

Abstract
The RhoGEF (Rho GTPase guanine-nucleotide-exchange factor) domain of AKAP-Lbc (A-kinase-anchoring protein-Lbc, also known as AKAP13) catalyses nucleotide exchange on RhoA and is involved in the development of cardiac hypertrophy. The RhoGEF activity of AKAP-Lbc has also been implicated in cancer. We have determined the X-ray crystal structure of the complex between RhoA–GDP and the AKAP-Lbc RhoGEF [DH (Dbl-homologous)–PH (pleckstrin homology)] domain to 2.1 Å (1 Å=0.1 nm) resolution. The structure reveals important differences compared with related RhoGEF proteins such as leukaemia-associated RhoGEF. Nucleotide-exchange assays comparing the activity of the DH–PH domain to the DH domain alone showed no role for the PH domain in nucleotide exchange, which is explained by the RhoA–AKAP-Lbc structure. Comparison with a structure of the isolated AKAP-Lbc DH domain revealed a change in conformation of the N-terminal ‘GEF switch’ region upon binding to RhoA. Isothermal titration calorimetry showed that AKAP-Lbc has only micromolar affinity for RhoA, which combined with the presence of potential binding pockets for small molecules on AKAP-Lbc, raises the possibility of targeting AKAP-Lbc with GEF inhibitors.
Keywords: AKAP13, AKAP-Lbc, GTPase, RhoGEF
Abbreviations: AKAP, A-kinase-anchoring protein; DH, Dbl-homologous; GEF, guanine-nucleotide-exchange factor; ITC, isothermal titration calorimetry; LARG, leukaemia-associated RhoGEF; MANT, N-methylanthraniloyl; MAPK, mitogen-activated protein kinase; PH, pleckstrin homology; PKA, protein kinase A; PRKD1, protein kinase D1; TEV, tobacco etch virus
Short abstract
The crystal structure of the RhoGEF domain of AKAP-Lbc in complex with RhoA combined with nucleotide exchange assays explain differences to related RhoGEF proteins and allow the possibility of targeting the AKAP-Lbc RhoGEF domain with small molecules.
INTRODUCTION
AKAP-Lbc (also known as AKAP13) is a member of the A-kinase-anchoring proteins [1,2]. AKAPs are diverse in sequence and domain composition, but have a common feature of binding the PKA (protein kinase A) regulatory subunit and controlling the localization of PKA [3–5]. The protein scaffolding mediated by AKAP-Lbc includes KSR1 (kinase suppressor of Ras 1), PKA, RAF, MEK [MAPK (mitogen-activated protein kinase)/ERK (extracellular-signal-regulated kinase) kinase], IKKβ [IκB (inhibitor of nuclear factor κB) kinase β] [6], protein kinases D, C [7,8] and N [9], SHP2 (Src homology 2 domain-containing protein tyrosine phosphatase 2) [10], Hsp20 (heat-shock protein 20) [11], p38 MAPK and 14-3-3 proteins [12–14]. AKAP-Lbc exists as at least three splice variants, however, all contain a conserved RhoGEF (Rho GTPase guanine-nucleotide-exchange factor) domain. RhoGEF domains decrease the affinity of a Rho GTPase for GDP so that GTP, present in higher concentrations in the cell, can bind. Thus RhoGEFs activate Rho GTPases allowing extracellular stimuli to promote Rho–GTP formation which initiates signalling pathways inside the cell.
RhoGEF domains are composed of a DH (Dbl-homologous) domain followed by a PH (pleckstrin homology) domain. This tandem DH–PH domain is a conserved unit in most RhoGEF proteins [15]. The DH domain is essential, but the role of the PH domain is variable. In some RhoGEFs, the PH domain controls protein localization through binding to phospholipids [16], or possibly in some cases to other molecules. However, for some RhoGEFs, the PH domain also contributes to the activity of the DH domain in catalysing nucleotide exchange, such as in the examples of PDZ-RhoGEF (ARHGEF11) [17,18], Dbs (MCF2L) [19–21], p115-RhoGEF [22] or leukaemia-associated RhoGEF (LARG and ARHGEF12) [23]. In the case of PDZ-RhoGEF, the PH domain was more important for nucleotide exchange with CDC42 than with RhoA [17]. In some cases, activated RhoA interacts with the PH domains as a form of positive feedback through a mechanism not involving alteration of the nucleotide-exchange rate [24].
AKAP-Lbc has a RhoGEF DH–PH domain between residues 1972 and 2342 (Figure 1A). Its PH domain was shown to be dispensable for activation of Rho, but important for the localization and transformative activity of AKAP-Lbc [25]. The region C-terminal to the DH–PH domain is involved in scaffolding PRKCH [also known as PKCη (protein kinase Cη)] and PRKD1 (protein kinase D1) to activate PRKD1 [8]. This C-terminal region is altered in oncogenic AKAP-Lbc [26].
Figure 1. Structure of AKAP-Lbc.

(A) Protein domains of AKAP-Lbc (AKAP13). The range of the construct used for structure determination is shown above the DH and PH domains. (B) Overview of the complex between RhoA–GDP and the DH–PH domains of AKAP-Lbc. RhoA is shown with red α-helices and yellow β-strands, and with the switch I and switch II loops in green. AKAP-Lbc is shown with its DH domain in blue and PH domain in green. (C) The binding of the RhoA switch I loop. The switch I loop is shown in yellow, with the AKAP-Lbc DH domain shown in green. (D) The binding of the RhoA switch II loop, coloured as for (C).
AKAP-Lbc has been shown to have a role in the development of cardiac hypertrophy [3,7,27,28]. In addition, a truncated form of AKAP-Lbc has been identified as an oncoprotein [29,30]. Evidence suggests that this oncogenic activity of AKAP-Lbc is through activation of the Rho pathway, via the RhoGEF activity of AKAP-Lbc [31,32]. An association between AKAP-Lbc genetic variants and high-risk familial breast cancer has also been detected [33]. These disease links involving the RhoGEF activity of AKAP-Lbc suggest that inhibitors of the RhoA–AKAP-Lbc interaction may be a worthwhile line of investigation for treatment of cardiac hypertrophy, pulmonary arterial hypertension [34] and possibly for cancers. For example, AKAP-Lbc is absent in normal liver, but expressed in hepatocarcinoma, where transformation was blocked by a Rho inhibitor [35]. The possibility in principle of targeting a GTPase–GEF interface has been demonstrated for Rac1–Tiam1 [36,37] and for RhoA–LARG [38]. To support these investigations on RhoA–AKAP-Lbc and to investigate the mechanism of activation of RhoA by AKAP-Lbc we have determined the X-ray crystal structure of RhoA–GDP in complex with the DH–PH tandem domain of AKAP-Lbc, compared it with the structure of the DH domain alone, and analysed the binding affinities and rates of nucleotide exchange in the RhoA–AKAP-Lbc complex.
MATERIALS AND METHODS
Cloning
DNA for RhoA (gi|10835049) or AKAP13 (gi|31563330, NP_006729.4, AKAP13 isoform 1) was PCR amplified and subcloned into an in-house pET-based vector pNIC28-Bsa4 [39] using ligation-independent cloning. The DNA template for RhoA was obtained from the Mammalian Gene Collection (IMAGE Consortium Clone ID 4102976). The resulting constructs expressed the desired proteins with N-terminal His6-tags and TEV (tobacco etch virus) protease tag cleavage sites (extension MHHHHHHSSGVDLGTENLYFQ*SM). AKAP13 DH domain point mutant constructs were created by PCR using the His6-tag DH–PH domain expression construct as a template. All mutations were verified by DNA sequencing.
Protein expression and purification
RhoA and AKAP13 DH–PH constructs were transformed into Escherichia coli BL21(DE3) competent cells containing the pRARE2 plasmid from the commercial Rosetta strain, and the transformants used to inoculate 50 ml of LB medium containing 50 μg/ml kanamycin and 34 μg/ml chloramphenicol. These cultures were incubated overnight at 37°C. These cultures were used to inoculate larger cultures of TB medium (RhoA) or LB medium (AKAP13) with 40 μg/ml kanamycin and grown at 37°C until an D600 of 1.0 (RhoA) or 0.4–0.5 (AKAP13) was reached. The temperature was then reduced to 20°C and the cultures induced with 0.5 mM IPTG. Expression was continued overnight. The cells were harvested by centrifugation, resuspended in binding buffer (500 mM NaCl, 50 mM Hepes, pH 7.5, 5% glycerol, 20 mM imidazole, pH 7.5, and 0.5 mM TCEP) and frozen until further use.
For purification, the cells were thawed and lysed by sonication on ice. PEI (polyethyleneimine) was added to a final concentration of 0.15% and the cell debris and precipitated DNA were spun down. RhoA and AKAP13 were each purified by passing the supernatants through a column of 5 ml of Ni2+-Sepharose resin (GE Healthcare). After washing, the protein was eluted with 25 ml of binding buffer containing 250 mM imidazole. The His6-tags were removed using TEV protease. AKAP13 was then mixed in equimolar ratio with RhoA, and the complex was concentrated and injected on to a S75 16/60 gel filtration column pre-equilibrated into GF buffer (50 mM Hepes, pH 7.5, 500 mM NaCl, 5% glycerol and 0.5 mM TCEP). After passing through a gravity column of 5 ml of Ni2+-Sepharose to remove impurities, the RhoA–AKAP13 complex was concentrated to 5.1 mg/ml. Protein concentrations were measured by UV absorbance, using the calculated molecular masses and estimated molar absorption coefficients, using a NanoDrop spectrophotometer (Thermo Scientific). The constructs of the AKAP13 DH domain and the DH domain point mutations were used to express and purify protein following a similar protocol.
Crystallization and data collection
RhoA–AKAP13 domain crystals grew by vapour diffusion at 4°C from a mixture of 50 nl of protein and 100 nl of a well solution containing 0.2 M ammonium sulfate, 0.1 M Bis-Tris, pH 5.5, and 25% (w/v) PEG 3350. Crystals were equilibrated into reservoir solution plus 25% ethylene glycol before freezing in liquid nitrogen. Data were collected at 100 K at the Diamond Synchrotron beamline I02. Data collection statistics can be found in Table 1.
Table 1. Data collection and refinement statistics.
Values within parentheses refer to the highest resolution shell.
| Parameter | RhoA–AKAP-Lbc DH–PH | AKAP-Lbc DH |
|---|---|---|
| PDB code | 4D0N | 4D0O |
| Space group | P212121 | P212121 |
| Number of molecules in the asymmetric unit | 1 | 2 |
| Unit cell dimensions a, b, c (Å) | 82.3, 86.6, 116.8 | 52.1, 94.8, 109.0 |
| Data collection | ||
| Resolution range (Å) | 82.32–2.10 (2.16–2.10) | 42.13–2.75 (2.90–2.75) |
| Unique observations | 49042 (3824) | 14660 (2101) |
| Average multiplicity | 3.4 (2.9) | 6.5 (6.5) |
| Completeness (%) | 99.3 (95.8) | 100.0 (100.0) |
| Rmerge | 0.06 (0.45) | 0.21 (1.18) |
| Mean (I)/σ(I) | 8.2 (1.6) | 7.2 (2.1) |
| Mean CC(1/2) | 0.998 (0.784) | 0.992 (0.530) |
| Refinement | ||
| R-value, Rfree (%) | 21.1, 23.9 | 24.0, 30.8 |
| RMSD from ideal bond length (Å) | 0.006 | 0.005 |
| RMSD from ideal bond angle (°) | 1.07 | 0.887 |
DH domain crystals grew under identical conditions [50 nl of protein and 100 nl of a well solution containing 0.2 M ammonium sulfate, 0.1 M Bis-Tris, pH 5.5, and 25% (w/v) PEG 3350] and were equilibrated into reservoir solution plus 20% ethylene glycol before freezing in liquid nitrogen. Data were collected at 100 K at the Diamond Synchrotron beamline I03.
Structure determination
The diffraction data were indexed and integrated using MOSFLM [40] and scaled using AIMLESS [41]. The structure was solved by molecular replacement using PHASER [42] and the structures of RhoA (PDB code 1XCG [17]) and p115-RhoGEF (PDB code 3ODW [43]) as search models. There was one molecule of the RhoA–AKAP-Lbc complex in the asymmetric unit. The model was built using Coot [44] and refined with REFMAC5 [45]. Rebuilding and refinement resulted in the final model. The model was validated using MOLPROBITY [46]. All structure figures were created using PyMOL (Schrödinger).
ITC (isothermal titration calorimetry)
Measurements were made on a MicroCal iTC200 (GE Healthcare) at 15°C. For measurements in the presence of GDP, all proteins were dialysed overnight into a buffer consisting of GF buffer with the addition of 50 μM GDP. For measurements in the absence of GDP, RhoA was incubated overnight with 5 mM ETDA and 6 units/ml alkaline phosphatase followed by gel filtration in GF buffer, and finally concentrated as before and dialysed overnight in GF buffer. For each measurement, the syringe was loaded with 0.6 mM RhoA (in the absence or presence of GDP) and 20×2 μl injections were made into the cell, which was filled with 0.04 mM of either AKAP-Lbc DH domain or AKAP-Lbc DH–PH domain. Data were analysed using MicroCal and Origin software. The results after fitting to a single-site model are shown in Table 2.
Table 2. ITC measurements.
Errors represent the error in the fit to the binding curve, not the overall error in the measurement.
| RhoA–GDP + DH | RhoA–GDP + DH–PH | RhoA + DH | RhoA + DH–PH | |
|---|---|---|---|---|
| N | 0.55 | 0.55 | 0.68 | 0.35 |
| ΔH (kcal/mol) | 20.2±2.0 | 7.6±0.9 | 12.7±0.5 | 49.6±10.2 |
| TΔS (kcal/mol) | 26.4 | 13.9 | 66.9 | 55.6 |
| Kd (μM) | 21.3±2.0 | 18.1±2.3 | 10.2±0.7 | 33.9±3.0 |
GDP/GTP exchange assays
RhoA was incubated with alkaline phosphatase to remove bound nucleotide and then purified by gel filtration under the same conditions as above. The nucleotide-free RhoA was incubated with MANT (N-methylanthraniloyl)-GDP (Life Technologies) for 15 min. The protein was then passed through a PD-10 column pre-equilibrated in GF buffer to remove excess MANT-GDP. Guanine-nucleotide-exchange assays were conducted in 20 μl aliquots in black 384-well low-volume plates (Greiner). The progress of the nucleotide exchange was monitored by fluorescence on a PheraStar plate reader (BMG Labtech) with excitation at 360 nm and detection at 440 nm. Exchange reactions were measured in GF buffer containing 10 mM MgCl2, with 0.5 μM RhoA–MANT-GDP and 50 μM unlabelled GMP-PNP, and in the presence or absence of varying concentrations of DH or DH–PH domain protein.
Druggability score calculations
Protein structures for binding-site analysis were prepared using the protein preparation function in Maestro (Schrödinger). All protein chains except the chain of interest were deleted. Hydrogen atoms were added and missing side chains were rebuilt using Prime (Schrödinger). Druggability scores were calculated using SiteMap (Schrödinger), using the default settings as implemented in Maestro, using the OPLS_2005 force field definition. For calculation of SiteMap scores from bromodomains, the conserved water molecules that form part of the acetyl-lysine binding pocket were retained in the structure used in the analysis.
RESULTS
Structure determination
Bacterial overexpression constructs were prepared for RhoA (residues 1–184) and the DH–PH domain of AKAP-Lbc (residues 1972–2342, residue numbers refer to AKAP13 isoform 1, Genbank ID NP_006729.4) (Figure 1A and Table 3). After initial purification the proteins were mixed and passed through a size-exclusion chromatography column together. RhoA and the DH–PH domain eluted at the same time from the size-exclusion column. Both the DH–PH domain alone and the complex of RhoA and DH–PH domain appeared to be monomeric during size-exclusion chromatography.
Table 3. Protein expression constructs.
| RhoA | AKAP-Lbc DH–PH domain | AKAP-Lbc DH domain | |
|---|---|---|---|
| Residue range* | Met1–Gly184 | Lys1973–Asp2342 | Glu1976–Lys2211 |
| Expression vector | pNIC28-Bsa4 | pNIC28-Bsa4 | pNIC28-Bsa4 |
| Molecular mass (kDa) | 20.9 | 43.4 | 27.9 |
*Sequences of the protein constructs that were used can be found in the Supplementary Online Material.
Crystals of the RhoA–DH–PH domain complex were obtained in the presence of GDP which allowed the structure to be determined by X-ray diffraction to 2.1 Å (1 Å=0.1 nm) resolution (Table 1). There was one molecule of a 1:1 RhoA(GDP)–DH–PH complex in the asymmetric unit (Figure 1B). The model was refined to a free R-factor of 23.8%. Residues 3–181 of RhoA and residues 1972–2340 of AKAP-Lbc (including the DH and PH domains) were resolved in the electron density.
AKAP-Lbc binds RhoA via its switch I and switch II loops
Overall, the structure resembles the general domain arrangement seen in previously published complexes between RhoA and other RhoGEFs. The AKAP-Lbc DH domain interacts with the switch I and switch II loops of RhoA, with a buried surface area at the interface of 1319 Å2 (Figure 2A). However, the closest relation to AKAP-Lbc that has been crystallized previously is p115-RhoGEF [43] with 31% sequence identity to AKAP-Lbc over the DH–PH domain (Supplementary Figure S1) and many of the residues involved in the interface with RhoA are not conserved in other RhoGEFs.
Figure 2. No contact between the AKAP-Lbc PH domain and RhoA.

(A and B) Two views of the separation between RhoA and the PH domain. Residues which in homologous proteins are involved in RhoA–PH domain interactions are shown as a stick representation. RhoA is coloured red, and the AKAP-Lbc DH and PH domains are coloured blue and green. (C and D) Two views, from equivalent orientations as (A) and (B), of the interaction between the PH domain of LARG and RhoA (PDB code 1X86). The LARG DH and PH domains are coloured brown and dark green. (E) Sequence alignment of the regions of the PH domain and DH domain potentially involved in RhoA–PH domain interactions, for AKAP-Lbc and three homologous RhoGEF domains, all three of which show an effect of the PH domain on nucleotide-exchange catalysis. Residues involved in the interface in LARG are indicated with asterisks below the alignment. For residue differences likely to be important for the lack of effect of the AKAP-Lbc PH domain, the asterisks are coloured red.
The RhoA switch I loop is bound between helices αA and αH of the DH domain (Figures 1B and 1C). At the C-terminal end of the loop, Glu40 forms a hydrogen bond with His2012 of the DH domain while the backbone carbonyl of Phe39 forms a hydrogen bond with the side chain of Lys2156. A central hydrophobic patch consisting of Thr2009, Val2159 and Leu2160 interacts with the RhoA residue Val38. At the N-terminal end of switch I, there is an extensive hydrogen-bonding network with the DH domain where the DH domain Glu2005 binds simultaneously the backbone nitrogen of RhoA Val38 and the side chains of Thr37 and Tyr34, while the DH domain Arg2163 also forms a hydrogen bond with Thr37 as well as the backbone carbonyl of Val35. An additional hydrophobic packing involves the aromatic ring of RhoA Tyr34 and the side chain of Glu2001.
The Rho switch II loop is bound in a similar pattern to switch I, with polar interactions between each end of the loop and the DH domain, and a hydrophobic packing in the middle (Figure 1D). At the C-terminal end, the DH domain Asp2193 forms hydrogen bonds with the backbone nitrogen and the side chain of RhoA Arg68. RhoA Leu69 packs in between DH domain residues Cys2105, Val2196, Ile2189 and Val2192, whereas the aromatic ring of Tyr66 packs against Ile2189 and the methyl group of Thr2155. At the N-terminal end of switch I, Gln2152 binds the backbone carbonyl of RhoA Asp59.
No contacts between the AKAP-Lbc PH domain and RhoA
For some RhoGEFs such as LARG (29% sequence identity with AKAP-Lbc over the DH–PH domain) the PH domain contributes to GEF activity. The AKAP-Lbc PH domain made no contacts with RhoA in the crystal structure (Figures 1B, 2A and 2B). Comparison with the structure of RhoA–LARG (PDB code 1X86) [23] reveals that residues crucial for the involvement of the PH domain in stabilizing the RhoA–RhoGEF complex are not conserved in AKAP-Lbc (Figures 2C, 2D and 2E). LARG, p115 and PRG all have a TXS(D/E) motif at the start of the PH domain helix (residues 1116–1119 in LARG) which interacts with RhoA Glu97. In AKAP-Lbc this is replaced by an SXEE sequence. The replacement of Ser2323 with glutamic acid in AKAP-Lbc is presumably unfavourable for binding the also negatively charged RhoA Glu97. Indeed, a LARG S1118D mutation cancelled the additional contribution of the LARG PH domain to nucleotide exchange, as did an E1023A mutation [23].
One proposal for the mechanism by which a RhoGEF PH domain can contribute to GEF activity is stabilization of the long C-terminal helix of the DH domain (αI). This helix forms crucial binding interactions with the GTPase switch II loop. Crucial residues for this stabilization are not conserved in AKAP-Lbc. In LARG, RhoA Arg68 is bound by Glu982 and Asn983 (Figures 2C and 2D). Asn983 is replaced with Lys2001 in AKAP-Lbc which cannot form the same binding interaction and RhoA Arg68 is instead bound to Asp2193, much closer to the N-terminus of αI (Figure 2B). Furthermore, the binding of Arg68 to the LARG DH domain is made possible by the curvature and stabilization of the αI C-terminus induced by close association to the PH domain (Figures 2D and 2E). This association is also required for proximity of the PH domain helix to RhoA Glu97. Although the crucial LARG residue Arg986 is conserved in AKAP-Lbc (Arg2204), the other residues are entirely non-conserved. For example, the LARG PH domain residues Tyr1039 and Glu1023 are replaced with Gln2255 and the shorter Asp2239 in AKAP-Lbc. The result is that the binding arrangement is quite different in AKAP-Lbc and results in an entirely straight long α-helix (αI) with four turns of this α-helix separating the residues interacting with switch II from the residues interacting with the PH domain (Figures 2A and 2B).
AKAP-Lbc PH domain is not required for catalysis of RhoA nucleotide exchange
To investigate whether for our constructs the AKAP-Lbc PH domain contributes to catalysis of nucleotide exchange on RhoA we cloned an expression construct containing just the DH domain (residues 1976–2211, terminating at the end of αI) for comparison with the combined DH–PH domain (Table 3). We performed nucleotide-exchange assays using the decrease in fluorescence when MANT-GDP moves into a solvent environment after being bound to a protein [47]. The rate of exchange of RhoA–MANT-GDP for RhoA–GTP was measured in the presence of 25 nM, 250 nM and 750 nM of either the AKAP-Lbc DH–PH domain or the DH domain alone (Figure 3). The rate increased with increasing concentrations of DH–PH or DH domain alone, but at all three concentrations measured the rate was unaffected by the presence of the PH domain.
Figure 3. The PH domain does not influence the DH-domain-induced GEF on RhoA.
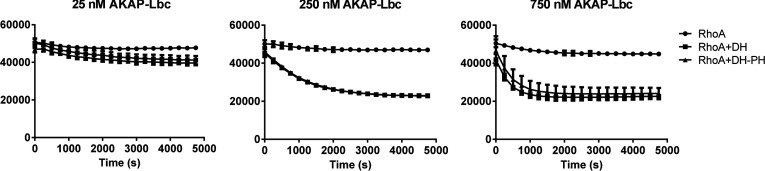
The rate of exchange of bound MANT-GDP for unlabelled GMP-PNP in the absence and presence of either the DH domain or the combined DH–PH domain of AKAP-Lbc. The decrease in fluorescence of MANT-GDP upon leaving RhoA was measured over time. The y-axis shows arbitrary fluorescence units. Measurements are plotted as the means±S.D. from three replicates.
To analyse further any possible role for the PH domain in the interactions with RhoA we compared the dissociation constants for the complexes of RhoA with the DH–PH domain or with the DH domain alone. We also wanted to measure dissociation constants with RhoA–GDP and with apo-RhoA to see whether AKAP-Lbc would have greater affinity for apo-RhoA as has been observed previously for other RhoGEFs [48]. Binding affinities were measured by ITC. For measurements in the presence of GDP, all proteins were dialysed beforehand against buffer containing GDP, and the measurements were performed in this buffer to counteract any effects of nucleotide binding. For measurements in the absence of GDP, any bound nucleotide was removed from RhoA beforehand by phosphatase treatment. The measurements revealed similar dissociation constants (Kd) within experimental error of ~20 μM for RhoA against either the DH–PH domain or the DH domain alone, in the presence or absence of GDP (Figure 4).
Figure 4. ITC measurements of the dissociation constants (Kd) between RhoA and the AKAP-Lbc DH–PH domain or the AKAP-Lbc DH domain, in the presence or absence of GDP.

Changes in the conformation of the DH domain upon binding to RhoA
The N-terminus of some DH domains contains a ‘GEF switch’ region [43], a short sequence motif just N-terminal to the first α-helix (αA) of the DH domain. For both LARG and p115-RhoGEF, this region was shown to be important for catalysis of nucleotide exchange [23,43]. Deletion of the GEF switch from LARG or mutation of the conserved tryptophan residue at the centre of the motif (LARG Trp769) to alanine or aspartate reduced GEF activity to 15–20% of wild-type [23]. Furthermore, LARG E790G also reduced GEF activity to 15–20%, suggesting that the critical factor is its interaction with RhoA Tyr34.
The GEF switch region of AKAP-Lbc's DH domain (Figure 5A) adopts a similar conformation to that of LARG (Figure 5C). RhoA Tyr34 packs against the conserved Glu2001 (LARG Glu790), whereas forming a hydrogen bond to the also conserved Glu2005 (LARG Glu794). The tryptophan appears to orient the backbone of the GEF switch region so that Glu2001 can form hydrogen bonds to two backbone nitrogens (Figures 5A and 5C). This presumably stabilizes the conformation of Glu2001 in a conformation favourable for interaction with RhoA Tyr34.
Figure 5. Comparison of the GEF switch region at the N-terminus of the AKAP-Lbc DH domain with the equivalent regions in LARG and p115-RhoGEF.

(A) The conformation of the AKAP-Lbc DH domain GEF switch region (coloured in magenta) when bound to RhoA. RhoA is coloured with red α-helices and yellow β-strands with its switch I region in green. The DH domain of AKAP-Lbc is coloured blue. (B) The conformation of the DH domain when not bound to RhoA. (C and D) The conformation of the equivalent regions in LARG when bound to RhoA (C) and p115-RhoGEF when not bound to RhoA (D). (E) Sequence alignment of the GEF switch regions of AKAP-Lbc and three homologous RhoGEF domains. Residues important for the packing of the GEF switch region are marked with asterisks below the alignment.
To analyse conformational changes in the DH domain on binding we crystallized the DH domain alone in the absence of RhoA (Table 1), revealing a significant movement of the GEF switch region (Figure 5B), which adopts an α-helical conformation with Trp1980 itself binding in a different pocket on the first DH domain α-helix. This we refer to as the inactive conformation, in which the position occupied by Trp1980 in the active conformation is now occupied by Ile1984 and Ile1983 (Figure 5B). We cannot rule out that the inactive α-helical conformation is induced by the N-terminal protein tag which is at the start of this α-helix. However, we note that the region around Trp1980 is weakly predicted to be α-helical by the PsiPred server [49], suggesting that a conformational exchange in and out of an α-helix induced by the absence or presence of RhoA is a realistic model. In this inactive conformation, the critical Glu2001 can no longer bind to RhoA (Figure 4B). In contrast with AKAP-Lbc, in the structure of p115-RhoGEF in the absence of RhoA [43], the GEF switch maintains an active conformation (Figure 5D).
Potential for targeting small molecules against the GEF activity of AKAP-Lbc
There are two shallow pockets (Figures 6A and 6B) on the surface of the DH domain which are involved in the binding of the RhoA switch I and switch II loops (Site 1 and Site 2). An inhibitor bound at either or both of these sites might prevent binding and activation of RhoA. To validate the functional consequences of small molecules binding at these sites we created 12 point mutations, either to alanine (smaller) or to tyrosine (larger) on the surface of the DH domain around the pockets (Figure 6D). Four of these mutant proteins (all replaced with a tyrosine residue) failed to generate soluble protein expression. The remaining eight were purified and analysed in the GEF assay (Figure 6C). The curves were fitted to an exponential equation to calculate a half-life for bound MANT-GDP at 750 nM AKAP-Lbc. Mutation R2163A (site 1) showed activity equivalent to wild-type. The remaining mutations to alanine all showed decreased exchange activity consistent with reduced affinity of binding, whereas the mutations to tyrosine all essentially abolished the GEF activity of AKAP-Lbc. This suggests that binding of small molecules at these sites would also abolish GEF activity.
Figure 6. Surface pockets on the AKAP-Lbc DH domain are potentially suitable for binding of protein–protein interface inhibitors.

(A) Site 1 which binds the switch I loop of RhoA. (B) Site 2 which binds the switch II loop of RhoA. The core of each pocket containing the residues in direct contact with RhoA is coloured dark orange, whereas the surrounding region of the pocket is coloured light orange. Sites of mutations made are indicated (C). Nucleotide-exchange assays for DH domain mutants compared with wild-type. (D) Approximate half-lives of RhoA–MANT-GDP based on fitting the curves in (C) to an exponential decay.
DISCUSSION
The RhoGEF activity of AKAP-Lbc is potentially interesting for therapeutic intervention. With only modest binding affinity for RhoA, development of a RhoA-competitive inhibitor should be possible. Furthermore, since the PH domain is not involved in nucleotide exchange, any compound screening programme need only be concerned with the DH domain. With the low sequence conservation between AKAP-Lbc and related RhoGEFs, it is likely that AKAP-Lbc-specific inhibitors could be generated.
We analysed the two potential binding pockets for small molecules using SiteMap (Schrödinger) which takes into account the volume of the pocket, degree of enclosure and degree of hydrophobicity. AKAP-Lbc had SiteMap Dscores of 0.74 and 0.83 respectively for the switch I and switch II binding pockets, with pocket volumes of 133 and 145 Å3. These were the top two sites (by Dscore) that were identified. By comparison with a well-known druggable protein with a larger binding side, using the same methodology the protein kinase Aurora A (PDB code 1MQ4) has a Dscore of 0.96 with a pocket volume of 591 Å3. Bromodomains for which small molecule ligands have been identified span a range of Dscores from >1.0 to 0.62 [50]. Within this range, two exemplars BRD4(1) and BAZ2B had median Dscores of 0.93 and 0.62 respectively [50]. For comparison, using the same SiteMap settings as for AKAP-Lbc, we calculated Dscores of 0.90 and 0.66 for BRD4(1) (PDB code 3MXF) and BAZ2B (PDB code 3G0L), with pocket volumes of 125 and 109 Å3. In conclusion, AKAP-Lbc appears at least as druggable as bromodomains for which many ligands have already been identified [51].
Finally, the analysis of the sequence and structural reasons for the lack of involvement of the AKAP-Lbc PH domain in GEF activity may allow useful predictions to be made for other RhoGEFs, of which there are approximately 72 human members having the DH–PH domain architecture [15].
Online data
AUTHOR CONTRIBUTION
Kamal Abdul Azeez purified the proteins, grew the protein crystals, performed GEF assays and measured the ITC data. Kamal Abdul Azeez and João Fernandes cloned AKAP13 mutants. Jonathan Elkins performed crystal mounting, solved and refined the crystal structures, performed the druggability score calculations, supervised the study and wrote the paper. Kamal Abdul Azeez and Jonathan Elkins analysed ITC and GEF assay data and prepared the Figures. Stefan Knapp and Enno Klussmann conceived the study and contributed to the paper.
FUNDING
K.R.A.A., S.K. and J.M.E. are supported by the Structural Genomics Consortium, a registered charity (number 1097737) that receives funds from AbbVie, Bayer, Boehringer Ingelheim, the Canada Foundation for Innovation, the Canadian Institutes for Health Research, Genome Canada, GlaxoSmithKline, Janssen, Lilly Canada, the Novartis Research Foundation, the Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda and the Wellcome Trust [grant number 092809/Z/10/Z]. E.K. was supported by the Deutsche Forschungsgemeinschaft (DFG) [grant number KL1415/4-2], the Else Kröner-Fresenius-Stiftung [grant number 2013_A145] and the German-Israeli Foundation [grant number I-1210-286.13/2012].
References
- 1.Diviani D., Soderling J., Scott J. D. AKAP-Lbc anchors protein kinase A and nucleates Gα12-selective Rho-mediated stress fiber formation. J. Biol. Chem. 2001;276:44247–44257. doi: 10.1074/jbc.M106629200. [DOI] [PubMed] [Google Scholar]
- 2.Klussmann E., Edemir B., Pepperle B., Tamma G., Henn V., Klauschenz E., Hundsrucker C., Maric K., Rosenthal W. Ht31: the first protein kinase A anchoring protein to integrate protein kinase A and Rho signaling. FEBS Lett. 2001;507:264–268. doi: 10.1016/S0014-5793(01)02995-7. [DOI] [PubMed] [Google Scholar]
- 3.Diviani D., Dodge-Kafka K. L., Li J., Kapiloff M. S. A-kinase anchoring proteins: scaffolding proteins in the heart. Am. J. Physiol. Heart Circ. Physiol. 2011;301:H1742–H1753. doi: 10.1152/ajpheart.00569.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scott J. D., Dessauer C. W., Taskén K. Creating order from chaos: cellular regulation by kinase anchoring. Ann. Rev. Pharm. Tox. 2013;53:187–210. doi: 10.1146/annurev-pharmtox-011112-140204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skroblin P., Grossmann S., Schäfer G., Rosenthal W., Klussmann E. Mechanisms of protein kinase A anchoring. Int. Rev. Cell Mol. Biol. 2010;283:235–330. doi: 10.1016/S1937-6448(10)83005-9. [DOI] [PubMed] [Google Scholar]
- 6.del Vescovo C. D., Cotecchia S., Diviani D. A-kinase-anchoring protein-Lbc anchors IκB kinase β to support interleukin-6-mediated cardiomyocyte hypertrophy. Mol. Cell. Biol. 2013;33:14–27. doi: 10.1128/MCB.00887-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carnegie G. K., Soughayer J., Smith F. D., Pedroja B. S., Zhang F., Diviani D., Bristow M. R., Kunkel M. T., Newton A. C., Langeberg L. K., Scott J. D. AKAP-Lbc mobilizes a cardiac hypertrophy signaling pathway. Mol. Cell. 2008;32:169–179. doi: 10.1016/j.molcel.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carnegie G. K., Smith F. D., McConnachie G., Langeberg L. K., Scott J. D. AKAP-Lbc nucleates a protein kinase D activation scaffold. Mol. Cell. 2004;15:889–899. doi: 10.1016/j.molcel.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 9.Cariolato L., Cavin S., Diviani D. A-kinase anchoring protein (AKAP)-Lbc anchors a PKN-based signaling complex involved in α1-adrenergic receptor-induced p38 activation. J. Biol. Chem. 2011;286:7925–7937. doi: 10.1074/jbc.M110.185645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burmeister B. T., Taglieri D. M., Wang L., Carnegie G. K. Src homology 2 domain-containing phosphatase 2 (Shp2) is a component of the A-kinase-anchoring protein (AKAP)-Lbc complex and is inhibited by protein kinase A (PKA) under pathological hypertrophic conditions in the heart. J. Biol. Chem. 2012;287:40535–40546. doi: 10.1074/jbc.M112.385641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edwards H. V, Scott J. D., Baillie G. S. The A-kinase-anchoring protein AKAP-Lbc facilitates cardioprotective PKA phosphorylation of Hsp20 on Ser16. Biochem. J. 2012;446:437–443. doi: 10.1042/BJ20120570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diviani D., Abuin L., Cotecchia S., Pansier L. Anchoring of both PKA and 14-3-3 inhibits the Rho-GEF activity of the AKAP-Lbc signaling complex. EMBO J. 2004;23:2811–2820. doi: 10.1038/sj.emboj.7600287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith F. D., Langeberg L. K., Cellurale C., Pawson T., Morrison D. K., Davis R. J., Scott J. D. AKAP-Lbc enhances cyclic AMP control of the ERK1/2 cascade. Nat. Cell Biol. 2010;12:1242–1249. doi: 10.1038/ncb2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pérez López I., Cariolato L., Maric D., Gillet L., Abriel H., Diviani D. A-kinase anchoring protein Lbc coordinates a p38 activating signaling complex controlling compensatory cardiac hypertrophy. Mol. Cell. Biol. 2013;33:2903–2917. doi: 10.1128/MCB.00031-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cook D. R., Rossman K. L., Der C. J. Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease. Oncogene. 2013;33:4021–4035. doi: 10.1038/onc.2013.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Viaud J., Gaits-Iacovoni F., Payrastre B. Regulation of the DH-PH tandem of guanine nucleotide exchange factor for Rho GTPases by phosphoinositides. Adv. Biol. Regul. 2012;52:303–314. doi: 10.1016/j.jbior.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 17.Derewenda U., Oleksy A., Stevenson A. S., Korczynska J., Dauter Z., Somlyo A. P., Otlewski J., Somlyo A. V., Derewenda Z. S. The crystal structure of RhoA in complex with the DH/PH fragment of PDZRhoGEF, an activator of the Ca2+ sensitization pathway in smooth muscle. Structure. 2004;12:1955–1965. doi: 10.1016/j.str.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 18.Bielnicki J. A., Shkumatov A. V., Derewenda U., Somlyo A. V., Svergun D. I., Derewenda Z. S. Insights into the molecular activation mechanism of the RhoA-specific guanine nucleotide exchange factor, PDZRhoGEF. J. Biol. Chem. 2011;286:35163–35175. doi: 10.1074/jbc.M111.270918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Snyder J. T., Worthylake D. K., Rossman K. L., Betts L., Pruitt W. M., Siderovski D. P., Der C. J., Sondek J. Structural basis for the selective activation of Rho GTPases by Dbl exchange factors. Nat. Struct. Mol. Biol. 2002;9:468–475. doi: 10.1038/nsb796. [DOI] [PubMed] [Google Scholar]
- 20.Rossman K. L., Worthylake D. K., Snyder J. T., Siderovski D. P., Campbell S. L., Sondek J. A crystallographic view of interactions between Dbs and Cdc42: PH domain-assisted guanine nucleotide exchange. EMBO J. 2002;21:1315–1326. doi: 10.1093/emboj/21.6.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rossman K. L., Cheng L., Mahon G. M., Rojas R. J., Snyder J. T., Whitehead I. P., Sondek J. Multifunctional roles for the PH domain of Dbs in regulating Rho GTPase activation. J. Biol. Chem. 2003;278:18393–18400. doi: 10.1074/jbc.M300127200. [DOI] [PubMed] [Google Scholar]
- 22.Wells C. D., Gutowski S., Bollag G., Sternweis P. C. Identification of potential mechanisms for regulation of p115 RhoGEF through analysis of endogenous and mutant forms of the exchange factor. J. Biol. Chem. 2001;276:28897–28905. doi: 10.1074/jbc.M102913200. [DOI] [PubMed] [Google Scholar]
- 23.Kristelly R., Gao G., Tesmer J. J. G. Structural determinants of RhoA binding and nucleotide exchange in leukemia-associated Rho guanine-nucleotide exchange factor. J. Biol. Chem. 2004;279:47352–47362. doi: 10.1074/jbc.M406056200. [DOI] [PubMed] [Google Scholar]
- 24.Medina F., Carter A. M., Dada O., Gutowski S., Hadas J., Chen Z., Sternweis P. C. Activated RhoA is a positive feedback regulator of the Lbc family of Rho guanine nucleotide exchange factor proteins. J. Biol. Chem. 2013;288:11325–11333. doi: 10.1074/jbc.M113.450056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olson M. F., Sterpetti P., Nagata K., Toksoz D., Hall A. Distinct roles for DH and PH domains in the Lbc oncogene. Oncogene. 1997;15:2827–31. doi: 10.1038/sj.onc.1201594. [DOI] [PubMed] [Google Scholar]
- 26.Sterpetti P., Hack A. A., Bashar M. P., Park B., Cheng S. D., Knoll J. H., Urano T., Feig L. A., Toksoz D. Activation of the Lbc Rho exchange factor proto-oncogene by truncation of an extended C terminus that regulates transformation and targeting. Mol. Cell. Biol. 1999;19:1334–1345. doi: 10.1128/mcb.19.2.1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Appert-Collin A., Cotecchia S., Nenniger-Tosato M., Pedrazzini T., Diviani D. The A-kinase anchoring protein (AKAP)-Lbc-signaling complex mediates alpha1 adrenergic receptor-induced cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 2007;104:10140–10145. doi: 10.1073/pnas.0701099104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diviani D. Modulation of cardiac function by A-kinase anchoring proteins. Curr. Opin. Pharm. 2008;8:166–173. doi: 10.1016/j.coph.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 29.Toksoz D., Williams D. A. Novel human oncogene lbc detected by transfection with distinct homology regions to signal transduction products. Oncogene. 1994;9:621–8. [PubMed] [Google Scholar]
- 30.Rubino D., Driggers P., Arbit D., Kemp L., Miller B., Coso O., Pagliai K., Gray K., Gutkind S., Segars J. Characterization of Brx, a novel Dbl family member that modulates estrogen receptor action. Oncogene. 1998;16:2513–26. doi: 10.1038/sj.onc.1201783. [DOI] [PubMed] [Google Scholar]
- 31.Zheng Y., Olson M. F., Hall A., Cerione R. A., Toksoz D. Direct involvement of the small GTP-binding protein Rho in lbc oncogene function. J. Biol. Chem. 1995;270:9031–9034. doi: 10.1074/jbc.270.2.626. [DOI] [PubMed] [Google Scholar]
- 32.Schwartz M. A., Toksoz D., Khosravi-Far R. Transformation by Rho exchange factor oncogenes is mediated by activation of an integrin-dependent pathway. EMBO J. 1996;15:6525–6530. [PMC free article] [PubMed] [Google Scholar]
- 33.Wirtenberger M., Tchatchou S., Hemminki K., Klaes R., Schmutzler R. K., Bermejo J. L., Chen B., Wappenschmidt B., Meindl A., Bartram C. R., Burwinkel B. Association of genetic variants in the Rho guanine nucleotide exchange factor AKAP13 with familial breast cancer. Carcinogenesis. 2006;27:593–598. doi: 10.1093/carcin/bgi245. [DOI] [PubMed] [Google Scholar]
- 34.Bear M. D., Li M., Liu Y., Giel-Moloney M. A., Fanburg B. L., Toksoz D. The Lbc Rho guanine nucleotide exchange factor α-catulin axis functions in serotonin-induced vascular smooth muscle cell mitogenesis and RhoA/ROCK activation. J. Biol. Chem. 2010;285:32919–32926. doi: 10.1074/jbc.M109.062513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sterpetti P., Marucci L., Candelaresi C., Toksoz D., Alpini G., Ugili L., Baroni G. S., Macarri G., Benedetti A. Cell proliferation and drug resistance in hepatocellular carcinoma are modulated by Rho GTPase signals. Am. J. Physiol. Gastrointest. Liver Physiol. 2006;290:G624–G632. doi: 10.1152/ajpgi.00128.2005. [DOI] [PubMed] [Google Scholar]
- 36.Gao Y., Dickerson J. B., Guo F., Zheng J., Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. U.S.A. 2004;101:7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruffoni A., Ferri N., Bernini S. K., Ricci C., Corsini A., Maffucci I., Clerici F., Contini A. 2-Amino-3-(phenylsulfanyl)norbornane-2-carboxylate: an appealing scaffold for the design of Rac1-Tiam1 protein–protein interaction inhibitors. J. Med. Chem. 2014;57:2953–2962. doi: 10.1021/jm401924s. [DOI] [PubMed] [Google Scholar]
- 38.Shang X., Marchioni F., Evelyn C. R., Sipes N., Zhou X., Seibel W., Wortman M., Zheng Y. Small-molecule inhibitors targeting G-protein-coupled Rho guanine nucleotide exchange factors. Proc. Natl. Acad. Sci. U.S.A. 2013;110:3155–3160. doi: 10.1073/pnas.1212324110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Savitsky P., Bray J., Cooper C. D. O., Marsden B. D., Mahajan P., Burgess-Brown N. A., Gileadi O. High-throughput production of human proteins for crystallization: the SGC experience. J. Struct. Biol. 2010;172:3–13. doi: 10.1016/j.jsb.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leslie A. G. W., Powell H. R. Processing diffraction data with mosflm. In: Read R. J., Sussman J. L., editors. Evolving Methods for Macromolecular Crystallography. Dordrecht: Springer; 2007. pp. 41–51. 245. [DOI] [Google Scholar]
- 41.Evans P. R. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr. 2011;D67:282–292. doi: 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. Phaser crystallographic software. J. Appl. Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen Z., Guo L., Sprang S. R., Sternweis P. C. Modulation of a GEF switch: autoinhibition of the intrinsic guanine nucleotide exchange activity of p115-RhoGEF. Protein Sci. 2011;20:107–117. doi: 10.1002/pro.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emsley P., Lohkamp B., Scott W. G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;D66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011;D67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen V. B., Arendall W. B., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;D66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hemsath L., Ahmadian M. R. Fluorescence approaches for monitoring interactions of Rho GTPases with nucleotides, regulators, and effectors. Methods. 2005;37:173–182. doi: 10.1016/j.ymeth.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 48.Cherfils J., Chardin P. GEFs: structural basis for their activation of small GTP-binding proteins. Trends Biochem. Sci. 1999;24:306–311. doi: 10.1016/S0968-0004(99)01429-2. [DOI] [PubMed] [Google Scholar]
- 49.Buchan D. W. A., Ward S. M., Lobley A. E., Nugent T. C. O., Bryson K., Jones D. T. Protein annotation and modelling servers at University College London. Nucleic Acids Res. 2010;38:W563–W568. doi: 10.1093/nar/gkq427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vidler L. R., Brown N., Knapp S., Hoelder S. Druggability analysis and structural classification of bromodomain acetyl-lysine binding sites. J. Med. Chem. 2012;55:7346–7359. doi: 10.1021/jm300346w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Filippakopoulos P., Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014;13:337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.