

Abstract
Proteins that traffic through the eukaryotic secretory pathway are commonly modified with N-linked carbohydrates. These bulky amphipathic modifications at asparagines intrinsically enhance solubility and folding energetics through carbohydrate-protein interactions. N-linked glycans can also extrinsically enhance glycoprotein folding by utilizing the glycoprotein homeostasis or “glycoproteostasis” network, comprising numerous glycan binding and/or modification enzymes or proteins that synthesize, transfer, sculpt and utilize N-linked glycans to direct folding vs. degradation, and trafficking of nascent N-glycoproteins through the cellular secretory pathway. If protein maturation is perturbed by misfolding and/or aggregation, stress pathways are often activated that result in transcriptional remodeling of the secretory pathway, in an attempt to alleviate the insult(s). The inability to achieve glycoproteostasis is linked to several pathologies, including amyloidoses, cystic fibrosis, and lysosomal storage diseases. Recent progress on genetic and pharmacologic adaptation of the glycoproteostasis network provides hope that drugs can be developed for these maladies in the near future.
INTRODUCTION
The maintenance of the proteome is central to organismal homeostasis. Since protein folding is an error prone process, making appropriate intracellular folding vs. degradation decisions is central to achieving protein homeostasis or proteostasis 1, 2. The misregulation of proteostasis is associated with a growing number of human diseases, including loss-of-function maladies caused by too much degradation and/or failure of a glycoprotein to reach its target environment and gain-of-toxic function disorders caused by misfolding and/or aggregation, often rooted in inappropriate folding vs. degradation, or quality control, decisions. The proteostasis network, comprising thousands of proteins that make up folding, degradation, and trafficking pathways, assists proteome folding and maintenance, sustaining the proteome even under conditions of stress (e.g., thermal, oxidative, gene multiplication, gene mutation etc.). Maintenance of the proteome under stress is enabled by activating stress-responsive signaling pathways that transcriptionally remodel the proteostasis network to maintain physiologically relevant folded and functional protein concentrations in the face of a variety of cellular challenges3, 4. Each eukaryotic subcellular compartment has a unique proteostasis network, made up of specialized and general components and regulated by a dedicated stress-responsive signaling pathway. The mammalian secretory pathway, which is the focus of this review owing to space limitations, is no exception. It has numerous specialized and common pathways and components, and features a multifaceted, three-arm stress-responsive signaling pathway for regulation of secretory pathway proteostasis.
Over 1/3 of the mammalian proteome undergoes biogenesis and maturation through the cellular secretory pathway–the majority of these proteins have glycans covalently attached to the amide side chain of Asn harbored within an N-glycosylation “sequon”, or Asn-Xxx-Ser/Thr sequence, where Xxx is any amino acid other than Pro5, 6. These so-called N-linked glycans are generally attached to largely unfolded nascent chains co-translationally in the lumen of the endoplasmic reticulum (ER) by the oligosaccharyltransferase (OST)7. As the ribosome directs the secreted and secretory pathway proteome through the translocon, OST-mediated N-glycosylation has important intrinsic effects on nascent glycoproteins. N-glycosylation can intrinsically enhance the folding energetics through native state carbohydrate-protein interactions8–10. N-glycosylated proteins are also more aggregation resistant, owing to the steric bulk and hydration of the glycan. The N-glycan installed in the ER is composed of three glucoses, nine mannoses and two N-acetylglucosamines (Glc3Man9GlcNAc2, Figure 1)–this core glycan is remodeled by enzymes including glycosidases and glycosyltransferases as glycoproteins move through the secretory pathway11. Thus, the N-linked glycan structure at a particular sequon within a particular protein is spatially dynamic, which provides information on the extent to which that protein has progressed through the secretory pathway, as well as its folding/trafficking fitness. Core N-glycan trimming also allows N-glycoproteins to take advantage of a specialized proteostasis network within the ER enabling folding vs. degradation, or quality control, decisions, while also regulating the trafficking of N-glycoproteins11. In this review, we will discuss the intrinsic and extrinsic role of N-glycans in maintaining glycoproteostasis.
Figure 1. The initial composition of an N-linked glycan.

The primary structure of the GlcNAc2Man9Glc3 glycan transferred to Asn residues in glycosylation sequons (Asn-Xxx-Ser/Thr sequences, where Xxx is any amino acid other than Pro). Symbols and colors for monosaccharides are those recommended by the Consortium for Functional Glycomics (http://www.functionalglycomics.org/). The modes of linkage for the residues of the glycan are indicated next to the lines joining the symbols (e.g., β4 indicates a β1–4 linkage, etc.).
Intrinsic influences of N-glycans on glycoproteostasis
Achieving glycoproteostasis is greatly facilitated when individual N-glycosylated proteins have favorable folding energetics and a low aggregation propensity2. It has been shown in some cases that the relatively rigid, largely extended and highly hydrated N-glycan intrinsically disfavors aggregation of N-glycoproteins10. It has also been proposed that N-glycosylation restricts the conformational entropy of the unfolded glycoprotein, increasing its free energy, which in turn would favor native folding (Fig. 2a)8, 12–15. This effect, however, is not general13, 16, 17. The random introduction of N-glycans into a glycosylation naïve protein, where specific glycan–protein interactions have not evolved and are therefore relatively unlikely, usually does not significantly stabilize a protein16, 17. Significant stabilization depends on the context of the N-glycosylation site, arguing against a generic excluded volume effect. When N-glycosylation of a particular protein leads to a more negative (and therefore more favorable) folding free energy relative to its non-glycosylated counterpart, emerging evidence suggests that this intrinsic favorable effect on protein folding energetics is a consequence of native state stabilizing interactions between the N-glycan and the protein. Experimental evidence demonstrates that hydrogen bonds18, 19, hydrophobic contacts15, 18, 20, and CH–π interactions between the glycan and the protein contribute to a more favorable free energy of folding18, 19, 21, as discussed in detail below.
Figure 2. Intrinsic effects of N-glycosylation on protein folding.

(a) A free energy diagram illustrating the change in energy of the unfolded (U) and native (N) states upon N-glycosylation. The energy of the unfolded state of the non-glycosylated protein (GU,ng) tends to increase upon N-glycosylation (GU,g), whereas the energy of the native state of the non-glycosylated protein (GN,ng) tends to decrease (GN,g). The effect of N-glycosylation on the free energy of folding (ΔGf,ng vs. ΔGf,g) is the sum of these effects, and can be on the order of several kcal mol−1 (see text). (b) Glycan-protein H-bonds in the mature, complex-type N-glycan in the Fc fragment of human IgG1 (PDB ID 1FC1). Note that the N-acetyl group of GlcNAc-1 is shown in the energetically unfavorable cis conformation; this may be a mis-assignment, since the electron densities of the acetyl methyl and carbonyl O groups are likely similar at this resolution. (c) Glycan-protein hydrophobic burial in human chorionic gonadotropin (PDB ID: 1HCN). The hydrophobic α-face of GlcNAc-1 is buried in a pocket formed by Pro24, Ile25, Leu26, while the N-acetyl methyl group is buried in an adjacent pocket formed by Ala23, Ile25, and Val76. (d) A glycan–protein CH–π interaction in the adhesion domain of the human protein CD2 (HsCD2ad; PDB ID 1GYA). The hydrogen atom on C5 of GlcNAc-1 interacts with the aromatic side chain of Phe63. The structural module shown is known as an “enhanced aromatic sequon” (see text). Only the first GlcNAc of the glycan is shown for clarity.
Hydrogen bonding
Given the abundance of hydrogen bond (H-bond) donors and acceptors contained in glycans and proteins, it is not surprising that H-bonds are commonly invoked to explain the native-state stabilizing effect of N-glycosylation. However, whether glycan–protein H-bonds contribute to stabilization depends on many factors, including the nature of the H-bond donors and acceptors involved in the H-bond, the gain in entropy from releasing bound water molecules into bulk solvent, and the environment in which the H-bond forms. This last factor is especially important since buried H-bonds tend to be stronger than solvent exposed H-bonds22. N-glycans are often highly solvent exposed, but in some cases, notably the N-glycan on the Fc fragment of antibodies, N-glycans can make desolvated H-bonds with proteins that are likely to contribute to native state stability (Figure 2b)23.
Hydrophobic effect
The notion that there are contributions from the hydrophobic effect to stabilizing native state protein-glycan interactions is perhaps surprising, given that the hydration and water solubility of carbohydrates are among their best-known features. Nevertheless, the surfaces of most carbohydrates are amphipathic, i.e., there are segregated non-polar and polar regions15, 18, 20. For example, in GlcNAc and other glucose-derived monosaccharides, the α-face of the pyranose ring is formed by the axial CH bonds, creating a non-polar surface (Figure 2c, d). Burial of this hydrophobic surface through interactions with hydrophobic protein side chains is energetically favorable, but glycan–protein hydrophobic interactions tend to be less favorable than protein–protein hydrophobic interactions because it is difficult to bury the non-polar surfaces without affecting the access of adjacent polar surfaces to water24, 25. As previously noted18, a good example of a stabilizing hydrophobic glycan–protein interaction is in human chorionic gonadotropin, wherein the α-face of the first GlcNAc residue (GlcNAc1) of the glycan is buried in a pocket formed by Pro24, Ile25, and Leu26 (Figure 2c)26. In addition, the hydrophobic methyl group of the N-acetyl group on GlcNAc1 is buried from water by interactions with the Ala23, Ile25, and Val76 side chains.
CH–π interactions
In Figure 2d, the interaction of the first GlcNAc residue of the N-glycan attached to the adhesion domain of the human protein CD2 (HsCD2ad) with the Phe side chain at the -2 position relative to the glycan attached to Asn is favorable not only because of the hydrophobic burial of the aryl ring against the axial CH bonds of the α-face of the GlcNAc, but also because of a stabilizing CH–π interaction between the axial H atoms on the α-face of the GlcNAc and the π-electron system of the Phe side chain. CH–π interactions between carbohydrates and protein aromatic rings have long been recognized as prominent features of glycan–protein interactions19, 21. For example, the binding sites of most lectins (carbohydrate binding proteins) are lined with aromatic side chains27. Furthermore, aromatic side chains as a group are highly enriched in the regions of protein surfaces that are close to glycosylation sites28, 29. While it is tempting to view these CH–π interactions as being due to electrostatic forces between the partial positive charge on the H atom of the polarized CH bonds of carbohydrates and the partial negative charge above aromatic rings due to their π-electron systems, theoretical30 and experimental24 studies suggest that CH–π interactions are actually primarily driven by van der Waals or induced dipole–induced dipole forces. For example, the strength of a CH–π interaction much like the one in Figure 2d in the context of a model system for glycan-protein interactions (the WW domain) was found to depend very weakly on the electron density of the aromatic ring24. This nearly complete suppression of electrostatic effects may be due to the changes in electrostatic forces having equal effects on CH–π interactions in the native state and water OH–π interactions in the denatured state. Based on this result, we expect there to be little preference for one type of aromatic amino acid over the others in glycan–protein interactions, except when steric considerations come into play. This hypothesis will be put to the test as more structures of glycosylated proteins are solved, enabling the enrichment of aromatic side chains near glycans to be better understood29.
The sum of the contributions from the individual effects is stabilizing: putting it all together
Although the native state stabilization free energy from each source covered above can be small, their sum is often considerable (Figure 2a). This is exemplified by the family of reverse-turn-based structural modules known as “enhanced aromatic sequons”9, 24, 31–33. In enhanced aromatic sequons, an example of which is shown in Figure 2d, interactions between the N-glycan and an aromatic side chain N-terminal to the glycosylation site are enforced by the geometry of the reverse turn. Glycosylation of the Asn in this enhanced aromatic sequon stabilizes HsCD2ad by −3 kcal mol−1.31 The hydrophobic effect and CH–π interactions between the Phe at -2 and the GlcNAc-1 attached to Asn contribute −1.8 kcal mol−1, while the Lys side chain at position -4 makes hydrophobic and possibly H-bond interactions with GlcNAc-2 that contributes the remainder of the free energy of stabilization. Glycan residues beyond the third carbohydrate ring attached to Asn (i.e., the first mannose residue) do not contribute to the folding kinetics or thermodynamics of HsCD2ad31. The stabilization resulting from the glycan–protein native state interactions in HsCD2ad is essential for its proper folding; in the absence of glycosylation, HsCD2ad is unable to fold in vitro under normal, native conditions31, 34. The effect of glycosylation on the proteostasis of HsCD2 in vivo is correspondingly profound. Loss of glycosylation causes the expression levels of HsCD2 to decrease to ~50% of the wild type expression levels34. The intrinsic effects of N-glycosylation are similarly important to the proteostasis of other N-glycoproteins, including the cystic fibrosis transmembrane conductance regulator (CFTR)35, and rhodopsin36 (although it should be noted that in each of these examples extrinsic effects of N-glycosylation also contribute to the proteostasis of these glycoproteins).
Extrinsic influence of N-glycans on glycoproteostasis
Another important function of N-glycans on secreted proteins is to allow these N-glycoproteins to utilize a proteostasis network reserved for them. Enzymatically trimmed core glycans allow N-glycoproteins to recruit an array of macromolecular folding assistants and quality control carbohydrate-binding proteins comprising the specialized glycoproteostasis network of the ER. The glycoproteostasis network assists N-glycoprotein folding, quality control and degradation. Removing the two terminal A-branch glucose residues allows N-glycoprotein interactions with the lectin chaperones, which facilitate N-glycoprotein folding (Figure 1). In contrast, removal of mannose residues from the core glycan targets N-glycoproteins for anterograde or retrograde exit from the ER11, 37.
Quality Control in the ER
ER glucosidases initially act co-translationally on the core glycan that is transferred to N-glycoprotein nascent chains as they are inserted into the ER lumen11. This yields monoglucosylated A-branch glycoproteins that are substrates for the membrane-integrated lectin chaperone, calnexin, and its soluble paralogue, calreticulin (Figure 3). Lectin chaperone binding and conformational cycling to promote folding is antagonized by removal of the final A-branch glucose by glucosidase II. In contrast, glucose readdition to the A-branch by UDP-Glc: glycoprotein glucosyltransferase 1 (UGT1; Figures 1 and 3) redirects improperly folded N-glycoproteins back into the calnexin/calreticulin folding pathway by a mechanism requiring recognition that the N-glycoprotein client is not properly folded38. Thus, UGT1 acts as a folding sensor and modifies non-natively folded N-glycoproteins that lack an A-branch glucose by re-adding an A-branch glucose, enabling reengagement by the lectin chaperones.
Figure 3. N-linked glycans as protein sorting tags.

N-linked glycans are synthesized by Alg genes and transferred to polypeptides by oligosaccharyltransferase (OST) to asparagine residues in the Asn-Xxx-Ser/Thr sequon. OST exists as two isoforms that differ in their catalytic subunits, STT3A and STT3B. Glucosidase I and II remove the first two glucose moieties (blue spheres), generating a monoglucosylated glycoform, which enables the glycoprotein to enter the lectin folding cycle by engaging calnexin (CNX) and calreticulin (CRT)11. Removal of the remaining glucose by glucosidase II releases the protein from CNX/CRT. If the glycoprotein requires additional folding cycles, a glucose moiety is added by UGT1 and the protein re-engages CNX and CRT. Proteins that have reached their native states (cube) are demannosylated by ER mannosidase I (ERManI). The mannose-trimmed glycans are recognized by ERGIC-53, VIPL and VIP36, which facilitate their packaging into Golgi-bound vesicles52. Proteins that do not fold into their native states undergo extensive mannose trimming by the mannosidase-like proteins EDEM1-3. Glycoforms generated by EDEM1-3 are recognized by the ERAD lectin receptors OS-9 and XTP3-B, which bring the misfolded proteins to the ERAD complex. The relative transcript levels of some of the proteins involved in glycoproteostasis are upregulated by UPR activation (highlighted in yellow)72. A number of enzyme inhibitors (red) have been employed to study the effects of glycan processing on protein trafficking and degradation.
The conserved globular β-sandwich domains of calnexin and calreticulin contain a single carbohydrate-binding site that interacts with the Glc1Man3 tetrasaccharide with a micromolar dissociation constant39, 40. A proline-rich arm (P-domain) extends away from the globular domain of calnexin and calreticulin. This arm comprises a co-factor interaction site at the tip that supports binding to the foldases ERp57 or cyclophilin B (CyPB)41, 42. ERp57 is an oxidoreductase that facilitates disulfide formation and/or rearrangement, and CyPB is a peptidyl proline isomerase (PPI) that catalyzes cis-trans proline isomerization.
The lectin chaperone–foldase complexes promote the folding and assembly of functional N-glycoproteins through several mechanisms43, 44. The complexes are localized to the ER and thus retain non-native N-glycoproteins in the optimal folding environment of the ER, providing additional opportunities for client N-glycoproteins to reach their native state. For example, UGT1 acts on N-glycoprotein clients that have likely been through one failed calnexin/calreticulin folding cycle. Chaperone binding also prevents the aggregation of N-glycoproteins for which the intrinsic aggregation-inhibiting effect of N-glycans is insufficient to maintain them in the soluble state, and slows the folding reaction45–47. Monoglucosylated N-glycoproteins that persistently bind calnexin/calreticulin as directed by UGT1 are proposed to fold slower than unglucosylated or unglycosylated proteins. Trapping folding substrates in the monoglucosylated state results in continual chaperone binding and arrests distal folding events as probed by oxidation48. Calnexin and calreticulin also appear to possess a protein interacting surface that prevents aggregation, as purified calnexin inhibits the misassembly of non-glycosylated substrates49. Recent FRET studies with calreticulin have identified open and closed P-domain conformations where the substrate is proposed to be sequestered in a cleft between the glycan-binding site and the P-domain50. ERp57 binding induced the closed conformation where the P-domain appears to clamp down on the substrate. Further studies are required to fully understand the mechanism by which ERp57 and CyPB work with the lectin chaperones to assist N-glycoprotein folding.
ER exit to the Golgi
ER mannosidase I (MAN1B1) removes mannose residues from properly folded proteins to mark them for anterograde trafficking (Figure 3). Depending on the N-glycoprotein cargo, ER exit can either simply result from the bulk flow of substrates that are no longer retained in the ER or the selective recognition by sorting receptors51. ERGIC-53, VIPL and VIP36 are examples of N-glycoprotein cargo receptors that recognize natively folded mannose-trimmed substrates and package them into COPII vesicles52. ERGIC-53 cycles between the ER and the ER-Golgi-intermediate compartment (ERGIC) supporting the trafficking of glycosylated substrates such as alpha-1-antitrypsin, coagulation factor V and VIII, and cathepsin Z37, 53. The pH and calcium sensitivity of ERGIC-53 are proposed to support substrate binding in the neutral pH and calcium-rich environment of the ER and release clients in the slightly more acidic ERGIC, assisting in the anterograde trafficking of properly folded N-glycoproteins.
Degradation
Proteins that fail to reach their native state exit the ER to the cytoplasm by a retrograde route where they are ubiquitinated and then degraded by the proteasome through a process known as ER-associated protein degradation or ERAD54, 55. Extensive demannosylation targets N-glycoproteins for degradation, as evidenced by mannosidase inhibitors delaying the degradation of glycosylated ERAD substrates. ERAD-directed glycoproteins possess Man5GlcNAc2 or Man6GlcNAc2 glycoforms56–58. Demannosylation by glycosylhydrolase 47 family members including ER mannosidase I, and possibly EDEM1-3, aids the ERAD process by reducing the size of the substrate to facilitate dislocation and removal of the client N-glycoproteins from the lectin chaperone binding and folding cycle. Removing mannose residues also creates glycoforms that are recognized by downstream ERAD carbohydrate-binding receptors. Of special note is the controversy over the function and localization of ER mannosidase I, as recent studies have localized it to the Golgi and questioned the involvement of its mannosidase activity in ERAD59–61.
The removal of a C-branch terminal α(1,2)-linked mannose exposes α(1,6)-linked mannose residues, generating substrates for the lectin ERAD receptors, OS-9 and XTP3-B (Figure 3). These carbohydrate-binding ERAD receptors contain one (OS-9) and two (XTP3-B) mannose 6-phosphate receptor homology domains (MRH). The MRH domain from OS-9 displays high affinity for glycoforms with exposed C-branch α(1,6)-linked mannoses62, while the carbohydrate binding specificity for XTP3-B is controversial63, 64. OS-9/XTP3-B deliver misfolded proteins to an ER membrane ERAD complex that contains the machinery required for dislocation and ubiquitination65. This supramolecular ERAD complex is nucleated by the membrane protein SEL1L. The luminal N-terminal domain of SEL1L contains eleven tetratricopeptide repeats (TPR) and five possible N-linked glycosylation sites that appear to work in concert to recruit the OS-9 or XTP3-B-substrate complex to the ERAD complex and to pass the substrate along to the dislocation and ubiquitination machinery. EDEM1 and EDEM3 also appear to serve this role as they both bind ERAD substrates and interact with SEL1L in a glycan dependent manner66, 67. ERAD substrates are prepared for dislocation by associated factors such as ERdj5 and BiP for EDEM1, and possibly GRP94 for OS-965, 68. Alternatively, these factors may recognize the aberrant structures using the traditional chaperone queries of exposed hydrophobic residues or free thiols for oxidoreductase searches. For this model, the associated lectin would directly bind to the ERAD complex by associating with the glycans of SEL1L11, 65, 67.
The regulation of glycoproteostasis networks
The accumulation of misfolded proteins in the ER disrupts the efficient maturation of additional entering nascent chains, leading to a breakdown in protein biogenesis within the secretory pathway. To circumvent such disturbances and maintain cellular homeostasis, the unfolded protein response (UPR) signaling pathway is activated, which regulates ER proteostasis3, 4. Enhancing ER proteostasis capacity by UPR activation helps to alleviate stress by upregulating the clearance of defective substrates, translational attenuation, and by increasing the capacity of the ER proteostasis network by transcriptional remodeling. If the stress persists, apoptosis or cell death is induced to preserve organismal homeostasis.
Activation of one or more of the three distinct ER-membrane integrated stress sensors induces signaling in the corresponding arm of the UPR. These stress sensors include the double-stranded RNA (PKR)–activated protein kinase-like eukaryotic initiation factor 2α kinase (PERK), the inositol-requiring transmembrane kinase/endoribonuclease 1 (IRE1), and the activating transcription factor-6 (ATF6)3, 4. The induction of each branch or arm of the UPR leads to the generation of a transcription factor that upregulates UPR genes involved in protein folding, quality control, degradation and lipid production. For detailed descriptions of the signaling pathways comprising the individual branches of UPR, readers are directed to reviews that concentrate on these topics3, 4. Here we will focus on how UPR activation influences the glycoproteostasis network.
The burden of accumulating misfolded or aggregated proteins within the ER can be reduced by translation attenuation, wherein the activity of the translation machinery is reduced. Transcript levels are also reduced by an enzymatic process69. Activated PERK phosphorylates and inhibits the translation initiation factor eIF2α, thereby inhibiting translation initiation70. By controlling translation and reducing the influx of cargo, ER chaperones and quality control factors are free to focus their attention on the current aberrations without adding additional client proteins to the ER.
Activation of b-ZIP transcription factors in the IRE1 and ATF6 adaptive signaling arms of the UPR upregulates UPR target genes that optimize the ER folding environment and enhance the efficient clearance of defective proteins through ERAD71. IRE1 induction leads to the increased transcription of genes encoding proteins involved in lipid synthesis, ER import, glycosylation, anterograde trafficking, ERAD (Figure 3), and the hexosamine biosynthetic pathway that synthesizes the N-glycan precursor GlcNAc3, 4, 72, 73. Augmentation of the ER volume dilutes defective proteins, whereas increasing the level of ERAD machinery facilitates rapid, efficient and less discriminating turnover of non-native cargo. A recent study has found that an increase in the hexosamine biosynthetic pathway enhanced ERAD, proteasomal activity and autophagy and resulted in an extension in the life span in C. elegans, providing an additional link between stress and the glycoproteostasis network73. ATF6 activation induces the transcription of key chaperones, co-chaperones and folding enzymes involved in protein maturation and quality control, including BiP and GRP9472. Together, the IRE1 and ATF6 transcription factors, emerging from the adaptive arms of the UPR, remodel the ER in an attempt to alleviate the stress created by the accumulation of defective client proteins.
Once the stress is diminished, the ER environment needs to be restored to its normal operational state for optimal protein production, a sort of organellar ‘rebooting.’ Raised levels of ERAD components can interfere with efficient protein maturation by prematurely targeting folding intermediates for degradation. The proper ER balance is re-established through a process termed ERAD tuning74. Many of the ERAD components are themselves subjected to rapid turnover, either through ERAD or autophagy. ERAD machinery is stabilized by the presence of misfolded cargo, as non-native proteins appear to serve as better targets for ubiquitination and destruction. Once the misfolded proteins are cleared, the rapid degradation of ERAD machinery helps to reinstate the proper ER balance.
If the misfolded protein load is not adequately cleared after prolonged UPR activation, a cell death program is initiated as an organismal defense mechanism against the accumulation of rogue cells and toxic misfolded or misassembled proteins3, 4. Persistent PERK activation starts one of the cascades leading to cell death, thus providing a mechanism to destroy cells that were unable to be rescued by UPR activation.
Proteostasis networks can also be regulated using mechanisms beyond alterations in transcript or protein levels. An example of this added layer of complexity is the tightly controlled activity and localization of calnexin (Figure 4). While the abundance of calnexin is modestly upregulated by UPR activation, its role in glycoproteostasis is further controlled by co-factors, co-chaperones, post-translational modifications and its localization within the ER. The ER is a large organelle composed of a continuous membrane that it is divided into a number of functional subcompartments75. Post-translational modification of the C-terminal cytoplasmic tail of calnexin directs its localization within the ER. Calnexin phosphorylation supports its increased association with ribosome-translocons, positioning the chaperone to aid in early glycoprotein maturation events76. Palmitoylation enriches calnexin localization to ribosome-translocons, as well as into the mitochondria-associated membranes (MAM) through its increased association with the sarcoplasmic reticulum Ca2+-ATPase (SERCA) calcium pump77. The substrate binding activity of calnexin is regulated by calcium binding78. The activity of calnexin is also influenced by co-chaperones or foldases. The localization of UGT1 to pre-Golgi intermediates by immunoelectron microscopy combined with the observation that UGT1 does not modify ribosome-arrested nascent chains, suggests that UGT1 modification and the rebinding of calnexin to reglucosylated substrates occurs near ER exit sites (ERES)79, 80. ERp57 association with calreticulin has recently been shown to support the closing of the P-domain on the substrate, perhaps enhancing its ability to aid in the folding process50. Calnexin, which possesses a longer P-domain, can be expected to function through a similar mechanism. Interestingly, stress treatments decrease calnexin palmitoylation levels and support the reorganization of calnexin and calreticulin into the ERQC77, 81, a quality control compartment that is enriched for ERAD machinery such as ER mannosidase I, EDEM1, Derlin-1, and OS-981. The lack of UGT1 and ERp57 in the ERQC suggests that the localization of calnexin into the ERQC might be a mechanism for ERAD substrate delivery to a dislocation and ubiquitination center, as calnexin substrate binding is expected to be weaker in the absence of ERp57 and UGT1.
Figure 4. Calnexin post-translational modifications modulate ER localization.

The localization of calnexin within the ER depends on post-translational modifications of its C-terminal tail. Palmitoylation of two cysteine residues located on the cytoplasmic tail of calnexin, causes an enrichment of the protein at mitochondria-associated membranes (MAMs) suggesting a role for calnexin in calcium regulation77. Palmitoylated calnexin is also enriched at the rough ER where it interacts with the translocon. Phosphorylation of the cytoplasmic tail enhances the interaction between calnexin and the ribosome-translocon complex76. ERp57 binding to CRT, and likely CNX, enhances the closing of the P-domain onto the folding substrate50. Unmodified calnexin is predominantly found at the ER exit site (ERES) and ER quality control (ERQC) compartments81. Calnexin functions in the later stages of folding by either accepting re-glucosylated substrates79, or releasing misfolded substrates for their subsequent degradation81.
Defects in glycoproteostasis are linked to pathology
Since N-glycoproteins are critical for many important physiological processes, defects in glycoproteostasis are associated with many diseases. For example, insertions or deletions in the calreticulin gene are found in patients suffering from chronic myeloid leukemia82. Mutations in the ER exit lectin ERGIC-53 are associated with blood coagulation diseases due to deficiencies in factors V and VIII53, and defects in UPR activation controlling secretory pathway proteostasis are linked to many maladies including bipolar disease and diabetes4. The impaired maturation of glycoconjugates is also associated with a long list of genetic disorders called “Congenital Disorders of Glycosylation” that display a wide array of symptoms owing to the vast spectrum of proteins impacted by glycosylation abnormalities6. Furthermore, mutations in N-glycoproteins commonly lead to misfolding, resulting in a loss-of-function due to enhanced ERAD clearance; cystic fibrosis and many lysosomal storage diseases are caused by such losses of function.
There are several traditional pharmacologic approaches that can be utilized to avoid disease-associated glycoproteostasis challenges (Table I). For example, the targeting of viral glycoproteins for destruction has been explored as an antiviral therapy. Since membrane envelopes of viruses such as influenza and HIV are comprised largely of glycoproteins, derailment of N-glycoprotein maturation by iminosugar inhibition of glycan processing enzymes has been used as a strategy for development of antiviral drugs83, 84. Glycoproteins such as hemagglutinin and neuraminidase for influenza and gp160 for HIV are essential for the viral life cycle, and the usurping of the ER for the robust production of these complicated glycoproteins taxes ER glycoproteostasis capacity. Glucosidase inhibitors such as N-butyl-deoxynojirimycin (NB-DNJ, miglustat or Zavesca) have been explored as possible antiviral therapies, as the accumulation of tri-glucosylated core glycans prevents the viral glycoproteins from availing themselves of the lectin chaperones calnexin and calreticulin for folding assistance83 (Figure 3). Glucosidase inhibition, which is surprisingly well tolerated in mammals85, 86, results in viral glycoprotein misfolding and the clearance through the ERAD pathway, thereby reducing the titer of infectious viral particles.
Table I.
Compounds tested as chemical regulators of glycoproteostasis
| Name | Structure | Mechanism of Action |
|---|---|---|
| Castanospermine (CST) |
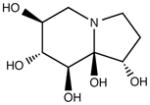
|
Alpha- and beta- glucosidase inhibitor |
| 1-Deoxymanojirimycin (DMJ) |
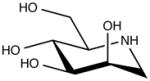
|
Alpha1,2-mannosidase inhibitor |
| 1-Deoxynojirimycin (DNJ) Derivatives: N-butyl-deoxynojirimycin (Miglustat, Zavesca), N-9-methoxynonyl-DNJ, UV-4. |
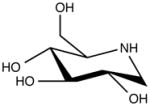
|
Alpha glucosidase inhibitor |
| Diltiazem |
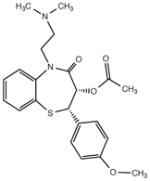
|
Calcium channel blocker |
| Kifunensin (KIF) |
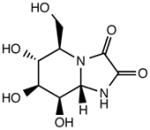
|
Class I glycoprotein processing alpha-mannosidase inhibitor |
| MG-132 |
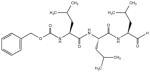
|
Reversible proteasome inhibitor |
| SAHA (suberoylanilide hydroxamic acid) |
|
Histone deacetylase inhibitor (Zinc ion chelator) |
| Tunicamycin (TUN) |
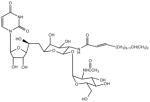
|
Blocks N-linked glycosylation by inhibiting transfer of N-acetylglucosamine-1-phosphate from UDP-N-acetylglucosamine to dolichol phosphate |
| Verapamil |
|
Calcium channel blocker |
Genetic or pharmacologic adaptation of the glycoproteostasis network is being aggressively explored as a strategy to provide effective therapies for a variety of human maladies, wherein defects are rooted in glycoproteostasis. Here, we will focus on efforts launched to develop small molecule proteostasis regulators that adapt or preemptively prepare the ER for an insult or stress, so as to boost the efficiency of N-glycoprotein maturation. Some proteostasis regulators under study increase the concentration of calcium in the ER, while others act as arm-selective UPR activators. Both strategies help to optimize the ER for maturation of properly folded substrates and the clearance of defective client proteins.
The ER is a site for Ca2+ storage and regulation87, and therefore it is not surprising that many ER glycoproteostasis network components are calcium-binding proteins, including calnexin and calreticulin. Since the ER regulates glycoproteostasis and calcium homeostasis, it is likely that their regulation is interdependent. Thus, it may be possible to achieve synergy in the rescue of glycoproteostasis by also altering ER Ca2+ levels. In fact, the influence that ER Ca2+ levels have on N-glycoprotein maturation appears to be protein specific. Inhibiting SERCA ER calcium influx channels has been found to increase the proper trafficking of the predominant mutant in CFTR (ΔF508) associated with cystic fibrosis88. The issue for CFTR appears to be overzealous quality control that retains in the ER a mutant N-glycoprotein having partial chloride channel activity, directing it for ERAD. ER retention of CFTR seems to be relaxed by a decrease in ER Ca2+ levels. Thus, decreasing ER Ca2+ levels could serve to get more CFTR to the plasma membrane, thereby enhancing the efficacy of Kalydeco, a channel potentiator introduced by Vertex Pharmaceuticals89,90, 91. Depletion of ER calcium has been shown to disrupt the ER proteostasis network by permitting the release of chaperones from the ER87. In contrast, the N-glycosylated mutant lysosomal enzymes associated with lysosomal storage disease need more attention from calnexin and calreticulin to fold properly, and increased ER Ca2+ levels provide an environment allowing more efficient folding and trafficking of these misfolding-prone enzymes. Thus, the Ca2+ channel blockers diltiazem and verapamil, which inhibit the ER ryanodine receptor calcium efflux channels and other targets, have been used to improve the cellular trafficking and activity of N-glycosylated mutant lysosomal enzymes associated with lysosomal storage diseases92–94. Altering ER Ca2+ levels in concert with direct perturbation of glycoproteostasis machinery is a promising therapeutic strategy, particularly as we learn more about how the mutant lysosomal enzyme–glycoproteostasis network component interactions change upon perturbation.
Small molecule stress-responsive signaling pathway activators can serve as proteostasis regulators to correct the trafficking of mutant N-glycoproteins associated with a number of diseases. Activation of the UPR using proteasome inhibitors such as MG-132 has been used to correct the trafficking of N-glycosylated mutant lysosomal enzymes associated with Gaucher and Tay-Sachs diseases92. Interestingly, enhanced glucocerebrosidase trafficking in MG-132–treated fibroblasts derived from patients with Gaucher disease was associated with down regulation of the expression level of the peptidyl proline isomerase FKBP10, suggesting that altering the finely choreographed interactions in the ER can be used to enhance mutant N-glycoprotein folding95. The histone deacetylase (HDAC) inhibitor suberoylanilide hydroxamic acid (SAHA) has been used to correct the cellular trafficking of mutant alpha-1-antitrypsin and GABAA receptors associated with epilepsy96, 97. While the mechanism for SAHA enhancement is uncertain, it appears to involve, in part, HDAC7 silencing and a calnexin sensitive pathway.
Summary and future directions
The importance of the intrinsic and extrinsic effects of N-glycosylation on glycoprotein folding is underscored by the nearly universal conservation of protein N-glycosylation across eukaryotes98, 99. Maintaining an effective glycoproteostasis network is pivotal for cellular proliferation and normal cell function. A number of mechanisms, involving ER, Golgi and cytoplasmic components, exist to keep the glycoproteostasis network in check. ER stress is one of the major factors contributing to the destabilization of glycoproteostasis, as glycoproteins are synthesized, modified, and folded in the ER.
The activation of UPR stress sensors is one of the first steps to alleviate the accumulation of misfolded or misassembled glycoproteins. Once the UPR is activated, a number of lectin chaperones, chaperones and glycan modifying enzymes are upregulated in order to re-balance ER proteostasis. Stress also contributes to the post-translational modification and altered localization of calnexin in the ER. Further investigations are required to determine how the organization of other components of the glycoproteostasis network is regulated by stress in the ER. Current efforts are also underway to develop methodologies to activate the individual arms of the UPR–the hypothesis being that more targeted therapies could be tailored to individual diseases while minimizing potential side effects72. Expanding our knowledge surrounding the parts list and the systems functions of the glycoproteostasis network and its regulation will lead to more sophisticated therapeutic strategies, as well as contribute to our understanding of how glycoproteostasis is achieved.
Acknowledgments
This work was supported by the National Institute of Health under award numbers GM086874 and GM094848 (to D. N. H.); DK075295, AG046495 and GM051105 (to E. T. P and J. W. K.); and a Chemistry-Biology Interface program training grant (GM08515 to L. L.).
Footnotes
Author contributions
D. N. H., L. L., E. T. P. and J. W. K. each contributed to the writing of this manuscript.
Competing financial interests
The authors declare no competing financial interests.
Contributor Information
Daniel N. Hebert, Email: dhebert@biochem.umass.edu.
Evan T. Powers, Email: epowers@scripps.edu.
Jeffery W. Kelly, Email: jkelly@scripps.edu.
References
- 1.Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Hartl FU. Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem. 2013;82:323–355. doi: 10.1146/annurev-biochem-060208-092442. [DOI] [PubMed] [Google Scholar]
- 2.Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- 3.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 4.Schroder M, Kaufman RJ. The Mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 5.Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta. 1999;1473:4–8. doi: 10.1016/s0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- 6.Freeze HH, Chong JX, Bamshad MJ, Ng BG. Solving Glycosylation Disorders: Fundamental Approaches Reveal Complicated Pathways. Am J Hum Genet. 2014;94:161–175. doi: 10.1016/j.ajhg.2013.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruiz-Canada C, Kelleher DJ, Gilmore R. Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell. 2009;136:272–283. doi: 10.1016/j.cell.2008.11.047. Identification of a new active site subunit for the OST that is capable of modifying proteins post-translationally. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Conner SE, Imperiali B. A molecular basis for glycosylation-induced conformational switching. Chem Biol. 1998;5:427–437. doi: 10.1016/s1074-5521(98)90159-4. [DOI] [PubMed] [Google Scholar]
- 9.Price JL, et al. N-glycosylation of enhanced aromatic sequons to increase glycoprotein stability. Biopolymers. 2012;98:195–211. doi: 10.1002/bip.22030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sola RJ, Griebenow K. Effects of glycosylation on the stability of protein pharmaceuticals. Journal of pharmaceutical sciences. 2009;98:1223–1245. doi: 10.1002/jps.21504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hebert DN, Molinari M. Flagging and docking: dual roles for N-glycans in protein quality control and cellular proteostasis. Trends Biochem Sci. 2012;37:404–410. doi: 10.1016/j.tibs.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoffmann D, Florke H. A structural role for glycosylation: lessons from the hp model. Fold Des. 1998;3:337–343. doi: 10.1016/S1359-0278(98)00046-7. [DOI] [PubMed] [Google Scholar]
- 13.Shental-Bechor D, Levy Y. Effect of glycosylation on protein folding: a close look at thermodynamic stabilization. Proc Natl Acad Sci U S A. 2008;105:8256–8261. doi: 10.1073/pnas.0801340105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wormald MR, et al. The conformational effects of N-glycosylation on the tailpiece from serum IgM. European journal of biochemistry/FEBS. 1991;198:131–139. doi: 10.1111/j.1432-1033.1991.tb15995.x. [DOI] [PubMed] [Google Scholar]
- 15.Yamaguchi H. Chaperone-like functions of N-glycans in the formation and stabilization of protein conformation. Trends Glycosci Glycotechnol. 2002;14:139–151. [Google Scholar]
- 16.Chen MM, et al. Perturbing the folding energy landscape of the bacterial immunity protein Im7 by site-specific N-linked glycosylation. Proc Natl Acad Sci U S A. 2010;107:22528–22533. doi: 10.1073/pnas.1015356107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Price JL, et al. Context-dependent effects of asparagine glycosylation on Pin WW folding kinetics and thermodynamics. J Am Chem Soc. 2010;132:15359–15367. doi: 10.1021/ja106896t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barb AW, Borgert AJ, Liu M, Barany G, Live D. Intramolecular glycan-protein interactions in glycoproteins. Methods Enzymol. 2010;478:365–388. doi: 10.1016/S0076-6879(10)78018-6. [DOI] [PubMed] [Google Scholar]
- 19.Quiocho FA. Protein-Carbohydrate Interactions - Basic Molecular-Features. Pure & Appl Chem. 1989;61:1293–1306. [Google Scholar]
- 20.Lemieux RU. How water provides the impetus for molecular recognition in aqueous solution. Acc Chem Res. 1996;29:373–380. [Google Scholar]
- 21.Asensio JL, Arda A, Canada FJ, Jimenez-Barbero J. Carbohydrate-aromatic interactions. Acc Chem Res. 2013;46:946–954. doi: 10.1021/ar300024d. [DOI] [PubMed] [Google Scholar]
- 22.Gao J, Bosco DA, Powers ET, Kelly JW. Localized thermodynamic coupling between hydrogen bonding and microenvironment polarity substantially stabilizes proteins. Nat Struct Mol Biol. 2009;16:684–690. doi: 10.1038/nsmb.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krapp S, Mimura Y, Jefferis R, Huber R, Sondermann P. Structural analysis of human IgG-Fc glycoforms reveals a correlation between glycosylation and structural integrity. J Mol Biol. 2003;325:979–989. doi: 10.1016/s0022-2836(02)01250-0. [DOI] [PubMed] [Google Scholar]
- 24.Chen W, et al. Structural and energetic basis of carbohydrate-aromatic packing interactions in proteins. J Am Chem Soc. 2013;135:9877–9884. doi: 10.1021/ja4040472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chavelas EA, Garcia-Hernandez E. Heat capacity changes in carbohydrates and protein-carbohydrate complexes. Biochem J. 2009;420:239–247. doi: 10.1042/BJ20082171. [DOI] [PubMed] [Google Scholar]
- 26.Wu H, Lustbader JW, Liu Y, Canfield RE, Hendrickson WA. Structure of human chorionic gonadotropin at 2.6 A resolution from MAD analysis of the selenomethionyl protein. Structure. 1994;2:545–558. doi: 10.1016/s0969-2126(00)00054-x. [DOI] [PubMed] [Google Scholar]
- 27.Weis WI, Drickamer K. Structural basis of lectin-carbohydrate recognition. Annu Rev Biochem. 1996;65:441–473. doi: 10.1146/annurev.bi.65.070196.002301. [DOI] [PubMed] [Google Scholar]
- 28.Petrescu AJ, Milac AL, Petrescu SM, Dwek RA, Wormald MR. Statistical analysis of the protein environment of N-glycosylation sites: implications for occupancy, structure, and folding. Glycobiology. 2004;14:103–114. doi: 10.1093/glycob/cwh008. Very informative surveys of the protein primary, secondary, and tertiary structural environments that surround N-linked glycans based on a database of glycoprotein crystal structures. [DOI] [PubMed] [Google Scholar]
- 29.Surleac MD, et al. The Structural Assessment of Glycosylation Sites Database - SAGS - An Overall View on N-Glycosylation. In: Petrescu SM, editor. Glycosylation. InTech; 2012. p. 3-20. Additional helpful survey of structural environment of N-linked glycans. [Google Scholar]
- 30.Nishio M. The CH/pi hydrogen bond in chemistry. Conformation, supramolecules, optical resolution and interactions involving carbohydrates. Physical chemistry chemical physics : PCCP. 2011;13:13873–13900. doi: 10.1039/c1cp20404a. [DOI] [PubMed] [Google Scholar]
- 31.Hanson SR, et al. The core trisaccharide of an N-linked glycoprotein intrinsically accelerates folding and enhances stability. Proc Natl Acad Sci U S A. 2009;106:3131–3136. doi: 10.1073/pnas.0810318105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Culyba EK, et al. Protein native-state stabilization by placing aromatic side chains in N-glycosylated reverse turns. Science. 2011;331:571–575. doi: 10.1126/science.1198461. This article introduced the “enhanced aromatic sequon”, an N-glycosylated structural module that stabilizes the proteins in which it is found and that appears to be glycosylated with unusually high efficiency by OST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Price JL, Powers DL, Powers ET, Kelly JW. Glycosylation of the enhanced aromatic sequon is similarly stabilizing in three distinct reverse turn contexts. Proc Natl Acad Sci U S A. 2011;108:14127–14132. doi: 10.1073/pnas.1105880108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wyss DF, et al. Conformation and function of the N-linked glycan in the adhesion domain of human CD2. Science. 1995;269:1273–1278. doi: 10.1126/science.7544493. [DOI] [PubMed] [Google Scholar]
- 35.Glozman R, et al. N-glycans are direct determinants of CFTR folding and stability in secretory and endocytic membrane traffic. J Cell Biol. 2009;184:847–862. doi: 10.1083/jcb.200808124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tam BM, Moritz OL. The role of rhodopsin glycosylation in protein folding, trafficking, and light-sensitive retinal degeneration. J Neurosci. 2009;29:15145–15154. doi: 10.1523/JNEUROSCI.4259-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nyfeler B, et al. Identification of ERGIC-53 as an intracellular transport receptor of alpha1-antitrypsin. J Cell Biol. 2008;180:705–712. doi: 10.1083/jcb.200709100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caramelo JJ, Parodi AJ. Getting in and out from calnexin/calreticulin cycles. J Biol Chem. 2008;283:10221–10225. doi: 10.1074/jbc.R700048200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kapoor M, et al. Interactions of substrate with calreticulin, an endoplasmic reticulum chaperone. J Biol Chem. 2003;278:6194–6200. doi: 10.1074/jbc.M209132200. [DOI] [PubMed] [Google Scholar]
- 40.Schrag JD, et al. The Structure of calnexin, an ER chaperone involved in quality control of protein folding. Mol Cell. 2001;8:633–644. doi: 10.1016/s1097-2765(01)00318-5. [DOI] [PubMed] [Google Scholar]
- 41.Frickel EM, et al. TROSY-NMR reveals interaction between ERp57 and the tip of the calreticulin P-domain. Proc Natl Acad Sci U S A. 2002;99:1954–1959. doi: 10.1073/pnas.042699099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kozlov G, et al. Structural basis of carbohydrate recognition by calreticulin. J Biol Chem. 2010;285:38612–38620. doi: 10.1074/jbc.M110.168294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hebert DN, Foellmer B, Helenius A. Calnexin and calreticulin promote folding, delay oligomerization and suppress degradation of influenza hemagglutinin in microsomes. Embo J. 1996;15:2961–2968. [PMC free article] [PubMed] [Google Scholar]
- 44.Vassilakos A, Cohen-Doyle MF, Peterson PA, Jackson MR, Williams DB. The molecular chaperone calnexin facilitates folding and assembly of class I histocompatibility molecules. EMBO J. 1996;15:1495–1506. [PMC free article] [PubMed] [Google Scholar]
- 45.Daniels R, Kurowski B, Johnson AE, Hebert DN. N-linked glycans direct the cotranslational folding pathway of influenza hemagglutinin. Mol Cell. 2003;11:79–90. doi: 10.1016/s1097-2765(02)00821-3. Describes a detailed model for the molecular choreography for nascent glycoprotein maturation and its interaction with ER lectin chaperones. [DOI] [PubMed] [Google Scholar]
- 46.Pearse BR, Hebert DN. Lectin chaperones help direct the maturation of glycoproteins in the endoplasmic reticulum. Biochim Biophys Acta. 2010;1803:684–693. doi: 10.1016/j.bbamcr.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pearse BR, et al. The role of UDP-Glc:glycoprotein glucosyltransferase 1 in the maturation of an obligate substrate prosaposin. J Cell Biol. 2010;189:829–841. doi: 10.1083/jcb.200912105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hebert DN, Foellmer B, Helenius A. Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell. 1995;81:425–433. doi: 10.1016/0092-8674(95)90395-x. [DOI] [PubMed] [Google Scholar]
- 49.Brockmeier A, Brockmeier U, Williams DB. Distinct contributions of the lectin and arm domains of calnexin to its molecular chaperone function. J Biol Chem. 2009;284:3433–3444. doi: 10.1074/jbc.M804866200. [DOI] [PubMed] [Google Scholar]
- 50.Wijeyesakere SJ, Rizvi SM, Raghavan M. Glycan-dependent and -independent interactions contribute to cellular substrate recruitment by calreticulin. J Biol Chem. 2013;288:35104–35116. doi: 10.1074/jbc.M113.507921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Warren G, Mellman I. Bulk flow redux? Cell. 1999;98:125–127. doi: 10.1016/s0092-8674(00)81006-5. [DOI] [PubMed] [Google Scholar]
- 52.Kamiya Y, et al. Molecular basis of sugar recognition by the human L-type lectins ERGIC-53, VIPL, and VIP36. J Biol Chem. 2008;283:1857–1861. doi: 10.1074/jbc.M709384200. [DOI] [PubMed] [Google Scholar]
- 53.Nichols WC, et al. Mutations in the ER-Golgi intermediate compartment protein ERGIC-53 cause combined deficiency of coagulation facyors V and VIII. Cell. 1998;93:61–70. doi: 10.1016/s0092-8674(00)81146-0. [DOI] [PubMed] [Google Scholar]
- 54.Hebert DN, Molinari M. In and Out of the ER: Protein Folding, Quality Control, Degradation, and Related Human Diseases. Physiol Rev. 2007;87:1377–1408. doi: 10.1152/physrev.00050.2006. [DOI] [PubMed] [Google Scholar]
- 55.Needham PG, Brodsky JL. How early studies on secreted and membrane protein quality control gave rise to the ER associated degradation (ERAD) pathway: the early history of ERAD. Biochim Biophys Acta. 2013;1833:2447–2457. doi: 10.1016/j.bbamcr.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ermonval M, Kitzmuller C, Mir AM, Cacan R, Ivessa NE. N-glycan structure of a short-lived variant of ribophorin I expressed in the MadIA214 glycosylation-defective cell line reveals the role of a mannosidase that is not ER mannosidase I in the process of glycoprotein degradation. Glycobiology. 2001;11:565–576. doi: 10.1093/glycob/11.7.565. [DOI] [PubMed] [Google Scholar]
- 57.Frenkel Z, Gregory W, Kornfeld S, Lederkremer GZ. Endoplasmic reticulum-associated degradation of mammalian glycoproteins involves sugar chain trimming to Man6-5GlcNAc2. J Biol Chem. 2003;278:34119–33424. doi: 10.1074/jbc.M305929200. [DOI] [PubMed] [Google Scholar]
- 58.Lederkremer GZ, Glickman MH. A window of opportunity: timing protein degradation by trimming of sugars and ubiquitins. Trends Biochem Sci. 2005;30:297–303. doi: 10.1016/j.tibs.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 59.Iannotti MJ, Figard L, Sokac AM, Sifers RN. A Golgi-localized mannosidase (MAN1B1) plays a non-enzymatic gatekeeper role in protein biosynthetic quality control. J Biol Chem. 2014;289:11844–11858. doi: 10.1074/jbc.M114.552091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pan S, Cheng X, Sifers RN. Golgi-situated endoplasmic reticulum alpha-1, 2-mannosidase contributes to the retrieval of ERAD substrates through a direct interaction with gamma-COP. Mol Biol Cell. 2013;24:1111–1121. doi: 10.1091/mbc.E12-12-0886. Provocative support for ER mannosidase I (Man1B1) actually residing in the Golgi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rymen D, et al. MAN1B1 deficiency: an unexpected CDG-II. PLoS genetics. 2013;9:e1003989. doi: 10.1371/journal.pgen.1003989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hosokawa N, Kamiya Y, Kamiya D, Kato K, Nagata K. Human OS-9, a lectin required for glycoprotein ERAD, recognizes mannose-trimmed N-glycans. J Biol Chem. 2009;284:17061–17068. doi: 10.1074/jbc.M809725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fujimori T, Kamiya Y, Nagata K, Kato K, Hosokawa N. Endoplasmic reticulum lectin XTP3-B inhibits endoplasmic reticulum-associated degradation of a misfolded alpha1-antitrypsin variant. The FEBS journal. 2013;280:1563–1575. doi: 10.1111/febs.12157. [DOI] [PubMed] [Google Scholar]
- 64.Yamaguchi D, Hu D, Matsumoto N, Yamamoto K. Human XTP3-B binds to alpha1-antitrypsin variant null(Hong Kong) via the C-terminal MRH domain in a glycan-dependent manner. Glycobiology. 2010;20:348–355. doi: 10.1093/glycob/cwp182. [DOI] [PubMed] [Google Scholar]
- 65.Christianson JC, Shaler TA, Tyler RE, Kopito RR. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1?SEL1L ubiquitin ligase complex for ERAD. Nat Cell Biol. 2008;10:272–282. doi: 10.1038/ncb1689. A tour-de-force analysis of the interactome for ERAD machinery complexes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Saeed M, et al. Role of the endoplasmic reticulum-associated degradation (ERAD) pathway in degradation of hepatitis C virus envelope proteins and production of virus particles. J Biol Chem. 2011;286:37264–37273. doi: 10.1074/jbc.M111.259085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cormier JH, Tamura T, Sunryd JC, Hebert DN. EDEM1 recognition and delivery of misfolded proteins to the SEL1L-containing ERAD complex. Mol Cell. 2009;34:627–633. doi: 10.1016/j.molcel.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ushioda R, et al. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science. 2008;321:569–572. doi: 10.1126/science.1159293. [DOI] [PubMed] [Google Scholar]
- 69.Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 70.Mori K. Signalling pathways in the unfolded protein response: development from yeast to mammals. J Biochem. 2009;146:743–750. doi: 10.1093/jb/mvp166. [DOI] [PubMed] [Google Scholar]
- 71.Shoulders MD, Ryno LM, Cooley CB, Kelly JW, Wiseman RL. Broadly applicable methodology for the rapid and dosable small molecule-mediated regulation of transcription factors in human cells. J Am Chem Soc. 2013;135:8129–8132. doi: 10.1021/ja402756p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shoulders MD, et al. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 2013;3:1279–1292. doi: 10.1016/j.celrep.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Denzel MS, et al. Hexosamine pathway metabolites enhance protein quality control and prolong life. Cell. 2014;156:1167–1178. doi: 10.1016/j.cell.2014.01.061. [DOI] [PubMed] [Google Scholar]
- 74.Bernasconi R, Molinari M. ERAD and ERAD tuning: disposal of cargo and of ERAD regulators from the mammalian ER. Curr Opin Cell Biol. 2011;23:176–183. doi: 10.1016/j.ceb.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Voeltz GK, Rolls MM, Rapoport TA. Structural organization of the endoplasmic reticulum. EMBO Rep. 2002;3:944–950. doi: 10.1093/embo-reports/kvf202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chevet E, et al. Phosphorylation by CK2 and MAPK enhances calnexin association with ribosomes. Embo J. 1999;18:3655–3666. doi: 10.1093/emboj/18.13.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lynes EM, et al. Palmitoylation is the Switch that Assigns Calnexin to Quality Control or ER Calcium Signaling. Journal of cell science. 2013 doi: 10.1242/jcs.125856. [DOI] [PubMed] [Google Scholar]
- 78.Brockmeier A, Williams DB. Potent lectin-independent chaperone function of calnexin under conditions prevalent within the lumen of the endoplasmic reticulum. Biochemistry. 2006;45:12906–12916. doi: 10.1021/bi0614378. [DOI] [PubMed] [Google Scholar]
- 79.Zuber C, et al. Immunolocalization of UDP-glucose:glycoprotein glucosyltransferase indicates involvement of pre-Golgi intermediates in protein quality control. Proc Natl Acad Sci U S A. 2001;98:10710–10715. doi: 10.1073/pnas.191359198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pearse BR, Gabriel L, Wang N, Hebert DN. A cell-based reglucosylation assay demonstrates the role of GT1 in the quality control of a maturing glycoprotein. J Cell Biol. 2008;181:309–320. doi: 10.1083/jcb.200712068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Frenkel Z, Shenkman M, Kondratyev M, Lederkremer GZ. Separate roles and different routing of calnexin and ERp57 in endoplasmic reticulum quality control revealed by interactions with asialoglycoprotein receptor chains. Mol Biol Cell. 2004;15:2133–2142. doi: 10.1091/mbc.E03-12-0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Klampfl T, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369:2379–2390. doi: 10.1056/NEJMoa1311347. [DOI] [PubMed] [Google Scholar]
- 83.Dalziel M, Crispin M, Scanlan CN, Zitzmann N, Dwek RA. Emerging principles for the therapeutic exploitation of glycosylation. Science. 2014;343:1235681. doi: 10.1126/science.1235681. [DOI] [PubMed] [Google Scholar]
- 84.Perry ST, et al. An iminosugar with potent inhibition of dengue virus infection in vivo. Antiviral Res. 2013;98:35–43. doi: 10.1016/j.antiviral.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 85.Butters TD, Dwek RA, Platt FM. Imino sugar inhibitors for treating the lysosomal glycosphingolipidoses. Glycobiology. 2005;15:43R–52R. doi: 10.1093/glycob/cwi076. [DOI] [PubMed] [Google Scholar]
- 86.Jeyakumar M, Dwek RA, Butters TD, Platt FM. Storage solutions: treating lysosomal disorders of the brain. Nat Rev Neurosci. 2005;6:713–725. doi: 10.1038/nrn1725. [DOI] [PubMed] [Google Scholar]
- 87.Booth C, Koch GL. Perturbation of cellular calcium induces secretion of luminal ER proteins. Cell. 1989;59:729–737. doi: 10.1016/0092-8674(89)90019-6. [DOI] [PubMed] [Google Scholar]
- 88.Egan ME, et al. Calcium-pump inhibitors induce functional surface expression of Delta F508-CFTR protein in cystic fibrosis epithelial cells. Nat Med. 2002;8:485–492. doi: 10.1038/nm0502-485. [DOI] [PubMed] [Google Scholar]
- 89.Van Goor F, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A. 2009;106:18825–18830. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rowe SM, Verkman AS. Cystic fibrosis transmembrane regulator correctors and potentiators. Cold Spring Harbor perspectives in medicine. 2013;3 doi: 10.1101/cshperspect.a009761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Balch WE, Roth DM, Hutt DM. Emergent properties of proteostasis in managing cystic fibrosis. Cold Spring Harbor perspectives in biology. 2011;3 doi: 10.1101/cshperspect.a004499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mu TW, Fowler DM, Kelly JW. Partial restoration of mutant enzyme homeostasis in three distinct lysosomal storage disease cell lines by altering calcium homeostasis. PLoS Biol. 2008;6:e26. doi: 10.1371/journal.pbio.0060026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mu TW, et al. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell. 2008;134:769–781. doi: 10.1016/j.cell.2008.06.037. Provides a powerful example of the potential utility for proteostasis regulators. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ong DS, Mu TW, Palmer AE, Kelly JW. Endoplasmic reticulum Ca2+ increases enhance mutant glucocerebrosidase proteostasis. Nat Chem Biol. 2010;6:424–432. doi: 10.1038/nchembio.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ong DS, et al. FKBP10 depletion enhances glucocerebrosidase proteostasis in Gaucher disease fibroblasts. Chem Biol. 2013;20:403–415. doi: 10.1016/j.chembiol.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bouchecareilh M, Hutt DM, Szajner P, Flotte TR, Balch WE. Histone deacetylase inhibitor (HDACi) suberoylanilide hydroxamic acid (SAHA)-mediated correction of alpha1-antitrypsin deficiency. J Biol Chem. 2012;287:38265–38278. doi: 10.1074/jbc.M112.404707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Di XJ, Han DY, Wang YJ, Chance MR, Mu TW. SAHA enhances Proteostasis of epilepsy-associated alpha1(A322D)beta2gamma2 GABA(A) receptors. Chem Biol. 2013;20:1456–1468. doi: 10.1016/j.chembiol.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cui J, Smith T, Robbins PW, Samuelson J. Darwinian selection for sites of Asn-linked glycosylation in phylogenetically disparate eukaryotes and viruses. Proc Natl Acad Sci U S A. 2009;106:13421–13426. doi: 10.1073/pnas.0905818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Banerjee S, et al. The evolution of N-glycan-dependent endoplasmic reticulum quality control factors for glycoprotein folding and degradation. Proc Natl Acad Sci U S A. 2007;104:11676–11681. doi: 10.1073/pnas.0704862104. [DOI] [PMC free article] [PubMed] [Google Scholar]