

Abstract
Multiple sclerosis (MS) is a chronic inflammatory disease of the CNS. While a broad range of therapeutics effectively reduce the incidence of focal white matter inflammation and plaque formation for patients with relapse-remitting forms of MS, a challenge within the field is to develop therapies that allow for axonal protection and remyelination. In the last decade, growing interest has focused on utilizing neural precursor cells (NPCs) to promote remyelination. To understand how NPCs function in chronic demyelinating environments, several excellent pre-clinical mouse models have been developed. One well accepted model is infection of susceptible mice with neurotropic variants of mouse hepatitis virus (MHV) that undergo chronic demyelination exhibiting clinical and histopathologic similarities to MS patients. Combined with the possibility that an environmental agent such as a virus could trigger MS, the MHV model of demyelination presents a relevant mouse model to assess the therapeutic potential of NPCs transplanted into an environment in which inflammatory-mediated demyelination is established.
Keywords: demyelination, multiple sclerosis, neural precursor cells, neural progenitor cells, remyelination, virus
Neural precursor cells (NPCs) represent heterogeneous, stem-like cells capable of self-renewal and multipotent differentiation potential within the developing and fully mature CNS [1]. Over 20 years ago, pioneering research demonstrated that NPCs could be isolated and cultured ex vivo from mitotically active regions within the embryonic rat and mouse and differentiate into neurons and glia [2,3]. Since then, stringent culturing protocols have been developed, enabling researchers to generate high-purity NPCs safely from various mammalian sources, including both embryonic and adult tissues [4–7]. The uncommitted nature of NPCs makes them a promising therapeutic candidate for the human demyelinating disease multiple sclerosis (MS). Not only do NPCs have the potential to replace damaged or nonfunctional cells within the CNS to promote repair and recovery, but they are also known to secrete immunomodulatory and neurotrophic factors, further expanding the therapeutic potential of the cells [8]. In order to assess the functional roles of engrafted NPCs, it is important to fully understand a broad range of inflammatory niches that may be supportive for NPC survival and function.
The mouse hepatitis virus (MHV) model of demyelination is a relevant MS model that differs from autoimmune-mediated demyelination including experimental autoimmune encephalomyelitis (EAE) as well as glial toxin models, for example, cuprizone, lysolecithin and ethidium bromide [9–12]. Mice infected with neurotrophic variants of MHV mice undergo chronic demyelination that is promoted through effector activity of virus-specific and nonspecific T cells [13,14]. Given the possibility of viral infection in initiating demyelination in humans as well as the fact that numerous neurotrophic viruses exist that are capable of persisting within the CNS, it is important to evaluate the therapeutic potential of engrafted NPCs in the presence of a persistent viral infection that is correlative with chronic neuroinflammation and demyelination [15].
Multiple sclerosis
MS is a complex disease of the CNS that is characterized by heterogeneous pathologies composed of both inflammatory and neurodegenerative components [16]. Although the identification of an etiological trigger of MS remains elusive, disease induction is thought to result from several features including both genetic predisposition and environmental factors, for example, microbial infection [17–22]. The most common histopathological feature at early stages of the disease includes intermittent episodes of acute inflammation within patches of white matter, resulting in demyelination [23]. Myelin is critical for maintaining efficient axonal conduction and oligodendrocytes, the myelin producer and maintainer of axonal health within the CNS, are damaged or destroyed in MS patients. Focal attacks during early disease are generally episodic, varying from 24 h to several weeks in length and are usually followed by near complete recovery of clinical symptoms, a disease course collectively referred to as relapse-remitting MS (RRMS) [24]. Spontaneous remission can be associated with waning inflammation and partial restoration of axonal conductivity due to remyelination [25,26]. Endogenous oligodendrocyte precursor cells (OPCs) are found to be universally dispersed within the human CNS and can be found in high density within some subacute lesions during early stages of MS [27]. Within a subacute demyelinating lesion, perivascular infiltrates composed of activated CD4+ and CD8+ T cells as well as macrophages are thought to act in concert with reactive microglia to release a milieu of proinflammatory factors that lead to oligodendrocyte dysregulation [23,28]. Additionally, clonally expanded class I restricted CD8+ T cells are found in close proximity to demyelinated axons and potentially target myelin epitopes [29]. Remyelination following OPC maturation leads to the formation of shadow plaques, in which patches of remyelinated white matter are composed of disproportionally thin myelin sheaths surrounding axons [27,30–35].
Although RRMS can last throughout the individual's lifespan, approximately 80% of patients with RRMS will develop a progressive disease within two decades following diagnosis, whereas 15% of individuals diagnosed with MS are classified as progressive patients [36]. In general, progressive MS is the latest stage of the disease, characterized by a gradual worsening of symptoms without remission. Severe neurological impairments dramatically reduce the quality of life for the individual, and this is mainly attributed to expanding cortical lesions impacting motor function. Pathologically, there is widespread axonal degeneration and grey matter neuropathy without a significant presence of the adaptive arm of immunity contributing to the immunopathology [37–40]. Rather, diffuse white and gray matter inflammation has been reported, correlating, in part, to global microglial activation as well as the presence of T cells, B cells and myelin-laden macrophages, which are restricted to the borders of preexisting lesions [28,41]. Furthermore, there is an overall failure of OPCs to efficiently remyelinate damaged white and gray matter areas, dramatically reducing the possibility for recovery [26,42].
With the use of preclinical mouse models of demyelination to study MS, researchers have identified both antigenic and cellular components of the disease that are believed to contribute to demyelination. This has led to the development of therapeutic advances that have generated favorable long-term prognoses for patients. Many of these US FDA-approved therapies show potent immunosuppressive properties that act to limit leukocyte infiltration into the brain, thereby reducing relapse events [43]. However, for patients with progressive disease, the therapeutic repertoire and treatment decision making is confounding. This is partially reflected by the reduced efficacy of many drugs whose mode of action is to inhibit leukocyte infiltration to the CNS, rather than target cells implicated in accelerating progressive disease, such as reactive astrocytes, microglia and degenerating neurons. In addition, an intact blood–brain barrier (BBB) during progressive MS may compartmentalize inflammatory leukocytes within the CNS, making many drugs inactive due to their inability to penetrate a reconstituted BBB [44]. Most importantly for patients with progressive MS is the fact that there are no treatment options that act to halt or even slow demyelination and axonal loss.
One therapeutic option to treat progressive MS would be to replenish or rejuvenate the pool of endogenous OPCs that show limited remyelination potential in the later stages of disease. Among the plethora of experimental approaches attempting to promote remyelination and repair, cellular replacement therapies using NPCs have emerged as a clinically relevant area of research. Recent successes demonstrating that bone marrow-derived mesenchymal neural precursor cells derived from mouse and human showed therapeutic potential for promoting recovery in preclinical mouse MS models resulted in FDA-approved Phase I safety-feasibility clinical trials for progressive MS patients using autologous bone marrow-derived mesenchymal neural precursor cells [45–48]. Moreover, transplantation of human fetal-derived NPCs into the frontal lobes of children with Pelizaeus–Merbacher disease, a rare dysmyelinating disorder in children, has revealed measurable gains in motor and/or cognition associated with remyelination [49]. Therefore, CNS delivery of hNPCs is a potentially viable and important approach to treat human demyelinating diseases such as MS.
A viral model of demyelination
Intracranial inoculation of C56BL/6 mice with the neurotrophic JHM strain of mouse hepatitis virus (JHMV) results in the dissemination of viral particles throughout the ventricular system before targeting ependymal cells lining the ventricles [50]. As virus penetrates further into the parenchyma of the brain and spinal cord, oligodendrocytes, astrocytes and microglia are susceptible to infection while neurons are sparred [51]. A robust innate host-defense response to JHM infection soon follows, characterized by the secretion of proinflammatory factors such as IFN-α, IFN-β, IL-1, IL-6, IL-12 and TNF-α [52,53]. Type I interferons have essential roles for protecting the host against JHM infection, as mice deficient in the IFN-α/β receptor show elevated viral load within the CNS and higher mortality, while exogenous treatment of mice with either Type I interferon limits dissemination of virus [54–56]. Furthermore, innate cellular components consisting of neutrophils, macrophages and natural killer (NK) cells respond to JHMV infection by rapidly migrating to the CNS to assist in permeabilizing the BBB [57–60]. BBB permeabilization promotes infiltration of sensitized JHMV-specific CD4+ and CD8+ T cells that can exert potent antiviral effector mechanisms [58,60,61].
Virus-specific CD4+ T cells function as a supporting cell for CD8+ T cells, promoting CD8+ T-cell expansion in the periphery and promoting survival and cytotoxicity within the CNS [62,63]. In addition, CD4+ T cells can control viral spread through their release of IFN-γ, which serves dual roles by inhibiting viral replication within oligodendrocytes and also inducing upregulation of MHC class II expression on microglia [62,64–67]. Depletion of CD4+ T cells alters CD8+ T cell-mediated control of viral replication within the CNS, mainly a result of reduced of IFN-γ expression and elevated CD8+ T-cell apoptosis [62]. Virus-specific CD8+ T cells are the primary cytolytic effector cell within the CNS during JHM infection and their peak accumulation coincides with viral clearance from glia [65,66,68]. ex vivo analysis of immunodominant JHMV-specific CD8+ T cells from JHMV-infected mice reveals elevated expression of IFN-γ and the cytolytic components granzyme B and perforin. In vivo, IFN-γ-mediated upregulation of MHC-I promotes perforin-mediated cytolysis of JHMV-infected astrocytes and microglia [64,68,69], while Fas/FasL and TNFR signaling on infected cells does not appear to contribute to viral clearance [70]. Oligodendrocytes are an initial reservoir for viral replication; however, they appear to be resistant to perforinmediated cytolysis by CD8+ T cells but rather control viral replication through IFN-γ receptor signaling. These findings are supported by Stohlman and colleagues [67], who demonstrated that expression of a dominant negative version of IFNGR on oligodendrocytes results in increased tropism and viral load within oligodendroglia throughout the brain and spinal cord.
Mice that survive acute JHMV infection progress into the immune-mediated chronic demyelinating phase of the disease, with clinical symptoms manifesting as ataxia and partial to complete hind limb paralysis, beginning 1 week following infection and peaking 2–3 weeks p.i. Histological cross sections of spinal cords from mice undergoing JHMV-induced demyelination demonstrate that oligodendrocyte dysfunction and loss of myelin integrity within white matter tracts is closely associated with the presence of both inflammatory leukocytes and presentation of viral antigen via MHC-I and MHC-II, rather than widespread apoptosis and/or necrosis of mature oligodendrocytes [71–73]. Moreover, a paucity of infectious viral particles within the CNS during chronic disease suggests that productive infection of new glial cells does not amplify demyelination. More likely, viral RNA quasispecies present within the CNS of persistently infected mice promote chronic inflammation and demyelination [74–76].
An effective way to visualize and quantify demyelination on a macroscopic scale is via luxol fast blue (LFB) staining. LFB contains a copper phthalocyanine dye that readily binds to lipoproteins found on myelin sheaths, resulting in a blue staining of myelin-rich regions and a lack of staining within demyelinating lesions. LFB analysis of spinal cord sections during persistent JHMV-infection reveals new lesion formation within the anterior funiculus of the spinal cord as early as day 7 p.i. [50]. As the severity neuroinflammation increases, new lesions are often observed within the lateral funiculus and posterior funiculus (Figure 1A) [50]. Additionally, axonopathy within the white matter tracts of the spinal cord is present as observed in the use of the SMI-32 or Bielschowsky's silver impregnation stain and initial observations suggested that this occurred concomitantly with demyelination, whereas axonal degeneration has been argued to precede oligodendrocyte dysregulation in MS [77,78].
Several studies have identified both T cells and macrophages as the main cellular component in inducing demyelination during chronic JHMV infection mice rather than viral-induced lysis of oligodendrocytes. This stems from studies showing that JHMV infection of RAG1 -/- immunodeficient mice (lacking functional T- and B-lymphocytes) results in extensive viral replication within oligodendrocytes, but limited demyelination [72,79]. Moreover, adoptive transfer of JHMV-sensitized splenocytes from wild-type mice into JHMV-infected RAG1-/- mice results in demyelination, while adoptive transfer of enriched CD8+ T cells obtained from JHMV-sensitized IFN-γ-deficient mice into JHMV-infected RAG1 -/- mice resulted in reduced severity of demyelination compared with recipients of IFN-γ-expressing CD8+ T cells, implicating IFN-γ expression from infiltrating CD8+ T cells as an important contributor to oligodendrocyte dysregulation [79]. Further studies utilizing CD4 -/- or CD8 -/- mice demonstrated the importance of both T-cell subsets in contributing to demyelination following JHMV infection [80]. With regard to IFN-γ impacting oligodendrocyte health, numerous reports have demonstrated enhanced sensitivity of OPCs to IFN-γ-induced apoptosis when compared with mature oligodendrocytes, suggesting that IFN-γ can induce cell death on oligodendrocyte-lineage cells depending on their maturation state [81–87]. This is supported by the finding that demyelination is observed within JHMV-infected mice whose selective ablation of the IFN-γ receptor on mature oligodendrocytes [67]. This could partially be explained by IFN-γ secretion by CD8+ T cells promoting the migration and accumulation of activated macrophages/microglia within white matter tracts of JHMV-infected mice [72,79]. Indeed, ultrastructural and immunofluorescence analysis of JHMV-induced demyelinating lesions features macrophages/microglia engulfing myelin near demyelinated axons [88–90]. However, discriminating between these cells types by immunophenotyping and light microscopy remains a challenge to determine the exact role each play in contributing to pathology during chronic neurological disease. More recently, Ransohoff and colleagues [91] have used electron microscopy to show a more pathogenic role for macrophages compared with microglia in EAE model of MS. Epitope spreading and the appearance of autoreactive T cells against host neuroantigens are not thought to contribute to demyelination during chronic JHMV infection. Altogether, these findings suggest that demyelination is multifaceted and numerous factors could contribute to pathology.
Numerous experimental models of CNS injury have demonstrated that endogenous OPCs can respond to ongoing demyelination through proliferation and maturation, restoring depleted pools of mature oligodendrocytes and actively participating in CNS remyelination [92–97]. Following JHMV infection, PDGFR-positive OPCs found within the spinal cord increase sixfold between days 6 and 14 p.i., suggesting that OPCs become mitotically active following the onset of demyelination [98]. Additionally, a rebound in the total frequency of mature oligodendrocytes to preinfection levels by 7 weeks p.i is also observed, presumably due to the maturation of the expanding OPC population [98]. Nevertheless, OPC differentiation does not lead to substantial clinical recovery and full remyelination within persistently infected mice as exposed axons are still detected months later following infection (Figure 1B) [98].
Figure 1. Histological characteristics of JHMV-induced neurologic disease.
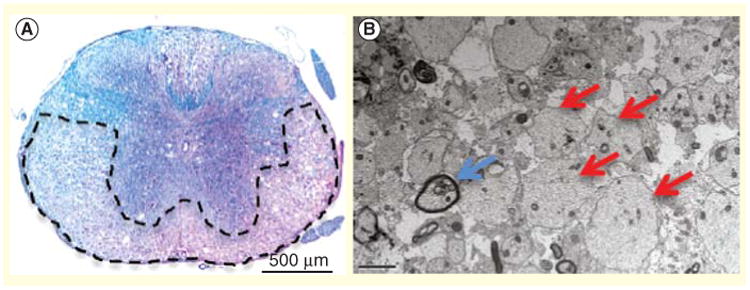
(A) A representative spinal cord was taken from an infected mouse at 5 weeks postinfection and stained with luxol fast blue to determine the extent of demyelination within the white matter tracts. Extensive demyelination is observed throughout the anterior and lateral regions of the white matter. (B) Electron micrograph images (1200×) showing demyelinated axons (red arrows) and remyelinated axons (blue arrow) at 5 weeks postinfection. Reproduced with permission from [119].
Several studies have identified several cytokines, chemokines and growth factors that can impact OPC proliferation and maturation in vivo [96,98–100]. For example, signaling through the CXCR2 chemokine receptor has been shown to enhance OPC proliferation and aid in the directional migration of OPCs within the developing mouse spinal cord, while its inhibition resulted in reduced proliferation and increased maturation in an autoimmune model of demyelination [99–101]. Using the A59 variant of MHV, Armstrong and colleagues [100] demonstrated that PDGF and FGF2 can regulate OPC biology by stimulating proliferation and limiting maturation through activation of the notch signaling pathway. Within the context of JHMV-induced neurological disease, the CXCR4/CXCL12 signaling axis may also play a crucial role in aiding OPC maturation. Indeed, blocking CXCL12's ability to bind to CXCR4 using the small molecule AMD3100 leads to an increase in PDGFRa-positive OPCs and a decrease in mature oligodendrocytes within the spinal cord, suggesting that CXCR4 signaling promotes OPC differentiation. These results are supported and extended by Klein and colleagues [102], who showed that activation of the CXCL12 scavenger receptor CXCR7 as well as CXCR4 results in OPC maturating during cuprizone-induced demyelination.
Neural precursor cell engraftment in MHV-infected mice
To better understand the potential therapeutic roles for NPCs intraspinally engrafted into a demyelinating environment, we chose day 14 post-JHMV infection as animals that survived the acute stage of disease are persistently infected with virus and clinical disease that is congruent with neuroinflammation and demyelination. To study the migratory and functional roles of NPCs without the possibility of rejection, early studies utilized a syngeneic transplant protocol whereby H-2b haplotype-matched mouse striatal NPCs from postnatal day 1 (P1) C56BL/6 mice were transplanted intraspinally into the T8 region of C57BL/6 recipient mice undergoing JHMV-induced demyelination [103]. Initial results demonstrated that transplanted NPCs readily migrated up to 12 mm both rostral and caudal from the transplant site as identified by colocalization of the oligodendrocyte marker CC1 and the proliferation marker Brdu [103]. Quantification of remyelinated axons resulted in up to 67% of axons remyelinated compared with 10% for nontransplanted controls, suggesting that NPCs can survive within the inflammatory niche and functionally incorporate throughout demyelinated white matter tracts following differentiation into mature oligodendrocytes [103]. Additional studies by Carbajal et al. [104] demonstrated that transplanted GFP-NPCs were shown to selectively colonize demyelinated white matter regions within the ventral and lateral funiculus regions of the spinal cord. Positional migration of NPCs was mediated, in part, by responding the CXC chemokine ligand CXCL12 via expression of the receptor CXCR4 expressed upon engrafted NPCs [104].
To determine whether reduction in clinical disease was due to the cells modifying the inflammatory microenvironment to reduce overall inflammation and increase recovery, mice that received mouse P1 NPCs were sacrificed at 10 and 14 days post-transplant and leukocyte infiltration was assessed by flow cytometry [105]. NPC transplantation did not alter the accumulation of T cells or macrophages within the CNS nor proinflammatory chemokine and cytokine gene expression, suggesting that the enhanced remyelination and recovery following transplantation was not a result of NPC bystander effects attenuating the inflammatory response [105].
An important question related to NPC transplantation is whether engrafted cells are capable of directly remyelinating axons or create a ‘nursing’ effect by secreting trophic factors capable of promoting maturation of endogenous OPCs into myelinating oligodendrocytes. We evaluated the contributions of the transcription factor OLIG1 on NPC differentiation and remyelination [106]. Under defined conditions, NPCs preferentially differentiate into oligodendroglia, whereas NPCs isolated from OLIG1-deficient (Olig1-/-) mice exhibit enhanced differentiation into astrocytes. Transplantation of Olig1-/- and Olig1 +/+ NPCs into JHMV-infected mice resulted in similar cell survival, proliferation and selective migration to areas of demyelination. However, only recipients of wild-type NPCs exhibited extensive remyelination compared with mice receiving Olig1-/- NPCs. In vivo characterization of NPCs revealed that Olig1+/+ NPCs preferentially differentiated into NG2-positive OPCs and formed processes expressing myelin basic protein that encircled axons. In contrast, the majority of transplanted Olig1-/- NPCs differentiated into GFAP-positive cells consistent with the astrocyte lineage. These findings reveal that OLIG1 function is required for the remyelination potential of NPCs after transplant, through specification and/or maintenance of oligodendroglial identity. In addition, we have recently employed two-photon microscopy to assess intercellular interactions of transplanted NPCs ex vivo [107]. Within this model, JHMV-infected Thy1-YFP mice, which express yellow fluorescent protein (YFP) from medium-to-large caliber axons within the spinal cord, received SVZ-derived NPCs that express GFP following their differentiation into oligodendrocytes (PLP-GFP). Several important observations were derived from this study including the finding that JHMV-infected Thy1-YFP mice displayed extensive axonal damage earlier than expected during JHMV-induced disease, suggesting that appearance of axonopathy precedes robust immune-mediated demyelination. This argues that axonal damage may be important in contributing to white matter damage and myelin loss. Whether viral infection of neurons and/or transport of viral proteins along axons is important in this process is currently not well defined [78]. In addition, two-photon imaging clearly showed that engrafted NPCs interacted with damaged axons and this resulted in remyelination and improved axonal integrity as determined by YFP expression (Figures 2A–2D) [107].
Figure 2. Axonal protection and remyelination transplantation of mouse neural precursor cells into JHMV-infected mice.
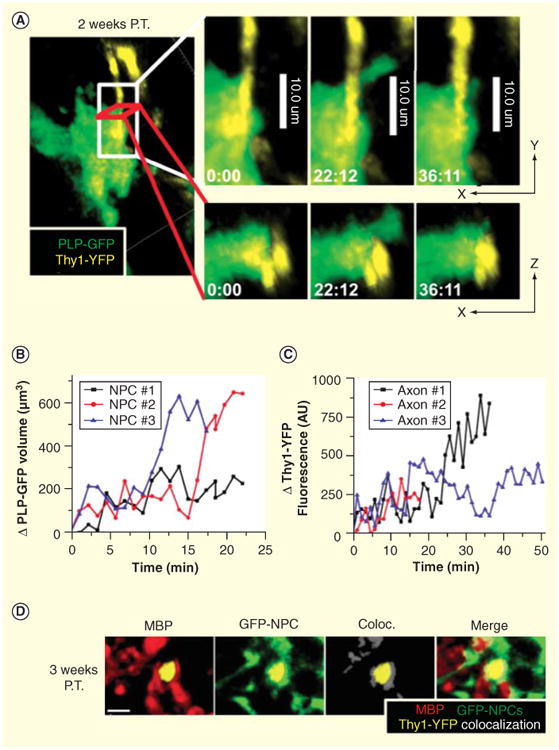
Transplanted NPCs expressing cytoplasmic GFP driven by the myelin proteolipid protein promoter wrap around damaged axons. (A) Representative image showing colocalization between PLP-GFP (green) and damaged axons (yellow) in the JHMV-infected Thy1-YFP spinal cord 15 days post-transfer. Panels on the right show enlarged time lapse images of a PLP-GFP-positive cell wrapping around an axon (minutes:seconds). The top panels depict x–y sections, the lower panels x–z sections. (B) Analysis of the change in volume of PLP-GFP fluorescence of three different NPCs during wrapping as determined by a time lapse 3D reconstruction of the 2P data and the Imaris ‘Surfaces’ tool. (C) Analysis of the change in Thy1-YFP fluorescence intensity during PLP-GFP wrapping presented as arbitrary fluorescent units (AU). (D) Immunostaining in a transverse section of a JHMV-infected Thy1-YFP spinal cord 21 days after GFP-NPC transplantation. MBP (red), YFP+ axons (yellow) and colocalization between overlapping GFP-NPC (green) and MBP fluorescence was determined using the Imaris colocalization tool (white). Merging of all three channels is shown on the right.
Scale bar = 4 μm.
NPC: Neural precursor cell.
Reproduced with permission from [107].
As an additional step to better understand the therapeutic potential of engraftment of NPCs in promoting clinical and histological recovery, we have transplanted MHC-mismatched mouse NPCs into JHMV-infected mice with established demyelination to determine whether allogeneic NPCs are recognized as foreign and rejected via immunological mechanisms. This is clinically relevant as transplantation of human NPCs into patients with Pelizaeus–Merbacher disease required administration of immunosuppressive drugs to limit potential rejection [49]. Similarly, transplantation of human embryonic stem cell (hESC)-OPCs into individuals with spinal cord injuries also was performed in conjunction with administration of immunosuppressive drugs [108,109]. Studies by Palmer and colleagues [110,111] have shown an important role for components of the innate immune response including NK cells in recognizing and rejecting MHC-mismatched NPCs following transplantation into the brains of mice. Similarly, we have demonstrated that engraftment of allogeneic NPCs into spinal cords of JHMV-infected mice results in rejection mediated, in part, by both T-lymphocytes as well as NK cells [112,113]. NPCs respond to both IFN-γ as well as viral infection in terms of expressing MHC class I and II as well as retinoic acid early precursor transcript-1 that allow for T-lymphocyte and NK recognition, respectively [112–114]. Collectively, these findings highlight that NPCs are recognized by cellular components of both the innate and adaptive immune response, indicating administration of immunosuppressive drugs must be considered in order to promote long-term survival and function.
Although syngeneic and allogeneic transplantation of mouse NPCs has generated valuable insights into mechanisms by which NPCs can function in an inflammatory CNS environment, another important research objective is to assess the therapeutic efficacy of engrafting human NPC or OPC-derived cells into JHMV-infected mice. We have previously transplanted high-purity predifferentiated human OPCs in mice undergoing JHMV-induced demyelination that resulted in limited clinical recovery [115]. Engrafted cells were rejected within 2 weeks post-transplantation even in the presence of immunosuppressive drugs targeting activated T-lymphocytes. Histologically, this resulted in only a slight increase in remyelination near the transplant site compared with HBSS transplanted mice [115]. This is in contrast to earlier studies using hESC-derived OPCs in a model of spinal cord injury in rat, where enhanced remyelination and improved motor function were observed following transplantation [116].
Alternatively, human NPCs have previously shown to exert neuroprotective effects in mouse and nonhuman primate models of EAE, suggesting they possess broader plasticity and function in vivo [117,118]. Indeed, we have recently demonstrated that engraftment of hESC-derived NPCs (hNPCs) into JHMV-infected mice with established demyelination resulted in clinical and histological improvement out to 6 months post-transplant although transplanted cells were rejected by day 8 following injection (Figure 3A & 3B) [119]. In contrast to mouse NPCs, the hNPCs neither migrated extensively from the site of injection nor appeared to differentiate into a neural lineage. hNPC-mediated recovery was associated with increased remyelination, but given that hNPCs were rejected within a relatively short period following injection it is unlikely these cells were directly contributing to remyelination [119]. Further, we do not believe remyelination was the result of acute inflammatory-mediated rejection as remyelinated axons were distributed both rostral and caudal to the implantation site rather than localized to the region of cell delivery. Mice transplanted with hNPCs showed reduced infiltration of CD4+ and CD8+ effector T cells into the spinal cords compared with transplant controls, while total numbers of CD4+CD25+FoxP3 regulatory T cells (Tregs) within the spinal cords were elevated (Figure 3C) [119]. Depletion of Tregs in hNPC-transplanted mice via anti-CD25 treatment inhibited the therapeutic benefits, highlighting the potential importance of these cells in contributing to hNPC-mediated recovery (Figure 3D). Cultured hNPCs secreted TGF-β1 and TGF-β2 compared with their undifferentiated hESC counterparts [119]. The roles of TGF- in impacting Treg maintenance and homeostasis are well established as previous work has shown that they promote FoxP3 expression in the peripheral Treg compartment, influencing their frequency and suppressive activity [120]. Therefore, one potential mechanism resulting in the enhanced recovery of these mice is via the immunomodulatory nature of the hNPCs, whereby expression of the anti-inflammatory cytokines TGF-β1 and TGF-β2 act in paracrine manner, influencing the local inflammatory environment within the spinal cord to promote accumulation of Tregs. An important role for Tregs during both acute and chronic JHMV infection has recently been demonstrated. IL-10-expressing virus-specific Tregs dampen proliferation of virus- specific effector CD4+ T cells and depletion of Tregs increases mortality, suggesting that during acute JHMV infection, Tregs limit immunopathological disease without negatively impacting viral clearance [121]. In addition, studies from Trandem et al. [122] have shown that adoptive transfer of Tregs to JHMV-infected mice attenuates clinical disease severity by dampening neuroinflammation and subsequently demyelination. These findings demonstrate the therapeutic potential of hNPCs in promoting sustained recovery through both promoting remyelination while limiting ongoing demyelination through muting neuroinflammation.
Figure 3. Intraspinal transplantation of hNPCs into JHMV-infected mice.
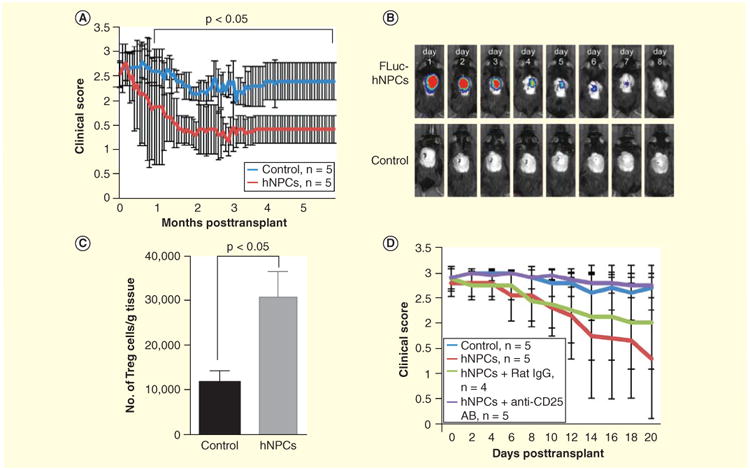
(A) Improved (p < 0.05) clinical recovery in hNPC-transplanted JHMV-infected mice was sustained out to 168 days post-transplantation (p.t.) compared with infected mice treated with vehicle alone. (B) Daily IVIS® imaging of luciferase-labeled hNPCs revealed that following intraspinal transplantation, cells are reduced to below the level of detection by day 8 post-transplantation; representative mice are shown. IVIS imaging was performed on vehicle-transplanted mice as a control. (C) Quantification of Treg numbers in spinal cords of mice indicated a significant (p < 0.05) increase in numbers of Tregs in hNPC-transplanted mice versus controls between 8 and 10 days post-transplantation. Data are representative of three independent experiments with a minimum of three mice per group; data are presented as average ± SEM. Mann-Whitney t tests were used to determine the p-values. (D) hNPC-transplanted mice receiving anti-CD25 antibody (purple line) did not display recovery in motor skills compared with either hNPC-treated mice (red line), hNPC-treated mice receiving isotype-matched control antibody (green line) or vehicle control mice (blue line).
Reproduced with permission from [119].
Expert commentary & five-year view
Mice infected with neurotrophic variants of MHV result in persistent infection that leads to chronic demyelination promoted by virus-specific and nonspecific T cells and macrophages. The histopathological similarities between MHV-infected mice with chronic neurological disease and MS make it an attractive model to understand the functions of NPCs in an inflammatory environment. In this review, we highlight recent advances regarding the use of both mouse and human NPCs in ameliorating the severity of neuroinflammation and demyelination within the context of a viral model of the human demyelinating MS. This article has also provided an overview on the potential of NPCs in promoting remyelination following transplantation. Transplanted mouse NPCs physically bind and remyelinate axons resulting in increased axonal integrity. In contrast, remyelination observed following hNPC transplantation must occur by resident oligodendroglia as hNPCs are rapidly rejected below the level of detection. Furthermore, these studies reveal clear differences based upon the cell type used for therapeutic intervention: while transplantation of both mouse and human NPCs led to remyelination, only treatment with human NPCs resulted in a dramatic reduction in neuroinflammation accompanied by an overall improvement in clinical disease compared with mouse NPC transplantation. These findings demonstrate both the immunomodulatory nature and therapeutic potential of hNPCs. Importantly, studies derived from our viral model of MS illustrate that survival of transplanted cells is not required for restoration of motor skills. These observations indicate that immune-mediated rejection of allogeneic NPCs may not be important and that administration of immunosuppressive drugs may not be necessary to promote long-term recovery. Future research characterizing soluble factors released from hNPCs that impact the remyelination and affecting neuroinflammation will greatly aid in defining the molecular and cellular underpinnings leading to recovery. Moreover, identification of such factors may preclude transplantation of hNPCs as it may be possible to deliver these molecules to promote CNS repair and clinical recovery. Future research will be required to confirm and extend these findings. Additional research will continue to characterize the therapeutic potential of NPCs derived from induced pluripotent stem cells. Advantages of this approach include eliminating ethical issues confronting studies utilizing ESCs, the possibility of limiting allorejection (if this remains important) as hNPCs would be MHC matched to the patient and eliminating the need for lifelong treatment with immunosuppressive drugs and increasing ease in culturing and differentiation. An important unmet clinical need for MS patients is an effective method to induce sustained remyelination and limit immune cell infiltration into the CNS, and this emphasizes the importance of further investigation into the therapeutic potential of hNPCs.
Key issues.
Identify mechanisms by which transplantation neural precursor cells (NPCs) improve clinical outcome through immunomodulation and promoting remyelination in different preclinical animal models of multiple sclerosis.
Continue to define mechanisms by which allogeneic NPCs are recognized as foreign; explore whether components of the innate and/or adaptive immune response participate in rejection.
Employ different preclinical models to confirm whether rejection of allogeneic NPCs affects clinical improvement.
Define the therapeutic benefit of iPSC-derived NPCs in affecting disease progression and remyelination.
Continue to interrogate the most effective route of NPC delivery, that is, intravenous, intrathecal or intraspinal injection.
Identify soluble factor(s) secreted by NPCs that influence both immune responses and glial biology, for example, oligodendrocyte precursor cells maturation.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants R01 NS041249 and R01 NS074987 (TEL) as well as National Multiple Sclerosis Society grant RG 4925 (TEL); NIH grant R01 GM-41514 (MDC); California Institute for Regenerative Medicine (CIRM) grants RM1-01717, CL1-00502 (JFL) and TR3-05603 (JFL).
Footnotes
Financial & competing interests disclosure: The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
References
- 1.Gage FH. Mammalian neural stem cells. Science. 2000;287(5457):1433–8. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- 2.Temple S. Division and differentiation of isolated CNS blast cells in microculture. Nature. 1989;340(6233):471–3. doi: 10.1038/340471a0. [DOI] [PubMed] [Google Scholar]
- 3.Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255(5052):1707–10. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- 4.Brustle O, Jones KN, Learish RD, et al. Embryonic stem cell-derived glial precursors: a source of myelinating transplants. Science. 1999;285(5428):754–6. doi: 10.1126/science.285.5428.754. [DOI] [PubMed] [Google Scholar]
- 5.Barberi T, Klivenyi P, Calingasan NY, et al. Neural subtype specification of fertilization and nuclear transfer embryonic stem cells and application in parkinsonian mice. Nat Biotechnol. 2003;21(10):1200–7. doi: 10.1038/nbt870. [DOI] [PubMed] [Google Scholar]
- 6.Onorati M, Camnasio S, Binetti M, et al. Neuropotent self-renewing neural stem (NS) cells derived from mouse induced pluripotent stem (iPS) cells. Mol Cell Neurosci. 2010;43(3):287–95. doi: 10.1016/j.mcn.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 7.Reubinoff BE, Itsykson P, Turetsky T, et al. Neural progenitors from human embryonic stem cells. Nat Biotechnol. 2001;19(12):1134–40. doi: 10.1038/nbt1201-1134. [DOI] [PubMed] [Google Scholar]
- 8.Kokaia Z, Martino G, Schwartz M, Lindvall O. Cross-talk between neural stem cells and immune cells: the key to better brain repair? Nat Neurosci. 2012;15(8):1078–87. doi: 10.1038/nn.3163. [DOI] [PubMed] [Google Scholar]
- 9.Bergmann CC, Lane TE, Stohlman SA. Coronavirus infection of the central nervous system: host-virus stand-off. Nat Rev Microbiol. 2006;4(2):121–32. doi: 10.1038/nrmicro1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yajima K, Suzuki K. Demyelination and remyelination in the rat central nervous system following ethidium bromide injection. Lab Invest. 1979;41(5):385–92. [PubMed] [Google Scholar]
- 11.Hall SM. The effect of injections of lysophosphatidyl choline into white matter of the adult mouse spinal cord. J Cell Sci. 1972;10(2):535–46. doi: 10.1242/jcs.10.2.535. [DOI] [PubMed] [Google Scholar]
- 12.Matsushima GK, Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001;11(1):107–16. doi: 10.1111/j.1750-3639.2001.tb00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lane TE, Buchmeier MJ. Murine coronavirus infection: a paradigm for virus-induced demyelinating disease. Trends Microbiol. 1997;5(1):9–14. doi: 10.1016/S0966-842X(97)81768-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Templeton SP, Perlman S. Pathogenesis of acute and chronic central nervous system infection with variants of mouse hepatitis virus, strain JHM. Immunol Res. 2007;39(1-3):160–72. doi: 10.1007/s12026-007-0079-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.von Herrath MG, Fujinami RS, Whitton JL. Microorganisms and autoimmunity: making t he barren field fertile? Nat Rev Microbiol. 2003;1(2):151–7. doi: 10.1038/nrmicro754. [DOI] [PubMed] [Google Scholar]
- 16.Steinman L. Immunology of relapse and remission in multiple sclerosis. Annu Rev Immunol. 2014;32:257–81. doi: 10.1146/annurev-immunol-032713-120227. [DOI] [PubMed] [Google Scholar]
- 17.International Multiple Sclerosis Genetics Consortium. Network-based multiple sclerosis pathway analysis with GWAS data from 15,000 cases and 30,000 controls. Am J Hum Genet. 2013;92(6):854–65. doi: 10.1016/j.ajhg.2013.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.International Multiple Sclerosis Genetics Consortium; Wellcome Trust Case Control Consortium. Sawcer S, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476(7359):214–19. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part I: the role of infection. Ann Neurol. 2007;61(4):288–99. doi: 10.1002/ana.21117. [DOI] [PubMed] [Google Scholar]
- 20.Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part II: noninfectious factors. Ann Neurol. 2007;61(6):504–13. doi: 10.1002/ana.21141. [DOI] [PubMed] [Google Scholar]
- 21.Haines JL, Terwedow HA, Burgess K, et al. Linkage of the MHC to familial multiple sclerosis suggests genetic heterogeneity. The Multiple Sclerosis Genetics Group. Hum Mol Genet. 1998;7(8):1229–34. doi: 10.1093/hmg/7.8.1229. [DOI] [PubMed] [Google Scholar]
- 22.Barcellos LF, Sawcer S, Ramsay PP, et al. Heterogeneity at the HLA-DRB1 locus and risk for multiple sclerosis. Hum Mol Genet. 2006;15(18):2813–24. doi: 10.1093/hmg/ddl223. [DOI] [PubMed] [Google Scholar]
- 23.Lassmann H, Bruck W, Lucchinetti CF. The immunopathology of multiple sclerosis: an overview. Brain Pathol. 2007;17(2):210–18. doi: 10.1111/j.1750-3639.2007.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Compston A, Coles A. Multiple sclerosis. Lancet. 2002;359(9313):1221–31. doi: 10.1016/S0140-6736(02)08220-X. [DOI] [PubMed] [Google Scholar]
- 25.Kornek B, Storch MK, Weissert R, et al. Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am J Pathol. 2000;157(1):267–76. doi: 10.1016/S0002-9440(10)64537-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang A, Tourtellotte WW, Rudick R, Trapp BD. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med. 2002;346(3):165–73. doi: 10.1056/NEJMoa010994. [DOI] [PubMed] [Google Scholar]
- 27.Chang A, Nishiyama A, Peterson J, et al. NG2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J Neurosci. 2000;20(17):6404–12. doi: 10.1523/JNEUROSCI.20-17-06404.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Traugott U, Reinherz EL, Raine CS. Multiple sclerosis: distribution of T cell subsets within active chronic lesions. Science. 1983;219(4582):308–10. doi: 10.1126/science.6217550. [DOI] [PubMed] [Google Scholar]
- 29.Neumann H, Medana IM, Bauer J, Lassmann H. Cytotoxic T lymphocytes in autoimmune and degenerative CNS diseases. Trends Neurosci. 2002;25(6):313–19. doi: 10.1016/s0166-2236(02)02154-9. [DOI] [PubMed] [Google Scholar]
- 30.Prineas JW, Kwon EE, Goldenberg PZ, et al. Multiple sclerosis. Oligodendrocyte proliferation and differentiation in fresh lesions. Lab Invest. 1989;61(5):489–503. [PubMed] [Google Scholar]
- 31.Lucchinetti C, Bruck W, Parisi J, et al. A quantitative analysis of oligodendrocytes in multiple sclerosis lesions. A study of 113 cases. Brain. 1999;122(Pt 12):2279–95. doi: 10.1093/brain/122.12.2279. [DOI] [PubMed] [Google Scholar]
- 32.Roy NS, Wang S, Harrison-Restelli C, et al. Identification, isolation, and promoter-defined separation of mitotic oligodendrocyte progenitor cells from the adult human subcortical white matter. J Neurosci. 1999;19(22):9986–95. doi: 10.1523/JNEUROSCI.19-22-09986.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lassmann H. Comparative neuropathology of chronic experimental allergic encephalomyelitis and multiple sclerosis. Schriftenr Neurol. 1983;25:1–135. [PubMed] [Google Scholar]
- 34.Schlesinger H. Zur Frage der akuten multiplen Sklerose und der encephalomyelitis disseminata im Kindesalter. Arb Neurol Inst [Wien] 1909;17:410–32. [Google Scholar]
- 35.Halfpenny C, Benn T, Scolding N. Cell transplantation, myelin repair, and multiple sclerosis. Lancet neurology. 2002;1(1):31–40. doi: 10.1016/s1474-4422(02)00004-2. [DOI] [PubMed] [Google Scholar]
- 36.Kremenchutzky M, Rice GP, Baskerville J, et al. The natural history of multiple sclerosis: a geographically based study 9: observations on the progressive phase of the disease. Brain. 2006;129(Pt 3):584–94. doi: 10.1093/brain/awh721. [DOI] [PubMed] [Google Scholar]
- 37.Antel J, Antel S, Caramanos Z, et al. Primary progressive multiple sclerosis: part of the MS disease spectrum or separate disease entity? Acta Neuropathol. 2012;123(5):627–38. doi: 10.1007/s00401-012-0953-0. [DOI] [PubMed] [Google Scholar]
- 38.Trapp BD, Peterson J, Ransohoff RM, et al. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338(5):278–85. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 39.Peterson JW, Bo L, Mork S, et al. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol. 2001;50(3):389–400. doi: 10.1002/ana.1123. [DOI] [PubMed] [Google Scholar]
- 40.Bjartmar C, Wujek JR, Trapp BD. Axonal loss in the pathology of MS: consequences for understanding the progressive phase of the disease. J Neurol Sci. 2003;206(2):165–71. doi: 10.1016/s0022-510x(02)00069-2. [DOI] [PubMed] [Google Scholar]
- 41.Prineas JW, Wright RG. Macrophages, lymphocytes, and plasma cells in the perivascular compartment in chronic multiple sclerosis. Lab Invest. 1978;38(4):409–21. [PubMed] [Google Scholar]
- 42.Wolswijk G. Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells. J Neurosci. 1998;18(2):601–9. doi: 10.1523/JNEUROSCI.18-02-00601.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hauser SL, Chan JR, Oksenberg JR. Multiple sclerosis: prospects and promise. Ann Neurol. 2013;74(3):317–27. doi: 10.1002/ana.24009. [DOI] [PubMed] [Google Scholar]
- 44.Hochmeister S, Grundtner R, Bauer J, et al. Dysferlin is a new marker for leaky brain blood vessels in multiple sclerosis. J Neuropathol Exp Neurol. 2006;65(9):855–65. doi: 10.1097/01.jnen.0000235119.52311.16. [DOI] [PubMed] [Google Scholar]
- 45.Cristofanilli M, Harris VK, Zigelbaum A, et al. Mesenchymal stem cells enhance the engraftment and myelinating ability of allogeneic oligodendrocyte progenitors in dysmyelinated mice. Stem Cells Dev. 2011;20(12):2065–76. doi: 10.1089/scd.2010.0547. [DOI] [PubMed] [Google Scholar]
- 46.Harris VK, Yan QJ, Vyshkina T, et al. Clinical and pathological effects of intrathecal injection of mesenchymal stem cell-derived neural progenitors in an experimental model of multiple sclerosis. J Neurol Sci. 2012;313(1-2):167–77. doi: 10.1016/j.jns.2011.08.036. [DOI] [PubMed] [Google Scholar]
- 47.Harris VK, Faroqui R, Vyshkina T, Sadiq SA. Characterization of autologous mesenchymal stem cell-derived neural progenitors as a feasible source of stem cells for central nervous system applications in multiple sclerosis. Stem cells translational medicine. 2012;1(7):536–47. doi: 10.5966/sctm.2012-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cristofanilli M, Cymring B, Lu A, et al. Cerebrospinal fluid derived from progressive multiple sclerosis patients promotes neuronal and oligodendroglial differentiation of human neural precursor cells in vitro. Neuroscience. 2013;250:614–21. doi: 10.1016/j.neuroscience.2013.07.022. [DOI] [PubMed] [Google Scholar]
- 49.Gupta N, Henry RG, Strober J, et al. Neural stem cell engraftment and myelination in the human brain. Sci Transl Med. 2012;4(155):155ra137. doi: 10.1126/scitranslmed.3004373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang FI, Hinton DR, Gilmore W, et al. Sequential infection of glial cells by the murine hepatitis virus JHM strain (MHV-4) leads to a characteristic distribution of demyelination. Lab Invest. 1992;66(6):744–54. [PubMed] [Google Scholar]
- 51.Fleming JO, Trousdale MD, Elzaatari FAK, et al. Pathogenicity of antigenic variants of murine coronavirus JHM selected with monoclonal-antibodies. J Virol. 1986;58(3):869–75. doi: 10.1128/jvi.58.3.869-875.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parra B, Hinton DR, Lin MT, et al. Kinetics of cytokine mRNA expression in the central nervous system following lethal and nonlethal coronavirus-induced acute encephalomyelitis. Virology. 1997;233(2):260–70. doi: 10.1006/viro.1997.8613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pearce BD, Hobbs MV, McGraw TS, Buchmeier MJ. Cytokine induction during T-cell-mediated clearance of mouse hepatitis virus from neurons in vivo. J Virol. 1994;68(9):5483–95. doi: 10.1128/jvi.68.9.5483-5495.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ireland DD, Stohlman SA, Hinton DR, et al. Type I interferons are essential in controlling neurotropic coronavirus infection irrespective of functional CD8 T cells. J Virol. 2008;82(1):300–10. doi: 10.1128/JVI.01794-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith AL, Barthold SW, Beck DS. Intranasally administered alpha/beta interferon prevents extension of mouse hepatitis virus, strain JHM, into the brains of BALB/cByJ mice. Antiviral Res. 1987;8(5-6):239–45. doi: 10.1016/S0166-3542(87)80002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Minagawa H, Takenaka A, Mohri S, Mori R. Protective effect of recombinant murine interferon beta against mouse hepatitis virus infection. Antiviral Res. 1987;8(2):85–95. doi: 10.1016/0166-3542(87)90079-9. [DOI] [PubMed] [Google Scholar]
- 57.Yong VW, Zabad RK, Agrawal S, et al. Elevation of matrix metalloproteinases (MMPs) in multiple sclerosis and impact of immunomodulators. J Neurol Sci. 2007;259(1-2):79–84. doi: 10.1016/j.jns.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 58.Hosking MP, Liu L, Ransohoff RM, Lane TE. A protective role for ELR+ chemokines during acute viral encephalomyelitis. PLoS Pathog. 2009;5(11):e1000648. doi: 10.1371/journal.ppat.1000648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Savarin C, Stohlman SA, Atkinson R, et al. Monocytes regulate T cell migration through the glia limitans during acute viral encephalitis. J Virol. 2010;84(10):4878–88. doi: 10.1128/JVI.00051-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou J, Stohlman SA, Atkinson R, et al. Matrix metalloproteinase expression correlates with virulence following neurotropic mouse hepatitis virus infection. J Virol. 2002;76(15):7374–84. doi: 10.1128/JVI.76.15.7374-7384.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou J, Stohlman SA, Hinton DR, Marten NW. Neutrophils promote mononuclear cell infiltration during viral-induced encephalitis. J Immunol. 2003;170(6):3331–6. doi: 10.4049/jimmunol.170.6.3331. [DOI] [PubMed] [Google Scholar]
- 62.Phares TW, Stohlman SA, Hwang M, et al. CD4 T Cells Promote CD8 T Cell Immunity at the Priming and Effector Site during Viral Encephalitis. J Virol. 2012;86(5):2416–27. doi: 10.1128/JVI.06797-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou J, Hinton DR, Stohlman SA, et al. Maintenance of CD8+ T cells during acute viral infection of the central nervous system requires CD4+ T cells but not interleukin-2. Viral Immunol. 2005;18(1):162–9. doi: 10.1089/vim.2005.18.162. [DOI] [PubMed] [Google Scholar]
- 64.Bergmann CC, Parra B, Hinton DR, et al. Perforin-mediated effector function within the central nervous system requires IFN-gamma-mediated MHC up-regulation. J Immunol. 2003;170(6):3204–13. doi: 10.4049/jimmunol.170.6.3204. [DOI] [PubMed] [Google Scholar]
- 65.Ramakrishna C, Stohlman SA, Atkinson RA, et al. Differential regulation of primary and secondary CD8(+) T cells in the central nervous system. J Immunol. 2004;173(10):6265–73. doi: 10.4049/jimmunol.173.10.6265. [DOI] [PubMed] [Google Scholar]
- 66.Parra B, Hinton DR, Marten NW, et al. IFN-gamma is required for viral clearance from central nervous system oligodendroglia. J Immunol. 1999;162(3):1641–7. [PubMed] [Google Scholar]
- 67.Gonzalez JM, Bergmann CC, Ramakrishna C, et al. Inhibition of interferon-gamma signaling in oligodendroglia delays coronavirus clearance without altering demyelination. Am J Pathol. 2006;168(3):796–804. doi: 10.2353/ajpath.2006.050496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lin MT, Stohlman SA, Hinton DR. Mouse hepatitis virus is cleared from the central nervous systems of mice lacking perforin-mediated cytolysis. J Virol. 1997;71(1):383–91. doi: 10.1128/jvi.71.1.383-391.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bergmann CC, Parra B, Hinton DR, et al. Perforin and gamma interferon-mediated control of coronavirus central nervous system infection by CD8 T cells in the absence of CD4 T cells. J Virol. 2004;78(4):1739–50. doi: 10.1128/JVI.78.4.1739-1750.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Parra B, Lin MT, Stohlman SA, et al. Contributions of Fas-Fas ligand interactions to the pathogenesis of mouse hepatitis virus in the central nervous system. J Virol. 2000;74(5):2447–50. doi: 10.1128/jvi.74.5.2447-2450.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stohlman SA, Hinton DR. Viral induced demyelination. Brain Pathol. 2001;11(1):92–106. doi: 10.1111/j.1750-3639.2001.tb00384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu GF, Perlman S. Macrophage infiltration, but not apoptosis, is correlated with immune-mediated demyelination following murine infection with a neurotropic coronavirus. J Virol. 1999;73(10):8771–80. doi: 10.1128/jvi.73.10.8771-8780.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Redwine JM, Buchmeier MJ, Evans CF. In vivo expression of major histocompatibility complex molecules on oligodendrocytes and neurons during viral infection. Am J Pathol. 2001;159(4):1219–24. doi: 10.1016/S0002-9440(10)62507-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fleming JO, Adami C, Pooley J, et al. Mutations associated with viral sequences isolated from mice persistently infected with MHV-JHM. Adv Exp Med Biol. 1995;380:591–5. doi: 10.1007/978-1-4615-1899-0_94. [DOI] [PubMed] [Google Scholar]
- 75.Rowe CL, Baker SC, Nathan MJ, Fleming JO. Evolution of mouse hepatitis virus: detection and characterization of spike deletion variants during persistent infection. J Virol. 1997;71(4):2959–69. doi: 10.1128/jvi.71.4.2959-2969.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Adami C, Pooley J, Glomb J, et al. Evolution of mouse hepatitis virus (MHV) during chronic infection: quasispecies nature of the persisting. MHV RNA Virology. 1995;209(2):337–46. doi: 10.1006/viro.1995.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dandekar AA, Wu GF, Pewe L, Perlman S. Axonal damage is T cell mediated and occurs concomitantly with demyelination in mice infected with a neurotropic coronavirus. J Virol. 2001;75(13):6115–20. doi: 10.1128/JVI.75.13.6115-6120.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Das Sarma J, Kenyon LC, Hingley ST, Shindler KS. Mechanisms of primary axonal damage in a viral model of multiple sclerosis. J Neurosci. 2009;29(33):10272–80. doi: 10.1523/JNEUROSCI.1975-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pewe L, Perlman S. Cutting edge: CD8 T cell-mediated demyelination is IFN-gamma dependent in mice infected with a neurotropic coronavirus. J Immunol. 2002;168(4):1547–51. doi: 10.4049/jimmunol.168.4.1547. [DOI] [PubMed] [Google Scholar]
- 80.Lane TE, Liu MT, Chen BP, et al. A central role for CD4(+) T cells and RANTES in virus-induced central nervous system inflammation and demyelination. J Virol. 2000;74(3):1415–24. doi: 10.1128/jvi.74.3.1415-1424.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Horiuchi M, Itoh A, Pleasure D, Itoh T. MEK-ERK signaling is involved in interferon-gamma-induced death of oligodendroglial progenitor cells. J Biol Chem. 2006;281(29):20095–106. doi: 10.1074/jbc.M603179200. [DOI] [PubMed] [Google Scholar]
- 82.Chew LJ, King WC, Kennedy A, Gallo V. Interferon-gamma inhibits cell cycle exit in differentiating oligodendrocyte progenitor cells. Glia. 2005;52(2):127–43. doi: 10.1002/glia.20232. [DOI] [PubMed] [Google Scholar]
- 83.Balabanov R, Strand K, Kemper A, et al. Suppressor of cytokine signaling 1 expression protects oligodendrocytes from the deleterious effects of interferon-gamma. J Neurosci. 2006;26(19):5143–52. doi: 10.1523/JNEUROSCI.0737-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Baerwald KD, Popko B. Developing and mature oligodendrocytes respond differently to the immune cytokine interferon-gamma. J Neurosci Res. 1998;52(2):230–9. doi: 10.1002/(SICI)1097-4547(19980415)52:2<230::AID-JNR11>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 85.Wang Y, Ren Z, Tao D, et al. STAT1/IRF-1 signaling pathway mediates the injurious effect of interferon-gamma on oligodendrocyte progenitor cells. Glia. 2010;58(2):195–208. doi: 10.1002/glia.20912. [DOI] [PubMed] [Google Scholar]
- 86.Lin WS, Kunkler PE, Harding HP, et al. Enhanced Integrated Stress Response Promotes Myelinating Oligodendrocyte Survival in Response to Interferon-gamma. Am J Pathol. 2008;173(5):1508–17. doi: 10.2353/ajpath.2008.080449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vartanian T, Li Y, Zhao M, Stefansson K. Interferon-gamma-induced oligodendrocyte cell death: implications for the pathogenesis of multiple sclerosis. Mol Med. 1995;1(7):732–43. [PMC free article] [PubMed] [Google Scholar]
- 88.Fleury HJA, Sheppard RD, Bornstein MB, Raine CS. Further ultrastructural observations of virus morphogenesis and myelin pathology in JHM virus encephalomyelitis. Neuropathol Appl Neurobiol. 1980;6(3):165–79. doi: 10.1111/j.1365-2990.1980.tb00288.x. [DOI] [PubMed] [Google Scholar]
- 89.Templeton SP, Kim TS, O'Malley K, Perlman S. Maturation and localization of macrophages and microglia during infection with a neurotropic murine coronavirus. Brain Pathol. 2008;18(1):40–51. doi: 10.1111/j.1750-3639.2007.00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Das Sarma J. Microglia-mediated neuroinflammation is an amplifier of virus-induced neuropathology. J Neurovirol. 2014;20(2):122–36. doi: 10.1007/s13365-013-0188-4. [DOI] [PubMed] [Google Scholar]
- 91.Yamasaki R, Lu H, Butovsky O, et al. J Exp Med. 2014;211(8):1533–49. doi: 10.1084/jem.20132477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Armstrong RC, Le TQ, Flint NC, et al. Endogenous cell repair of chronic demyelination. J Neuropathol Exp Neurol. 2006;65(3):245–56. doi: 10.1097/01.jnen.0000205142.08716.7e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Keirstead HS, Blakemore WF. The role of oligodendrocytes and oligodendrocyte progenitors in CNS remyelination. Adv Exp Med Biol. 1999;468:183–97. doi: 10.1007/978-1-4615-4685-6_15. [DOI] [PubMed] [Google Scholar]
- 94.Blakemore WF, Keirstead HS. The origin of remyelinating cells in the central nervous system. J Neuroimmunol. 1999;98(1):69–76. doi: 10.1016/s0165-5728(99)00083-1. [DOI] [PubMed] [Google Scholar]
- 95.Liu L, Darnall L, Hu T, et al. Myelin repair is accelerated by inactivating CXCR2 on nonhematopoietic cells. J Neurosci. 2010;30(27):9074–83. doi: 10.1523/JNEUROSCI.1238-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Murtie JC, Zhou YX, Le TQ, et al. PDGF and FGF2 pathways regulate distinct oligodendrocyte lineage responses in experimental demyelination with spontaneous remyelination. Neurobiol Dis. 2005;19(1-2):171–82. doi: 10.1016/j.nbd.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 97.McTigue DM, Tripathi RB. The life, death, and replacement of oligodendrocytes in the adult. CNS J Neurochem. 2008;107(1):1–19. doi: 10.1111/j.1471-4159.2008.05570.x. [DOI] [PubMed] [Google Scholar]
- 98.Carbajal KS, Miranda JL, Tsukamoto MR, Lane TE. CXCR4 signaling regulates remyelination by endogenous oligodendrocyte progenitor cells in a viral model of demyelination. Glia. 2011;59(12):1813–21. doi: 10.1002/glia.21225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tsai HH, Frost E, To V, et al. The chemokine receptor CXCR2 controls positioning of oligodendrocyte precursors in developing spinal cord by arresting their migration. Cell. 2002;110(3):373–83. doi: 10.1016/s0092-8674(02)00838-3. [DOI] [PubMed] [Google Scholar]
- 100.Armstrong RC, Le TQ, Frost EE, et al. Absence of fibroblast growth factor 2 promotes oligodendroglial repopulation of demyelinated white matter. J Neurosci. 2002;22(19):8574–85. doi: 10.1523/JNEUROSCI.22-19-08574.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Robinson S, Tani M, Strieter RM, et al. The chemokine growth-regulated oncogene-alpha promotes spinal cord oligodendrocyte precursor proliferation. J Neurosci. 1998;18(24):10457–63. doi: 10.1523/JNEUROSCI.18-24-10457.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Williams JL, Patel JR, Daniels BP, Klein RS. Targeting CXCR7/ACKR3 as a therapeutic strategy to promote remyelination in the adult central nervous system. J Exp Med. 2014;211(5):791–9. doi: 10.1084/jem.20131224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Totoiu MO, Nistor GI, Lane TE, Keirstead HS. Remyelination, axonal sparing, and locomotor recovery following transplantation of glial-committed progenitor cells into the MHV model of multiple sclerosis. Exp Neurol. 2004;187(2):254–65. doi: 10.1016/j.expneurol.2004.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Carbajal KS, Schaumburg C, Strieter R, et al. Migration of engrafted neural stem cells is mediated by CXCL12 signaling through CXCR4 in a viral model of multiple sclerosis. Proc Natl Acad Sci USA. 2010;107(24):11068–73. doi: 10.1073/pnas.1006375107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hardison JL, Nistor G, Gonzalez R, et al. Transplantation of glial-committed progenitor cells into a viral model of multiple sclerosis induces remyelination in the absence of an attenuated inflammatory response. Exp Neurol. 2006;197(2):420–9. doi: 10.1016/j.expneurol.2005.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Whitman LM, Blanc CA, Schaumburg CS, et al. Olig1 function is required for remyelination potential of transplanted neural progenitor cells in a model of viral-induced demyelination. Exp Neurol. 2012;235(1):380–7. doi: 10.1016/j.expneurol.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Greenberg ML, Weinger JG, Matheu MP, et al. Two-photon imaging of remyelination of spinal cord axons by engrafted neural precursor cells in a viral model of multiple sclerosis. Proc Natl Acad Sci USA. 2014;111(22):E2349–55. doi: 10.1073/pnas.1406658111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Faulkner J, Keirstead HS. Human embryonic stem cell-derived oligodendrocyte progenitors for the treatment of spinal cord injury. Transpl Immunol. 2005;15(2):131–42. doi: 10.1016/j.trim.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 109.Asterias. Safety Study of GRNOPC1 in Spinal Cord Injury. 2010 Available from: http://clinicaltrials.gov/ct2/show/NCT01217008.
- 110.Chen Z, Phillips LK, Gould E, et al. MHC mismatch inhibits neurogenesis and neuron maturation in stem cell allografts. PLoS One. 2011;6(3):e14787. doi: 10.1371/journal.pone.0014787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Phillips LK, Gould EA, Babu H, et al. Natural killer cell-activating receptor NKG2D mediates innate immune targeting of allogeneic neural progenitor cell grafts. Stem Cells. 2013;31(9):1829–39. doi: 10.1002/stem.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Weinger JG, Weist BM, Plaisted WC, et al. C mismatch results in neural progenitor cell rejection following spinal cord transplantation in a model of viral-induced demyelination. Stem Cells. 2012;30(11):2584–95. doi: 10.1002/stem.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Weinger JG, Plaisted WC, Maciejewski SM, et al. Activating Receptor NKG2D Targets RAE-1-expressing allogeneic neural precursor cells in a viral model of multiple sclerosis stem cells. Stem Cells. 2014 doi: 10.1002/stem.1760. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Plaisted WC, Weinger JG, Walsh CM, Lane TE. T cell mediated suppression of neurotropic coronavirus replication in neural precursor cells. Virology. 2014;449:235–43. doi: 10.1016/j.virol.2013.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hatch MN, Schaumburg CS, Lane TE, Keirstead HS. Endogenous remyelination is induced by transplant rejection in a viral model of multiple sclerosis. J Neuroimmunol. 2009;212(1-2):74–81. doi: 10.1016/j.jneuroim.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 116.Keirstead HS, Nistor G, Bernal G, et al. Human embryonic stem cell-derived oligodendrocyte progenitor cell transplants remyelinate and restore locomotion after spinal cord injury. J Neurosci. 2005;25(19):4694–705. doi: 10.1523/JNEUROSCI.0311-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Aharonowiz M, Einstein O, Fainstein N, et al. Neuroprotective effect of transplanted human embryonic stem cell-derived neural precursors in an animal model of multiple sclerosis. PLoS One. 2008;23(9):e3145. doi: 10.1371/journal.pone.0003145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pluchino S, Gritti A, Blezer E, et al. Human neural stem cells ameliorate autoimmune encephalomyelitis in non-human primates. Ann Neurol. 2009;66(3):343–54. doi: 10.1002/ana.21745. [DOI] [PubMed] [Google Scholar]
- 119.Chen L, Coleman R, Leang R, et al. Human neural precursor cells promote neurologic recovery in a viral model of multiple sclerosis. Stem Cell Reports. 2014;2(6):825–37. doi: 10.1016/j.stemcr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. 2005;201(7):1061–1067. doi: 10.1084/jem.20042276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Anghelina D, Zhao J, Trandem K, Perlman S. Role of regulatory T cells in coronavirus-induced acute encephalitis. Virology. 2009;385(2):358–67. doi: 10.1016/j.virol.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Trandem K, Anghelina D, Zhao J, Perlman S. Regulatory T cells inhibit T cell proliferation and decrease demyelination in mice chronically infected with a coronavirus. J Immunol. 2010;184(8):4391–400. doi: 10.4049/jimmunol.0903918. [DOI] [PMC free article] [PubMed] [Google Scholar]