

Abstract
Tissue hypoxia not only occurs under pathological conditions but is also an important microenvironmental factor that is critical for normal embryonic development. Hypoxia-inducible factors HIF-1 and HIF-2 are oxygen-sensitive basic helix-loop-helix transcription factors, which regulate biological processes that facilitate both oxygen delivery and cellular adaptation to oxygen deprivation. HIFs consist of an oxygen-sensitive α-subunit, HIF-α, and a constitutively expressed β-subunit, HIF-β, and regulate the expression of genes that are involved in energy metabolism, angiogenesis, erythropoiesis and iron metabolism, cell proliferation, apoptosis, and other biological processes. Under conditions of normal PO2, HIF-α is hydroxylated and targeted for rapid proteasomal degradation by the von Hippel-Lindau (VHL) E3-ubiquitin ligase. When cells experience hypoxia, HIF-α is stabilized and either dimerizes with HIF-β in the nucleus to form transcriptionally active HIF, executing the canonical hypoxia response, or it physically interacts with unrelated proteins, thereby enabling convergence of HIF oxygen sensing with other signaling pathways. In the normal, fully developed kidney, HIF-1α is expressed in most cell types, whereas HIF-2α is mainly found in renal interstitial fibroblast-like cells and endothelial cells. This review summarizes some of the most recent advances in the HIF field and discusses their relevance to renal development, normal kidney function and disease.
Keywords: erythropoiesis, renal ischemia-reperfusion injury, renal fibrosis, renal cell cancer
OVERVIEW OF HIF SIGNALING
Tissue Hypoxia, Aside from being a frequently encountered clinical problem as a result of pulmonary or cardiovascular conditions, is also an important microenvironmental factor that is critical for the regulation of normal embryonic development and stem cell maintenance (1, 2, 21, 22, 86, 123, 140). Over the last decade, major advances have been made in deciphering the molecular mechanisms that allow cells to respond and to adapt to low PO2. As key mediators in cellular oxygen homeostasis, hypoxia-inducible factor-1 and -2 (HIF-1 and HIF-2) facilitate both oxygen delivery and adaptation to oxygen deprivation by regulating the expression of gene products that are involved in cellular energy metabolism and glucose transport, angiogenesis, erythropoiesis and iron metabolism, pH regulation, apoptosis, and cell proliferation as well as cell-cell and cell-matrix interactions (112, 136). Examples of classic HIF target genes are phosphoglycerate kinase-1 (PGK), glucose transporter-1 (GLUT1), vascular endothelial growth factor (VEGF), and erythropoietin (EPO) (for detailed reviews, see Refs. 112 and 136).
HIF-1 and HIF-2 (here collectively referred to as HIF) are members of the Per-ARNT-Sim (PAS) family of heterodimeric basic helix-loop-helix (bHLH) transcription factors and consist of an oxygen-sensitive α-subunit and a constitutively expressed β-unit, also known as the aryl hydrocarbon receptornuclear translocator (ARNT) or simply HIF-β (112, 136). Direct transcriptional regulation occurs through the binding of HIF heterodimers to hypoxia-response elements (HREs), which are present in regulatory regions of hypoxia-sensitive genes (Fig. 1) (136). With regard to their ability to transcriptionally regulate specific hypoxia-responsive genes, HIF-1 and HIF-2 have distinct functions and only partially overlap. For example, glycolytic genes appear to be predominantly regulated by HIF-1 (50), whereas our group and others have suggested that HIF-2 is the main regulator of hypoxic VEGF and EPO induction in tissues that express both HIF-1 and HIF-2 (85, 93, 135). Oct-4, a transcription factor important in regulating stem cell fate, appears to be a specific HIF-2 target (21). Target gene selectivity between HIF-1 and HIF-2 may be the result of tissue-specific interactions with other nuclear factors, differential interactions with transcriptional cofactors, or be a reflection of tissue- and cell type-dependent differences in the ratios of HIF-α protein levels (for a recent review on this issue, see Ref. 104).
Fig. 1.

Canonical and noncanonical hypoxic signaling through hypoxia-inducible factor (HIF). Under normoxia, hydroxylation of HIF-α-subunits by HIF prolyl-4-hydroxylases is required for binding to the pVHL-E3-ubiquitin ligase complex. After polyubiquitination, HIF-α is degraded by the proteasome. During hypoxia when prolyl-hydroxylases are inactive, HIF-α-subunit degradation is inhibited. HIF-α translocates to the nucleus, where it binds to HIF-β. HIF-α/β heterodimers then bind to the HIF-DNA consensus binding site, RCGTG, and increase transcription of HIF-target genes, e.g., erythropoietin (EPO), VEGF, and glucose transporter-1 (GLUT1). Factor-inhibiting HIF (FIH) is an asparagine (Asn) hydroxylase that modulates cofactor recruitment to the HIF transcriptional complex via asparagine hydroxylation of the HIF-α COOH-terminal transactivation domain. FIH activity is oxygen dependent. Noncanonical HIF signaling occurs through biochemical interaction with other proteins, such as the Notch intracellular domain (ICD; for a more complete overview of HIF-α-interacting proteins, see Ref. 136). Nitric oxide (NO), reactive oxygen species (ROS), the Krebs cycle metabolites succinate and fumarate, cobalt chloride (CoCl2), and Fe chelators such as desferroxamine are known to inhibit HIF prolyl-4-hydroxylases in the presence of oxygen. PHI, prolyl-hydroxylase inhibitor; Pro, proline.
In addition to heterodimerization with HIF-β resulting in the formation of a bHLH transcription factor, which mediates the canonical hypoxia response, HIF-α subunits also regulate biological processes through direct protein-protein interaction with other factors. These include, among others, tumor suppressor protein p53 and the c-Myc protooncogene (67, 98). A more recent example is the ability of HIF-1α to biochemically associate with the intracellular domain of Notch (Notch ICD), thereby increasing Notch signaling through upregulation of Notch target genes, such as Hey and Hes (39). How HIF-α and Notch interact exactly and whether other cofactors are involved are presently unclear, but the observation that HIF-1α modulates Notch signaling through a direct protein-protein interaction underscores the importance of HIF-α as a regulator of important intracellular pathways, independent of its role in HRE-mediated transcription.
HIF activation is dependent on stabilization of the oxygen-sensitive α-subunit and its subsequent translocation to the nucleus, where it dimerizes with HIF-β and recruits transcriptional cofactors such as CBP and p300 (112, 136). Normally, under conditions of adequate oxygen supply, hydroxylated HIF-α binds to the von Hippel-Lindau tumor suppressor protein (pVHL), which is part of an E3-ubiquitin ligase complex that targets HIF-α for proteasomal degradation (Fig. 1). All three known HIF-α-subunits, i.e., HIF-1α, HIF-2α, and HIF-3α, have been shown to bind to pVHL (the role of HIF-3α in hypoxic signaling is unclear; a HIF-3α splice variant, IPAS, may be inhibitory) (75). The pVHL/HIF-α interaction is highly conserved between species and requires iron- and oxygen-dependent hydroxylation of specific proline residues (Pro402 and Pro564 in human HIF-1α; Pro405 and Pro531 in human HIF-2α ) within the oxygen-dependent degradation domain (ODD) of HIF-α. Prolyl-hydroxylation by prolyl-4-hydroxylases and binding to pVHL are absolutely required for the execution of HIF proteolysis under normoxia (for a review on this topic, see Ref. 112). During hypoxia, prolyl-hydroxylases are inactive and HIF-α degradation is inhibited. In cell culture, HIF-α -subunits typically accumulate when oxygen concentrations fall below 5% (55). Three major mammalian HIF prolyl-hydroxylases have been identified, i.e., prolyl-hydroxylase domain (PHD)1, PHD2, and PHD3, all of which are expressed in renal epithelial cells (117). Whereas PHD2 appears to be the hydroxylase that is essential for HIF-α degradation under normoxia (9), PHD3 seems to be important for hydroxylation of HIF-α during reoxygenation (6). Furthermore, differential effects of individual PHDs on HIF-1α and HIF-2α hydroxylation have been reported, suggesting that stability of individual HIF-α subunits and thus target gene expression may be affected by tissue- and cell type-specific differences in PHD expression and activity levels (6). The activity of PHDs and thus hypoxic stabilization of HIF-α subunits can be modulated by mitochondrial reactive oxygen species (ROS) (Fig. 1), implicating mitochondria in oxygen sensing. Although debated for many years (3, 16, 17, 25, 118, 131), recent studies using a combination of genetic, pharmacological, and small-interference RNA-based approaches provided further evidence that ROS generated by mitochondrial complex III are required for hypoxic HIF-α stabilization independent of mitochondrial oxygen consumption (13, 40, 79), supporting the initial claim made by Chandel et al. (16) that mitochondria function as oxygen sensors. ROS have been shown to inhibit PHD activity, probably by changing the redox state of enzyme-bound iron that is required for catalytic activity (34). An additional model of mitochondrial oxygen sensing was proposed by Hagen et al. (43), in which an intracellular shift of oxygen resulting from decreased mitochondrial oxygen consumption (nitric oxide-mediated inhibition of the mitochondrial respiratory chain) increased substrate availability and thus HIF prolyl-hydroxylation under moderate to severe hypoxia.
A second hypoxic switch operates in the COOH-terminal transactivation domain of HIF-α with the hydroxylation of an asparagine residue. During hypoxia, asparagine hydroxylation is blocked and CBP/p300 recruitment is facilitated, enabling increased levels of transcription (Fig. 1). Factor-inhibiting-HIF (FIH) hydroxylates the asparagine residue at position Asn803 in human HIF-1α, which corresponds to asparagine Asn851 in human HIF-2α. FIH is expressed in renal tubular epithelial cells and glomeruli, as shown by immunohistochemical methods (117). Inhibition of FIH results in increased HIF target gene expression even under severe hypoxia or in certain cell lines that are unable to degrade HIF-α(122).
Besides hypoxic activation, a nonhypoxic increase in HIF transcriptional activity has been shown to be mediated by nitric oxide and TNF-α(111), interleukin 1 (45, 121), angiotensin II (99), and a variety of growth factors including epidermal growth factor, insulin, and insulin-like growth factors (28, 54, 121, 129, 142). Nitric oxide, ROS, and certain oncogenes such as v-Src and activated Ras have been shown to inhibit HIF prolyl-hydroxylation (for reviews on this topic, see Refs. 56 and 112). In contrast, HIF activation induced through the phosphoinositide 3-kinase/Akt-1/mammalian target of rapamycin pathway appears to be mediated through increased HIF-α protein translation (32, 70, 144). Thus it is easy to envision that HIF activation is likely to occur in a variety of different renal disease settings even in the absence of significant hypoxia.
Whereas HIF-1α is ubiquitously expressed, HIF-2α expression is more restricted. HIF-2α has been found in hepatocytes, cardiomyocytes, glial cells, type II pneumocytes, and endothelial cells (137). HIF-regulated gene expression in normal, nontransformed primary renal tubular epithelial cells appears to be solely controlled by HIF-1, as shown by our laboratory and others (47, 103). In the adult rodent, HIF-2α was largely found in renal interstitial fibroblasts and renal endothelial cells under conditions of carbon monoxide poisoning or renal ischemia (103, 137). In this context, it is of interest that VHL-deficient renal cancer cells of the clear cell type, which are derived from the renal epithelium, reexpress HIF-2α in more advanced lesions, whereas HIF-1α expression seems to decrease (77, 97). Although the increase in HIF-2α and decrease in HIF-1α expression are most likely a reflection of a progression in oncogenic transformation, the molecular mechanisms underlying this phenomenon are not well understood.
The list of HIF-regulated genes (either directly or indirectly regulated by HIF) has grown rapidly (136). HIF is involved in the regulation of a multitude of biological processes that are relevant to kidney function under physiological and pathological conditions. These include glucose and energy metabolism, angiogenesis, erythropoiesis and iron homeostasis, cell migration, and cell-cell and cell-matrix interactions. Factors that are directly regulated by HIF and have relevance to the pathogenesis of acute and chronic kidney diseases include heme oxygenase-1 (HO-1), VEGF, plasminogen activator inhibitor-1 (PAI-1), tissue inhibitor of metalloproteinase-1 (TIMP-1), connective tissue growth factor (CTGF), EPO, Wilms’ tumor suppressor (WT-1), and others (112, 136) (Fig. 2) (see HIF SIGNALING AND RENAL INJURY).
Fig. 2.

Examples of direct transcriptional HIF targets with relevance to kidney function. Shown are selected direct HIF target genes and their classification into functional groups. For a comprehensive list of HIF target genes, the reader is referred to Wenger et al. (136). Some of the HIF targets listed here appear to be preferentially regulated by HIF-2 (e.g., VEGF and EPO). In contrast to HIF-2α, HIF-1α is expressed in most renal epithelial cells, whereas HIF-2α is mainly found in endothelial cells and renal interstitial fibroblast-like cells. HIF-1α is also expressed in papillary and inner medullary interstitial and endothelial cells but was not detected in interstitial and endothelial cells of the cortex and outer medulla (103). ANP, atrial natriuretic peptide; Bnip-3, BCL2/adenovirus E1B 19-kDa-interacting protein 3 (proapoptotic BH3 domain; only BCL-2 family member); c-Met, tyrosine kinase receptor for scatter factor/hepatocyte growth factor (SF/HGF); CXCR4, chemokine receptor 4; CTGF, connective tissue growth factor; EC, endothelial cell; ECM, extracellular matrix; eNOS, endothelial nitric oxide synthase; FLT-1, fetal liver tyrosine kinase-1 (VEGF receptor-1); IC, interstitial cell; IGFBP-1, insulin growth factor binding protein-1; iNOS, inducible nitric oxide synthase; PAI-1, plasminogen activator inhibitor-1; RTEC, renal tubular epithelial cell; TIMP-1, tissue inhibitor of metalloproteinase-1.
MOUSE MODELS OF DEFECTIVE HIF SIGNALING
Tissue hypoxia occurs not only under pathological conditions but also during normal embryogenesis under physiological conditions, indicating an important role for HIF in development. In fact, inactivation of either HIF-1α, HIF-2α, HIF-β, or the VHL tumor suppressor in the murine germ line results in embryonic or perinatal lethality. Mice homozygously deficient for HIF-1α die in utero between embryonic (E) days 8 and 11 from neural tube defects, cardiovascular malformations, and increased cell death in the cephalic mesenchyme associated with tissue hypoxia (52, 105). HIF-β (ARNT)-deficient mice are not viable beyond E10.5 and die from defective vasculogenesis of the yolk sac and branchial arches (76). Different phenotypes have been published for HIF-2α germ line knockout mice, most likely reflecting variations in genetic background. Most HIF-2α homozygous knockout mice die in utero or perinatally unless bred as heterozygotes in a C57/BL6J and 129S6/SvEv background (113). HIF-2α-deficient mice 1) developed problems with catecholamine synthesis in the organ of Zuckerkandl, resulting in heart failure and midgestational death (128); 2) had problems with VEGF-mediated lung maturation, resulting in perinatal death (20); 3) developed severe vascular defects in the yolk sac and embryo proper, resulting in death between E9.5 and 13.5 (91); and 4) exhibited defective ROS scavenging, resulting in hepatic steatosis, cardiac hypertrophy, skeletal myopathy, and hypocellular bone marrow and mitochondrial abnormalities (113). Inactivation of pVHL results in an increase in HIF-1 and HIF-2 transcriptional activity, and mice deficient in pVHL die during midgestation from abnormal placental vasculogenesis, indicating that tight regulation of HIF-α proteolysis is critical for normal development (35).
To overcome embryonic lethality associated with germ line inactivation, conditional alleles have been generated that now allow tissue- and cell type-specific targeting of HIF-1α, HIF-2α, HIF-β, and pVHL (41, 106, 134). To inactivate pVHL, HIF-1α, HIF-2α, and HIF-β in renal proximal tubule cells, our laboratory has generated Cre transgenic mice using a mutated version of the rat phosphoenolpyruvate carboxykinase (PEPCK) promoter (95). Whereas PEPCK-Cre-mediated inactivation of HIF-1α or HIF-β alone did not result in an abnormal kidney phenotype, inactivation of pVHL resulted in HIF-dependent development of tubular and glomerular cysts (Fig. 3) (95). Whether HIFs play a more general role in renal cyst development, e.g., in the setting of autosomal dominant polycystic kidney disease, is unclear and remains to be investigated. Podocyte-specific inactivation of pVHL resulted in diverse pathological changes reported by our group and by others. These include foot process effacement, resulting in proteinuria, as well as crescent formation, leading to rapidly progressive renal failure (12, 24, 120).
Fig. 3.
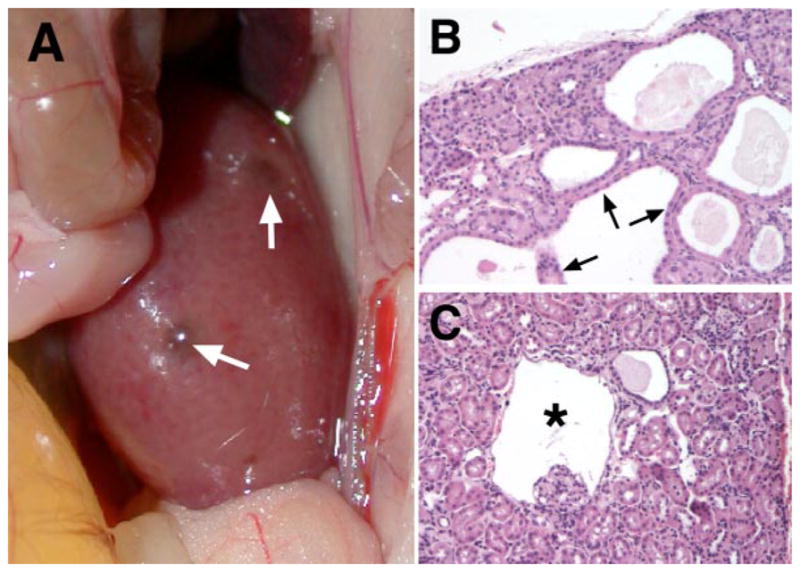
Consequences of von Hippel-Lindau (VHL) gene inactivation in the kidney. Renal cyst development in mice with inactivation of pVHL in proximal renal tubule cells using the PEPCK-Cre transgene (95) is shown. A: macroscopically visible renal cysts in a pVHL-deficient kidney (white arrows). B: multiple renal cysts lined by cuboidal, eosinophilic epithelial cells. Hematoxylin and eosin stain, magnification ×200. C: glomerular cyst development in pVHL-deficient kidneys. Shown is a glomerular cyst (*) with the glomerular tuft located at the cyst basis. Studies with the ROSA26-lacZ Cre-reporter indicated recombination activity in Bowman’s capsule, suggesting that Bowman’s capsule of this cyst is pVHL deficient. Hematoxylin and eosin stain, magnification ×200.
HIF IN RENAL DEVELOPMENT
Hypoxia occurs physiologically during embryogenesis, and stabilization of HIF-α-subunits has been demonstrated during nephrogenesis in vitro and in vivo (8, 31). However, the exact role of HIF signaling in renal development is largely unexplored. To date, a developmental phenotype in the kidney has not been described for either pVHL or HIF knockout mice. Although the role of HIF signaling in renal development is unclear, HIF-α-subunits exhibit a cell type- and stage-specific expression pattern during nephrogenesis. This correlated with the expression of important angiogenic factors, such as VEGF and endoglin, supporting the notion that HIF signaling has a regulatory role in the developing kidney (8). HIF-1α expression was predominantly found in the cortical and medullary collecting ducts, S-shaped bodies, and glomerular cells (8). The expression of HIF-2α was detectable in podocytes, as well as in cortical and medullary endothelial and interstitial cells, but was absent in the fully developed kidney (8, 31). Furthermore, a distinct role for HIF-1α and HIF-2α in glomerular development has been proposed based on the finding that S- or comma-shaped bodies expressed only HIF-1α, whereas more mature glomeruli expressed HIF-2α (8). Although these studies support the notion that hypoxia plays a functional role during nephrogenesis, mechanistic insights from in vitro studies and from genetic approaches with conditional knockout mice are currently pending and are needed to define the role of individual HIF transcription factors during renal development.
HIF AND ERYTHROPOIESIS
In the regulation of erythropoiesis, the kidney serves as the most important physiological oxygen sensor and responds to systemic hypoxia with a rapid increase in EPO production by renal interstitial fibroblast-like cells (for recent reviews on the regulation and tissue expression of EPO, see Refs. 27 and 53). Other tissues, such as the liver, also have the capacity to produce EPO in an oxygen-dependent manner. However, non-renal EPO production in the setting of end-stage renal disease is not able to compensate for the loss of renal EPO, resulting in anemia that requires treatment with systemically administered recombinant EPO. Although HIF-1 was initially identified as the factor that induces EPO during hypoxia, we and others have proposed that HIF-2 is the more important regulator of hypoxic EPO induction in hepatocytes and retinal cells (85, 93, 135). It is also likely that HIF-2 has the same dominance in renal EPO-regulation, as HIF-2α expression in the kidney colocalizes with EPO-producing renal interstitial fibroblast-like cells (103, 114). However, to provide a definitive answer, genetic studies are needed that compare the functional consequences of HIF-2α vs. HIF-1α ablation in renal interstitial cells.
The ability of cells to efficiently target HIF-α for proteasomal degradation under normoxia is essential for normal erythropoiesis and has clinical importance with regard to EPO production. Patients with congenital Chuvash polycythemia are homozygous for a specific mutation in the VHL tumor suppressor commonly found at amino acid position Arg200 (Arg200Trp) (5, 90). Chuvash patients have raised red blood cell (RBC) counts from increased EPO production as a result of elevated HIF activity; however, they do not develop the tumors that are typically seen in patients with VHL disease, such as CNS hemangioblastomas, renal cell cancer of the clear cell type, and pheochromocytomas (36). Furthermore, mutations in PHD2, the most abundant PHD protein, result in a rare form of familial polycythemia from the inability to properly degrade HIF-α under normoxia (92).
In mice, we have shown that inactivation of pVHL in hepatocytes results in polycythemia due to an increase in hepatic HIF-2α (42, 94). In this model, pVHL was inactivated in a subset of hepatocytes and resulted in a 20- to 40-fold increase in serum EPO levels that was associated with profound elevation of hemoglobin values and RBC counts (93). In contrast to the liver, EPO production in kidneys from mutant mice was suppressed, as expected. Simultaneous inactivation of pVHL and HIF-1α did not change hemoglobin or RBC values, whereas simultaneous inactivation of pVHL and HIF-2α restored erythropoiesis to normal levels (93, 94), illustrating the importance of HIF-2 in hypoxic EPO regulation.
More importantly, our results support the notion that inhibition of HIF-α degradation in nonrenal tissues is sufficient to substantially raise systemic EPO levels and thus may be useful for the treatment of anemia. Indeed, pharmacological targeting of HIF-α degradation by prolyl-hydroxylase inhibition increased serum EPO levels in humans (71) and improved anemia of chronic disease and inflammation in animal models (69). In the latter studies, inhibition of HIF-α hydroxylation not only increased serum EPO levels but also improved iron uptake and metabolism. This is not surprising, as transferrin and its receptor had been previously found to be direct transcriptional targets of HIF (73, 100, 124). Interestingly, inhibition of prolyl-hydroxylation also resulted in a suppression of hepatic hepcidin, which is known to occur during hypoxia or in the presence of anemia, suggesting a role for HIF in its regulation. Hepcidin inhibits iron transport in the intestinal epithelium, the placenta, and macrophages through its effects on ferroportin protein stability, thus negatively impacting iron metabolism. It is upregulated during inflammation and is a key factor in the development of anemia of chronic disease (for recent reviews, see Refs. 30 and 33). Inhibition of prolyl-hydroxylation therefore has the potential to be a powerful pharmacological tool for the treatment of anemia in general and, in particular, anemia that is refractory to treatment with EPO.
HIF SIGNALING AND RENAL INJURY
HIF-α stabilization has been demonstrated in vivo in several acute and chronic renal injury models (57, 68, 81, 101–103, 125–127, 137, 139). While acutely injured kidneys appear to benefit from the protective effects of HIF-regulated biological processes, chronic hypoxia, mediated in part through HIF-1, can contribute to increased extracellular matrix production and epithelial-to-mesenchymal transition (EMT), thereby potentially promoting renal fibrosis and the progression of renal disease. The following section summarizes recent data on the biological effects of increased HIF signaling during acute and chronic hypoxic renal injury.
Acute Hypoxic Injury
During acute renal ischemia when HIF-α proteolysis is inhibited, HIF-1α can be detected in the nucleus of renal tubular epithelial cells, where it dimerizes with HIF-1β to form transcriptionally active HIF-1. In contrast, HIF-2α is undetectable in this cell type but is strongly expressed in renal interstitial fibroblasts and endothelial cells, supporting the notion that HIF-1 is the key mediator of hypoxic HIF signaling in nontransformed renal epithelia (101–103, 137, 139). As a global regulator of cellular adaptation to hypoxia, HIF regulates critical biological processes important for the survival of acutely hypoxic cells, such as anaerobic glycolysis (Pasteur effect), protein translation, cellular proliferation, and apoptosis. The role of HIF in the regulation of mitochondrial signaling, hypoxic cell death, and recovery from ischemia-reperfusion injury is controversial and most likely dependent on the cell type examined and the experimental conditions used (for a recent discussion on this topic, see Refs. 10, 37, and 44). Genetic studies with conditional knockout mice, for example, showed that HIF-1-deficient rodent brains (neuron-specific knockout) can either be protected from (46) or become more susceptible to (19) cerebral ischemic injury depending on the experimental model used, supporting the notion that “cytoprotective” effects of HIF-1 may be context dependent.
With regard to acute ischemic renal injury, we have shown that when glucose availability is limited, HIF-1 regulates the onset of hypoxia-induced cell death through its effects on glucose uptake and metabolism (10). Under hypoxic conditions, HIF-1-competent cells demonstrated increased glucose uptake and consumption, due to upregulation of glucose transporters and glycolytic enzymes, resulting in faster depletion of glucose resources and thus earlier cell death compared with HIF-1-deficient cells. We furthermore found that HIF-1, in the presence of glucose, was not required for renal epithelial cell survival under hypoxia and did not play a role in the execution of hypoxia-mediated cell death (10). Nevertheless, the role of HIF-1 in acute ischemia-reperfusion injury in vivo may be different. In vivo studies with kidney-specific HIF-1α conditional knockout mice are currently ongoing in our laboratory to address this question.
Despite its controversial role in acute hypoxic cell death, HIF-1 is known to upregulate factors that have been shown to be cytoprotective in hypoxic renal injury, including VEGF (59, 61), HO-1 (4, 49, 84, 133), and EPO (132). Pretreatment of rodents with a HIF prolyl-hydroxylase inhibitor (7, 38) or cobalt chloride (68, 81, 125), a chelating agent known to inhibit HIF-α proteolysis, resulted in improved glomerular filtration rate after ischemia and in models of acute nephrotoxic and glomerular injury associated with hypoxia, providing evidence that HIF-1 signaling is involved in “ischemic preconditioning” of the kidney, as has been proposed for other organs (14, 96). Although it is unclear at the moment which signaling pathways and which renal cell types mediate this protective effect, inhibition of HIF prolyl-hydroxylation may be a powerful strategy in improving clinical outcome of ischemia-reperfusion injuries.
Chronic Hypoxic Injury
Chronic hypoxia has long been thought to be a major factor in the progression of chronic renal diseases irrespective of the underlying cause (29), as renal “scarring” is associated with loss of microvasculature. Moreover, recent work has suggested that discrepancies between oxygen demand and supply can even occur “early” in diseased kidneys before visible scarring is detected (82). Work by our laboratory and others supports the notion that HIF signaling could potentially promote the development of renal fibrosis by at least three mechanisms: 1) direct regulation of fibrogenic factors and synergy with transforming growth factor-β1 (TGF-β1), which is a potent profibrotic factor; 2) its potential role in EMT; and 3) its role in inflammation. In contrast to its potential profibrotic role, HIF may also have a protective function through its pro-angiogenic and cytoprotective effects under certain chronic renal disease conditions (126, 127, 139).
HIF and profibrotic gene expression
Hypoxia induces collagen I, decreases matrix-metallopeptidase 2, and increases TIMP-1 in renal epithelial cells (89). Hypoxia can also act synergistically with TGF-β1 in the regulation of certain hypoxia-responsive genes such as VEGF (108), endoglin (109), and EPO (110). Synergistic effects between hypoxia and TGF-β1 have furthermore been demonstrated with regard to the production of collagens (26, 107). These observations and the finding that several genes which play critical roles in renal fibrogenesis are direct HIF-1 targets [e.g., TIMP-1 (88), PAI-1 (60), CTGF (47)] suggest that increased HIF activity is likely to play an important role in the pathogenesis of tubulointerstitial fibrosis through direct transcriptional regulation of specific profibrotic genes and/or through enhancement of TGF-β1 signaling. Synergistic interaction between SMAD3, a downstream effector of TGF-β1, and HIF-1 has been suggested by Sanchez-Elsner et al. (108) as a possible mechanism in the transcriptional regulation of VEGF. Although a direct role for HIF has yet to be demonstrated, hypoxia also increases SMAD3 mRNA levels and promotes the thrombospondin-dependent release of latent TGF-β, thus activating TGF-β signaling (143). Whereas the concept of direct regulation of profibrotic genes by HIF-1 is straightforward and consistent with the canonical hypoxia response, the interplay of HIF-1 and TGF-β signaling appears to be complex and is more difficult to understand.
HIF and EMT
Elegant in vivo studies have shown that EMT contributes significantly to the development of renal fibrosis (58). EMT is characterized by the disassembly of intercellular contacts, such as E-cadherin adherens junctions, leading to cell-cell separations associated with an increase in motility and reorganization of the actin cytoskeleton. This eventually results in the generation of fibroblast-like cells that express mesenchymal markers and display increased motility and invasiveness (58). We and others shown that hypoxia increased the percentage of transitioned renal epithelial cells in a HIF-dependent fashion in vitro and in vivo (48, 62, 78), supporting a role for HIF-1 in the dedifferentiation and transition of renal epithelial to mesenchymal fibroblast-like cells. The observation that hypoxia, through HIF, influences the differentiation state of cells has also been made in other biological systems (1, 2, 21, 22, 86, 123, 140). Although the underlying molecular mechanisms may differ between cell types, an increase in Notch signaling as a result of direct biochemical interaction between HIF-1α and the Notch ICD may be one of the mechanisms by which cells are maintained in an undifferentiated state, as has been suggested by Gustaffsson et al. (39). How important this interaction will be for the hypoxic induction of EMT in the context of renal fibrosis remains to be investigated.
HIF and inflammation
A third mechanism by which HIF may impact the pathogenesis of tubulointerstitial disease is through the regulation of inflammatory responses. Microenvironmental changes, such as hypoxia, strongly impact inflammatory cell recruitment (66) and function (23). Elegant studies with tissue-specific HIF knockout mice showed that HIF-1 is essential for myeloid cell-mediated inflammation mainly through its effects on cellular ATP generation. Inactivation of HIF-1 resulted in a profound impairment of myeloid cell aggregation, motility, and invasiveness, whereas forced expression of HIF-1 had the opposite effect (66). Furthermore, hypoxia and HIF-1 in lymphocytes have been shown to modulate lymphocytic function and T cell receptor signaling (15, 63, 87, 115). Thus, given these observations, it is easy to conceive that alterations in HIF signaling in inflammatory cells may also play a significant role in renal inflammation and subsequent fibrosis and thus in the progression of chronic renal disease.
HIF AND RENAL CANCER
The most common form of kidney cancer is renal cell cancer of the clear cell type (CC-RCC). A molecular hallmark of sporadic CC-RCC and hereditary CC-RCC associated with the von Hippel-Lindau familial tumor syndrome are mutations in the VHL tumor suppressor pVHL. Loss of pVHL function results in oxygen-independent HIF-α stabilization, increased HIF transcriptional activity, and constitutive upregulation of HIF target genes. While patients with sporadic CC-RCC are characterized by somatic inactivation of both VHL gene copies in renal epithelial cells, patients with the VHL tumor syndrome transmit germ line mutations of the VHL gene. VHL patients are predisposed to develop an autosomal dominant familial tumor syndrome that follows Knudson’s two-hit hypothesis (loss of the remaining wild-type allele in tumors). VHL disease is characterized by the development of highly vascularized, benign and malignant neoplasms in multiple organs. Typical disease manifestations include renal cysts, which sometimes can mimic autosomal dominant polycystic kidney disease (18), renal cell carcinomas, hemangioblastomas of the retina and central nervous system, and pheochromocytomas (74). Although the highly vascular nature of VHL-deficient tumors is easily explained by increased VEGF production as a result of increased HIF transcriptional activity, VHL-associated renal carcinogenesis is more difficult to understand and most likely requires multiple other genetic events in addition to loss of pVHL function. Besides regulating the degradation of HIF-α-subunits, pVHL has been shown to have additional biological functions, which may or may not be critical for renal tumorigenesis (41).
Aside from a regulatory role in tumor angiogenesis, HIF plays a key role in the regulation of factors that are important for the development and invasiveness of CC-RCC. These include, among others, TGF-α, a potent renal epithelial mitogen, cell cycle regulator cyclin D1 (CCND1), and chemokine receptor CXCR4 (11, 97, 116, 119, 138, 141). With regard to the individual contribution of HIF-1 and HIF-2 to renal tumor development, it is of interest that a substantial number of VHL-defective CC-RCC cell lines do not express HIF-1α, but express HIF-2α (83). This is in contrast to normal, nontransformed renal epithelial cells, in which HIF-2α is not detectable during ischemia (103). A bias toward HIF-2α expression was also found in clinical CC-RCC samples (130). Thus VHL-associated tumor development may depend on a shift in the ratio of HIF-1α vs. HIF-2α levels toward an increase in HIF-2α. In support of this hypothesis, VHL-reconstituted 786-O CC-RCC cells transfected with a nondegradable form of HIF-2α were still able to form tumors in nude mice, thereby overriding pVHL’s tumor suppressor function (65). By contrast, expression of nondegradable HIF-1α in a similar experimental setting did not have a tumor-promoting effect (80). Consistent with these findings, inactivation of HIF-2α by RNA interference suppressed tumor formation in a VHL-deficient background (64, 145). Taken together, these reports suggest that HIF-1 and HIF-2 have diverse functions with regard to VHL renal tumorigenesis. HIF-2 has been proposed to preferentially regulate signaling pathways critical for renal cell growth, such as signaling through the TGF-α /epidermal growth factor receptor pathway and through cell cycle regulator cyclin D1 (11, 97, 116, 138, 141).
In addition to VHL-associated CC-RCC, HIF-α stabilization can be found in renal cell cancers that are associated with mutations of the tuberous sclerosis tumor suppressor TSC-2 (72) and in rare leiomyomatosis-associated renal cancers. The latter form of renal cancer is characterized by fumarate hydratase deficiency, the inability to convert fumarate to malate, which results in HIF prolyl-hydroxylase inhibition; fumarate acts as a competitive inhibitor of HIF prolyl-hydroxylation (51) (Fig. 1). Whether an increase in HIF-1 and HIF-2 activity in these rare forms of renal cancer has the same biological effects as in VHL-negative CC-RCC is unclear and awaits further investigation.
SUMMARY
In this review, I have summarized the most recent findings in HIF signaling and have tried to provide the reader with a perspective on how recent advances in HIF biology may affect our understanding of renal development and disease, including renal cancer. This review was by no means intended to cover all possible aspects of HIF biology in the kidney, but instead it focuses on selected areas that have high potential for impacting current concepts of pathogenesis and treatment of kidney diseases. The reader is furthermore provided with an appreciation of the broad spectrum of cell type- and context-dependent effects of canonical and noncanonical HIF signaling, which in certain settings can be quite contrasting (e.g., its proapoptotic vs. its antiapoptotic effects). In keeping with this notion, HIF’s beneficial cytoprotective effect in acute ischemia-reperfusion injury is contrasted to its role in inflammation and its potentially profibrotic role in chronic renal hypoxia, which needs further study. These considerations are particularly important with regard to pharmacological inhibition of HIF prolyl-hydroxylases, which offers enormous potential for the treatment of anemia and acute hypoxic injuries. Rigorous investigations of HIF’s effects on metabolism, inflammation, cell growth, and differentiation are therefore required before this treatment strategy can be safely exploited in clinical practice.
Acknowledgments
I thank the National Cancer Institute, the National Institute of Diabetes and Digestive and Kidney Diseases, the National Kidney Foundation, and the American Heart Association for supporting the work in the laboratory. Furthermore, I thank Mangatt Biju, Debra Higgins, Michael Madaio, and Erinn Rankin for a critical reading of the manuscript.
Footnotes
DISCLOSURES
To remain within the scope of this review, I have limited the number of references and apologize to those colleagues whose original work was not cited. In those cases, the reader is referred to selected review articles.
References
- 1.Adelman DM, Gertsenstein M, Nagy A, Simon MC, Maltepe E. Placental cell fates are regulated in vivo by HIF-mediated hypoxia responses. Genes Dev. 2000;14:3191–3203. doi: 10.1101/gad.853700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adelman DM, Maltepe E, Simon MC. Multilineage embryonic hematopoiesis requires hypoxic ARNT activity. Genes Dev. 1999;13:2478–2483. doi: 10.1101/gad.13.19.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agani FH, Pichiule P, Chavez JC, LaManna JC. The role of mitochondria in the regulation of hypoxia-inducible factor 1 expression during hypoxia. J Biol Chem. 2000;275:35863–35867. doi: 10.1074/jbc.M005643200. [DOI] [PubMed] [Google Scholar]
- 4.Agarwal A, Nick HS. Renal response to tissue injury: lessons from heme oxygenase-1 gene ablation and expression. J Am Soc Nephrol. 2000;11:965–973. doi: 10.1681/ASN.V115965. [DOI] [PubMed] [Google Scholar]
- 5.Ang SO, Chen H, Hirota K, Gordeuk VR, Jelinek J, Guan Y, Liu E, Sergueeva AI, Miasnikova GY, Mole D, Maxwell PH, Stockton DW, Semenza GL, Prchal JT. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nature Genet. 2002;32:614–621. doi: 10.1038/ng1019. [DOI] [PubMed] [Google Scholar]
- 6.Appelhoff RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW, Ratcliffe PJ, Gleadle JM. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279:38458–38465. doi: 10.1074/jbc.M406026200. [DOI] [PubMed] [Google Scholar]
- 7.Bernhardt WM, Kany S, Campean V, Juergensen JS, Weidemann A, Warnecke C, Klaus S, Guenzler V, Amann K, Willam C, Wiesener M, Eckardt KU. Preconditional activation of HIF ameliorates ischemic acute renal failure. ASN Annual Meeting, Philadelphia, PA (Abstract) J Am Soc Nephrol. 2005;16:195A. doi: 10.1681/ASN.2005121302. [DOI] [PubMed] [Google Scholar]
- 8.Bernhardt WM, Schmitt R, Rosenberger C, Munchenhagen PM, Grone HJ, Frei U, Warnecke C, Bachmann S, Wiesener MS, Willam C, Eckardt KU. Expression of hypoxia-inducible transcription factors in developing human and rat kidneys. Kidney Int. 2006;69:114–122. doi: 10.1038/sj.ki.5000062. [DOI] [PubMed] [Google Scholar]
- 9.Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1α in normoxia. EMBO J. 2003;22:4082–4090. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Biju MP, Akai Y, Shrimanker N, Haase VH. Protection of HIF-1-deficient primary renal tubular epithelial cells from hypoxia-induced cell death is glucose dependent. Am J Physiol Renal Physiol. 2005;289:F1217–F1226. doi: 10.1152/ajprenal.00233.2005. [DOI] [PubMed] [Google Scholar]
- 11.Bindra RS, Vasselli JR, Stearman R, Linehan WM, Klausner RD. VHL-mediated hypoxia regulation of cyclin D1 in renal carcinoma cells. Cancer Res. 2002;62:3014–3019. [PubMed] [Google Scholar]
- 12.Brukamp K, Jim B, Moeller MJ, Holzman LB, Haase VH. VHL deficiency in novel podocyte cell lines results in survival disadvantage. ASN Annual Meeting, Philadelphia, PA (Abstract) J Am Soc Nephrol. 2005;16:157A. [Google Scholar]
- 13.Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M, Scarpulla RC, Chandel NS. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005;1:409–414. doi: 10.1016/j.cmet.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 14.Cai Z, Manalo DJ, Wei G, Rodriguez ER, Fox-Talbot K, Lu H, Zweier JL, Semenza GL. Hearts from rodents exposed to intermittent hypoxia or erythropoietin are protected against ischemia-reperfusion injury. Circulation. 2003;108:79–85. doi: 10.1161/01.CIR.0000078635.89229.8A. [DOI] [PubMed] [Google Scholar]
- 15.Caldwell CC, Kojima H, Lukashev D, Armstrong J, Farber M, Apasov SG, Sitkovsky MV. Differential effects of physiologically relevant hypoxic conditions on T lymphocyte development and effector functions. J Immunol. 2001;167:6140–6149. doi: 10.4049/jimmunol.167.11.6140. [DOI] [PubMed] [Google Scholar]
- 16.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1α during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000;275:25130–25138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 18.Chatha RK, Johnson AM, Rothberg PG, Townsend RR, Neumann HP, Gabow PA. Von Hippel-Lindau disease masquerading as autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2001;37:852–858. doi: 10.1016/s0272-6386(01)80136-0. [DOI] [PubMed] [Google Scholar]
- 19.Chavez JC, Baranova O, LanManna JC, Ratan RR. Brain specific deletion of the HIF-1α gene increases ischemic injury in a mouse model of focal cerebral ischemia (Hypoxia and Development, Physiology and Disease, Breckenridge, CO, Keystone Symposia). Silverthorne, CO. Keystone Symposia on Molecular and Cellular Biology; 2006. p. 67. [Google Scholar]
- 20.Compernolle V, Brusselmans K, Acker T, Hoet P, Tjwa M, Beck H, Plaisance S, Dor Y, Keshet E, Lupu F, Nemery B, Dewerchin M, Van Veldhoven P, Plate K, Moons L, Collen D, Carmeliet P. Loss of HIF-2α and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med. 2002;8:702–710. doi: 10.1038/nm721. [DOI] [PubMed] [Google Scholar]
- 21.Covello KL, Kehler J, Yu H, Gordan JD, Arsham AM, Hu CJ, Labosky PA, Simon MC, Keith B. HIF-2α regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006;20:557–570. doi: 10.1101/gad.1399906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cowden Dahl KD, Fryer BH, Mack FA, Compernolle V, Maltepe E, Adelman DM, Carmeliet P, Simon MC. Hypoxia-inducible factors 1α and 2α regulate trophoblast differentiation. Mol Cell Biol. 2005;25:10479–10491. doi: 10.1128/MCB.25.23.10479-10491.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS. HIF-1α is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ding M, Cui S, Binnie M, Jothy S, Li C, Nagy A, Steer BM, Marsden P, Rastaldi MP, Haase VH, Cohen CD, Kretzler M, Quaggin SE. Loss of the tumor suppressor VHL leads to rapidly progressive glomerulonephritis and pulmonary hemorrhage in mice. ASN Annual Meeting, Philadelphia, PA (Abstract) J Am Soc Nephrol. 2005;16:107A. [Google Scholar]
- 25.Enomoto N, Koshikawa N, Gassmann M, Hayashi J, Takenaga K. Hypoxic induction of hypoxia-inducible factor-1α and oxygen-regulated gene expression in mitochondrial DNA-depleted HeLa cells. Biochem Biophys Res Commun. 2002;297:346–352. doi: 10.1016/s0006-291x(02)02186-1. [DOI] [PubMed] [Google Scholar]
- 26.Falanga V, Zhou L, Yufit T. Low oxygen tension stimulates collagen synthesis and COL1A1 transcription through the action of TGF-β1. J Cell Physiol. 2002;191:42–50. doi: 10.1002/jcp.10065. [DOI] [PubMed] [Google Scholar]
- 27.Fandrey J. Oxygen-dependent and tissue-specific regulation of erythropoietin gene expression. Am J Physiol Regul Integr Comp Physiol. 2004;286:R977–R988. doi: 10.1152/ajpregu.00577.2003. [DOI] [PubMed] [Google Scholar]
- 28.Feldser D, Agani F, Iyer NV, Pak B, Ferreira G, Semenza GL. Reciprocal positive regulation of hypoxia-inducible factor 1α and insulin-like growth factor 2. Cancer Res. 1999;59:3915–3918. [PubMed] [Google Scholar]
- 29.Fine LG, Bandyopadhay D, Norman JT. Is there a common mechanism for the progression of different types of renal diseases other than proteinuria? Towards the unifying theme of chronic hypoxia. Kidney Int. 2000;57(Suppl 75):S22–S26. [PubMed] [Google Scholar]
- 30.Fleming RE, Bacon BR. Orchestration of iron homeostasis. N Engl J Med. 2005;352:1741–1744. doi: 10.1056/NEJMp048363. [DOI] [PubMed] [Google Scholar]
- 31.Freeburg PB, Robert B, St John PL, Abrahamson DR. Podocyte expression of hypoxia-inducible factor (HIF)-1 and HIF-2 during glomerular development. J Am Soc Nephrol. 2003;14:927–938. doi: 10.1097/01.asn.0000059308.82322.4f. [DOI] [PubMed] [Google Scholar]
- 32.Fukuda R, Hirota K, Fan F, Jung YD, Ellis LM, Semenza GL. Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J Biol Chem. 2002;277:38205–38211. doi: 10.1074/jbc.M203781200. [DOI] [PubMed] [Google Scholar]
- 33.Ganz T, Nemeth E. Iron imports. IV. Hepcidin and regulation of body iron metabolism. Am J Physiol Gastrointest Liver Physiol. 2006;290:G199–G203. doi: 10.1152/ajpgi.00412.2005. [DOI] [PubMed] [Google Scholar]
- 34.Gerald D, Berra E, Frapart YM, Chan DA, Giaccia AJ, Mansuy D, Pouyssegur J, Yaniv M, Mechta-Grigoriou F. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118:781–794. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 35.Gnarra JR, Ward JM, Porter FD, Wagner JR, Devor DE, Grinberg A, Emmert-Buck MR, Westphal H, Klausner RD, Linehan WM. Defective placental vasculogenesis causes embryonic lethality in VHL-deficient mice. Proc Natl Acad Sci USA. 1997;94:9102–9107. doi: 10.1073/pnas.94.17.9102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gordeuk VR, Sergueeva AI, Miasnikova GY, Okhotin D, Voloshin Y, Choyke PL, Butman JA, Jedlickova K, Prchal JT, Polyakova LA. Congenital disorder of oxygen sensing: association of the homozygous Chuvash polycythemia VHL mutation with thrombosis and vascular abnormalities but not tumors. Blood. 2004;103:3924–3932. doi: 10.1182/blood-2003-07-2535. [DOI] [PubMed] [Google Scholar]
- 37.Greijer AE, van der Wall E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J Clin Pathol. 2004;57:1009–1014. doi: 10.1136/jcp.2003.015032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo G, Lin A, Guenzler V, Liu DY, Klaus S, Arend M, Flippin L, Langsetmo I, Witschi C, Wang QJ. Improvement of kidney function in a rat model of renal ischemia-reperfusion injury by treatment with a novel HIF prolyl-hydroxylase inhibitor. ASN Annual Meeting, St. Louis, MO (Abstract) J Am Soc Nephrol. 2004;15:460A. [Google Scholar]
- 39.Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, Ruas JL, Poellinger L, Lendahl U, Bondesson M. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell. 2005;9:617–628. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 40.Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 41.Haase VH. The VHL tumor suppressor in development and disease: functional studies in mice by conditional gene targeting. Semin Cell Dev Biol. 2005;16:564–574. doi: 10.1016/j.semcdb.2005.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haase VH, Glickman JN, Socolovsky M, Jaenisch R. Vascular tumors in livers with targeted inactivation of the von Hippel-Lindau tumor suppressor. Proc Natl Acad Sci USA. 2001;98:1583–1588. doi: 10.1073/pnas.98.4.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hagen T, Taylor CT, Lam F, Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1α. Science. 2003;302:1975–1978. doi: 10.1126/science.1088805. [DOI] [PubMed] [Google Scholar]
- 44.Harris AL. Hypoxia—a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 45.Hellwig-Burgel T, Rutkowski K, Metzen E, Fandrey J, Jelkmann W. Interleukin-1β and tumor necrosis factor-β stimulate DNA binding of hypoxia-inducible factor-1. Blood. 1999;94:1561–1567. [PubMed] [Google Scholar]
- 46.Helton R, Cui J, Scheel JR, Ellison JA, Ames C, Gibson C, Blouw B, Ouyang L, Dragatsis I, Zeitlin S, Johnson RS, Lipton SA, Barlow C. Brain-specific knock-out of hypoxia-inducible factor-1α reduces rather than increases hypoxic-ischemic damage. J Neurosci. 2005;25:4099–4107. doi: 10.1523/JNEUROSCI.4555-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Higgins DF, Biju MP, Akai Y, Wutz A, Johnson RS, Haase VH. Hypoxic induction of Ctgf is directly mediated by Hif-1. Am J Physiol Renal Physiol. 2004;287:F1223–F1232. doi: 10.1152/ajprenal.00245.2004. [DOI] [PubMed] [Google Scholar]
- 48.Higgins DF, Johnson RS, Haase VH. Hypoxic signaling enhances renal fibrosis (Hypoxia and Development, Physiology and Disease, Breckenridge, CO, Keystone Symposia). Silverthorne, CO. Keystone Symposia on Molecular and Cellular Biology; 2006. p. 74. [Google Scholar]
- 49.Horikawa S, Yoneya R, Nagashima Y, Hagiwara K, Ozasa H. Prior induction of heme oxygenase-1 with glutathione depletor ameliorates the renal ischemia and reperfusion injury in the rat. FEBS Lett. 2002;510:221–224. doi: 10.1016/s0014-5793(01)03270-7. [DOI] [PubMed] [Google Scholar]
- 50.Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1α (HIF-1α) and HIF-2α in hypoxic gene regulation. Mol Cell Biol. 2003;23:9361–9374. doi: 10.1128/MCB.23.24.9361-9374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung YL, Merino M, Trepel J, Zbar B, Toro J, Ratcliffe PJ, Linehan WM, Neckers L. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8:143–153. doi: 10.1016/j.ccr.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 52.Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jelkmann W. Molecular biology of erythropoietin. Intern Med. 2004;43:649–659. doi: 10.2169/internalmedicine.43.649. [DOI] [PubMed] [Google Scholar]
- 54.Jiang BH, Jiang G, Zheng JZ, Lu Z, Hunter T, Vogt PK. Phosphatidylinositol 3-kinase signaling controls levels of hypoxia-inducible factor 1. Cell Growth Differ. 2001;12:363–369. [PubMed] [Google Scholar]
- 55.Jiang BH, Semenza GL, Bauer C, Marti HH. Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am J Physiol Cell Physiol. 1996;271:C1172–C1180. doi: 10.1152/ajpcell.1996.271.4.C1172. [DOI] [PubMed] [Google Scholar]
- 56.Kaelin WG. Proline hydroxylation and gene expression. Annu Rev Biochem. 2005;74:115–128. doi: 10.1146/annurev.biochem.74.082803.133142. [DOI] [PubMed] [Google Scholar]
- 57.Kairaitis LK, Wang Y, Gassmann M, Tay YC, Harris DC. HIF-1α expression follows microvascular loss in advanced murine adriamycin nephrosis. Am J Physiol Renal Physiol. 2005;288:F198–F206. doi: 10.1152/ajprenal.00244.2003. [DOI] [PubMed] [Google Scholar]
- 58.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kang DH, Hughes J, Mazzali M, Schreiner GF, Johnson RJ. Impaired angiogenesis in the remnant kidney model. II. Vascular endothelial growth factor administration reduces renal fibrosis and stabilizes renal function. J Am Soc Nephrol. 2001;12:1448–1457. doi: 10.1681/ASN.V1271448. [DOI] [PubMed] [Google Scholar]
- 60.Kietzmann T, Roth U, Jungermann K. Induction of the plasminogen activator inhibitor-1 gene expression by mild hypoxia via a hypoxia response element binding the hypoxia-inducible factor-1 in rat hepatocytes. Blood. 1999;94:4177–4185. [PubMed] [Google Scholar]
- 61.Kim YG, Suga SI, Kang DH, Jefferson JA, Mazzali M, Gordon KL, Matsui K, Breiteneder-Geleff S, Shankland SJ, Hughes J, Kerjaschki D, Schreiner GF, Johnson RJ. Vascular endothelial growth factor accelerates renal recovery in experimental thrombotic microangiopathy. Kidney Int. 2000;58:2390–2399. doi: 10.1046/j.1523-1755.2000.00422.x. [DOI] [PubMed] [Google Scholar]
- 62.Kimura K, Iwano M, Akai Y, Nakatani K, Harada K, Yamaguchi Y, Saito Y, Johnson RS, Neilson EG, Haase VH. HIF-1α is a key molecule for the progression of EMT-mediated renal fibrosis. ASN Annual Meeting, Philadelphia, PA (Abstract) J Am Soc Nephrol. 2005;16:656A. [Google Scholar]
- 63.Kojima H, Gu H, Nomura S, Caldwell CC, Kobata T, Carmeliet P, Semenza GL, Sitkovsky MV. Abnormal B lymphocyte development and autoimmunity in hypoxia-inducible factor 1α-deficient chimeric mice. Proc Natl Acad Sci USA. 2002;99:2170–2174. doi: 10.1073/pnas.052706699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kondo K, Kim WY, Lechpammer M, Kaelin WG., Jr Inhibition of HIF2α is sufficient to suppress pVHL-defective tumor growth (Abstract) PLoS Biol. 2003;1:E83. doi: 10.1371/journal.pbio.0000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kondo K, Klco JM, Nakamura E, Lechpammer M, Kaelin WG. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell. 2002;1:237–246. doi: 10.1016/s1535-6108(02)00043-0. [DOI] [PubMed] [Google Scholar]
- 66.Kong T, Eltzschig HK, Karhausen J, Colgan SP, Shelley CS. Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of β2 integrin gene expression. Proc Natl Acad Sci USA. 2004;101:10440–10445. doi: 10.1073/pnas.0401339101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC, Huang LE. HIF-1α induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004;23:1949–1956. doi: 10.1038/sj.emboj.7600196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kudo Y, Kakinuma Y, Mori Y, Morimoto N, Karashima T, Furihata M, Sato T, Shuin T, Sugiura T. Hypoxia-inducible factor-1α is involved in the attenuation of experimentally induced rat glomerulonephritis. Nephron Exp Nephrol. 2005;100:e95–e103. doi: 10.1159/000084575. [DOI] [PubMed] [Google Scholar]
- 69.Langsetmo I, Nichols B, Seeley T, Stephenson B, Klaus S, Lin A, Liu DY. FG-2216 corrects anemia and improves iron utilization in a rat model of anemia of chronic disease: comparison to darbepoetin. ASN Annual Meeting, Philadelphia, PA (Abstract) J Am Soc Nephrol. 2005;16:481A. [Google Scholar]
- 70.Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1α (HIF-1α) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol. 2001;21:3995–4004. doi: 10.1128/MCB.21.12.3995-4004.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu DY, Neff TB, Guenzler V, Langsetmo I, Fourney P, Nichols B, Muthukrishnan E, Lin A, Klaus S. Novel and beneficial pharmacodynamic properties of endogenous EPO and complete erythropoiesis induced by selective HIF prolyl hydroxylase inhibitors. ASN Annual Meeting, Philadelphia, PA (Abstract) J Am Soc Nephrol. 2005;16:761A. [Google Scholar]
- 72.Liu MY, Poellinger L, Walker CL. Up-regulation of hypoxia-inducible factor 2α in renal cell carcinoma associated with loss of Tsc-2 tumor suppressor gene. Cancer Res. 2003;63:2675–2680. [PubMed] [Google Scholar]
- 73.Lok CN, Ponka P. Identification of a hypoxia response element in the transferrin receptor gene. J Biol Chem. 1999;274:24147–24152. doi: 10.1074/jbc.274.34.24147. [DOI] [PubMed] [Google Scholar]
- 74.Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, Oldfield EH. von Hippel-Lindau disease. Lancet. 2003;361:2059–2067. doi: 10.1016/S0140-6736(03)13643-4. [DOI] [PubMed] [Google Scholar]
- 75.Makino Y, Cao R, Svensson K, Bertilsson G, Asman M, Tanaka H, Cao Y, Berkenstam A, Poellinger L. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature. 2001;414:550–554. doi: 10.1038/35107085. [DOI] [PubMed] [Google Scholar]
- 76.Maltepe E, Schmidt JV, Baunoch D, Bradfield CA, Simon MC. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature. 1997;386:403–407. doi: 10.1038/386403a0. [DOI] [PubMed] [Google Scholar]
- 77.Mandriota SJ, Turner KJ, Davies DR, Murray PG, Morgan NV, Sowter HM, Wykoff CC, Maher ER, Harris AL, Ratcliffe PJ, Maxwell PH. HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron. Cancer Cell. 2002;1:459–468. doi: 10.1016/s1535-6108(02)00071-5. [DOI] [PubMed] [Google Scholar]
- 78.Manotham K, Tanaka T, Matsumoto M, Ohse T, Inagi R, Miyata T, Kurokawa K, Fujita T, Ingelfinger JR, Nangaku M. Transdifferentiation of cultured tubular cells induced by hypoxia. Kidney Int. 2004;65:871–880. doi: 10.1111/j.1523-1755.2004.00461.x. [DOI] [PubMed] [Google Scholar]
- 79.Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT, Simon MC. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-α activation. Cell Metab. 2005;1:393–399. doi: 10.1016/j.cmet.2005.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Maranchi JK, Vasselli JR, Riss J, Bonifacio JS, Linehan WM, Klausner RD. The contribution of VHL substrate binding and HIF-1α to the phenotype of vhl loss in renal cell carcinoma. Cancer Cell. 2002;1:247–253. doi: 10.1016/s1535-6108(02)00044-2. [DOI] [PubMed] [Google Scholar]
- 81.Matsumoto M, Makino Y, Tanaka T, Tanaka H, Ishizaka N, Noiri E, Fujita T, Nangaku M. Induction of renoprotective gene expression by cobalt ameliorates ischemic injury of the kidney in rats. J Am Soc Nephrol. 2003;14:1825–1832. doi: 10.1097/01.asn.0000074239.22357.06. [DOI] [PubMed] [Google Scholar]
- 82.Matsumoto M, Tanaka T, Yamamoto T, Noiri E, Miyata T, Inagi R, Fujita T, Nangaku M. Hypoperfusion of peritubular capillaries induces chronic hypoxia before progression of tubulointerstitial injury in a progressive model of rat glomerulonephritis. J Am Soc Nephrol. 2004;15:1574–1581. doi: 10.1097/01.asn.0000128047.13396.48. [DOI] [PubMed] [Google Scholar]
- 83.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 84.Morimoto K, Ohta K, Yachie A, Yang Y, Shimizu M, Goto C, Toma T, Kasahara Y, Yokoyama H, Miyata T, Seki H, Koizumi S. Cytoprotective role of heme oxygenase (HO)-1 in human kidney with various renal diseases. Kidney Int. 2001;60:1858–1866. doi: 10.1046/j.1523-1755.2001.01000.x. [DOI] [PubMed] [Google Scholar]
- 85.Morita M, Ohneda O, Yamashita T, Takahashi S, Suzuki N, Nakajima O, Kawauchi S, Ema M, Shibahara S, Udono T, Tomita K, Tamai M, Sogawa K, Yamamoto M, Fujii-Kuriyama Y. HLF/ HIF-2α is a key factor in retinopathy of prematurity in association with erythropoietin. EMBO J. 2003;22:1134–1146. doi: 10.1093/emboj/cdg117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Morrison SJ, Csete M, Groves AK, Melega W, Wold B, Anderson DJ. Culture in reduced levels of oxygen promotes clonogenic sympathoadrenal differentiation by isolated neural crest stem cells. J Neurosci. 2000;20:7370–7376. doi: 10.1523/JNEUROSCI.20-19-07370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Neumann AK, Yang J, Biju MP, Joseph SK, Johnson RS, Haase VH, Freedman BD, Turka LA. Hypoxia inducible factor 1α regulates T cell receptor signal transduction. Proc Natl Acad Sci USA. 2005;102:17071–17076. doi: 10.1073/pnas.0506070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Norman JT, Clark IM, Garcia PL. Hypoxia promotes fibrogenesis in human renal fibroblasts. Kidney Int. 2000;58:2351–2366. doi: 10.1046/j.1523-1755.2000.00419.x. [DOI] [PubMed] [Google Scholar]
- 89.Orphanides C, Fine LG, Norman JT. Hypoxia stimulates proximal tubular cell matrix production via a TGF-β1-independent mechanism. Kidney Int. 1997;52:637–647. doi: 10.1038/ki.1997.377. [DOI] [PubMed] [Google Scholar]
- 90.Pastore Y, Jedlickova K, Guan Y, Liu E, Fahner J, Hasle H, Prchal JF, Prchal JT. Mutations of von Hippel-Lindau tumor-suppressor gene and congenital polycythemia. Am J Hum Genet. 2003;73:412–419. doi: 10.1086/377108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Peng J, Zhang L, Drysdale L, Fong GH. The transcription factor EPAS-1/hypoxia-inducible factor 2α plays an important role in vascular remodeling. Proc Natl Acad Sci USA. 2000;97:8386–8391. doi: 10.1073/pnas.140087397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Percy MJ, Zhao Q, Flores A, Harrison C, Lappin TR, Maxwell PH, McMullin MF, Lee FS. A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc Natl Acad Sci USA. 2006;103:654–659. doi: 10.1073/pnas.0508423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rankin EB, Higgins DF, Walisser JA, Johnson RS, Bradfield CA, Haase VH. Inactivation of the arylhydrocarbon receptor nuclear translocator (Arnt) suppresses von Hippel-Lindau disease-associated vascular tumors in mice. Mol Cell Biol. 2005;25:3163–3172. doi: 10.1128/MCB.25.8.3163-3172.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rankin EB, Keith B, Simon C, Haase VH. Inactivation of HIF-2α suppresses the VHL associated phenotype in the liver (Hypoxia and Development, Physiology and Disease, Breckenridge, Co. Keystone Symposia). Silverthorne, CO. Keystone Symposia on Molecular and Cellular Biology; 2006. p. 92. [Google Scholar]
- 95.Rankin EB, Tomaszewski JE, Haase VH. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res. 2006;66:2576–2583. doi: 10.1158/0008-5472.CAN-05-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ratan RR, Siddiq A, Aminova L, Lange PS, Langley B, Ayoub I, Gensert J, Chavez J. Translation of ischemic preconditioning to the patient: prolyl hydroxylase inhibition and hypoxia inducible factor-1 as novel targets for stroke therapy. Stroke. 2004;35:2687–2689. doi: 10.1161/01.STR.0000143216.85349.9e. [DOI] [PubMed] [Google Scholar]
- 97.Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, Pugh CW, Maxwell PH, Harris AL, Ratcliffe PJ. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol. 2005;25:5675–5686. doi: 10.1128/MCB.25.13.5675-5686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, Dillehay LE, Madan A, Semenza GL, Bedi A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1α. Genes Dev. 2000;14:34–44. [PMC free article] [PubMed] [Google Scholar]
- 99.Richard DE, Berra E, Pouyssegur J. Nonhypoxic pathway mediates the induction of hypoxia-inducible factor 1α in vascular smooth muscle cells. J Biol Chem. 2000;275:26765–26771. doi: 10.1074/jbc.M003325200. [DOI] [PubMed] [Google Scholar]
- 100.Rolfs A, Kvietikova I, Gassmann M, Wenger RH. Oxygen-regulated transferrin expression is mediated by hypoxia-inducible factor-1. J Biol Chem. 1997;272:20055–20062. doi: 10.1074/jbc.272.32.20055. [DOI] [PubMed] [Google Scholar]
- 101.Rosenberger C, Griethe W, Gruber G, Wiesener M, Frei U, Bachmann S, Eckardt KU. Cellular responses to hypoxia after renal segmental infarction. Kidney Int. 2003;64:874–886. doi: 10.1046/j.1523-1755.2003.00159.x. [DOI] [PubMed] [Google Scholar]
- 102.Rosenberger C, Heyman SN, Rosen S, Shina A, Goldfarb M, Griethe W, Frei U, Reinke P, Bachmann S, Eckardt KU. Up-regulation of HIF in experimental acute renal failure: evidence for a protective transcriptional response to hypoxia. Kidney Int. 2005;67:531–542. doi: 10.1111/j.1523-1755.2005.67110.x. [DOI] [PubMed] [Google Scholar]
- 103.Rosenberger C, Mandriota S, Jurgensen JS, Wiesener MS, Horstrup JH, Frei U, Ratcliffe PJ, Maxwell PH, Bachmann S, Eckardt KU. Expression of hypoxia-inducible factor-1α and -2α in hypoxic and ischemic rat kidneys. J Am Soc Nephrol. 2002;13:1721–1732. doi: 10.1097/01.asn.0000017223.49823.2a. [DOI] [PubMed] [Google Scholar]
- 104.Ruas JL, Poellinger L. Hypoxia-dependent activation of HIF into a transcriptional regulator. Semin Cell Dev Biol. 2005;16:514–522. doi: 10.1016/j.semcdb.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 105.Ryan HE, Lo J, Johnson RS. HIF-1 α is required for solid tumor formation and embryonic vascularization. EMBO J. 1998;17:3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ryan HE, Poloni M, McNulty W, Elson D, Gassmann M, Arbeit JM, Johnson RS. Hypoxia-inducible factor-1α is a positive factor in solid tumor growth. Cancer Res. 2000;60:4010–4015. [PubMed] [Google Scholar]
- 107.Saed GM, Zhang W, Chegini N, Holmdahl L, Diamond MP. Alteration of type I and III collagen expression in human peritoneal mesothelial cells in response to hypoxia and transforming growth factor-α 1. Wound Repair Regen. 1999;7:504–510. doi: 10.1046/j.1524-475x.1999.00504.x. [DOI] [PubMed] [Google Scholar]
- 108.Sanchez-Elsner T, Botella LM, Velasco B, Corbi A, Attisano L, Bernabeu C. Synergistic cooperation between hypoxia and transforming growth factor-β pathways on human vascular endothelial growth factor gene expression. J Biol Chem. 2001;276:38527–38535. doi: 10.1074/jbc.M104536200. [DOI] [PubMed] [Google Scholar]
- 109.Sanchez-Elsner T, Botella LM, Velasco B, Langa C, Bernabeu C. Endoglin expression is regulated by transcriptional cooperation between the hypoxia and transforming growth factor-β pathways. J Biol Chem. 2002;277:43799–43808. doi: 10.1074/jbc.M207160200. [DOI] [PubMed] [Google Scholar]
- 110.Sanchez-Elsner T, Ramirez JR, Sanz-Rodriguez F, Varela E, Bernabeu C, Botella LM. A cross-talk between hypoxia and TGF-β orchestrates erythropoietin gene regulation through SP1 and Smads. J Mol Biol. 2004;336:9–24. doi: 10.1016/j.jmb.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 111.Sandau KB, Zhou J, Kietzmann T, Brune B. Regulation of the hypoxia-inducible factor 1α by the inflammatory mediators nitric oxide and tumor necrosis factor-α in contrast to desferroxamine and phenyl-arsine oxide. J Biol Chem. 2001;276:39805–39811. doi: 10.1074/jbc.M107689200. [DOI] [PubMed] [Google Scholar]
- 112.Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 113.Scortegagna M, Ding K, Oktay Y, Gaur A, Thurmond F, Yan LJ, Marck BT, Matsumoto AM, Shelton JM, Richardson JA, Bennett MJ, Garcia JA. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1−/− mice. Nat Genet. 2003;35:331–340. doi: 10.1038/ng1266. [DOI] [PubMed] [Google Scholar]
- 114.Scortegagna M, Ding K, Zhang Q, Oktay Y, Bennett MJ, Bennett M, Shelton JM, Richardson JA, Moe O, Garcia JA. HIF-2α regulates murine hematopoietic development in an erythropoietin-dependent manner. Blood. 2005;105:3133–3140. doi: 10.1182/blood-2004-05-1695. [DOI] [PubMed] [Google Scholar]
- 115.Sitkovsky M, Lukashev D. Regulation of immune cells by local-tissue oxygen tension: HIF1α and adenosine receptors. Nat Rev Immunol. 2005;5:712–721. doi: 10.1038/nri1685. [DOI] [PubMed] [Google Scholar]
- 116.Smith K, Gunaratnam L, Morley M, Franovic A, Mekhail K, Lee S. Silencing of epidermal growth factor receptor suppresses hypoxia-inducible factor-2-driven VHL−/− renal cancer. Cancer Res. 2005;65:5221–5230. doi: 10.1158/0008-5472.CAN-05-0169. [DOI] [PubMed] [Google Scholar]
- 117.Soilleux EJ, Turley H, Tian YM, Pugh CW, Gatter KC, Harris AL. Use of novel monoclonal antibodies to determine the expression and distribution of the hypoxia regulatory factors PHD-1, PHD-2, PHD-3 and FIH in normal and neoplastic human tissues. Histopathology. 2005;47:602–610. doi: 10.1111/j.1365-2559.2005.02280.x. [DOI] [PubMed] [Google Scholar]
- 118.Srinivas V, Leshchinsky I, Sang N, King MP, Minchenko A, Caro J. Oxygen sensing and HIF-1 activation does not require an active mitochondrial respiratory chain electron-transfer pathway. J Biol Chem. 2001;276:21995–21998. doi: 10.1074/jbc.C100177200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature. 2003;425:307–311. doi: 10.1038/nature01874. [DOI] [PubMed] [Google Scholar]
- 120.Steenhard B, Isom K, Stroganova L, St John PL, Freeburg PB, Holzman LB, Abrahamson DR. Podocyte-selective deletion of von Hippel-Lindau (VHL) protein causes albuminuria. ASN Annual Meeting, Philadelphia, PA (Abstract) J Am Soc Nephrol. 2005;16:667A. [Google Scholar]
- 121.Stiehl DP, Jelkmann W, Wenger RH, Hellwig-Burgel T. Normoxic induction of the hypoxia-inducible factor 1α by insulin and interleukin-1β involves the phosphatidylinositol 3-kinase pathway. FEBS Lett. 2002;512:157–162. doi: 10.1016/s0014-5793(02)02247-0. [DOI] [PubMed] [Google Scholar]
- 122.Stolze IP, Tian YM, Appelhoff RJ, Turley H, Wykoff CC, Gleadle JM, Ratcliffe PJ. Genetic analysis of the role of the asparaginyl hydroxylase factor inhibiting hypoxia-inducible factor (HIF) in regulating HIF transcriptional target genes. J Biol Chem. 2004;279:42719–42725. doi: 10.1074/jbc.M406713200. [DOI] [PubMed] [Google Scholar]
- 123.Studer L, Csete M, Lee SH, Kabbani N, Walikonis J, Wold B, McKay R. Enhanced proliferation, survival, and dopaminergic differentiation of CNS precursors in lowered oxygen. J Neurosci. 2000;20:7377–7383. doi: 10.1523/JNEUROSCI.20-19-07377.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tacchini L, Bianchi L, Bernelli-Zazzera A, Cairo G. Transferrin receptor induction by hypoxia. HIF-1-mediated transcriptional activation and cell-specific post-transcriptional regulation. J Biol Chem. 1999;274:24142–24146. doi: 10.1074/jbc.274.34.24142. [DOI] [PubMed] [Google Scholar]
- 125.Tanaka T, Kojima I, Ohse T, Inagi R, Miyata T, Ingelfinger JR, Fujita T, Nangaku M. Hypoxia-inducible factor modulates tubular cell survival in cisplatin nephrotoxicity. Am J Physiol Renal Physiol. 2005;289:F1123–F1133. doi: 10.1152/ajprenal.00081.2005. [DOI] [PubMed] [Google Scholar]
- 126.Tanaka T, Kojima I, Ohse T, Ingelfinger JR, Adler S, Fujita T, Nangaku M. Cobalt promotes angiogenesis via hypoxia-inducible factor and protects tubulointerstitium in the remnant kidney model. Lab Invest. 2005;85:1292–1307. doi: 10.1038/labinvest.3700328. [DOI] [PubMed] [Google Scholar]
- 127.Tanaka T, Matsumoto M, Inagi R, Miyata T, Kojima I, Ohse T, Fujita T, Nangaku M. Induction of protective genes by cobalt ameliorates tubulointerstitial injury in the progressive Thy1 nephritis. Kidney Int. 2005;68:2714–2725. doi: 10.1111/j.1523-1755.2005.00742.x. [DOI] [PubMed] [Google Scholar]
- 128.Tian H, Hammer RE, Matsumoto AM, Russell DW, McKnight SL. The hypoxia-responsive transcription factor EPAS1 is essential for catecholamine homeostasis and protection against heart failure during embryonic development. Genes Dev. 1998;12:3320–3324. doi: 10.1101/gad.12.21.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Treins C, Giorgetti-Peraldi S, Murdaca J, Semenza GL, Van Obberghen E. Insulin stimulates hypoxia-inducible factor 1 through a phosphatidylinositol 3-kinase/target of rapamycin-dependent signaling pathway. J Biol Chem. 2002;277:27975–27981. doi: 10.1074/jbc.M204152200. [DOI] [PubMed] [Google Scholar]
- 130.Turner KJ, Moore JW, Jones A, Taylor CF, Cuthbert-Heavens D, Han C, Leek RD, Gatter KC, Maxwell PH, Ratcliffe PJ, Cranston D, Harris AL. Expression of hypoxia-inducible factors in human renal cancer: relationship to angiogenesis and to the von Hippel-Lindau gene mutation. Cancer Res. 2002;62:2957–2961. [PubMed] [Google Scholar]
- 131.Vaux EC, Metzen E, Yeates KM, Ratcliffe PJ. Regulation of hypoxia-inducible factor is preserved in the absence of a functioning mitochondrial respiratory chain. Blood. 2001;98:296–302. doi: 10.1182/blood.v98.2.296. [DOI] [PubMed] [Google Scholar]
- 132.Vesey DA, Cheung C, Pat B, Endre Z, Gobe G, Johnson DW. Erythropoietin protects against ischaemic acute renal injury. Nephrol Dial Transplant. 2004;19:348–355. doi: 10.1093/ndt/gfg547. [DOI] [PubMed] [Google Scholar]
- 133.Vogt BA, Shanley TP, Croatt A, Alam J, Johnson KJ, Nath KA. Glomerular inflammation induces resistance to tubular injury in the rat. A novel form of acquired, heme oxygenase-dependent resistance to renal injury. J Clin Invest. 1996;98:2139–2145. doi: 10.1172/JCI119020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Walisser JA, Bunger MK, Glover E, Harstad EB, Bradfield CA. Patent ductus venosus and dioxin resistance in mice harboring a hypomorphic Arnt allele. J Biol Chem. 2004;279:16326–16331. doi: 10.1074/jbc.M400784200. [DOI] [PubMed] [Google Scholar]
- 135.Warnecke C, Zaborowska Z, Kurreck J, Erdmann VA, Frei U, Wiesener M, Eckardt KU. Differentiating the functional role of hypoxia-inducible factor (HIF)-1α and HIF-2α (EPAS-1) by the use of RNA interference: erythropoietin is a HIF-2α target gene in Hep3B and Kelly cells. FASEB J. 2004;18:1462–1464. doi: 10.1096/fj.04-1640fje. [DOI] [PubMed] [Google Scholar]
- 136.Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE (Abstract) Sci STKE. 2005;2005:re12. doi: 10.1126/stke.3062005re12. [DOI] [PubMed] [Google Scholar]
- 137.Wiesener MS, Jurgensen JS, Rosenberger C, Scholze CK, Horstrup JH, Warnecke C, Mandriota S, Bechmann I, Frei UA, Pugh CW, Ratcliffe PJ, Bachmann S, Maxwell PH, Eckardt KU. Widespread hypoxia-inducible expression of HIF-2α in distinct cell populations of different organs. FASEB J. 2003;17:271–273. doi: 10.1096/fj.02-0445fje. [DOI] [PubMed] [Google Scholar]
- 138.Wykoff CC, Sotiriou C, Cockman ME, Ratcliffe PJ, Maxwell P, Liu E, Harris AL. Gene array of VHL mutation and hypoxia shows novel hypoxia-induced genes and that cyclin D1 is a VHL target gene. Br J Cancer. 2004;90:1235–1243. doi: 10.1038/sj.bjc.6601657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yuan HT, Li XZ, Pitera JE, Long DA, Woolf AS. Peritubular capillary loss after mouse acute nephrotoxicity correlates with down-regulation of vascular endothelial growth factor-A and hypoxia-inducible factor-1α. Am J Pathol. 2003;163:2289–2301. doi: 10.1016/s0002-9440(10)63586-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yun Z, Maecker HL, Johnson RS, Giaccia AJ. Inhibition of PPAR gamma 2 gene expression by the HIF-1-regulated gene DEC1/ Stra13: a mechanism for regulation of adipogenesis by hypoxia. Dev Cell. 2002;2:331–341. doi: 10.1016/s1534-5807(02)00131-4. [DOI] [PubMed] [Google Scholar]
- 141.Zatyka M, da Silva NF, Clifford SC, Morris MR, Wiesener MS, Eckardt KU, Houlston RS, Richards FM, Latif F, Maher ER. Identification of cyclin D1 and other novel targets for the von Hippel-Lindau tumor suppressor gene by expression array analysis and investigation of cyclin D1 genotype as a modifier in von Hippel-Lindau disease. Cancer Res. 2002;62:3803–3811. [PubMed] [Google Scholar]
- 142.Zelzer E, Levy Y, Kahana C, Shilo BZ, Rubinstein M, Cohen B. Insulin induces transcription of target genes through the hypoxia-inducible factor HIF-1α /ARNT. EMBO J. 1998;17:5085–5094. doi: 10.1093/emboj/17.17.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zhang H, Akman HO, Smith EL, Zhao J, Murphy-Ullrich JE, Batuman OA. Cellular response to hypoxia involves signaling via Smad proteins. Blood. 2003;101:2253–2260. doi: 10.1182/blood-2002-02-0629. [DOI] [PubMed] [Google Scholar]
- 144.Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, Simons JW, Semenza GL. Modulation of hypoxia-inducible factor 1α expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000;60:1541–1545. [PubMed] [Google Scholar]
- 145.Zimmer M, Doucette D, Siddiqui N, Iliopoulos O. Inhibition of hypoxia-inducible factor is sufficient for growth suppression of VHL −/− tumors. Mol Cancer Res. 2004;2:89–95. [PubMed] [Google Scholar]