

Abstract
Purinergic signaling comprises one key pathway in modulating bladder smooth muscle (BSM) contractility, disorders of which become highly prevalent with aging. ADP was first observed to modulate BSM contractility >40 yr ago, yet the underlying molecular mechanism still remains unclear. Here, we demonstrate, using myography, that ADP and ADPβS dose-dependently induce mouse BSM contraction, and ADP-induced BSM contraction is blocked by a selective P2Y12 receptor (P2Y12R) antagonist, PSB 0739 (25 μM), but is unaffected by P2Y1 and P2Y13 receptor antagonists. P2Y12R in BSM exhibits distinct pharmacological properties that are different from P2Y12R in platelets. After an immediate contraction, prolonged exposure to ADP causes BSM to become refractory to further ADP-mediated contraction. However, in mice lacking ectonucleotidases Entpd1 (ATP→ADP→AMP) or Nt5e (AMP→adenosine), or by inhibiting adenosine signaling, the refractory response was altered, resulting in repeated BSM contractions in response to repeated ADP (0.1-1 mM) stimulation. Our data indicate that P2Y12R undergoes slow desensitization; ADP-P2Y12 signaling is tightly regulated by Entpd1/Nt5e activity and adenosine receptors; and ADP-adenosine signaling play an important role in modulating P2X-mediated BSM contraction. The identification of P2Y12R in BSM, and the current clinical availability of P2Y12R inhibitors, such as clopidogrel, offers potentially novel treatment strategies for bladder contractility disorders.—Yu, W., Sun, X., Robson, S. C., Hill, W. G. ADP-induced bladder contractility is mediated by P2Y12 receptor and temporally regulated by ectonucleotidases and adenosine signaling.
Keywords: detrusor, urinary tract, myography, micturition, mechanotransduction
Lower urinary tract symptoms (LUTS), including bladder overactivity, urinary urgency and frequency, urinary incontinence, and bladder pain, afflict millions of people and severely compromise quality of life and independence. A recent study reported that LUTS were highly prevalent among men and women aged >40 yr, with >70% experiencing some symptoms (1). The etiology of LUTS is extremely poorly understood, and treatment options are few and of limited efficacy.
Bladder smooth muscle (BSM) plays an essential role during urinary filling and voiding, with highly coordinated phases of relaxation and contraction. Consequently, myogenic disorders are frequently involved in LUTS, as may be seen with the development of significant noncholinergic and non-P2X1 receptor-mediated contraction in diabetic bladder dysfunction (2), yet knowledge of the underlying mechanisms remains poorly understood (3).
BSM contractility is, in part, regulated by purinergic signaling. Extracellular purines like ATP and UTP can be released either as neurotransmitters at neuromuscular junctions or from somatic cells in response to environmental stress, and they bind to P2X (1–7) and P2Y (1, 2, 4, 6, 11–14) receptors to elicit downstream signaling. ATP/UTP can be further sequentially converted into ADP/UDP and ultimately to adenosine by ectonucleotidases to activate P2Y receptors and adenosine receptors (P1) correspondingly. Thus, ectonucleotidases temporally and spatially regulate the availability of purines to activate P1 and P2 receptors (4).
The functional significance of purinergic signaling in regulating bladder and other organs had been shown >40 yr ago, in experiments with neuronal release or exogenously applied purines causing smooth muscle contraction or relaxation (5, 6). Since then, significant effort has been spent on decoding this mechanism. It is now generally accepted that P2X1 receptors mediate ATP signaling by contracting BSM (7), and adenosine A2 receptors have been pharmacologically shown to relax BSM (8, 9). The magnitude of P2X1-mediated force generation is believed to be species dependent, with a relatively small contribution in humans; however, in disease and in aging in human bladders, this neurotransmitter pathway is up-regulated at the expense of cholinergic stimulation (10–15) and can account for up to 65% of the total contraction force (16). Furthermore, it has been shown that chronic administration of anticholinergics, the leading medication class for patients, results in an up-regulation of purinergic responsiveness, which may explain why they lose efficacy (17). Recently, we identified P2Y6 receptor in BSM, which is a UDP receptor that, on activation, can modulate BSM tone to potentiate P2X1-mediated contraction force (18). We also recently showed that ectonucleoside triphosphate diphosphohydrolase 1 (Entpd1) and ecto-5′-nucleotidase (Nt5e) are both expressed in mouse BSM (19). These findings further confirm the importance of purinergic signaling in modulating BSM contractility and also indicate new and previously unrecognized complexity in these pathways.
Despite recent progress, significant uncertainties still exist, and one of these is the long-standing question surrounding ADP-induced effects on BSM contractility. Remarkably, since the initial observation ∼1970, that ADP could induce significant BSM contractility, different laboratories have tried to understand this phenomenon, but the receptor responsible has not been identified (20–29). The prevailing consensus has been that in addition to the ATP/P2X1 signaling, there is also an ADP or ADPβS-sensitive contractile P2Y receptor in BSM. However, other studies have reported ADP-induced relaxation of BSM (22, 25, 28), which would appear to be contradictory. Overall, the current literature contributes to a lack of certainty about the effects of ADP and the pathways involved. This uncertainty is further compounded by experiments in which mucosa-intact bladder strips are studied, and the interpretation often invokes a role for release of urothelial effectors, including ATP. Our aim in this study was to introduce clarity by focusing on BSM contractility absent the confounding effects of urothelium.
To solve this long-standing puzzle, we sought to define the identity of the ADP receptor in BSM, and also to understand how this receptor could be involved in modulating BSM contractility. Following pharmacological screening of P2Y receptor family members, we identified a distinct P2Y12 receptor with a unique pharmacological profile in BSM. P2Y12R mediates ADP-induced effects and modulates BSM contractility through crosstalk with adenosine receptors, made possible by the coordinated activity of two purine converting enzymes: Entpd1 and Nt5e.
MATERIALS AND METHODS
Materials
Unless otherwise specified, all chemicals were obtained from Sigma-Aldrich (St. Louis, MO, USA) and were of reagent grade or better.
Animals
C57BL/6J, Entpd1−/− and Nt5e−/− mice (3–4 mo old) were used in this study. Mice were euthanized by inhalation of 100% CO2. After euthanasia and thoracotomy, the bladders were rapidly excised and processed as described below. Entpd1−/− and Nt5e−/− transgenic mice were described in detail previously (30, 31), and they are all in C57BL/6J background.
All animal studies were performed in adherence to U.S. National Institutes of Health guidelines for animal care and use and with the approval of the Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee.
Agonists and antagonists
Atropine and ADP were purchased from Sigma-Aldrich. Adenosine-5′-(β-thio)-diphosphate lithium salt (ADPβS) was purchased from Jena Bioscience (Jena, Germany). All other agonists and antagonists were purchased from R&D Systems (Minneapolis, MN, USA), including P2X1 and P2X3 receptor agonist α,β-methyleneadenosine 5′-triphosphate trisodium salt (α,β-meATP); P2Y1 receptor agonist MRS 2365 and antagonist MRS 2500; P2Y2 receptor agonist MRS 2768; P2Y6 receptor agonist MRS 2693 and antagonist MRS 2578; P2Y12 receptor antagonists AR-C 66096 and PSB 0739; P2Y13 receptor antagonist MRS 2211; P2Y1, P2Y12, and P2Y13 receptor agonist 2-methylthioadenosine diphosphate trisodium salt (2-MesADP); and adenosine receptor agonist NECA and antagonist CGS 15943. Dose response of agonists and antagonists was analyzed using the GraphPad Prism 6 built-in nonlinear curve fitting program (GraphPad, San Diego, CA, USA).
Myography
Briefly, bladders were pinned on a small Sylgard block, and muscle was dissected free of the mucosal tissue, as described previously (18). BSM strips were then cut longitudinally (2–3 mm wide and 5–7 mm long) and mounted in an SI-MB4 tissue bath system (World Precision Instruments, Sarasota, FL, USA). Force sensors were connected to a TBM 4M transbridge (World Precision Instruments), and the signal was amplified by PowerLab (ADInstruments, Colorado Springs, CO, USA) and monitored through Chart software (AD Instruments). Contraction force was monitored dynamically with a sampling rate of 2000/s. BSM strips were gently prestretched to get optimized force and equilibrated for ≥1 h before any experiments. All experiments were conducted at 37°C in physiological saline solution, with continuous bubbling of 95% O2 and 5% CO2.
Electrical field stimulation (EFS)
EFS is performed with an S48 field stimulator (Grass Technologies, Quincy, RI, USA) using standard protocols, as described previously (32). The setting of stimulation parameters was modified from the Sibley (32) report and experimentally determined as follows: voltage: 50 V; stimulus duration: 0.05 ms; trains of stimuli: 3 s; frequencies: 1, 2, 5, 10, 20, and 50 Hz; single train of stimuli for every frequency with 3-min interval. These basic settings are used for EFS-induced contraction force.
Statistical analysis
The number (n) of samples for all experiments was 4 ≤ n ≤ 12. All data are expressed as means ± sd. To determine significance for simple treatment effect, paired (whenever possible), or unpaired Student's t tests were performed. For multiple comparisons, analysis of variance was first performed, and if the value of P was <0.05, the Bonferroni t test was applied. Tests were considered significant at P < 0.05.
RESULTS
ADP and ADPβS induce BSM contraction
Myography on bladder strips without urothelium was used to study ADP-induced effects on BSM. Doses of ADP ranging from 0.1 to 5000 μM were tested. At concentrations below 1 μM, there were no effects observed. However, at concentrations starting from 10 to 100 μM, BSM contractions were observed immediately after the addition of ADP, with clear dose dependence and EC50 ∼ 395 μM (Fig. 1A–C). ADP induced a rapid force increase followed by a quick decline to the baseline after peak force generation (Fig. 1A). ADPβS, a nonhydrolyzable ADP, was also tested. At concentrations starting from 1 to 5 μM, ADPβS stimulated clear dose-dependent BSM contraction with EC50 ∼ 40 μM (Fig. 1D–F). ADPβS-induced response is different from that of ADP-induced response in several ways. First, ADPβS requires much less dose (at least 10 times less) to induce BSM contraction. Second, ADPβS induces stronger contraction than ADP. Third, the ADP-induced contraction returns quickly (5–10 s) to a level often below the baseline of before contraction, but ADPβS-induced contraction lasts for a long time (10–15 min) with a slow decay (Figs. 1D and 6B). These interesting phenomena imply complex signaling might be involved in regulating ADP-mediated responses.
Figure 1.

ADP and ADPβS induce BSM contraction with dose dependence. A, D) Representative traces of BSM responses to indicated concentrations of ADP (A) and ADPβS (D). ADP or ADPβS was quickly washed out 3 times after each response, and there was a 15-min interval between each addition. B, E) Summary of force change (peak force minus baseline) with increasing ADP (B) or ADPβS (E) concentrations. *P < 0.05. C, F) Nonlinear regression of ADP (C) and ADPβS (F) dose and contraction force response using the GraphPad Prism dose-response-stimulation program.
Figure 6.
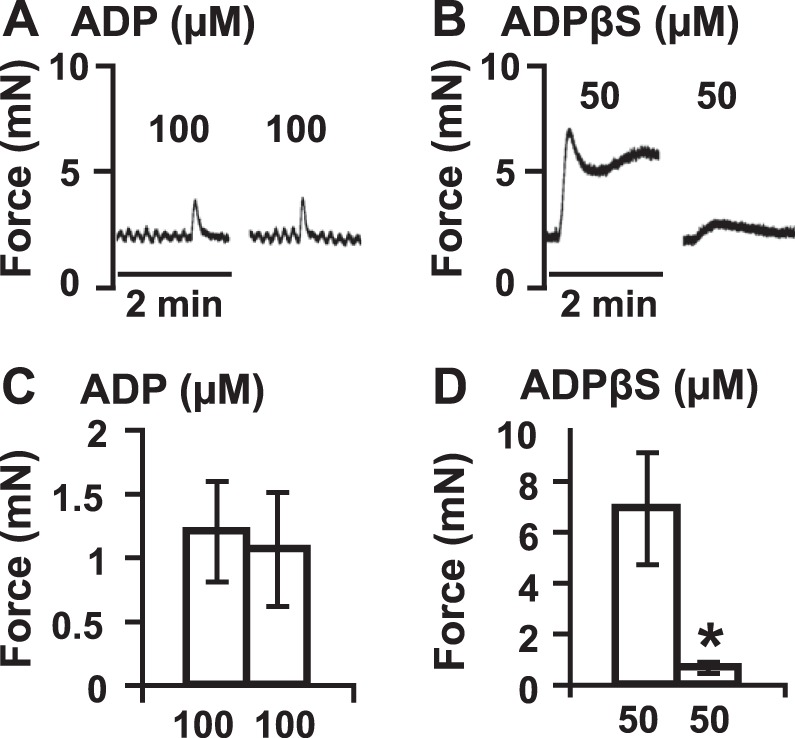
BSM P2Y12R undergoes slow desensitization. A) Addition of 100 μM ADP induces a quick BSM contraction. When it is washed out 3 times within 5–10 min, further addition of 100 μM ADP can induce another BSM contraction with similar force intensity. B) ADPβS (50 μM) induces a strong BSM contraction (pretreated with atropine and 10 μM α,β-meATP). After a 30-min incubation, the same concentration of ADPβS induces only a small BSM contraction (10%), indicating a significant desensitization. Note that the ADP-induced contraction has a quick increase and return (5–10 s), while ADPβS, a nonhydrolysis ADP analog, induces a strong contraction and a very slow return (∼15 min). C, D) Quantification of data from panels A, B, respectively. *P < 0.05.
ADP-induced BSM contraction can be blocked by a P2Y12R antagonist: PSB 0739 with dose dependence
Selective P2Y receptor agonists and antagonists were used to define the identity of the ADP receptor. P2Y1, P2Y12, and P2Y13 are all ADP receptors. MRS 2365 (EC50 0.4 nM), a highly potent, selective P2Y1 receptor agonist, is ineffective at eliciting any contraction force from BSM at a concentration up to 10 μM (Fig. 2A), and this is confirmed by MRS 2500, a potent and selective antagonist of P2Y1 receptor, which inhibits ADP-induced aggregation of human platelets with an IC50 value of 0.95 nM (33). Pretreatment of BSM with 2 μM MRS 2500 for 15 min does not inhibit ADP induced BSM contraction (Fig. 2A). This is also true for MRS 2211, a competitive P2Y13 receptor antagonist with pIC50 value of 5.97, which does not block ADP-induced BSM contraction at a concentration up to 1 μM (Fig. 2E). These data indicate that P2Y1 and P2Y13 receptors are not the receptors responsible for ADP-induced contraction of BSM. However, PSB 0739, a highly potent P2Y12 receptor antagonist (Ki 24.9 nM) in human platelets, fully blocks ADP-induced BSM contraction force (Fig. 2D), indicating that P2Y12R mediates the response. We also tested a selective P2Y2R agonist, MRS 2768 (EC50 1.89 μM) at 20 μM, but no contraction was observed (Fig. 2B). A selective P2Y6R antagonist MRS 2578 (IC50 98 nM) at 5 μM also did not inhibit ADP-induced contraction. We did not try P2Y4R agonist/antagonists, because we were unable to detect significant expression of this receptor in the bladder by PCR (not shown). The almost total abolition of measurable contractile force by PSB 0739 (Fig. 2D) also indicates that other receptors do not contribute.
Figure 2.
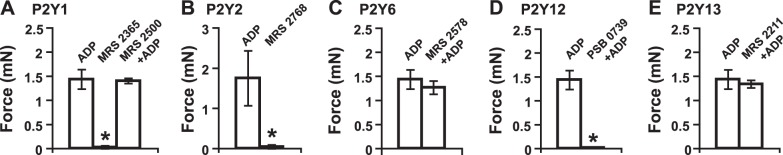
P2Y12R selective antagonist blocks ADP-induced BSM contraction. In all panels, ADP was used at 100 μM. A) Effects of selective P2Y1 receptor agonist (10 μM MRS 2365) and antagonist (2 μM MRS 2500) on BSM contractility. B) Effects of selective P2Y2 receptor agonist (20 μM MRS 2768) on BSM contractility. C) Effects of selective P2Y6 receptor antagonist (5 μM MRS 2578) on BSM contractility. D) Effects of selective P2Y12 receptor antagonist (25 μM PSB 0739) on BSM contractility. E) Effects of selective P2Y13 receptor antagonist (1 μM MRS 2211) on BSM contractility. Only selective P2Y12 receptor antagonist PSB 0739 blocks ADP-induced BSM contraction. Antagonists were added to BSM 15 min before ADP stimulation. *P < 0.05.
We further determined the dose response of PSB 0739 on ADP-induced BSM contraction (Fig. 3). To avoid possible complications of adenosine receptor signaling pathway, BSM strips were pretreated with 2.5 μM CGS 15943 (adenosine receptor antagonist), and 0–25 μM PSB 0739 showed dose-dependent inhibition of 500 μM ADP-induced BSM contraction with IC50 ∼ 8 μM (Fig. 3). To explore whether P2X1 receptors were involved in ADP-induced response, we pretreated BSM strips with 10 μM α,β-meATP for 15 min, which fully desensitizes P2X1 receptors mediated contraction in BSM. When exposed to 50 μM ADPβS, strong BSM contraction was still observed (Fig. 6B), confirming a non-P2X1 receptor mediated, but an independent P2Y12R-mediated response. In separate experiments, PSB 0739 only slightly decreased, but did not abolish 10 μM α, β-meATP-induced BSM contraction, which is mediated by P2X1 receptors (Fig. 3B). We conclude, therefore, that P2Y12R is responsible for ADP-induced BSM contraction, and P2Y12R might be able to modulate P2X1R-mediated BSM contraction.
Figure 3.

PSB 0739 inhibits ADP-induced BSM contraction with dose dependence. A) Representative traces of BSM responses to indicated pharmacological application. To avoid possible complication of adenosine receptor-mediated effect, BSM strips were pretreated with 2.5 μM of CGS 15943 in this experiment. Left trace is a 500 μM ADP-induced quick BSM contraction. Right trace indicates when pretreated with 25 μM PSB 0739, the 500-μM ADP-induced BSM contraction was abolished. B) Left trace is the control α,β-meATP-induced BSM contraction. Right trace indicates that pretreatment with 25 μM PSB 0739 does not abolish P2X1 receptor-mediated BSM contraction, which is induced by 10 μM α, β-meATP. Right panel summarizes the data, which indicate the pretreatment of PSB 0739 only slightly inhibits α,β-meATP-induced BSM contraction (∼15%). This is possibly due to the involvement of P2Y12R in normal BSM muscle tension development. C) Summary of force inhibition (peak force minus baseline) on ADP-induced contraction with increasing PSB 0739 concentrations. Right panel shows nonlinear regression of PSB 0739 dose and contraction force response using the GraphPad Prism dose-response-inhibition program. *P < 0.05. D) Normalized version of C, with each force change normalized as a percentage change (compared to 500 μM ADP response as 1) with increasing PSB 0739 concentrations. Right panel shows nonlinear regression of PSB 0739 dose and contraction force response using the GraphPad Prism dose-response-inhibition program. *P < 0.05.
BSM P2Y12R has distinct pharmacological properties
There are currently no highly selective agonists for P2Y12R, but there are compounds that activate several P2Y receptors, including P2Y1R, P2Y12R, and P2Y13R. ADP analog ADPβS is a potent agonist that induces BSM contraction (21), a finding that we confirmed (Figs. 1D–F and 6B). In addition to ADP and ADPβS, 2-MesADP is another potent agonist for P2Y12R, with a pEC50 value of 9.05 for human platelets. However, it also activates P2Y1 and P2Y13 receptors. Surprisingly, it showed only weak potency in inducing BSM contraction, with no obvious effect below 10 μM and dose-dependent contractions at 50 and 200 μM, similar to that of native agonist ADP (Fig. 4A). This effect was fully abolished by pretreating BSM with PSB 0739, indicating a P2Y12R-mediated contraction (Fig. 4B). Even more interestingly, AR-C 66096, a potent and selective P2Y12R antagonist, which inhibits ADP-induced aggregation of washed human platelets with a pIC50 value of 8.16 (34), showed strong potency as an agonist in provoking BSM contraction. This drug exhibited clear force development at 1 μM and significant dose-dependent increases in BSM contraction force at higher concentrations (Fig. 4C). Likewise, the effect of AR-C 66096 can also be fully blocked by PSB 0739, indicating P2Y12R signaling (Fig. 4D). These data indicate the P2Y12Rs in BSM might have distinct pharmacological properties.
Figure 4.
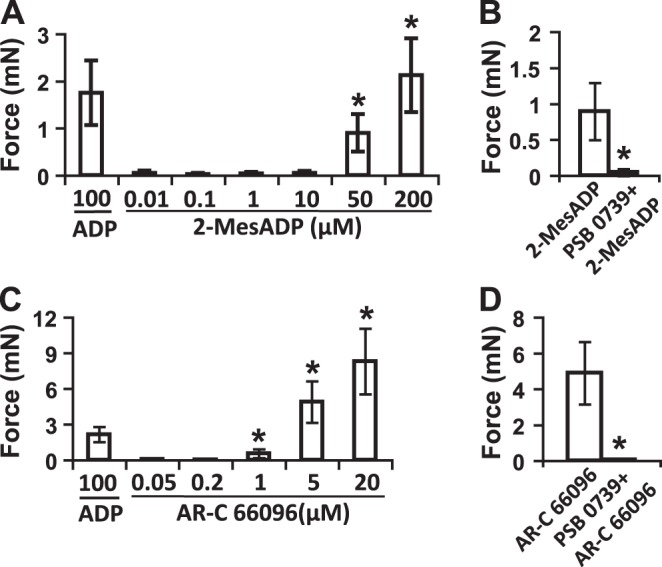
Pharmacological properties of 2-MesADP and AR-C 66096 on BSM P2Y12R. A) Quantitative force responses of BSM on indicated concentrations of 2-MesADP. Only at 50 and 200 μM did 2-MesADP induce significant BSM contraction forces. ADP (100 μM)-induced BSM contraction force is included for comparison. B) 2-MesADP (50 μM)-induced BSM contraction was fully blocked by P2Y12R selective antagonist PSB 0739. BSM was pretreated with 25 μM PSB 0739 for 15 min before 2-MesADP stimulation. C) Quantitative force responses of BSM on indicated concentrations of AR-C 66096. AR-C 66096 induced significant BSM contraction forces starting from 1 μM with dose dependence. ADP (100 μM)-induced BSM contraction force is included for comparison. D) AR-C 66096 (5 μM)-induced BSM contraction was fully blocked by P2Y12R selective antagonist PSB 0739. BSM was pretreated with 25 μM PSB 0739 for 15 min before AR-C 66096 stimulation. *P < 0.05.
ADP is converted to adenosine, which activates adenosine receptor, thereby causing subsequent relaxation
The differential responses of BSM to ADP and ADPβS in Fig. 1 indicate that complex signaling might be involved in regulating ADP-mediated responses, since ADP can be readily degraded to AMP and adenosine. We recently defined the expression of Entpd1 and Nt5e on BSM (19) and, thus, hypothesized that ADP might be undergoing conversion to adenosine by the concerted action of these two enzymes. If true, then activation of adenosine receptor might relax BSM, and this could explain some of the anomalous results described in the literature of ADP-mediated relaxation effects (22, 25, 28) and might also explain the quick return of the ADP-induced contraction to below baseline (Fig. 1A). To test this hypothesis, 0.1 mM ADP was added to BSM strips, which induced an initial contraction. Strips were incubated for 30 min without ADP washout, and then another dose of 0.1 mM ADP was added, which does not induce significant contraction (Fig. 5A, E). A further addition of 1 mM ADP also had no effect, suggesting ADP, or ADP metabolites may have relaxed BSM and made it refractory to further contraction.
Figure 5.
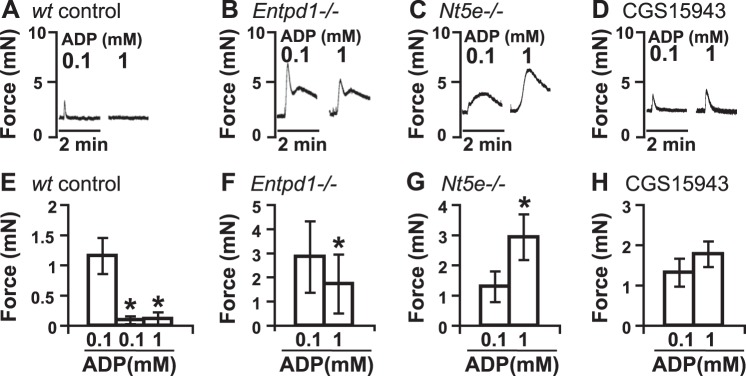
ADP-induced signaling in BSM is modulated by Entpd1, Nt5e, and adenosine receptors. A–D) Representative traces of BSM responses to 0.1 and 1 mM ADP are shown in wt control (A), Entpd1−/− (B), Nt5e−/− (C), and CGS 15943 (D) pretreated samples. ADP is not washed out, and there is a 30-min interval between two ADP stimulations. E–H) Quantification of data from panels A–D, respectively. *P < 0.05.
We next performed three different experiments aimed at perturbing this ADP-adenosine-converting pathway or interfering with its receptors. First, we tested whether ENTPD1, an enzyme present on BSM, which converts ADP to AMP, is required to mediate this relaxation response, using bladders from Entpd1-knockout mice. Interestingly, 0.1 mM ADP induces a very strong contraction of Entpd1−/− BSM (Fig. 5B, F). Furthermore, after 30 min of incubation with added ADP, the addition of 1 mM ADP induced another strong contraction that was ∼30% less (Fig. 5B, F), indicating that loss of the ability to convert ADP to downstream metabolites means BSM retains contractile function. This mandates an important role of Entpd1 in the ADP-mediated relaxation effect. NT5E, an enzyme required to convert AMP to adenosine, also played a significant role in ADP-mediated BSM relaxation. After a 30-min incubation with 0.1 mM ADP, Nt5e−/− BSM exhibits a 100% increase in contraction force in response to 1 mM ADP compared to the first response (Fig. 5C, G). Not surprisingly, the inability of the tissue to generate extracellular adenosine had dramatic consequences consistent with inhibition of the relaxation pathway. To further examine whether Entpd1 and Nt5e convert ADP to adenosine, activating adenosine receptors, BSM was pretreated with CGS 15943 (2.5 μM), a potent adenosine receptor antagonist. In this experiment, 30 min of incubation with 0.1 mM ADP does not fully relax BSM, as happened in the control experiment (Fig. 5A), instead showing a strong contraction in response to subsequent 1 mM ADP addition (Fig. 5D, H). In summary, these results consistently indicate that Entpd1 (Fig. 5B) and Nt5e (Fig. 5C) actively and coordinately convert ADP to adenosine, which mediates relaxation effects on BSM through the activation of adenosine receptors (Fig. 5D). These data are also consistent with ADPβS-induced response previously (21) and in the following data (Fig. 6), in which nonhydrolyzable ADPβS causes prolonged BSM contraction, indicating a contractile function of P2Y12R and a critical role of Entpd1/Nt5e and adenosine in modulating this function. These data indicate an important role of the balance of ADP and adenosine signaling.
BSM P2Y12R undergoes slow desensitization, which may contribute to ADP-mediated BSM relaxation
P2Y12R plays an important role in platelet aggregation, and undergoes quick desensitization in response to autocrine ADP stimulation. It has also been shown that pretreatment of platelets with ADP results in decreased aggregation induced by collagen and thrombin (35). The question of whether P2Y12R desensitizes quickly in BSM, and, in this way, could contribute to the ADP-mediated BSM relaxation is unknown. In our previous experiments, we noticed that Entpd1−/− BSM, which lacks an ADP conversion enzyme, exhibits smaller contraction force in response to secondary 1 mM ADP stimulation (Fig. 5B). However, in Nt5e−/− BSM, which lacks the AMP conversion enzyme but has normal ADP→AMP activity, we noticed a significantly stronger contraction force in response to 1 mM ADP compared to the first 0.1 mM ADP stimulation (Fig. 5C). Thus, in the Entpd1−/− setting, ADP likely persists in the extracellular space with increased receptor exposure time, thereby causing significant P2Y12R desensitization (shown by decreased contraction in response to the second ADP stimulation, even at much high concentration of agonist ADP). In the Nt5e−/− setting, Entpd1 converts ADP→AMP quickly, so ADP concentrations in the extracellular space are reduced, while AMP levels increase and remain high. In this case, P2Y12R will not be desensitized, and adenosine receptor will remain inactive because of lacking adenosine, thus BSM contraction shows a significantly elevated dose response to 1 mM ADP. While in the case of inhibiting adenosine receptor with CGS 15943, a short temporary contraction was observed in response to second ADP stimulation but could not maintain an elevated contracting status like those of Entpd1−/− and Nt5e−/− BSM (Fig. 5D), because normal Entpd1 and Nt5e are present in BSM, and ADP concentrations in the extracellular space are reduced quickly.
These data imply that P2Y12R in BSM might undergo a slow desensitization rather than the rapid response seen in platelets. Consistent with this, we have observed a prolonged ADP-induced contraction in Entpd1−/− and Nt5e−/− BSM (Fig. 5B, C), and which is also consistent with the previously reported BSM response to ADPβS that induced a strong, yet prolonged, elevation of BSM contraction (21). To confirm our observation and examine our hypothesis further, we pretreated BSM with 100 μM ADP and quickly washed it out within 5–10 min. Immediately afterward, another dose of 100 μM ADP was added to BSM, which induced a comparable contraction force to the first dose, indicating no obvious desensitization of P2Y12R in BSM (Fig. 6A, C). In another experimental setting, ADPβS (50 μM) induced a strong and prolonged BSM contraction (as long as 15 min), and when BSM was incubated for 30 min, further stimulation with the same concentration of ADPβS (50 μM) only induced 10% of the initial contraction force, indicating significant desensitization of P2Y12Rs (Fig. 6B, D).
ADP-adenosine signaling modulates EFS-induced BSM purinergic contraction
BSM resting length or muscle tension is a key determinant for BSM force generation, and this phenomenon has been known for decades (36). We have recently found that UDP activation of P2Y6R increases BSM tone and thus potentiates BSM purinergic contraction in response to EFS (18). Therefore, we examined whether ADP-adenosine signaling may also be involved in the regulation of BSM purinergic contraction, which is primarily believed to be P2X-mediated. EFS induces rapid neuronal release of neurotransmitters to cause BSM contraction, thereby mimicking in vivo neuromuscular innervation (32). To perform these experiments, BSM was pretreated with 0.5 μM atropine to block muscarinic contraction and, thus, leave BSM purinergic contraction intact (Fig. 7A). BSM was then treated with increasing concentrations of ADP (1–1000 μM) for 15 min before EFS. Results show that pretreatment with ADP significantly inhibits BSM purinergic contraction and is dose dependent (Fig. 7B), indicating an important role of ADP-adenosine signaling in regulating purinergic neurotransmitter signals to BSM. To confirm this, Entpd1−/− and Nt5e−/− BSM strips were pretreated with 0.1–1 mM ADP. EFS-induced purinergic contractions were significantly increased in these tissues compared to the controls (Fig. 7C, D), indicating that Entpd1 and Nt5e play a role in regulating BSM purinergic contraction by performing extracellular purine conversion and limiting their availability. Furthermore, pretreatment with CGS 15943 also significantly blocks ADP-adenosine induced relaxation on purinergic contraction (Fig. 7E), indicating the involvement of adenosine receptors. This conclusion was further confirmed by use of the potent adenosine receptor agonist, NECA (Fig. 7F), which, at high concentrations, fully abrogated the ability of EFS to contract the muscle. To examine whether direct inhibition of P2Y12R signaling will modulate EFS-induced BSM purinergic contraction, we pretreated BSM with a P2Y12R selective antagonist PSB 0739, which significantly inhibited purinergic contraction, indicating a direct involvement of P2Y12R signaling in modulating BSM contractility (Fig. 7G). Interestingly, pretreatment with AR-C 66096 followed by EFS of bladder strips also inhibited purinergic contraction (Fig. 7H), and the inhibiting effect was partially blocked by pretreatment with CGS15943. Therefore, it appears that AR-C 66096 undergoes enzymatic conversion to a compound able to activate adenosine receptor.
Figure 7.

EFS-induced purinergic BSM contraction is modulated by P2Y12R, Entpd1, Nt5e, and adenosine receptors. A) Representative traces of BSM purinergic contractions in response to 1-, 2-, 5-, 10-, 20-, and 50-Hz frequency of EFS. BSM is pretreated with 0.5 μM of atropine to inhibit muscarinic contraction in all panels. There are 3-min intervals between each stimulation. B) Pretreatment (15 min) with ADP inhibits BSM purinergic contraction induced by EFS with dose dependence. Pretreatment with 1 μM of ADP does not inhibit BSM purinergic contraction significantly. When pretreated with ≥10 μM ADP, significant inhibition is observed. C–E) In BSM lacking Entpd1 (C), lacking Nt5e (D), or pretreated with CGS 15943 (E), the relaxation effect of ADP on purinergic contraction is significantly inhibited. F) Adenosine receptor agonist NECA inhibits BSM purinergic contraction. G) Blocking P2Y12R with 15-min pretreatment with 25 μM PSB 0739 significantly inhibits BSM purinergic contraction. H) Pretreatment (15 min) with 10 μM AR-C 66096 significantly inhibits BSM purinergic contraction, which is partially blocked by CGS 15943.
DISCUSSION
P2Y12R was identified a decade ago (37–39). Long before its cloning, it was pharmacologically described as an ADP receptor in platelets and as a molecular target of the antiplatelet drugs like ticlopidine and clopidogrel (Plavix). P2Y12R was initially identified in platelets and brain, and further studies revealed its expression in microglial cells, leukocytes, dendritic cells, and vascular smooth muscle cells (40). Recent studies have shown that P2Y12R is highly expressed in vascular smooth muscle cells, where activation causes vasoconstriction. The receptor is also involved in vascular inflammation, atherosclerosis, and mitogenesis (41–45). The ability of ADP to induce BSM contraction has long been known, yet the receptor responsible for this function has remained unknown for >40 yr. Recent development of potent and selective agonists and antagonists for P2Y receptors makes it possible for us to unravel this puzzle. In our hands, ADP and ADPβS induce dose-dependent BSM contraction force responses, which is consistent with previous reports (Fig. 1). Through pharmacological screening (Figs. 2 and 3), ADP-induced BSM contraction can only be abolished by the selective P2Y12R antagonist PSB 0739, but not by other selective P2Y receptor antagonists, and, in particular, antagonists for the other two ADP receptors, P2Y1 and P2Y13 receptors. Thus, we conclude that P2Y12R is the molecule that mediates ADP-induced contraction in BSM.
2-MesADP is a potent agonist in activating P2Y12R in several systems, with EC50 at nanomolar levels (38, 39, 46). But in BSM, 2-MesADP only shows weak stimulation (Fig. 4A), and this has also been observed previously in vascular smooth muscle, which requires 10 μM 2-MesADP to induce both human and mouse vasculature to contract (42, 43). This suggests there may be a distinct P2Y12R isoform in smooth muscle systems. More interestingly, an ATP analog AR-C 66096, which is deemed to be a potent and selective P2Y12 antagonist in platelets (34), consistently exhibits strong agonist potency on inducing BSM contraction, and this effect is totally abolished by selective P2Y12R antagonist PSB 0739 (Fig. 4C, D). This finding is unexpected, since ATP derivatives reportedly antagonize the platelet Gi-linked receptor. However, we have noticed that ATP derivatives show agonistic function on P2Y12R in other systems. For example, ATPαS behaves as a weak agonist on P2Y12R expressed in oocytes (38), and 2-MesATP, 2-Cl-ATP, and ATPγS also behave as agonists on P2Y12R expressed in Chinese hamster ovary (CHO) cells (39). This phenomenon has also been noticed for another ADP receptor, P2Y1R, which responds differentially to ATP (as agonist) in different expression systems (47, 48). The underlying mechanisms are not yet known but might be due to different microenvironments, including a differential repertoire of ectonucleotidases. Another possibility could lie in its interactions with other proteins, resulting in tissue-specific protein complexes; P2Y12R might also have different glycosylation in BSM, which could be essential for signal transduction (49). Genetic polymorphisms or variants of P2Y12R in different species/tissues might be another possibility. Indeed, such polymorphisms have been shown to be associated with differential platelet responsiveness to ADP (50, 51).
It is well known that after being exposed to ADP, platelets become unresponsive to a second stimulation with ADP (35, 40). Strikingly, BSM shows the same response (Fig. 5A). Desensitization is a possible mechanism, but consensus has not been reached on whether platelet P2Y12R really undergoes a rapid desensitization. In vivo studies have indicated that platelet P2Y12R remains functional under repeated ADP stimulation, but it is also reported that P2Y12R undergoes quick desensitization, mediated by G-protein-coupled receptor kinases (GRKs). Our data indicate that P2Y12R in BSM is refractory to rapid down-regulation by ADP or ADPβS stimulation. ADP and ADPβS can repeatedly induce BSM contraction whenever it is quickly washed out after exposure (Figs. 1 and 6A). ADP also induces repeated BSM contraction when Entpd1 or Nt5e proteins are deleted or when adenosine receptors are blocked (Fig. 5B–D). Even following ADPβS (50 μM) stimulation for 30 min, it retains ∼10% contractility (Fig. 6B, D). Collectively, these findings suggest that P2Y12R in BSM undergoes slow desensitization, even when it is strongly stimulated.
The P2Y12R in platelets is coupled to a Giα2 protein to inhibit adenylyl cyclase activity. The downstream signaling is not fully understood but might include PI3 kinases and cAMP-dependent protein kinase A (PKA; ref. 52). It also might include crosstalk between P2Y12R (Gi) and P2Y1R (Gq) through diacylglycerol metabolism (53). While we have not studied P2Y12R-mediated downstream signaling in BSM, we expect that P2Y12R in BSM is coupled to Gi, too, whereupon activation will inhibit adenylyl cyclase to induce BSM contraction. It has been convincingly shown in this study that adenosine receptors are involved in the regulation of BSM contractility. Adenosine A2a and A2b receptors have been suggested to mediate BSM relaxation in previous reports, although the exact receptor type and the signaling pathways are not fully known (8, 9). A2a/A2b receptors are usually coupled to Gs protein, which, on activation, will increase adenylyl cyclase activity. Thus, it seems that adenylyl cyclase is likely the key protein to mediate crosstalk between P2Y12R and A2a/A2b receptors, which might positively and negatively regulate BSM contractility through the same downstream signaling pathways (Fig. 8). Entpd1 and Nt5e are two other key proteins that are necessary participants in the crosstalk between P2Y12R and A2a/A2b receptors. They convert ADP/AMP to adenosine, thus temporally regulating purine dynamics in the extracellular space, which dynamically modulates the crosstalk between P2Y12R and A2a/A2b receptors. This conclusion is strongly supported by our data on differentiated responses on ADP and ADPβS, in knockout mice showing that deletion of these enzymes, and inhibition of A2a/A2b receptors, alters BSM contractile responses (Figs. 1, 5, 6, 7).
Figure 8.

Proposed working model. In addition to activating P2X1 receptor on BSM, parasympathetically released ATP during micturition will also be converted to ADP quickly, which further activates P2Y12R, and inhibits adenylyl cyclase activity to decrease intracellular cAMP level. This will cause BSM contraction by an unknown mechanism and may also potentiate P2X-mediated BSM contraction force, possibly by inhibiting adenosine-mediated relaxation. ADP will be further converted by Entpd1 and Nt5e on BSM to adenosine, which binds to adenosine receptors and activates adenylyl cyclase to increase intracellular cAMP level, and eventually relaxes BSM after urinary voiding. Thus, adenylyl cyclase may function as a key protein and common pathway for the crosstalk between P2Y12R and adenosine receptors, and Entpd1 and Nt5e may serve as the temporal regulators for the crosstalk between P2Y12R and adenosine receptors. The dynamic interplay of these positive and negative signals may play a crucial role in modulating BSM purinergic contractility and could result in disordered bladder contractility when disrupted.
P2Y12-A2a/A2b signaling not only functions in the setting of exogenously added ADP, but also modulates EFS-induced BSM contractions, where BSM contractility is changed when either of P2Y12R and A2a/A2b receptors are activated or blocked, or Entpd1 and Nt5e are deleted (Fig. 7). EFS induces neuronal release of neurotransmitters, which mimics in vivo synaptic firing; thus, our data suggest P2Y12R might play an important functional role in modulating bladder function in vivo. So far, the molecular detail on how P2Y12-A2a/A2b-mediated signaling modulates BSM contractility remains unclear. It could modulate BSM muscle tone to regulate BSM contractility, since the muscle tone is a predeterminant for maximal BSM contraction force (36). It could also further interact with P2X-mediated signaling (54). This is supported by in vitro block of P2Y12Rs (PSB 0739) inhibits α, β-meATP induced BSM contraction (Fig. 1D–F). These questions need be answered in future studies.
The pharmacological significance of P2Y12R in hemostasis and thrombosis has long been recognized due to the potent antithrombotic thienopyridine compounds ticlopidine and clopidogrel, metabolites of which selectively inhibit P2Y12R in platelets. Recently, P2Y12R has also been proposed as a possible treatment target for other disorders like vasospasm (42–44). In this study, we have identified functional expression of P2Y12R in BSM, which is responsible for the long sought after ADP-mediated BSM response. Defining this distinct P2Y12R pathway in BSM, in combination with new insights into ectonucleotidase-mediated crosstalk with adenosine receptors, sheds important new light on the purinergic regulation of BSM contractility. These molecules also provide potential new drug targets, in particular, P2Y12R, which has multiple clinical medications available for antithrombotic treatment. Further studies are needed to understand how P2Y12R is involved in both animal bladder disease models and human bladder disorders like LUTS, which will potentially suggest new treatment avenues for bladder disorders.
Acknowledgments
This work was supported by U.S. National Institute of Diabetes and Digestive and Kidney Diseases/National Institutes of Health (http://nih.gov/) grants DK-095922 (to W.Y.), DK-083299 (to W.G.H.), and AI045897 and CA164970 (to S.C.R.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors declare no conflicts of interest.
Footnotes
- α,β-meATP
- α,β-methyleneadenosine 5′-triphosphate trisodium salt
- ADPβS
- adenosine-5′-(β-thio)-diphosphate lithium salt
- BSM
- bladder smooth muscle
- EFS
- electrical field stimulation
- ENTPD1
- ectonucleoside triphosphate diphosphohydrolase 1
- LUTS
- lower urinary tract symptoms
- 2MesADP
- 2-methylthioadenosine diphosphate trisodium salt
- Nt5e
- 5′-nucleotidase
REFERENCES
- 1. Coyne K. S., Sexton C. C., Thompson C. L., Milsom I., Irwin D., Kopp Z. S., Chapple C. R., Kaplan S., Tubaro A., Aiyer L. P., Wein A. J. (2009) The prevalence of lower urinary tract symptoms (LUTS) in the U. S. A., the UK and Sweden: results from the Epidemiology of LUTS (EpiLUTS) study. BJU Int. 104, 352–360 [DOI] [PubMed] [Google Scholar]
- 2. Liu G., Daneshgari F. (2005) Alterations in neurogenically mediated contractile responses of urinary bladder in rats with diabetes. Am. J. Physiol. Renal Physiol. 288, F1220–F1226 [DOI] [PubMed] [Google Scholar]
- 3. Ford A. P., Cockayne D. A. (2011) ATP and P2X purinoceptors in urinary tract disorders. Handb. Exp. Pharmacol. 202,485–526 [DOI] [PubMed] [Google Scholar]
- 4. Burnstock G. (2011) Introductory overview of purinergic signalling. Front. Biosci. 3, 896–900 [DOI] [PubMed] [Google Scholar]
- 5. Burnstock G., Satchell D. G., Smythe A. (1972) A comparison of the excitatory and inhibitory effects of non-adrenergic, non-cholinergic nerve stimulation and exogenously applied ATP on a variety of smooth muscle preparations from different vertebrate species. Br. J. Pharmacol. 46, 234–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burnstock G., Dumsday B., Smythe A. (1972) Atropine resistant excitation of the urinary bladder: the possibility of transmission via nerves releasing a purine nucleotide. Br. J. Pharmacol. 44, 451–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mulryan K., Gitterman D. P., Lewis C. J., Vial C., Leckie B. J., Cobb A. L., Brown J. E., Conley E. C., Buell G., Pritchard C. A., Evans R. J. (2000) Reduced vas deferens contraction and male infertility in mice lacking P2X1 receptors. Nature 403, 86–89 [DOI] [PubMed] [Google Scholar]
- 8. Dixon A. K., Gubitz A. K., Sirinathsinghji D. J., Richardson P. J., Freeman T. C. (1996) Tissue distribution of adenosine receptor mRNAs in the rat. Br. J. Pharmacol. 118, 1461–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gopalakrishnan S. M., Buckner S. A., Milicic I., Groebe D. R., Whiteaker K. L., Burns D. J., Warrior U., Gopalakrishnan M. (2002) Functional characterization of adenosine receptors and coupling to ATP-sensitive K+ channels in Guinea pig urinary bladder smooth muscle. J. Pharmacol. Exp. Ther. 300, 910–917 [DOI] [PubMed] [Google Scholar]
- 10. Palea S., Artibani W., Ostardo E., Trist D. G., Pietra C. (1993) Evidence for purinergic neurotransmission in human urinary bladder affected by interstitial cystitis. J. Urol. 150, 2007–2012 [DOI] [PubMed] [Google Scholar]
- 11. Bayliss M., Wu C., Newgreen D., Mundy A. R., Fry C. H. (1999) A quantitative study of atropine-resistant contractile responses in human detrusor smooth muscle, from stable, unstable and obstructed bladders. J. Urol. 162, 1833–1839 [PubMed] [Google Scholar]
- 12. O'Reilly B. A., Kosaka A. H., Chang T. K., Ford A. P., Popert R., McMahon S. B. (2001) A quantitative analysis of purinoceptor expression in the bladders of patients with symptomatic outlet obstruction. BJU Int. 87, 617–622 [DOI] [PubMed] [Google Scholar]
- 13. Yoshida M., Homma Y., Inadome A., Yono M., Seshita H., Miyamoto Y., Murakami S., Kawabe K., Ueda S. (2001) Age-related changes in cholinergic and purinergic neurotransmission in human isolated bladder smooth muscles. Exp. Gerontol. 36, 99–109 [DOI] [PubMed] [Google Scholar]
- 14. Yoshida M., Miyamae K., Iwashita H., Otani M., Inadome A. (2004) Management of detrusor dysfunction in the elderly: changes in acetylcholine and adenosine triphosphate release during aging. Urology 63(Suppl. 1), 17–23 [DOI] [PubMed] [Google Scholar]
- 15. Ruggieri M. R., Sr. (2006) Mechanisms of disease: role of purinergic signaling in the pathophysiology of bladder dysfunction. Nat. Clin. Pract. Urol. 3, 206–215 [DOI] [PubMed] [Google Scholar]
- 16. Andersson K. E., Arner A. (2004) Urinary bladder contraction and relaxation: physiology and pathophysiology. Physiol. Rev. 84, 935–986 [DOI] [PubMed] [Google Scholar]
- 17. Uvin P., Boudes M., Menigoz A., Franken J., Pinto S., Gevaert T., Verplaetse R., Tytgat J., Vennekens R., Voets T., De Ridder D. (2013) Chronic administration of anticholinergics in rats induces a shift from muscarinic to purinergic transmission in the bladder wall. Eur. Urol. 64, 502–510 [DOI] [PubMed] [Google Scholar]
- 18. Yu W., Sun X., Robson S. C., Hill W. G. (2013) Extracellular UDP enhances P2X-mediated bladder smooth muscle contractility via P2Y(6) activation of the phospholipase C/inositol trisphosphate pathway. FASEB J. 27, 1895–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yu W., Robson S. C., Hill W. G. (2011) Expression and distribution of ectonucleotidases in mouse urinary bladder. PLoS One 6, e18704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Burnstock G., Cusack N. J., Meldrum L. A. (1984) Effects of phosphorothioate analogues of ATP, ADP and AMP on guinea-pig taenia coli and urinary bladder. Br. J. Pharmacol. 82, 369–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Palea S., Corsi M., Pietra C., Artibani W., Calpista A., Gaviraghi G., Trist D. G. (1994) ADP beta S induces contraction of the human isolated urinary bladder through a purinoceptor subtype different from P2X and P2Y. J. Pharmacol. Exp. Ther. 269, 193–197 [PubMed] [Google Scholar]
- 22. Suzuki H., Kokubun S. (1994) Subtypes of purinoceptors in rat and dog urinary bladder smooth muscles. Br. J. Pharmacol. 112, 117–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hashimoto M., Kokubun S. (1995) Contribution of P2-purinoceptors to neurogenic contraction of rat urinary bladder smooth muscle. Br. J. Pharmacol. 115, 636–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Naramatsu M., Yamashita T., Kokubun S. (1997) The signalling pathway which causes contraction via P2-purinoceptors in rat urinary bladder smooth muscle. Br. J. Pharmacol. 122, 558–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McMurray G., Dass N., Brading A. F. (1998) Purinoceptor subtypes mediating contraction and relaxation of marmoset urinary bladder smooth muscle. Br. J. Pharmacol. 123, 1579–1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wu C., Sui G. P., Fry C. H. (2004) Purinergic regulation of guinea pig suburothelial myofibroblasts. J Physiol 559, 231–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kennedy C., Tasker P. N., Gallacher G., Westfall T. D. (2007) Identification of atropine- and P2X1 receptor antagonist-resistant, neurogenic contractions of the urinary bladder. J. Neurosci. 27, 845–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aronsson P., Andersson M., Ericsson T., Giglio D. (2010) Assessment and characterization of purinergic contractions and relaxations in the rat urinary bladder. Basic Clin. Pharmacol. Toxicol. 107, 603–613 [DOI] [PubMed] [Google Scholar]
- 29. Fry C. H., Young J. S., Jabr R. I., McCarthy C., Ikeda Y., Kanai A. J. (2012) Modulation of spontaneous activity in the overactive bladder: the role of P2Y agonists. Am. J. Physiol. Renal Physiol. 302, F1447–F1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Enjyoji K., Sévigny J., Lin Y., Frenette P. S., Christie P. D., Esch J. S., 2nd, Imai M., Edelberg J. M., Rayburn H., Lech M., Beeler D. L., Csizmadia E., Wagner D. D., Robson S. C., Rosenberg R. D. (1999) Targeted disruption of cd39/ATP diphosphohydrolase results in disordered hemostasis and thromboregulation. Nat. Med. 5, 1010–1017 [DOI] [PubMed] [Google Scholar]
- 31. Thompson L. F., Eltzschig H. K., Ibla J. C., Van De Wiele C. J., Resta R., Morote-Garcia J. C., Colgan S. P. (2004) Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J. Exp. Med. 200, 1395–1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sibley G. N. (1984) A comparison of spontaneous and nerve-mediated activity in bladder muscle from man, pig and rabbit. J. Physiol. 354, 431–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cattaneo M., Lecchi A., Ohno M., Joshi B. V., Besada P., Tchilibon S., Lombardi R., Bischofberger N., Harden T. K., Jacobson K. A. (2004) Antiaggregatory activity in human platelets of potent antagonists of the P2Y 1 receptor. Biochem. Pharmacol. 68, 1995–2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Humphries R. G., Tomlinson W., Ingall A. H., Cage P. A., Leff P. (1994) FPL 66096: a novel, highly potent and selective antagonist at human platelet P2T-purinoceptors. Br. J. Pharmacol. 113, 1057–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hardy A. R., Conley P. B., Luo J., Benovic J. L., Poole A. W., Mundell S. J. (2005) P2Y1 and P2Y12 receptors for ADP desensitize by distinct kinase-dependent mechanisms. Blood 105, 3552–3560 [DOI] [PubMed] [Google Scholar]
- 36. Finkbeiner A. E., Bissada N. K. (1980) Effect of detrusor muscle length and tension on its response to pharmacologic and electrical stimulation. Part II. In vitro study. Urology 16, 650–655 [DOI] [PubMed] [Google Scholar]
- 37. Foster C. J., Prosser D. M., Agans J. M., Zhai Y., Smith M. D., Lachowicz J. E., Zhang F. L., Gustafson E., Monsma F. J., Jr, Wiekowski M. T., Abbondanzo S. J., Cook D. N., Bayne M. L., Lira S. A., Chintala M. S. (2001) Molecular identification and characterization of the platelet ADP receptor targeted by thienopyridine antithrombotic drugs. J. Clin. Invest. 107, 1591–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hollopeter G., Jantzen H. M., Vincent D., Li G., England L., Ramakrishnan V., Yang R. B., Nurden P., Nurden A., Julius D., Conley P. B. (2001) Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature 409, 202–207 [DOI] [PubMed] [Google Scholar]
- 39. Zhang F. L., Luo L., Gustafson E., Lachowicz J., Smith M., Qiao X., Liu Y. H., Chen G., Pramanik B., Laz T. M., Palmer K., Bayne M., Monsma F. J., Jr. (2001) ADP is the cognate ligand for the orphan G protein-coupled receptor SP1999. J. Biol. Chem. 276, 8608–8615 [DOI] [PubMed] [Google Scholar]
- 40. Gachet C. (2012) P2Y(12) receptors in platelets and other hematopoietic and non-hematopoietic cells. Purinergic Signal. 8, 609–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Evans D. J., Jackman L. E., Chamberlain J., Crosdale D. J., Judge H. M., Jetha K., Norman K. E., Francis S. E., Storey R. F. (2009) Platelet P2Y(12) receptor influences the vessel wall response to arterial injury and thrombosis. Circulation 119, 116–122 [DOI] [PubMed] [Google Scholar]
- 42. Wihlborg A. K., Wang L., Braun O. O., Eyjolfsson A., Gustafsson R., Gudbjartsson T., Erlinge D. (2004) ADP receptor P2Y12 is expressed in vascular smooth muscle cells and stimulates contraction in human blood vessels. Arterioscler. Thromb. Vasc. Biol. 24, 1810–1815 [DOI] [PubMed] [Google Scholar]
- 43. Hogberg C., Svensson H., Gustafsson R., Eyjolfsson A., Erlinge D. (2010) The reversible oral P2Y12 antagonist AZD6140 inhibits ADP-induced contractions in murine and human vasculature. Int. J. Cardiol. 142, 187–192 [DOI] [PubMed] [Google Scholar]
- 44. Rauch B. H., Rosenkranz A. C., Ermler S., Böhm A., Driessen J., Fischer J. W., Sugidachi A., Jakubowski J. A., Schrör K. (2010) Regulation of functionally active P2Y12 ADP receptors by thrombin in human smooth muscle cells and the presence of P2Y12 in carotid artery lesions. Arterioscler. Thromb. Vasc. Biol. 30, 2434–2442 [DOI] [PubMed] [Google Scholar]
- 45. Lee C. W., Hwang I., Park C. S., Lee H., Park D. W., Kang S. J., Lee S. W., Kim Y. H., Park S. W., Park S. J. (2011) Comparison of differential expression of P2Y(1)(2) receptor in culprit coronary plaques in patients with acute myocardial infarction versus stable angina pectoris. Am. J. Cardiol. 108, 799–803 [DOI] [PubMed] [Google Scholar]
- 46. Macfarlane D. E., Srivastava P. C., Mills D. C. (1983) 2-Methylthioadenosine[β-32P]diphosphate. An agonist and radioligand for the receptor that inhibits the accumulation of cyclic AMP in intact blood platelets. J. Clin. Invest. 71, 420–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Filippov A. K., Brown D. A., Barnard E. A. (2000) The P2Y(1) receptor closes the N-type Ca2+ channel in neurones, with both adenosine triphosphates and diphosphates as potent agonists. Br. J. Pharmacol. 129, 1063–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Palmer R. K., Boyer J. L., Schachter J. B., Nicholas R. A., Harden T. K. (1998) Agonist action of adenosine triphosphates at the human P2Y1 receptor. Mol. Pharmacol. 54, 1118–1123 [PubMed] [Google Scholar]
- 49. Zhong X., Kriz R., Seehra J., Kumar R. (2004) N-linked glycosylation of platelet P2Y12 ADP receptor is essential for signal transduction but not for ligand binding or cell surface expression. FEBS Lett. 562, 111–117 [DOI] [PubMed] [Google Scholar]
- 50. Fontana P., Dupont A., Gandrille S., Bachelot-Loza C., Reny J. L., Aiach M., Gaussem P. (2003) Adenosine diphosphate-induced platelet aggregation is associated with P2Y12 gene sequence variations in healthy subjects. Circulation 108, 989–995 [DOI] [PubMed] [Google Scholar]
- 51. Staritz P., Kurz K., Stoll M., Giannitsis E., Katus H. A., Ivandic B. T. (2009) Platelet reactivity and clopidogrel resistance are associated with the H2 haplotype of the P2Y12-ADP receptor gene. Int. J. Cardiol. 133, 341–345 [DOI] [PubMed] [Google Scholar]
- 52. Geiger J., Brich J., Hönig-Liedl P., Eigenthaler M., Schanzenbächer P., Herbert J. M., Walter U. (1999) Specific impairment of human platelet P2Y(AC) ADP receptor-mediated signaling by the antiplatelet drug clopidogrel. Arterioscler. Thromb. Vasc. Biol. 19, 2007–2011 [DOI] [PubMed] [Google Scholar]
- 53. Guidetti G. F., Lova P., Bernardi B., Campus F., Baldanzi G., Graziani A., Balduini C., Torti M. (2008) The Gi-coupled P2Y12 receptor regulates diacylglycerol-mediated signaling in human platelets. J. Biol. Chem. 283, 28795–28805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Berridge M. J. (2008) Smooth muscle cell calcium activation mechanisms. J Physiol 586, 5047–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]