

Abstract
Low birth weight and rapid postnatal growth increases risk of cardiovascular-disease (CVD); however, underlying mechanisms are poorly understood. Previously, we demonstrated that rats exposed to a low-protein diet in utero that underwent postnatal catch-up growth (recuperated) have a programmed deficit in cardiac coenzyme Q (CoQ) that was associated with accelerated cardiac aging. It is unknown whether this deficit occurs in all tissues, including those that are clinically accessible. We investigated whether aortic and white blood cell (WBC) CoQ is programmed by suboptimal early nutrition and whether postweaning dietary supplementation with CoQ could prevent programmed accelerated aging. Recuperated male rats had reduced aortic CoQ [22 d (35±8.4%; P<0.05); 12 m (53±8.8%; P<0.05)], accelerated aortic telomere shortening (P<0.01), increased DNA damage (79±13% increase in nei-endonucleaseVIII-like-1), increased oxidative stress (458±67% increase in NAPDH-oxidase-4; P<0.001), and decreased mitochondrial complex II-III activity (P<0.05). Postweaning dietary supplementation with CoQ prevented these detrimental programming effects. Recuperated WBCs also had reduced CoQ (74±5.8%; P<0.05). Notably, WBC CoQ levels correlated with aortic telomere-length (P<0.0001) suggesting its potential as a diagnostic marker of vascular aging. We conclude that early intervention with CoQ in at-risk individuals may be a cost-effective and safe way of reducing the global burden of CVDs.—Tarry-Adkins, J. L., Fernandez-Twinn, D. S., Chen, J.-H., Hargreaves, I. P., Martin-Gronert, M. S., McConnell, J. M., Ozanne, S. E. Nutritional programming of coenzyme Q: potential for prevention and intervention?
Keywords: telomeres, cardiovascular disease, aging, vascular disease
It has been known for several years that low birth weight is strongly associated with increased risk of cardiovascular disease (CVD) in later life (1, 2). Furthermore, risk of CVD and its associated metabolic dysfunction is exacerbated in low-birth-weight babies who experienced rapid postnatal growth (3–5). Animal models have provided valuable insights into potential underlying molecular mechanisms that link suboptimal maternal exposures to later outcomes of cardiovascular health. These include reductions in cardiomyocyte numbers at birth (6); structural changes, including alterations in aortic wall thickness (7); and increases in cardiac fibrosis (8) and increased cardiac oxidative stress (9).
Excessive reactive oxygen species (ROS) is known to damage cellular macromolecules such as proteins, lipids and DNA, if cellular antioxidant defenses are insufficient to maintain redox homeostasis. In particular, ROS accelerate telomere shortening in somatic cells (10, 11) by preferentially damaging the guanine-rich repeat sequences within telomeric DNA. Telomeres shorten after every somatic cell division, and in many species, including birds (12, 13), mice (13), and humans (13), telomere length has been correlated with longevity. Furthermore, telomere length plays a pivotal role in the onset, development, and prognosis of CVD (14). In humans, the aorta is a major site of telomere attrition (15), and aortic telomere length has been shown to be negatively correlated with age and atherosclerotic grade (15, 16). Using a well-established rat model of nutritionally induced low birth weight followed by accelerated postnatal growth (recuperated or catch-up growth model), we have previously demonstrated that these growth patterns are associated with reduced longevity compared with controls (17) and significant reductions in kidney (17), aortic (18), pancreatic islet (19), and cardiac (20) telomere length. The reductions in renal and cardiac telomere length were linked to a programmed deficit in renal (21) and cardiac coenzyme Q9 (CoQ9) levels (20) in later life.
The CoQ (or ubiquinone) molecule consists of a benzoquinone ring linked to an isoprenoid side chain, the length of which varies between species. In humans, the most common form is CoQ10, containing 10 isoprenoid units, whereas CoQ9 (containing 9 isoprenoid units) is the most common isoform in rodents (although rodents can convert dietary CoQ10 into CoQ9). CoQ acts as an electron carrier, shuttling electrons between complexes I and III and complexes II and III of the mitochondrial electron transport chain (ETC). In its reduced form (ubiquinol), it is a potent antioxidant, preventing initiation and propagation of lipid peroxidation (22). We have recently demonstrated that dietary supplementation with CoQ10 can ameliorate indexes of cardiac aging in rats exposed to catch-up growth by preventing accelerated cardiac telomere shortening, premature induction of p21 and p53 (mediators of cell senescence), and induction of apoptotic markers (20). Notably, CoQ10 supplementation exhibited no detrimental effects on control offspring (20).
Our previous studies have thus demonstrated that suboptimal nutrition in early life can lead to a programmed deficit in renal and cardiac CoQ9 in later life. However, it is unknown whether this deficit is tissue specific or is present in all tissues, most notably including those that are clinically accessible. Furthermore, it is unknown whether CoQ deficiency is a very early consequence of a suboptimal early environment and therefore likely to be a causative factor in mediating detrimental consequences in the offspring. Therefore, this study aimed to investigate the effects of poor maternal nutrition followed by rapid postnatal catch-up growth on CoQ9 levels and molecular markers of aging in aortic tissue at weaning; determine whether supplementation of a clinically relevant dose of dietary CoQ10 could restore any observed deficit in aortic CoQ9 and therefore correct molecular indexes of accelerated aging in later postnatal life; and establish whether levels of CoQ9 were also programmed in white blood cells (WBCs) and therefore identify its potential value as a diagnostic tool for assessing CVD susceptibility in later life and provide rationale for intervention in high risk individuals.
MATERIALS AND METHODS
Animal experimental groups
All procedures involving animals were conducted under the British Animals (Scientific Procedures) Act (1986). Pregnant Wistar rats were maintained on a 20% protein (control) diet or an isocaloric low-protein (LP; 8%) diet fed ad libitum, as described previously (23). Both diets were purchased from Arie Blok (Woerden, The Netherlands). The day of birth was recorded as d 1 of postnatal life. Pups born to LP-diet-fed dams were cross fostered to control-fed mothers on postnatal d 3 to create a recuperated litter. Each recuperated litter was culled to 4 male pups at random to maximize their plane of nutrition. The control group was the offspring of mothers fed the 20% protein diet and suckled by dams fed the 20% protein diet. Each control litter was culled to 8 pups. To prevent any stress to the animals when cross fostered, pups were transferred with some of their own bedding. Body weights were recorded at postnatal d 3, 7, 14, and 21 and at 12 mo. At 21 d, 2 males/litter were weaned onto standard laboratory chow (Special Diet Services, Witham, UK) and the other 2 were weaned onto the same diet supplemented with CoQ10 to give a dose of 1 mg/kg body weight/d. Animals were maintained on these diets until 12 mo of age. A further cohort of animals (control and recuperated offspring without CoQ10 supplementation) was weaned at 21 d of age, denied access to food overnight, and killed at 22 d of age. All animals were killed by CO2 asphyxiation. At postmortem, aortic tissue was removed, weighed, and snap-frozen in liquid nitrogen and then stored at −80°C until analysis. For all measurements, 1 pup/litter was used; thus, n represents number of litters throughout. Only male animals were used in this study.
CoQ10 diet preparation
A dose of CoQ10 (1 mg/kg body weight/d; refs. 24–27) was used in this study. This was achieved by appropriate CoQ10 supplementation of laboratory chow, as we have described previously (20). Diet was prepared 2×/wk throughout the study.
CoQ9 and CoQ10 measurement
Total tissue ubiquinone (CoQ9 and CoQ10) status was quantified in whole aortic tissue by reverse-phase HPLC with ultraviolet (UV) detection at 275 nm as described previously (20). CoQ10 was separated on an HPLC column (Techsphere ODS; 5 μm, 150×4.6 mm; Capital Analytical Ltd., Leeds, UK). The mobile phase consisted of ethanol:methanol:60% (v/v) perchloric acid; 700:300:1.2 (v/v) to which was added 7 g of sodium perchlorate (20). The flow rate was maintained at 0.7 ml/min, (20).
WBC isolation
Samples of whole blood (10 ml) were obtained via cardiac puncture and added to tubes containing 1% 0.5 M EDTA (pH 8.0) and shaken. Samples were then divided equally into 4 tubes. Five volumes of red blood cell (RBC) lysis buffer [150 mM ammonium chloride, 10 mM potassium bicarbonate, and 0.1 mM EDTA (500 mM); pH 8.0] was added to each tube. Samples were incubated at room temperature for 5 min, vortexed, and then centrifuged at 4°C for 10 min at 10,000 g. Supernatant containing RBCs was removed and discarded to leave WBC pellets. The pellets were then cleaned by adding 2 vol of RBC buffer, mixed, and centrifuged as above. After removal of the supernatant, 1 ml of RBC buffer was added to the pellets and mixed. The WBCs were counted using a cell counter (Countess Automated Cell Counter; Invitrogen, Paisley, UK). WBC pellets were then centrifuged, and the supernatant was removed, snap-frozen, and stored at −80°C until analysis.
Mitochondrial complex activities
All mitochondrial complex activities were measured at 30°C on the Uvikon XL spectrophotometer (Kontron Instruments, Ltd., Watford, UK). Before assay, all sample homogenates of whole aortic tissue were subjected to 3 freeze-thaw cycles to disrupt the mitochondrial membranes and allow substrates access to the active sites of the enzymes. Activities of complex I (NADH:ubiquinone reductase; EC 1.6.5.3), complex II–III (succinate:cytochrome c reductase; EC 1.3.5.1+EC 1.10.2.2), and complex IV as well as citrate synthase (CS; EC 1.1.1.27) activity were assayed as described previously (9). As CS is a mitochondrial marker enzyme, all complex activities were expressed as a ratio to CS to compensate for differences in mitochondrial enrichment in the cell samples.
Telomere length analysis
High-molecular-weight DNA was extracted from whole aortic tissue using the Wizard Genomic DNA Isolation kit (Promega, Southampton, UK) according to the manufacturer's instructions. DNA quantity and purity were determined using a Nanodrop spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA). DNA (1.2 μg) was digested by HinfI and Rsa1 restriction enzymes, separated by pulsed field gel electrophoresis (PFGE). The restricted DNA samples were quenched with 5× SDS loading buffer and loaded onto agarose gels containing SYBR safe stain (Invitrogen). Gels were checked for nonspecific degradation of an undigested DNA control and complete digestion of the enzyme-restricted DNA by visualizing the stained gels under UV light using Gel Doc visualization software (Syngene, Cambridge, UK). The separated DNA was transferred onto nylon membranes by Southern blotting. Telomere length was measured using Telo TAGGG telomere length assays (Roche Diagnostics, Mannheim, Germany; ref. 9). Telomere signals were analyzed using Adobe Photoshop (Adobe Systems, Inc., San Jose, CA, USA) and MacBas software (Fujifilm UK, Bedford, UK). Telomere length was quantified as described previously (9).
Markers of oxidative stress and antioxidant defense capacity
Western blotting analysis of whole aortic tissue was used to determine protein expression of nei endonuclease VIII-like 1 (NEIL-1), nicotinamide adenine dinucleotide diphosphate (NADPH) oxidase 4 (NOX-4), xanthine oxidase (XO), manganese superoxidase-dismutase (MnSOD), and catalase. Protein was extracted and assayed as described previously (9), and 20 μg protein was loaded onto 10, 12, or 15% polyacrylamide gels, dependent on the molecular weight of the protein to be measured. The samples were then electrophoresed and transferred to polyvinylidene fluoride membranes (9), using the following concentrations: NEIL-1, 1:500 (Novus Biologicals, Littleton, CO, USA); MnSOD, 1:10,000 (Upstate Biochemicals, Watford, UK); and catalase, 1:10,000 (Abcam, Cambridge, Cambridgeshire, UK) using anti-rabbit IgG secondary antibodies. XO (1:200; Santa Cruz Biotechnology, Heidelberg, Germany) was detected using anti-mouse IgGs. NOX-4 (1:200; Santa Cruz Biotechnology) was detected using anti-goat IgGs. Equal protein loading was confirmed by staining electrophoresed gels with Coomassie blue to visualize total protein.
Statistical analysis
Where appropriate, data were analyzed either using a 3-way ANOVA with maternal diet, CoQ10 supplementation, and age as the independent variables. Otherwise, a 2-way ANOVA was used with maternal diet and age as the independent variables. Data are represented as means ± sem. A value of P < 0.05 was considered statistically significant. All statistical analyses were performed using Statistica 7 software (Statsoft, Inc., Milton Keynes, UK). In all cases, n refers to the number of litters.
RESULTS
Recuperated animals were born small and underwent rapid postnatal growth
At postnatal d 3 and 7, recuperated pups were significantly (P<0.001) lighter compared with control offspring. However, by postnatal d 21, this group had undergone accelerated postnatal growth; therefore, the body weights were similar between groups (Fig. 1). At 12 mo of age, body weight remained similar between groups (Table 1).
Figure 1.
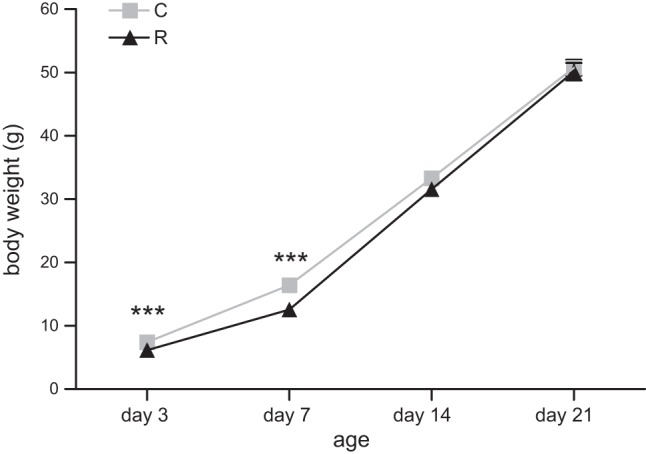
Effect of in utero protein restriction and accelerated postnatal growth on preweaning body weights. Results are expressed as means ± sem. ***P < 0.001 for recuperated (R) vs. control (C); n=10/group.
Table 1.
Group body weights
| Group | Body weight (g) |
|---|---|
| Control | 977 ± 26.7 |
| Recuperated | 937 ± 29.8 |
| Control CoQ | 1002 ± 38.0 |
| Recuperated CoQ | 954 ± 30.7 |
Aortic and WBC CoQ9 levels were reduced in recuperated offspring
At 22 d of age, aortic CoQ9 concentration was significantly reduced (P<0.05) in the recuperated group compared with controls (Fig. 2A). There was also an effect of aging such that CoQ9 levels in 12 mo aortas were significantly (P<0.05) lower than those observed at 22 d of age. At 12 mo of age, both aortic and WBC levels of CoQ9 were significantly reduced (P<0.05) in the recuperated group compared with control animals (Fig. 2A). CoQ10 supplementation had no effect on aortic CoQ9 levels in control animals (203±39 vs. 196±35 pmol/mg protein) nor in recuperated animals (119±15 vs. 158±30 pmol/mg protein). Likewise, CoQ9 levels in WBCs were also unaffected by CoQ10 supplementation (control: 161±32; control CoQ: 140±17; recuperated: 111±4; recuperated CoQ: 108±12 pmol/mg protein). Interestingly however, a strong positive correlation (P<0.0001; r2=0.84) was observed between aortic and WBC CoQ9 concentrations at 12 mo of age in control and recuperated offspring (Fig. 2B).
Figure 2.

Effect of in utero protein restriction and accelerated postnatal growth on aortic and WBC CoQ9 levels in 22 d and 12 mo rats (A), correlation between aortic and WBC CoQ9 concentrations in 12 mo male rats (B), aortic linked complex II-III activity in 22 d and 12 mo male rats (C), and correlation between aortic CoQ9 levels and linked complex II-III activity in 12 mo male rats (D). Results are expressed as means ± sem. *P < 0.05; n = 6–8/group.
Recuperated offspring had a deficit in linked complex II-III enzyme activity
Consistent with the CoQ9 deficit, a significant (P<0.05) reduction in linked complex II-III activity was observed in the recuperated group compared with controls at 22 d of age (Fig. 2C), whereas there was no difference in complex I (0.2±0.03 vs. 0.2±0.04 ratio to CS activity) or complex IV activity (0.01±0.001 vs. 0.01±0.001 ratio to CS activity). A deficit in linked complex II-III activity was still present at 12 mo of age (Fig. 2C) and CoQ9 levels were positively correlated with linked complex II-III activity; (P=0.004; r2=0.29) in control and recuperated offspring, (Fig. 2D). CoQ10 supplementation had no effect on complex II-III activity in the recuperated group (0.02±0.003 in unsupplemented vs. 0.02±0.003 in supplemented group; expressed as ratio to CS activity), however CoQ10 supplementation resulted in a significant (P<0.001) reduction in linked complex II-III activity in the control group (0.04±0.005 in unsupplemented vs. 0.02±0.003 in the supplemented group; ratio to CS activity).
Indexes of oxidative stress were ameliorated by CoQ10 supplementation
At 12 mo of age, there was no significant effect of maternal diet on aortic XO protein expression; however, CoQ10 supplementation significantly (P<0.01) reduced XO levels (Fig. 3A). NOX-4 protein levels were markedly (P<0.001) increased in the recuperated group compared with controls (Fig. 3B), an effect that was prevented by CoQ10 supplementation (P<0.001; Fig. 3B).
Figure 3.
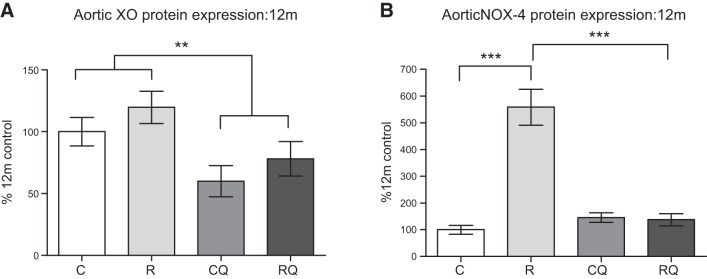
Effect of in utero protein restriction, accelerated postnatal growth, and CoQ10 supplementation on XO (A) and NOX-4 (B) protein expression in 12 mo male rats. C, control; CQ, control CoQ; R, recuperated; RQ, recuperated CoQ. Results are expressed as means ± sem. **P < 0.05, ***P < 0.001; n = 6/group.
CoQ10 supplementation prevents aortic telomere shortening in recuperated offspring
At 12 mo of age, recuperated animals had shorter aortic telomeres compared with controls, as reflected by significantly (P<0.01) fewer long (145–48.5 kb) and significantly (P<0.01) more short (4.2–1.3 kb) telomeres (Fig. 4A). CoQ10 supplementation prevented the increased telomere shortening in the recuperated group (Fig. 4A). There was a positive correlation between WBC CoQ9 concentration and the proportion of the longest (145-48.5 kb) telomere fragments (r2=0.42; P<0.05; Fig. 4B) and a negative correlation with the proportion of the shortest (4.2-1.3 kb) telomere fragments (r2=0.37; P<0.05; Fig. 4C).
Figure 4.

Effect of in utero protein restriction and accelerated postnatal growth on telomere length (A), correlation between aortic telomere length and WBC CoQ9 concentrations in 12 mo male rats (B, C), and NEIL-1 (D) protein expression in 12 mo male rat aortas. C, control; CQ, control CoQ; R, recuperated; RQ, recuperated CoQ. Results are expressed as means ± sem. **P < 0.01, ***P < 0.01; n = 6/group.
CoQ10 supplementation abrogates up-regulation of NEIL-1, a base excision repair (BER) enzyme
At 22 d of age, NEIL-1 protein levels were significantly (P<0.05) increased in the recuperated group compared with controls (264±54 vs. 100±25%). Elevated NEIL-1 protein levels were maintained in the recuperated group at 12 mo of age (P<0.001; Fig. 4D). CoQ10 supplementation was able to prevent (P<0.001) this increase (Fig. 4D). Protein expression of Nthl endonuclease III-like-1 (NTHL-1) and 8 oxoguanine DNA glycosylase 1 (OGG-1) were undetectable at both ages.
Antioxidant-defense capacity is altered in recuperated offspring and can be ameliorated by CoQ10 supplementation
At 22 d of age, MnSOD and catalase protein levels were significantly (P<0.001) increased in the recuperated group compared with controls (Fig. 5A). At 12 mo of age, MnSOD levels were significantly reduced in the recuperated group; however, this decrease was not ameliorated by CoQ10 supplementation (Fig. 5B). Catalase levels (P<0.05) remained significantly elevated in the recuperated group compared with controls at 12 mo of age. However, CoQ10 supplementation significantly (P<0.001) decreased catalase protein expression (Fig. 5C).
Figure 5.
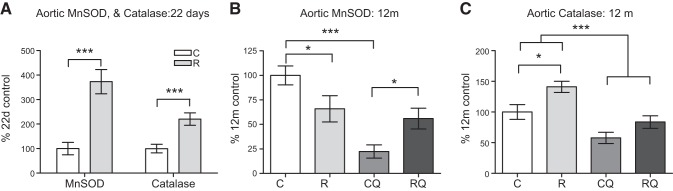
Effect of in utero protein restriction and accelerated postnatal growth on antioxidant protein expression in 22 d (A) and 12 mo (B, C) male rat aortas. C, control; CQ, control CoQ; R, recuperated; RQ, recuperated CoQ. Results are expressed as means ± sem. *P < 0.05, ***P < 0.001; n = 6/group.
DISCUSSION
In this study, we demonstrated for the first time that an exposure to suboptimal nutrition during early life resulted in a deficit of aortic CoQ9 across the life course, which is associated with accelerated aortic telomere shortening. The normal life span for the rats used in this study is between 13 and 15 mo (28); therefore, the study of animals at 12 mo of age gives valuable insight into changes occurring toward the end of life. At 12 mo of age, aortic CoQ9 levels were ∼10 times lower than those previously reported in the heart (20) and lower in renal tissue (21). It is known that all tissues, with the exception of RBCs can synthesize CoQ; however, the levels of synthesis can vary greatly between tissues and this is thought to be largely dependent on how metabolically active and mitochondrially rich the tissue is. Therefore, lower aortic CoQ9 levels may reflect lower metabolic requirements of the aorta compared with the heart and may explain why CoQ10 supplementation was unable to significantly increase aortic CoQ9 status in either group.
Oxidative stress is a common feature observed in a number of models of developmental programming, including maternal hypoxia, maternal obesity, maternal protein restriction, and placental insufficiency (29). Oxidative stress can result from mitochondrial dysfunction and NOX and XO up-regulation (30) and has been strongly implicated in the pathogenesis of CVD (30). Poor maternal nutrition followed by rapid postnatal growth resulted in decreased linked complex II-III activity in aortic tissue, which was associated with increased levels of NOX-4, suggestive of a prooxidative phenotype in recuperated aortas. Increased oxidative stress is known to accelerate telomere shortening by preferentially damaging the guanosine-rich DNA sequences of telomeric DNA (31), and as a response to oxidatively damaged DNA, enzymes in the BER DNA damage pathway can be activated (32). Consistent with the observed increase in oxidative stress, we observed increased indexes of DNA damage in recuperated offspring, including increased levels of the BER enzyme NEIL-1 and accelerated aortic telomere shortening. Since the CoQ9 deficit is evident before telomere shortening (18), this supports a role for CoQ9 levels as an early programming mediator of telomere shortening, aging, and disease of the aorta.
Damage to DNA and other cellular macromolecules can occur if there is an imbalance between ROS generation and subsequent antioxidant defense capacity. At 22 d of age, expression of MnSOD [a mitochondrial specific antioxidant enzyme that is responsible for the conversion of the major cellular ROS. superoxide anion (O2−), into hydrogen peroxide (H2O2)] in recuperated offspring was significantly increased, however, by 12 mo of age, this effect was reversed, perhaps signifying that mitochondrial antioxidant defenses are compromised in older recuperated animals and aging is instrumental in the loss of MnSOD expression. A deficiency in MnSOD has been shown to increase mitochondrial oxidative stress and aggravate age-dependent vascular relaxation (33), and loss of this enzyme is a common phenotype of vasculature dysfunction (34, 35). Thus, the age-dependent loss of MnSOD may facilitate the observed oxidative stress in recuperated offspring. Levels of catalase (a nonmitochondrial antioxidant enzyme that catalyzes H2O2 into H2O and O2) remained elevated in recuperated offspring, which suggests that this compensatory response to oxidative stress is maintained. Indeed, it has previously been reported that this enzyme is up-regulated in sites of aortic coarctation, in the presence of oxidative stress (36). Taken together with the increased NOX-4 protein expression, reduced linked complex II-III activity, and CoQ9 deficit, this suggests a specific mitochondrial dysfunction in the aortas of recuperated offspring associated with an increase in oxidative stress.
CoQ10 supplementation was able to decrease oxidative stress by reducing NOX-4 and XO protein levels, restoring NEIL-1 protein to control levels, and critically, preventing accelerated telomere attrition. These findings support a direct role for oxidative stress in accelerated telomere attrition in recuperated offspring. Further support for this role comes from studies showing that MitoQ (a CoQ analog) can counteract fibroblast telomere shortening under mild oxidative stress conditions (37). CoQ10 supplementation did not, however, alter NOX-4, NEIL-1, or telomere length in the control group, which implies that CoQ10 supplementation is capable of ameliorating accelerated aortic aging only where a CoQ9 deficit exists. Notably, there was no adverse effect where CoQ9 levels were normal. While CoQ10 supplementation did not alter MnSOD levels in the recuperated group, catalase was reduced, which is likely due to its overall beneficial effects on the lowering of ROS levels (as evidenced by reduced XO and NOX-4).
Our studies have now shown that a CoQ deficit and compromised mitochondria occur in renal, heart, and aortic tissues of recuperated animals. Our initial simple in in vitro studies suggested that if CoQ levels were normalized then mitochondrial function would be fully restored (21). In the current in vivo work, where rats were supplemented with dietary CoQ, our findings suggest a complex and more indirect beneficial effect of high serum levels of CoQ. Premature aging is reversed by dietary CoQ, but tissue concentrations and mitochondrial activity are not corrected. Instead CoQ appears to induce the expression of additional beneficial antioxidant defenses.
Human meta-analyses have demonstrated that CoQ10 supplementation (doses ranging from 60 to 300 mg/d) can improve clinical outcome in patients with heart failure (38–40). Most notably, safety studies indicate that CoQ10 is well tolerated, has low toxicity, and does not induce serious adverse effects in humans. Risk assessments for CoQ10 based on various clinical trial data indicate that the observed safety level for CoQ10 in humans is between 900 and 1200 mg/d/person. Overall, these data from preclinical and clinical studies confirm that CoQ10 is safe for use as a dietary supplement (41, 42). In our study, we utilized a dose of 1 mg/kg body weight/d, a dose far lower than the reported maximum safe dose and one that has been previously tolerated without side effects (24–27).
Globally, CVD is responsible for more deaths than any other disease, claiming an estimated 17.3 million lives in 2008, a number that is predicted to grow to >23.3 million by 2030 (43). These statistics impress a critical need for the development of early biomarkers for CVD risk. For a biomarker to be feasible, it must be present in clinically accessible tissue. By measuring CoQ9 status in WBCs from control and recuperated rats, we demonstrated a significant CoQ9 deficiency in the WBCs of recuperated offspring. Notably, this strongly correlated to aortic CoQ9 levels. Furthermore, we also showed a highly significant relationship between CoQ9 levels and aortic telomere length in WBCs, suggesting that low WBC CoQ9 levels can predict short aortic telomeres and therefore susceptibility to aortic disease. These studies in rodents therefore have identified a biomarker in a clinically accessible tissue that has the potential to be used as a tool to identify individuals at risk of cardiovascular disease. A next step will be to establish whether these findings can be confirmed in humans and therefore make their prognostic potential a realistic possibility.
In summary, we have demonstrated, for the first time to our knowledge, that nutritionally induced low birth weight and catch-up growth leads to a programmed deficit in aortic CoQ9 that is coupled to increased DNA damage and telomere shortening. This accelerated aortic cellular aging can be prevented with dietary CoQ10 postweaning. The fact that CoQ9 levels were also programmed and detectable in blood raises the exciting possibility that WBC CoQ measurements could be prioritized as a marker of vascular aging and risk of CVD in later life. Early intervention with CoQ10 in identified at-risk individuals could therefore represent a safe and cost-effective treatment for cardiovascular disease.
Acknowledgments
This work was supported by the British Heart Foundation (PG/09/037/27387, FS/09/029/27902) and Medical Research Council (MC_UU_12012/4). S.E.O. is a British Heart Foundation Senior Fellow and a member of the Medical Research Council Metabolic Diseases Unit. I.P.H. is supported by the UK Department of Health's National Institute for Health Research Biomedical Research Centres funding scheme at University College London Hospital (University College London).
Footnotes
- BER
- base excision repair
- CVD
- cardiovascular disease
- CoQ
- coenzyme Q
- CS
- citrate synthase
- ETC
- electron transport chain
- MnSOD
- manganese superoxide dismutase
- NEIL-1
- nei endonuclease VIII-like 1
- NOX
- nicotinamide adenine dinucleotide diphosphate oxidase
- RBC
- red blood cell
- ROS
- reactive oxygen species
- WBC
- white blood cell
- XO
- xanthine oxidase
REFERENCES
- 1. Barker D. J., Winter P. D., Osmond C., Margetts B., Simmons S. J. (1989) Weight in infancy and death from ischaemic heart disease. Lancet 2, 577–580 [DOI] [PubMed] [Google Scholar]
- 2. Rich-Edwards J. W., Stampfer M. J., Manson J. E., Rosner B., Hankinson S. E., Colditz G. A., Willets W. C., Hennekens C. H. (1997) Birth weight and risk of cardiovascular disease in a cohort of women followed up since 1976. BMJ 315, 396–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Eriksson J. G., Forsen T., Tuomilehto J., Winter P. D., Osmond C., Barker D. J. (1999) Catch-up growth in childhood and death from coronary heart disease: longitudinal study. BMJ 318, 427–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nobili V., Alisi A., Panera N., Agostini C. (2008) Low birth weight and catch-up growth associated with metabolic syndrome: a ten year systemic review. Pedriatr. Endocrinol. Rev 6, 241–247 [PubMed] [Google Scholar]
- 5. Huxley R. R., Shiell A. W., Law C. M. (2000) The role of size at birth and postnatal catch-up growth in determining systolic blood pressure: a systemic review of the literature. J. Hypertens. 18, 815–831 [DOI] [PubMed] [Google Scholar]
- 6. Corstius H. B., Zimanyi M. A., Maka N., Herath T., Thomas W., van der Laarse A., Wreford N. G., Black M. J. (2005). Effect of intrauterine growth restriction on the number of cardiomyocytes in rat hearts. Pediatr. Res 57, 796–800 [DOI] [PubMed] [Google Scholar]
- 7. Skilton M. R., Gosby A. K., Wu B. J., Ho L. M., Stocker R., Caterson I. D., Celermajer D. S. (2006) Maternal undernutrition reduces aortic wall thickness and elastin content in offspring of rats without altering endothelial function. Clin. Sci. (Lond.) 111, 281–287 [DOI] [PubMed] [Google Scholar]
- 8. Lim K., Zimanyi M. A., Black M. J. (2006) Effect of maternal protein restriction on rats on cardiac fibrosis and capilliarization in adulthood. Pediatr. Res. 60, 83–87 [DOI] [PubMed] [Google Scholar]
- 9. Tarry-Adkins J. L., Martin-Gronert M. S., Fernandez-Twinn D. S., Hargreaves I., Alfaradhi M. Z., Land J. M., Aiken C. E., Ozanne S. E. (2013) Poor maternal nutrition followed by accelerated postnatal growth leads to alterations in DNA damage and repair, oxidative and nitrosative stress and antioxidative defense capacity. FASEB J. 27, 379–390 [DOI] [PubMed] [Google Scholar]
- 10. Richter T., von Zglinicki T. (2007) A continuous correlation between oxidative stress and telomere length in fibroblasts. Exp. Gerontol. 11, 1039–1042 [DOI] [PubMed] [Google Scholar]
- 11. von Zglinicki T. Oxidative stress shortens telomeres. (2002) Trends Biochem. Sci. 7, 339–344 [DOI] [PubMed] [Google Scholar]
- 12. Heidinger B. J., Blount D. J., Boner W., Griffiths K., Metcalfe N. B., Monaghan P. (2012) Telomere length in early life predicts lifespan. Proc. Natl. Acad. Sci. U. S. A. 109, 1743–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Haussmann M. F., Winkler D. W., O'Reilly K. M., Huntington C. E., Nisbet I. C., Vleck C. M. (2003) Telomeres shorten more slowly in long-lived birds and mammals than in short-lived ones. Proc. Biol. Soc. 270, 1387–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Minamino T., Komuro I. (2008) Role of vascular senescence. Front. Biosci. 1, 2971–2979 [DOI] [PubMed] [Google Scholar]
- 15. Okuda K., Khan M. Y., Skurnik J., Kimura A., Aviv H., Aviv A. (2000) Telomere attrition of the human abdominal aorta: relationships with age and atherosclerosis. Atherosclerosis 152, 391–398 [DOI] [PubMed] [Google Scholar]
- 16. Aviv H., Khan M. Y., Skurnick J., Kimura M., Gardner J., Priolo L., Aviv A. (2001) Age dependent aneuploidy and telomere length of the human vascular endothelium. Atherosclerosis 159 281–287 [DOI] [PubMed] [Google Scholar]
- 17. Jennings B. J., Ozanne S. E., Dorling M., Hales C. N. (1999) Early growth determines longevity in male rats and may be related to telomere shortening in the kidney. FEBS Lett. 448, 4–8 [DOI] [PubMed] [Google Scholar]
- 18. Tarry-Adkins J. L., Martin-Gronert M. S., Chen J. H., Cripps R. L., Ozanne S. E. (2008) Maternal diet influences DNA damage, aortic telomere length, oxidative stress and antioxidant defense capacity in rats. FASEB J. 22, 2037–2044 [DOI] [PubMed] [Google Scholar]
- 19. Tarry-Adkins J. L., Chen J. H., Smith N. S., Jones R. H., Cherif H., Ozanne S. E. (2009) Poor maternal nutrition followed by accelerated postnatal growth leads to telomere shortening and increased markers of cell senescence in rat islets. FASEB J. 23, 1521–1528 [DOI] [PubMed] [Google Scholar]
- 20. Tarry-Adkins J. L., Blackmore H. L., Martin-Gronert M. S., Fernandez-Twinn D. S., McConnell J. M., Hargreaves I. P., Giussani D. A., Ozanne S. E. (2013) Coenzyme Q prevents accelerated cardiac aging in a rat model of poor maternal nutrition and accelerated postnatal growth. Mol. Metab. 2, 480–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shelley P., Tarry-Adkins J., Martin-Gronert M., Poston L., Heales S., Clark J., Ozanne S., McConnell J. (2007) Rapid neonatal weight gain in rats results in a renal ubiquinone (CoQ) deficiency associated with premature death. Mech. Ageing Dev. 128, 681–688 [DOI] [PubMed] [Google Scholar]
- 22. Turunen M., Olssen J., Dallner J. G. (2004) Metabolism and function of coenzyme Q. Biochim. Biophys. Acta 1660, 171–184 [DOI] [PubMed] [Google Scholar]
- 23. Snoeck A., Remacle C., Reusens B., Hoett J. J. (1990) Effect of low protein diet during pregnancy on the fetal rat endocrine pancreas. Biol. Neonate 57, 107–118 [DOI] [PubMed] [Google Scholar]
- 24. Santoz-Gonzalez M., Gomez-Diaz C., Nava P., Villalba J. M. (2007) Modifications of plasma proteome in long-lived rats fed on a coenzyme Q10-supplemented diet. Exp. Gerontol. 42, 798–806 [DOI] [PubMed] [Google Scholar]
- 25. Bello R. I., Gomez-Diaz C., Buron M. I., Alcain F. J., Navas P., Villalba J. M. (2005). Enhanced antioxidant protection of liver membranes in long lived rats fed on a coenzyme Q10-supplemented diet. Exp. Gerontol. 40, 694–706 [DOI] [PubMed] [Google Scholar]
- 26. Lonnrot K., Holm P., Lagerstedt A., Huhtala H., Alho H. (1998) The effects of lifelong ubiquinone supplementation on the Q9 and Q10 tissue concentrations and life span of male rats and mice. Biochem. Mol. Biol. Int. 44, 727–737 [DOI] [PubMed] [Google Scholar]
- 27. Quiles J. L., Ochoa J. J., Huertas J. R., Mataix J. (2004) Coenzyme Q supplementation protects from age-related DNA double-strand breaks and increases lifespan in rats fed on a PUFA-rich diet. Exp. Gerontol. 39, 189–194 [DOI] [PubMed] [Google Scholar]
- 28. Hales C. N., Desai M., Ozanne S. E., Crowther N. J. (1996) Fishing in the stream of diabetes: from measuring insulin to the control of fetal organogenesis. Biochem. Soc. Trans. 24, 341–350 [DOI] [PubMed] [Google Scholar]
- 29. Tarry-Adkins J. L., Ozanne S. E. (2014) The impact of early nutrition on the ageing trajectory. Proc. Nutr. Soc. 73, 289–301 [DOI] [PubMed] [Google Scholar]
- 30. Sugamura K., Keaney J. F. (2011) Reactive oxygen species in cardiovascular disease. Free Radic. Biol. Med. 51, 978–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Oikawa S., Kawanishi S. (1999) Site-specific DNA damage at GGG sequence by oxidative stress may accelerate telomere shortening. FEBS Lett. 453, 365–368 [DOI] [PubMed] [Google Scholar]
- 32. Parsons J. L., Dianov G. L. (2013) Co-ordination of base excision repair and genome instability. DNA Damage (Amst.) 12, 326–333 [DOI] [PubMed] [Google Scholar]
- 33. Wenzel P., Schuhmacher S., Kienhofer J., Muller J., Hortman M., Oleze M., Schluz E., Treiber N., Kawamoto T., Scharffetter-Kochanek K., Munzel T., Burkle A., Bachschmid M. M., Diaber A. A. (2008) Manganese superoxide dismutase and aldehyde dehydrogenase deficiency increase mitochondrial oxidative stress and aggravate age-dependent vascular dysfunction. Cardiovasc. Res. 80, 280–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Asimakis G. K., Lick S., Patterson C. (2002) Postischemic recovery of contractile function is impaired in SOD2(-/-) but not SOD1(-/-) mouse hearts. Circulation 105, 981–986 [DOI] [PubMed] [Google Scholar]
- 35. Chen Z. Y., Siu B., Ho Y. S., Vincent R., Chua C. C., Hamdy R. C., Chua B. H. (1998) Overexpression of MnSOD protects against myocardial ischemia/reperfusion injury in transgenic mice. J. Mol. Cell. Cardiol. 30, 2281–2289 [DOI] [PubMed] [Google Scholar]
- 36. Sindhu R. K., Robert C. K., Ehdaie A., Zhan C. D., Vazari N. D. (2005) Effects of aortic coarctation on aortic antioxidant enzymes and NAPDH oxidase protein expression. Life Sci. 76, 945–953 [DOI] [PubMed] [Google Scholar]
- 37. Saretzki G., Murphy M. P., von Zglinicki T. (2003) MitoQ counteracts telomere length and elongates lifespan of fibroblasts. Aging Cell 2, 141–143 [DOI] [PubMed] [Google Scholar]
- 38. Molyneux S. L., Florkowski C. M., Richards A. M., Lever M., Young J. M., George P. M. (2009) Coenzyme Q10; an adjuctive therapy for for congestive heart failure? N. Z. Med. J. 122, 74–79 [PubMed] [Google Scholar]
- 39. Fotino A,D., Thompson-Paul A. M., Bazzano L. A. (2013) Coenzyme Q supplementation on heart failure: a meta-analysis. Am. J. Clin. Nutr. 97, 268–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sander S., Coleman C. I., Patel A. A., Kluger J., White C. M. (2006) The impact of coenzyme Q10 on systolic function in patients with chronic heart failure. J. Card. Fail. 12, 464–472 [DOI] [PubMed] [Google Scholar]
- 41. Hidaka T., Fuji K., Funahashi I., Fukutomi N., Hoseo K. (2008) Safety assessment of coenzyme Q10 (CoQ10). Biofactors 32, 199–208 [DOI] [PubMed] [Google Scholar]
- 42. Ikematsu H., Nakamura K., Harashima S., Fujii K., Fukutomi N. (2006) Safety assessment of coenzyme Q10 (Kaneka Q10) in healthy subjects: A double-blind, randomized placebo-controlled trial. Regul. Toxicol. Pharmacol. 44, 212–218 [DOI] [PubMed] [Google Scholar]
- 43. World Health Organization. (2011) Global Status Report on Noncommunicable Diseases 2010, World Health Organization, Geneva, Switzerland [Google Scholar]