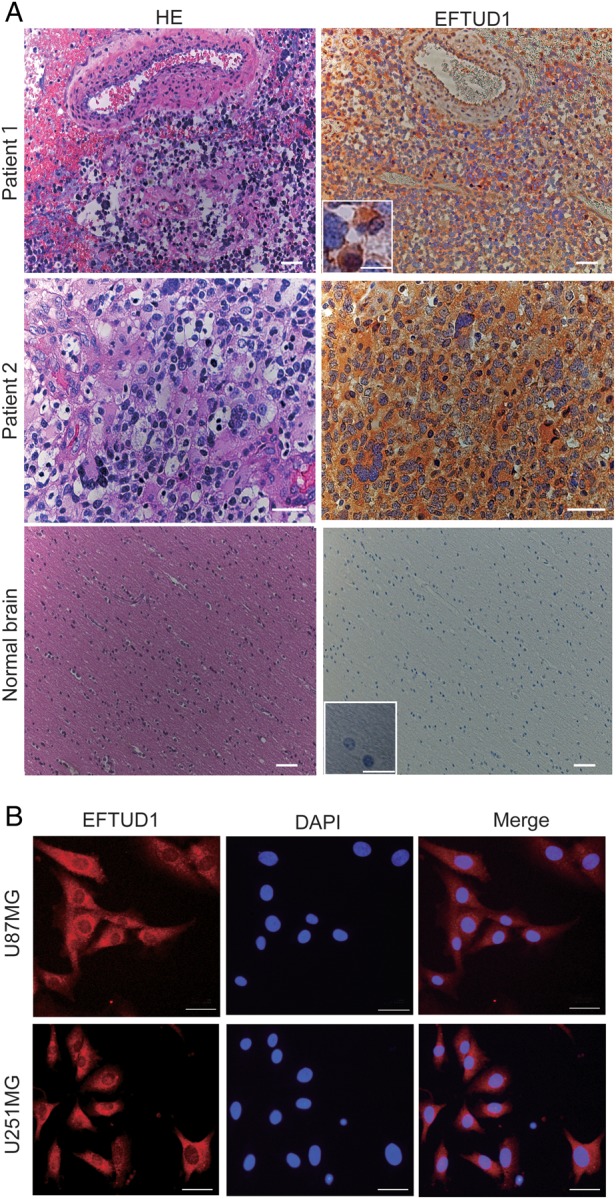
EFTUD1 expression pattern in glioma cells and tissue analyzed by immunohistochemistry (A) and immunofluorescence (B). (A) Immunohistochemical analysis of EFTUD1 expression in glioma tissue and normal brain tissue. Representative immunohistochemical sections of 2 GBM patient tissues (Patient 1, magnification 10×; Patient 2, magnification 40×) and normal brain tissue (magnification 10×) using an EFTUD1 antibody are shown. The sections were counterstained with hematoxylin. Scale bar, 50 μm. The boxed area shows higher magnification images. Scale bar in the boxed area, 10 μm. (B) Immunofluorescent cell staining of glioma cell lines (U87MG and U251MG) using an EFTUD1 antibody and a rhodamine-conjugated secondary antibody. Cell nuclei were stained with DAPI. Scale bar, 50 μm.
Inhibition of Glioma Cell Growth by Downregulation of EFTUD1 In Vitro and In Vivo
U87MG and U251MG glioma cell lines exhibited high expression of EFTUD1 (Fig. 1B) and were therefore selected for further testing. Transfection with 2 different siRNAs targeting EFTUD1 downregulated EFTUD1 expression in these glioma cells at 1 and 6 days after transfection (see Supplemental Fig. S4).
In order to reveal the role of EFTUD1 in glioma cell growth, cell viability during EFTUD1 downregulation was examined using the Cell Titer Glo luminescent cell viability assay. Compared with control cells, EFTUD1 siRNA-treatment significantly suppressed the proliferation of 2 glioma cell lines 6 days after transfection (Fig. 3A). Growth suppression, caused by downregulation of EFTUD1, was also demonstrated in other glioma cell lines with different p53 status (see Supplemental Fig. S5). The clonogenicity assay revealed that the number of colonies formed by EFTUD1 siRNA-transfected U87MG glioma cells was significantly lower than those formed by control glioma cells (Fig. 6B). Additionally, we investigated the effect of EFTUD1 knockdown on U87MG cell growth in a mouse xenograft tumor model using lentivirus-mediated shRNAs (see Supplemental Fig. S6A and B). In the EFTUD1 knockdown group, the growth of U87MG xenograft tumors was significantly suppressed, and gene silencing of EFTUD1 by lentivirus treatment in tumor samples was confirmed in vivo. (see Supplemental Fig. S6C and D). These data indicated that EFTUD1 plays a role in glioma cell growth and cell viability.
Fig. 3.

Inhibition of glioma cell proliferation by EFTUD1 downregulation. (A) Effect of EFTUD1 siRNAs on cell proliferation in U87MG and U251MG glioma cells. Cell viability was analyzed 0, 2, 4, and 6 days after transfection. Results are presented as fold relative to the cell viability in mock-treated cells at day 0. Data are mean ± S.D. (bars) of 3 experiments. **P < 0.01 using 1-way ANOVA. (B) Cell-cycle analysis of EFTUD1 siRNA-transfected glioma cells with and without nocodazol (0.1 mg/mL for 12 h). Cells were fixed with 70% ethanol 4 days after transfection, treated with RNAse, and stained with propidium iodide. Cell-cycle distributions were analyzed by flow cytometry. Data are shown as the percentage of cells in various phases of the cell cycle. (C) Effect of EFTUD1 siRNAs on apoptosis in U87MG and U251MG glioma cells. Caspase 3/7 activity in glioma cells was analyzed 6 days after siRNA transfection. Caspase 3/7 activity was normalized to viable cell signals measured by the CellTiter Glo luminescent cell-viability assay. Relative caspase 3/7 activity is presented as fold relative to this normalized value found in control siRNA-transfected cells. Results are mean ± S.D. (bars) of 3 experiments. *P < .05, **P < .01 using 1-way ANOVA. (D) Western blot analysis of p53 and p21 in U87MG and U251MG 3 days after siRNA transfection. β-Actin was used as a loading control.
Fig. 6.

Antitumor effect by combined inhibition of EFTUD1 with an autophagy blocker. (A) Effect of combined treatment (EFTUD1 siRNAs and CQ) on glioma cell growth. CQ (50 μM) was added to siRNA-transfected cells 24 hours before analysis. Cell viability analysis was performed 0, 2, 4, and 6 days after transfection. Results are presented as fold relative to the cell viability in control siRNA-treated cells at day 0. Data are mean ± S.D. (bars) of 3 experiments. **P < 0.01 using 1-way ANOVA. (B) Effect of combined treatment on clonogenicity in U87MG glioma cells. Twenty-four hours after transfection, cells were treated with CQ (50 μM) for 3 hours (combined treatment) and then plated out to grow on soft agar in 6-well plates at 2 × 104 cells/well. Colonies stained with MTT were counted 4 weeks after siRNA transfection. Results are mean ± S.D. (bars) of 3 experiments. n.s. not significant, *P < .05, **P < .01 using 1-way ANOVA. (C) Effect of combined treatment on apoptosis in U87MG glioma cells. CQ (50 μM) was added 24 hours before analysis (combined treatment). Caspase 3/7 activity in U87MG glioma cells was analyzed 6 days after siRNA transfection. Caspase 3/7 activity was normalized to viable cell signals measured by the CellTiter Glo luminescent cell-viability assay. Relative caspase-3/7 activity is presented as fold relative to this normalized value found in control siRNA-transfected cells. Results are mean ± S.D. (bars) of 3 experiments. n.s. not significant, *P < .05, **P < .01 using 1-way ANOVA.
G1 Arrest and Apoptosis in Glioma Cells by EFTUD1 Downregulation
The cell cycle during EFTUD1 downregulation in U87MG and U251MG cells was examined by DNA content analysis (Fig. 3B). Compared with controls, inhibition of EFTUD1 expression induced an increase in the proportion of G1-phase cells and a corresponding decrease in S-phase cells. To further confirm that EFTUD1 inhibition leads to G1 arrest, we also evaluated the cell-cycle analysis in the additional presence of nocodazol, which has been previously reported to synchronize all cycling cells at metaphase.25 The cell cycle of control glioma cells showed a significant increase in the G2/M phase and a decrease in the G1 phase. In contrast, EFTUD1 siRNA-transfected glioma cells kept a higher peak of G1 phase than G2/M phase, even in the presence of nocodazol. These results clearly indicate that the downregulation of EFTUD1 triggered G1 arrest in the cell cycle of glioma cells.
Caspase 3/7 activity was measured 6 days after siRNA transfection. Compared with controls, EFTUD1 siRNA treatment induced significant apoptosis in U87MG and U251MG glioma cells (Fig. 3C). Additionally, analysis of the p53-p21 axis during EFTUD1 downregulation revealed that the expression of both p53 and p21 was suppressed in EFTUD1 siRNA-transfected glioma cells (Fig. 3D).
Redistribution of eIF6 to Perinuclear Regions and Cytoplasm by Downregulation of EFTUD1
We speculated that the downregulation of EFTUD1 would affect ribosome biogenesis. To address this issue, we analyzed the molecular behavior of eIF6. eIF6 plays an essential role in the nuclear export and cytoplasmic maturation of ribosome subunits and is then recycled to the nucleus10 (see Supplemental Fig. S2). Previous reports demonstrated that mutations in either EFTUD1 or SBDS resulted in retention of eIF6 on nascent subunits and redistribution of eIF6 to the cytoplasm.4,13,26 We therefore investigated the localization of eIF6 during EFTUD1 downregulation. As shown in Fig. 4A, eIF6 was mainly localized in the nucleus (specifically in the nucleolus) of U87MG glioma cells treated with control siRNA. In contrast, U87MG glioma cells treated with EFTUD1 siRNAs for 3 days showed a greater redistribution of eIF6 to the perinuclear regions and cytoplasm (Fig. 4A and B), which is consistent with a previous report13 indicating that EFTUD1 downregulation impaired ribosome biogenesis.
Fig. 4.

Impairment of ribosome biogenesis by EFTUD1downregulation. (A) Immunofluorescent cell staining of eIF6 in U87MG glioma cells treated with EFTUD1 siRNAs 3 days after transfection. U87MG cells treated with control siRNA and EFTUD1 siRNAs were immunoprobed using an eIF6 antibody, followed by fluorescein isothiocyanate-conjugated secondary antibody. Nuclei were stained with DAPI. Scale bar, 50 μm. (B) Quantitative analysis of cells with impaired ribosome biogenesis. The number of cells showing high eIF6 expression in the perinuclear regions of the cytoplasm in the merge pictures was counted visually. Mean ± S.D. (bars) for at least 10 independent random fields are shown. **P < 0.01, using 1-way ANOVA.
Autophagy Induced by Glioma Cells in Response to EFTUD1 Downregulation
Given that ribosome biogenesis was impaired by EFTUD1inhibition, protein synthesis should be also reduced in glioma cells. Eukaryotic cells adapt to starvation and low protein synthesis by digesting their own cytoplasmic materials into amino acids and fatty acids to maintain the cellular energy level, which is a process called autophagy.27,28 Furthermore, various cancers, including glioma, have been shown to induce autophagy in response to nutrient deprivation, low protein synthesis, hypoxia, and anticancer treatments.29–36 To reveal whether autophagy was induced in glioma cells during EFTUD1 downregulation, we investigated the expression of the autophagy-associated proteins, light chain 3 (LC3) and p62 (SQSTM1/sequestosome 1), in U87MG and U251MG glioma cells. During nonselective autophagy, the cytosolic form of LC3-I is converted to its autophagosome membrane-associated form, LC3-II. LC3 is ultimately degraded by autolysosomal enzymes, which results in degradation of the total LC3 amount (LC3-I plus LC3-II).29,37,38 During selective autophagy, autophagic degradation of specific proteins is associated with the degradation of p62.37 Here, autophagy-associated changes after EFTUD1 siRNA transfection were observed in both U87MG and U251MG, specifically as the degradation of total LC3 and p62 (Fig. 5). Compared with controls, another autophagy-associated change, conversion from LC3- I to LC3-II (LC3-II/LC3-I ratio), was also observed during the early period after EFTUD1-siRNAs treatment. Although the increasing expression of LC3-II progressed over time in control siRNA-transfected glioma cells, this could have been due to metabolite accumulation consistent with basal autophagy rather than promotion of autophagy.39 Thus, the glioma cells induced both nonselective and selective autophagy in response to EFTUD1 downregulation.
Fig. 5.

Autophagy induced by glioma cells as an adaptive response to EFTUD1 downregulation. Western blot analysis of autophagy-associated proteins, LC3 (I and II) and p62, in U87MG and U251 glioma cells after siRNA transfection. β-Actin was used as a loading control.
Enhancement of Antitumor Effect by Inhibiting Protective Autophagy in EFTUD1 siRNA- transfected Glioma Cells
Autophagy can play either a cytoprotective or cytocidal role under various stressful conditions.30,40–44 Cytoprotective autophagy is induced by cancer cells as an adaptive response to unfavorable conditions when cells are exposed to hypoxia, ionizing radiation, and chemotherapeutic agents.29–36 On the other hand, a cytocidal role of autophagy has also been proposed to be autophagic cell death because accumulation of autophagosomes was observed in dying cells in response to anticancer therapy.45–47 To reveal the role of autophagy in EFTUD1 siRNA-transfected glioma cells, we investigated how autophagy blockade affected viability of U87MG and U251MG glioma cells treated with EFTUD1 siRNA. An autophagy blocker, CQ, was added to EFTUD1 siRNA-transfected glioma cells 24 hours before cell-viability analysis. As shown in Fig. 6A, compared with single treatment (either CQ or EFTUD1 siRNA alone), combined treatment (EFTUD1 siRNA plus CQ) significantly suppressed the proliferation of 2 glioma cell lines 6 days after transfection, suggesting that the autophagic response to EFTUD1 downregulation is cytoprotective. Treatment with an autophagy blocker against tumor cells, which are surviving by means of protective autophagy, has been shown to be therapeutically effective for several cancers including glioma.29,33,40,48,49 The antitumor effect of combined treatment (EFTUD1 siRNA and CQ) was also assessed in a clonogenicity assay and an apoptosis assay. The addition of CQ resulted in reduced colony formation by siRNA-transfected glioma cells (Fig. 6B). Furthermore, caspase 3/7 activity was measured 6 days after siRNA transfection with CQ. Combined treatment resulted in more apoptosis than either individual treatment alone (CQ or EFTUD1 siRNA alone in U87MG) (Fig. 6C).
Discussion
SEREX has been applied to a variety of human tumor types and has successfully identified novel tumor antigens.50–52 We have previously used SEREX to isolate several candidate glioma antigens including Sox6.2,3 Here, we identified a novel glioma antigen, EFTUD1, that was highly expressed in gliomas but not in normal brain tissue. According to in silico analyses (see Supplemental Fig. S1), EFTUD1 was highly expressed in malignant gliomas. These findings suggest that EFTUD1 might be a therapeutic target for glioma.
EFTUD1 plays a role in ribosome biogenesis and translational activation of ribosomes. The rate of ribosome biogenesis controls cellular growth and proliferation.14,19,20 Ribosome biogenesis is therefore tightly regulated in mammalian cells and is activated in response to extracellular stimuli such as nutrient requests and stress. This tight regulation between extracellular signaling and ribosome biogenesis is disrupted in cancers; cancer cells produce excessive ribosomes, which are necessary for the protein synthesis associated with aberrant cancer growth. Recently, the cross interaction among EFTUD1, SBDS, and eIF6 in ribosome biogenesis has been reported,4–8,10–12,26 and several reports have shown overexpression of eIF6 in various cancers.53,54 Gandin et al reported that eIF6 can affect tumorigenesis by showing that that mouse embryonic fibroblasts from eIF6 heterozygous (eIF6+/−) mice formed fewer transformed colonies in soft agar when infected with oncogene-carrying retroviruses as compared with those from wild-type mice.9 Similarly, Miluzio et al also demonstrated that eIF6 inactivation delayed tumorigenesis and reduced tumor growth in mice.55 These reports suggest that EFTUD1 may also become unregulated in cancers. Here, we demonstrated that EFTUD1 was highly expressed in gliomas and that its downregulation suppressed glioma cell growth both in vitro and in vivo. In contrast, EFTUD1 was rarely expressed in normal brain tissue in this study. However, proliferative cells in the normal brain, such as neural stem and progenitor cells, may display active ribosome biogenesis. It would be important to investigate the effects of EFTUD1 inhibition on these cells in a future study.
Previous reports have shown cross interaction among EFTUD1, SBDS, and eIF6 in ribosome biogenesis.4,5,7,10 Shwachman-Diamond syndrome, which is caused by defects in the SBDS gene, is a ribosomopathy resulting from failed release of eIF6 from 60S-ribosome subunits due to loss in cooperative activity between SBDS and EFTUD1 during ribosome maturation.4 Because Finch et al demonstrated that EFTUD1 inactivation impaired ribosome biogenesis in the same manner as in Shwachman-Diamond syndrome,4 we analyzed the molecular behavior of eIF6 in glioma cells during EFTUD1 inhibition. As the previous papers reported,4,13 we demonstrated the cytoplasmic accumulation of eIF6 in EFTUD1 siRNA-transfected glioma cells indicating that EFTUD1 downregulation impairs ribosome biogenesis.
Chemotherapeutic agents blocking ribosome biogenesis have been shown to induce cell-cycle arrest and apoptosis via ribosomal stress.56–58 Here, EFTUD1 downregulation also induced cell cycle arrest and apoptosis in glioma cells. Although cell-growth suppression due to ribosomal stress often occurs via the p53-dependent pathway,15,16 ribosomal stress via the p53-independent pathway has also been reported.23,59–61 Regardless of p53 status, EFTUD1 inhibition reduced tumor growth in several glioma cell lines with different p53 status. EFTUD1 inhibition also induced cell-cycle arrest and apoptosis in U87MG (p53-wild type) and U251MG (p53-mutant type) glioma cell lines. These results therefore suggested that EFTUD1 inhibition might trigger ribosomal stress via a p53-independent pathway. On the other hand, as shown in Fig. 3, EFTUD1 downregulation decreased the expression of p53 and p21. This result suggested that EFTUD1 inhibition might trigger the loss of p53 stability and subsequent decrease of p21 expression. However, it is unlikely that these results were due to the regulation of upstream signaling to p53 because they were observed in U87MG (p53-wild type) and U251MG (p53-mutant type) glioma cells. We speculated that the decreased expression of p53 and p21 might arise secondarily because of decreased global translation by ribosome biogenesis inhibition. In fact, the mTOR inhibitor (RAD001), which controls ribosome biogenesis and protein synthesis, was shown to decrease p53 and p21 expression because of the inhibition of global translation.62
The induction of autophagy was also demonstrated in EFTUD1 siRNA-transfected glioma cells. Autophagy can play dual roles against cancer treatments. Recently, the therapeutic strategy to modulate autophagy has shown efficacy for various cancers.29,30,40,41,43,44,48,49,63 Thus, understanding the role of autophagy against EFTUD1 inhibition is useful for determining therapeutic strategy. In this study, an autophagy blocker enhanced the growth suppression of glioma cells treated with EFTUD1 siRNA, suggesting that glioma cells induced protective autophagy as an adaptive response to EFTUD1 downregulation. Many studies have shown that protective autophagy contributes to a resistance mechanism in cancer cells treated with conventional DNA-damaging chemotherapy, molecularly targeted therapy, and antiangiogenic therapy.29–31,33,34,41,43 Tumor cells under protective autophagy can be sensitized to anticancer drugs using an autophagy blocker.29,44,45,48–50 Protein-synthesis inhibitors have also been shown to enhance the antitumor effect of treatment when combined with an autophagy blocker in several cancers, including gliomas.49,64,65 These findings provide the molecular rationale for combining an autophagy blocker with EFTUD1 inhibition as the therapeutic strategy. CQ, traditionally used as an antimalarial drug, is the only US Food and Drug Administration-approved autophagy inhibitor. Although CQ was shown to have a concentration-dependent antitumor effect in glioma cells,66 CQ dose level in the present study was not high enough to produce the antitumor effect with a single treatment. Here, we demonstrated that combined therapy with EFTUD1 siRNA and CQ produced a significantly greater antitumor effect than either agent given alone. Thus, EFTUD1 inhibition in combination with autophagy blockade is expected to represent an effective therapeutic strategy for the management of gliomas.
Conclusions
Here, we demonstrated that EFTUD1 is highly expressed in gliomas and plays a role in the cell proliferation of these tumors. Downregulation of EFTUD1 induced cell-cycle arrest and apoptosis in gliomas by impairing ribosome biogenesis. Additionally, protective autophagy was induced by glioma cells as an adaptive response to EFTUD1 downregulation. Combination therapy consisting of EFTUD1 downregulation with an autophagy blocker enhanced the antitumor effect. Although further analysis is required, these findings indicate that EFTUD1 is a promising therapeutic target and that EFTUD1 inhibition with autophagy blockade is an effective therapeutic strategy for gliomas.
Supplementary material
Funding
This research was supported by Japan Society for the Promotion of Science Grants-in-Aid for Scientific Research (B), Grant number 22390283.
Supplementary Material
Acknowledgments
We thank Ms. Y. Aikawa, Ms. N. Tsuzaki, Ms. K. Koide, and Ms. S. Teramoto for technical assistance, and Dr. T. Tsujikawa for database analysis.
Conflict of interest statement: None declared.
References
- 1.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi S, Fusaki N, Ohta S, et al. Downregulation of KIF23 suppresses glioma proliferation. J Neurooncol. 2012;106(3):519–529. doi: 10.1007/s11060-011-0706-2. [DOI] [PubMed] [Google Scholar]
- 3.Ueda R, Iizuka Y, Yoshida K, et al. Identification of a human glioma antigen, SOX6, recognized by patients’ sera. Oncogene. 2004;23(7):1420–1427. doi: 10.1038/sj.onc.1207252. [DOI] [PubMed] [Google Scholar]
- 4.Finch AJ, Hilcenko C, Basse N, et al. Uncoupling of GTP hydrolysis from eIF6 release on the ribosome causes Shwachman-Diamond syndrome. Genes Dev. 2011;25(9):917–929. doi: 10.1101/gad.623011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graindorge JS, Rousselle JC, Senger B, et al. Deletion of EFL1 results in heterogeneity of the 60 S GTPase-associated rRNA conformation. J Mol Biol. 2005;352(2):355–369. doi: 10.1016/j.jmb.2005.07.037. [DOI] [PubMed] [Google Scholar]
- 6.Lo KY, Li Z, Bussiere C, et al. Defining the pathway of cytoplasmic maturation of the 60S ribosomal subunit. Mol Cell. 2010;39(2):196–208. doi: 10.1016/j.molcel.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Panse VG, Johnson AW. Maturation of eukaryotic ribosomes: acquisition of functionality. Trends Biochem Sci. 2010;35(5):260–266. doi: 10.1016/j.tibs.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ceci M, Gaviraghi C, Gorrini C, et al. Release of eIF6 (p27BBP) from the 60S subunit allows 80S ribosome assembly. Nature. 2003;426(6966):579–584. doi: 10.1038/nature02160. [DOI] [PubMed] [Google Scholar]
- 9.Gandin V, Miluzio A, Barbieri AM, et al. Eukaryotic initiation factor 6 is rate-limiting in translation, growth and transformation. Nature. 2008;455(7213):684–688. doi: 10.1038/nature07267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miluzio A, Beugnet A, Volta V, et al. Eukaryotic initiation factor 6 mediates a continuum between 60S ribosome biogenesis and translation. EMBO Rep. 2009;10(5):459–465. doi: 10.1038/embor.2009.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanvito F, Piatti S, Villa A, et al. The beta4 integrin interactor p27(BBP/eIF6) is an essential nuclear matrix protein involved in 60S ribosomal subunit assembly. J Cell Biol. 1999;144(5):823–837. doi: 10.1083/jcb.144.5.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ganapathi KA, Austin KM, Lee CS, et al. The human Shwachman-Diamond syndrome protein, SBDS, associates with ribosomal RNA. Blood. 2007;110(5):1458–1465. doi: 10.1182/blood-2007-02-075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menne TF, Goyenechea B, Sanchez-Puig N, et al. The Shwachman-Bodian-Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat Genet. 2007;39(4):486–495. doi: 10.1038/ng1994. [DOI] [PubMed] [Google Scholar]
- 14.Belin S, Beghin A, Solano-Gonzalez E, et al. Dysregulation of ribosome biogenesis and translational capacity is associated with tumor progression of human breast cancer cells. PLoS One. 2009;4(9):e7147. doi: 10.1371/journal.pone.0007147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chakraborty A, Uechi T, Kenmochi N. Guarding the 'translation apparatus’: defective ribosome biogenesis and the p53 signaling pathway. Wiley Interdiscip Rev RNA. 2011;2(4):507–522. doi: 10.1002/wrna.73. [DOI] [PubMed] [Google Scholar]
- 16.Deisenroth C, Zhang Y. Ribosome biogenesis surveillance: probing the ribosomal protein-Mdm2-p53 pathway. Oncogene. 2010;29(30):4253–4260. doi: 10.1038/onc.2010.189. [DOI] [PubMed] [Google Scholar]
- 17.Sezgin G, Henson AL, Nihrane A, et al. Impaired growth, hematopoietic colony formation, and ribosome maturation in human cells depleted of Shwachman-Diamond syndrome protein SBDS. Pediatr Blood Cancer. 2012;60(2):281–286. doi: 10.1002/pbc.24300. [DOI] [PubMed] [Google Scholar]
- 18.Drygin D, Siddiqui-Jain A, O'Brien S, et al. Anticancer activity of CX-3543: a direct inhibitor of rRNA biogenesis. Cancer Res. 2009;69(19):7653–7661. doi: 10.1158/0008-5472.CAN-09-1304. [DOI] [PubMed] [Google Scholar]
- 19.Montanaro L, Trere D, Derenzini M. Nucleolus, ribosomes, and cancer. Am J Pathol. 2008;173(2):301–310. doi: 10.2353/ajpath.2008.070752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat Rev Cancer. 2003;3(3):179–192. doi: 10.1038/nrc1015. [DOI] [PubMed] [Google Scholar]
- 21.Rujkijyanont P, Watanabe K, Ambekar C, et al. SBDS-deficient cells undergo accelerated apoptosis through the Fas-pathway. Haematologica. 2008;93(3):363–371. doi: 10.3324/haematol.11579. [DOI] [PubMed] [Google Scholar]
- 22.Drygin D, Lin A, Bliesath J, et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011;71(4):1418–1430. doi: 10.1158/0008-5472.CAN-10-1728. [DOI] [PubMed] [Google Scholar]
- 23.Li J, Yu L, Zhang H, et al. Down-regulation of pescadillo inhibits proliferation and tumorigenicity of breast cancer cells. Cancer Sci. 2009;100(12):2255–2260. doi: 10.1111/j.1349-7006.2009.01325.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lai MD, Xu J. Ribosomal proteins and colorectal cancer. Curr Genomics. 2007;8(1):43–49. doi: 10.2174/138920207780076938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arima Y, Nitta M, Kuninaka S, et al. Transcriptional blockade induces p53-dependent apoptosis associated with translocation of p53 to mitochondria. J Biol Chem. 2005;280(19):19166–19176. doi: 10.1074/jbc.M410691200. [DOI] [PubMed] [Google Scholar]
- 26.Wong CC, Traynor D, Basse N, et al. Defective ribosome assembly in Shwachman-Diamond syndrome. Blood. 2011;118(16):4305–4312. doi: 10.1182/blood-2011-06-353938. [DOI] [PubMed] [Google Scholar]
- 27.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6(4):463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 28.Reggiori F, Klionsky DJ. Autophagy in the eukaryotic cell. Eukaryot Cell. 2002;1(1):11–21. doi: 10.1128/EC.01.1.11-21.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu YL, DeLay M, Jahangiri A, et al. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012;72(7):1773–1783. doi: 10.1158/0008-5472.CAN-11-3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu YL, Jahangiri A, Delay M, et al. Tumor cell autophagy as an adaptive response mediating resistance to treatments such as antiangiogenic therapy. Cancer Res. 2012;72(17):4294–4299. doi: 10.1158/0008-5472.CAN-12-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palumbo S, Comincini S. Autophagy and ionizing radiation in tumors: the “survive or not survive” dilemma. J Cell Physiol. 2013;228(1):1–8. doi: 10.1002/jcp.24118. [DOI] [PubMed] [Google Scholar]
- 32.Zois CE, Koukourakis MI. Radiation-induced autophagy in normal and cancer cells: towards novel cytoprotection and radio-sensitization policies? Autophagy. 2009;5(4):442–450. doi: 10.4161/auto.5.4.7667. [DOI] [PubMed] [Google Scholar]
- 33.Fan QW, Cheng C, Hackett C, et al. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci Signal. 2010;3(147):ra81. doi: 10.1126/scisignal.2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fan QW, Weiss WA. Autophagy and Akt promote survival in glioma. Autophagy. 2011;7(5):536–538. doi: 10.4161/auto.7.5.14779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Humbert M, Medova M, Aebersold DM, et al. Protective autophagy is involved in resistance towards MET inhibitors in human gastric adenocarcinoma cells. Biochem Biophys Res Commun. 2013;431(2):264–269. doi: 10.1016/j.bbrc.2012.12.120. [DOI] [PubMed] [Google Scholar]
- 36.Lin JF, Tsai TF, Liao PC, et al. Benzyl isothiocyanate induces protective autophagy in human prostate cancer cells via inhibition of mTOR signaling. Carcinogenesis. 2012;34(2):406–414. doi: 10.1093/carcin/bgs359. [DOI] [PubMed] [Google Scholar]
- 37.Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8(4):445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3(6):542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 39.Wu H, Yang JM, Jin S, et al. Elongation factor-2 kinase regulates autophagy in human glioblastoma cells. Cancer Res. 2006;66(6):3015–3023. doi: 10.1158/0008-5472.CAN-05-1554. [DOI] [PubMed] [Google Scholar]
- 40.Corcelle EA, Puustinen P, Jaattela M. Apoptosis and autophagy: Targeting autophagy signalling in cancer cells –'trick or treats’? FEBS J. 2009;276(21):6084–6096. doi: 10.1111/j.1742-4658.2009.07332.x. [DOI] [PubMed] [Google Scholar]
- 41.Hait WN, Jin S, Yang JM. A matter of life or death (or both): understanding autophagy in cancer. Clin Cancer Res. 2006;12(7 Pt 1):1961–1965. doi: 10.1158/1078-0432.CCR-06-0011. [DOI] [PubMed] [Google Scholar]
- 42.Kondo Y, Kanzawa T, Sawaya R, et al. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5(9):726–734. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- 43.Wu WK, Coffelt SB, Cho CH, et al. The autophagic paradox in cancer therapy. Oncogene. 2012;31(8):939–953. doi: 10.1038/onc.2011.295. [DOI] [PubMed] [Google Scholar]
- 44.Yang ZJ, Chee CE, Huang S, et al. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2011;10(9):1533–1541. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bursch W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 2001;8(6):569–581. doi: 10.1038/sj.cdd.4400852. [DOI] [PubMed] [Google Scholar]
- 46.Lomonaco SL, Finniss S, Xiang C, et al. Cilengitide induces autophagy-mediated cell death in glioma cells. Neuro Oncol. 2011;13(8):857–865. doi: 10.1093/neuonc/nor073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsujimoto Y, Shimizu S. Another way to die: autophagic programmed cell death. Cell Death Differ. 2005;12(Suppl 2):1528–1534. doi: 10.1038/sj.cdd.4401777. [DOI] [PubMed] [Google Scholar]
- 48.Boya P, Gonzalez-Polo RA, Casares N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25(3):1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Loehberg CR, Strissel PL, Dittrich R, et al. Akt and p53 are potential mediators of reduced mammary tumor growth by cloroquine and the mTOR inhibitor RAD001. Biochem Pharmacol. 2012;83(4):480–488. doi: 10.1016/j.bcp.2011.11.022. [DOI] [PubMed] [Google Scholar]
- 50.Forgber M, Trefzer U, Sterry W, et al. Proteome serological determination of tumor-associated antigens in melanoma. PLoS One. 2009;4(4):e5199. doi: 10.1371/journal.pone.0005199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kagaya A, Shimada H, Shiratori T, et al. Identification of a novel SEREX antigen family, ECSA, in esophageal squamous cell carcinoma. Proteome Sci. 2011;9(1):31. doi: 10.1186/1477-5956-9-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miles AK, Rogers A, Li G, et al. Identification of a novel prostate cancer-associated tumor antigen. Prostate. 2007;67(3):274–287. doi: 10.1002/pros.20520. [DOI] [PubMed] [Google Scholar]
- 53.Rosso P, Cortesina G, Sanvito F, et al. Overexpression of p27BBP in head and neck carcinomas and their lymph node metastases. Head Neck. 2004;26(5):408–417. doi: 10.1002/hed.10401. [DOI] [PubMed] [Google Scholar]
- 54.Sanvito F, Vivoli F, Gambini S, et al. Expression of a highly conserved protein, p27BBP, during the progression of human colorectal cancer. Cancer Res. 2000;60(3):510–516. [PubMed] [Google Scholar]
- 55.Miluzio A, Beugnet A, Grosso S, et al. Impairment of cytoplasmic eIF6 activity restricts lymphomagenesis and tumor progression without affecting normal growth. Cancer Cell. 2011;19(6):765–775. doi: 10.1016/j.ccr.2011.04.018. [DOI] [PubMed] [Google Scholar]
- 56.Lindstrom MS, Zhang Y. Ribosomal protein S9 is a novel B23/NPM-binding protein required for normal cell proliferation. J Biol Chem. 2008;283(23):15568–15576. doi: 10.1074/jbc.M801151200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Morgado-Palacin L, Llanos S, Serrano M. Ribosomal stress induces L11- and p53-dependent apoptosis in mouse pluripotent stem cells. Cell Cycle. 2012;11(3):503–510. doi: 10.4161/cc.11.3.19002. [DOI] [PubMed] [Google Scholar]
- 58.Suzuki A, Kogo R, Kawahara K, et al. A new PICTure of nucleolar stress. Cancer Sci. 2012;103(4):632–637. doi: 10.1111/j.1349-7006.2012.02219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yazbeck VY, Buglio D, Georgakis GV, et al. Temsirolimus downregulates p21 without altering cyclin D1 expression and induces autophagy and synergizes with vorinostat in mantle cell lymphoma. Exp Hematol. 2008;36(4):443–450. doi: 10.1016/j.exphem.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 60.Donati G, Brighenti E, Vici M, et al. Selective inhibition of rRNA transcription downregulates E2F-1: a new p53-independent mechanism linking cell growth to cell proliferation. J Cell Sci. 2011;124(Pt 17):3017–3028. doi: 10.1242/jcs.086074. [DOI] [PubMed] [Google Scholar]
- 61.Donati G, Montanaro L, Derenzini M. Ribosome biogenesis and control of cell proliferation: p53 is not alone. Cancer Res. 2012;72(7):1602–1607. doi: 10.1158/0008-5472.CAN-11-3992. [DOI] [PubMed] [Google Scholar]
- 62.Beuvink I, Boulay A, Fumagalli S, et al. The mTOR inhibitor RAD001 sensitizes tumor cells to DNA-damaged induced apoptosis through inhibition of p21 translation. Cell. 2005;120(6):747–759. doi: 10.1016/j.cell.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 63.Yang ZJ, Chee CE, Huang S, et al. Autophagy modulation for cancer therapy. Cancer Biol Ther. 2011;11(2):169–176. doi: 10.4161/cbt.11.2.14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Enzenmuller S, Gonzalez P, Debatin KM, et al. Chloroquine overcomes resistance of lung carcinoma cells to the dual PI3 K/mTOR inhibitor PI103 by lysosome-mediated apoptosis. Anticancer Drugs. 2013;24(1):14–19. doi: 10.1097/CAD.0b013e32835a36db. [DOI] [PubMed] [Google Scholar]
- 65.Seitz C, Hugle M, Cristofanon S, et al. The dual PI3 K/mTOR inhibitor NVP-BEZ235 and chloroquine synergize to trigger apoptosis via mitochondrial-lysosomal cross-talk. Int J Cancer. 2012;132(11):2682–2693. doi: 10.1002/ijc.27935. [DOI] [PubMed] [Google Scholar]
- 66.Kim EL, Wustenberg R, Rubsam A, et al. Chloroquine activates the p53 pathway and induces apoptosis in human glioma cells. Neuro Oncol. 2010;12(4):389–400. doi: 10.1093/neuonc/nop046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.French PJ, Swagemakers SM, Nagel JH, et al. Gene expression profiles associated with treatment response in oligodendrogliomas. Cancer Res. 2005;65(24):11335–11344. doi: 10.1158/0008-5472.CAN-05-1886. [DOI] [PubMed] [Google Scholar]
- 68.Sun L, Hui AM, Su Q, et al. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell. 2006;9(4):287–300. doi: 10.1016/j.ccr.2006.03.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.