

Abstract
Superoxide dismutases (SODs, EC 1.15.1.1) are ubiquitous enzymes that efficiently catalyze the dismutation of superoxide radical anions to protect biological molecules from oxidative damage. The crystal structure of nickel-containing SOD (NiSOD) from Streptomyces seoulensis was determined for the resting, x-ray-reduced, and thiosulfate-reduced enzyme state. NiSOD is a homohexamer consisting of four-helix-bundle subunits. The catalytic center resides in the N-terminal active-site loop, where a Ni(III) ion is coordinated by the amino group of His-1, the amide group of Cys-2, two thiolate groups of Cys-2 and Cys-6, and the imidazolate of His-1 as axial ligand that is lost in the chemically reduced state as well as after x-ray-induced reduction. This structure represents a third class of SODs concerning the catalytic metal species, subunit structure, and oligomeric organization. It adds a member to the small number of Ni-metalloenzymes and contributes with its Ni(III) active site to the general understanding of Ni-related biochemistry. NiSOD is shown to occur also in bacteria other than Streptomyces and is predicted to be present in some cyanobacteria.
Oxygen metabolizing organisms have to face the toxicity of superoxide radicals (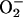 ) that are generated by a single electron transfer to dioxygen. Superoxide dismutases (SODs) are dedicated to keep the concentration of
) that are generated by a single electron transfer to dioxygen. Superoxide dismutases (SODs) are dedicated to keep the concentration of 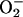 in controlled low limits, thus protecting biological molecules from oxidative damage (1). SODs are generally classified according to the metal species which acts as redox-active center to catalyze the dismutation reaction
in controlled low limits, thus protecting biological molecules from oxidative damage (1). SODs are generally classified according to the metal species which acts as redox-active center to catalyze the dismutation reaction 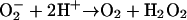 .
.
Until recently, three metal species have been found (2): manganese or iron in manganese- or iron-containing SODs (MnSOD and FeSOD), and copper as the catalytically active metal in copper- and zinc-containing SODs (Cu,ZnSOD), the latter deriving from a distinct and independent evolutionary line. Cu,ZnSOD is found in all eukaryotic species and is also widely distributed in prokaryotes (3). MnSOD is present in many bacteria, mitochondria, and chloroplasts, as well as in the cytosol of eukaryotic cells (4). FeSOD is found in bacteria and several higher plants (5, 6). Regarding the tertiary structure, SODs known so far are grouped into two structural organizations: a flattened eight-stranded Greek-key β-barrel adopted by Cu,ZnSODs and a two-domain organization comprising mainly α-helices typical for both Mn- and FeSODs.
Recently, a SOD with only nickel in the active site (NiSOD) was purified from several aerobic soil bacteria of the Streptomyces species (7–9). All clinical and soil isolates of Streptomyces reported to date possess this cytoplasmic NiSOD, and for some strains the additional presence of an Fe(Zn)SOD was reported (7, 10, 11). NiSOD is distinct from the Mn-, Fe-, or Cu,ZnSODs on the basis of amino acid sequence, immunological crossreactivity, and spectroscopic properties. The gene for NiSOD (sodN) shows no apparent sequence similarity to other SODs nor to other known proteins. The sodN genes cloned from Streptomyces seoulensis and Streptomyces coelicolor share 92% amino acid sequence homology, making NiSOD another class of SODs (7–9, 12, 13). Despite the dissimilarity of NiSOD to known SODs, the catalytic rate constant of NiSOD at ≈109 M–1·s–1 per metal center (12) is on the same high level of Cu,ZnSODs. Production of active NiSOD requires N-terminal proteolytic processing and accessory proteins as concluded from low efficiency of overexpression in Escherichia coli (14). Ni(II)-ions play a regulatory role in NiSOD gene transcription and in posttranslational processing, thus combining Ni(II) availability with the amount of active NiSOD (14).
There are nine nickel enzymes known to date (15): urease, NiFe-hydrogenase, CO-dehydrogenase, CO dehydrogenase/acetyl-CoA synthase, methyl-coenzyme M reductase, glyoxalase I, aci-reductone dioxygenase, NiSOD, and methylenediurease. Except for the latter two, the enzyme structures were determined to molecular level revealing distinct metallocenter environments.
We present the crystal structure of NiSOD from S. seoulensis (16) refined in two crystal forms (“small-cell” and “big-cell”) to the resolution of 1.68 Å and 1.6 Å, respectively. Crystal structures of the x-ray- and thiosulfate-reduced enzyme are also presented here. In addition, we address the occurrence of NiSODs in the bacterial realm.
Materials and Methods
Expression and Mutagenesis of NiSOD. Native NiSOD was expressed as described (13). The QuikChange site-directed mutagenesis kit (Stratagene) was used for the switching of amino acids. The pGEMsodN (pGEM-T easy vector containing the sodN coding region and the promoter region of sodN) was used as template and the PCR-amplified DNA containing the desired mutations was transformed into E. coli ET12567. Nonmethylated plasmid DNA was prepared and sequence fidelity was checked by DNA sequencing. SacI and SphI fragments containing mutated sodN locus were ligated with Streptomyces expression vector pIJ702 and the resulting ligate was transformed into protoplasts of Streptomyces lividans TK24, S. coelicolor A3 (2), or S. lividans TK24 ΔsodN (strain with nonfunctional sodN gene derived from S. lividans TK24).
Protein Purification and Crystallization. NiSOD was purified as described (13). NiSOD crystals were grown as described (17) from 1.85 M ammonium sulfate, 0.1 M sodium acetate, pH 5.25, and 10% glycerol (small-cell crystal form) and from 2 M ammonium sulfate and 5% 2-propanol (big-cell crystal form), respectively. Crystals of reduced NiSOD in the big-cell crystal form were obtained by soaking in 2 M ammonium sulfate, 5% 2-propanol, and 100 mM sodium thiosulfate for 24 h before data collection, loosing almost completely their characteristic yellow-brown color. In an x-ray fluorescence experiment on reduced NiSOD crystals, the Ni K-edge energy was shifted by 0.8 eV to lower energy with respect to the resting form, in agreement with the formal transition of Ni(III) to Ni(II).
EPR Spectroscopy. Continuous-wave EPR spectra were recorded at 100 K with a Bruker EMX X-band spectrometer equipped with a BVT 3000 cryostat. Simulations and data processing were performed by using a winepr program from Bruker (Rheinstetten, Germany).
Sedimentation Equilibrium by Analytical Ultracentrifugation. Analytical ultracentrifugation experiments were performed in a Beckman Optima XL-1. Sedimentation equilibrium experiments were carried out at 293 K with a rotor speed of 10,000 rpm on a sample volume of 100 μl. Absorbance data were collected at 280 and 380 nm. The partial specific volume of NiSOD was calculated as 0.738 ml/g. Data were analyzed by nonlinear, least-squares analysis, assuming a single thermodynamic component.
X-Ray Data Collection and Processing. Multiple-wavelength anomalous dispersion data at the Ni K-edge and high-resolution diffraction data (high-dose data) were collected for both crystal forms. Subsequently, data of the thiosulfate-reduced enzyme in the big-cell form were collected. Statistics of data processing (performed with mosflm/scala; ref. 18) and phasing were reported in detail (17) and are summarized in Table 3, and Data Sets 1–5, which are published as supporting information on the PNAS web site.
Phasing and Refinement. Ni positions were located by using multiple-wavelength anomalous dispersion data of the small-cell crystal form (17), followed by calculation of initial phases with cns (19). A total of 660 of 702 residues were modeled into the electron density map by arp/warp (20), and the remaining residues were modeled with the program o (21). Positional and isotropic B factor refinement was performed by using cns, treating the hexamer's subunits independently, and monitored by geometry quality and by the Rfree (Table 3). The resulting model was then placed twice in the asymmetric unit of the big-cell crystal form and refined as described above. The structure of the big-cell form served as starting model for refinement of the thiosulfate-reduced NiSOD employing the same protocol. Throughout all calculations, Ni to ligand distances were left unrestrained. A three-wavelengths multiple-wavelength anomalous dispersion experiment (17) on the big-cell form (480 frames per data set) was used to investigate x-ray-induced reduction of resting NiSOD. The first 320 frames (80° oscillation angle) of the initial data set collected at the Ni K-edge (low dose) and all data collected last at the remote wavelength (intermediate dose) were used in independent refinements employing the high-dose crystal structure as starting model. Conventional and simulated annealing maps computed with models from which His-1 and Ni were omitted, were inspected. For the low-dose structure, His-1 was modeled into the difference density map and refined with refmac5 (22), applying a Nδ-to-Ni ion target distance of 2.1 Å (23). The resulting Ni–Nδ distance was not supported by 2 Fo – Fc electron density, hence His-1 Nδ was allowed to drift back into density by releasing the restraint. For the intermediate-dose structure, the imidazolate was fitted into difference density without applying restraints on the Nδ–Ni distance.
Accessibility and Electrostatics Analysis. Solvent accessibility of active-site atoms was calculated with naccess (24) by using a probe radius of 1.25 Å. Solvent-accessible surfaces and electrostatic potential properties were calculated by using grasp (25).
Results and Discussion
Overall Structure. The oligomeric state of NiSOD was reported (7–9) as a homotetramer composed of 13.4-kDa subunits, as concluded by gel filtration experiments. Analytical ultracentrifugation (Fig. 5, which is published as supporting information on the PNAS web site) coupled with MS, however, reveals that the enzyme is a homohexamer in solution. The hexamer exhibits a globular shape in which all protein atoms lie in a hollow sphere with outer diameter of 72 Å and inner diameter of 23 Å (Fig. 1A). The interior of the hexamer is solvent-accessible through three narrow channels and contains water molecules and sulfate ions from the crystallization liquor, indicating the exchange of solvent molecules between the enzyme's interior and exterior, as well as inherent structural flexibility between couples of subunits that form the channels. Ni ions are arranged at vertices of a distorted octahedron with Ni–Ni distances ranging from 23.4 to 27.7 Å. Subunit interactions are mainly of hydrophobic character with two-thirds of interface residues being apolar. The subunits are furthermore held together by polar interactions of their N-terminal active-site loop (see below) and α-helices as well as between α-helices. Approximately 35% of the subunit surface is buried in interfaces to four neighboring subunits.
Fig. 1.

Overall structure of NiSOD. (A) The solvent-accessible surface of NiSOD viewed along the hexamer's threefold symmetry axis. The outer surface (black mesh) is sliced to allow the view to the inner solvent-filled space (in orange) and the protein backbone trace (chain A, yellow; chain B, blue; chain C, red; chain D, magenta; chain E, cyan; chain F, green; Ni ions, salmon-colored spheres). Arrows indicate the three twofold symmetry axes and the entrance to channels that render the inner space accessible to solvent molecules. (B) Ribbon representation of a NiSOD subunit. The N-terminal loop hosting the Ni ion protrudes from the body of the four-helix bundle. Residues involved in aromatic stacking are shown as a ball-and-stick representation. (C) Residues linking the active-site loop (subunit A) to neighboring chains by polar interactions. His-1 Nε takes part in a hydrogen-bonding triangle with Glu-17 and Arg-47 of subunit C. Main-chain oxygen atoms of Asp-3 and Leu-4 hydrogen bond to the side chain of Arg-39 in subunit C. The side-chain oxygen atoms of Asp-3 hydrogen bond to the side chains of Lys-52, Ser-86, and Lys-89 of subunit F.
The mature NiSOD molecule comprises 117 residues and adopts a four-helix bundle in the all-antiparallel topology (Fig. 1B). The subunit's hydrophobic core consists of 17 aliphatic residues with an exception of Phe-63. This residue participates, together with Tyr-9, Tyr-62, Phe-111, and Trp-112, to aromatic stacking at the N-terminal side of the four-helix bundle, connecting the beginning of the first helix with the ends of the second and the C-terminal helices. A short α-helical loop from Lys-65 to Tyr-70 connects the second and third helix. The active site is hosted in a loop formed by eight consecutive N-terminal residues of which Pro-5 in cis-conformation is critical for its conformation. The requirement for proteolytic cleavage of 14 N-terminal amino acids to produce active NiSOD (14) finds a plausible structural explanation: these residues would either clash into the neighboring subunits during hexamer assembly or prevent the formation of the Ni coordination.
The four-helix body of a subunit is not involved in stabilizing the respective N-terminal loop; instead, the N-terminal activesite loop of e.g., subunit A interacts by means of hydrogen bonds with two neighboring subunits, here C and F (Fig. 1C). Mutagenesis studies on residues His-1, Asp-3, Glu-17, Arg-39, and Arg-47 (Table 1) are in line with the observation that the interactions between the subunits, rather than within subunits are crucial for stabilization of the active site, and hence for enzyme activity.
Table 1. Activities of NiSOD mutants expressed in S. lividans TK24 ΔsodN.
| Mutated residue | Mutated to | Activity in cell extracts,* % |
|---|---|---|
| Wild type† | 100 | |
| His-1 | Ala, Cys, Asp, Lys, Asn, Gln, Arg, Trp, and Tyr | ND |
| Asp-3 | Ala | 23 |
| Tyr-9 | Ala | ND |
| Phe | 78 | |
| Lys | ND | |
| Gln | ND | |
| Trp | 45 | |
| Glu-17 | Ala | ND |
| Arg-39 | Ala | ND |
| Arg-47 | Ala | 89 |
ND, not detected (<5%).
SOD activity was measured by standard cytochrome c assay. Activity of S. lividans TK24 ΔsodN harboring pIJ702 was subtracted from total SOD activity in cell extract.
S. lividans TK24 ΔsodN harboring pIJsodN (pIJ702 vector containing the sodN coding region and the promoter region of sodN gene from S. seoulensis).
Ni Coordination. The evidence for a Ni ion in the active site came first from EPR spectra (7). A subsequent 61Ni isotope substitution experiment (Fig. 6, which is published as supporting information on the PNAS web site) gave unequivocal identification of the rhombic EPR signal to Ni. Our previous x-ray absorption spectroscopy data (12) showed that the Ni in resting enzyme (as purified) is five- or six-coordinated, where one or two N or O donors are lost upon reduction with dithionite, resulting in a planar four-coordinated Ni site. Moreover, EPR spectra suggested the presence of an axial N-ligand. However, both our initial x-ray structure of resting NiSOD and of thiosulfate-reduced NiSOD showed square-planar Ni coordination by the amino group of His-1, the amide group of Cys-2, and the thiolate group of Cys-2 and of Cys-6 (Fig. 2 C and E). The side chain of His-1 does not coordinate the Ni ion but was rather involved in hydrogen bonds to the main-chain oxygen atom of Val-8 through Nδ and to Glu-17 of a neighboring subunit through Nε (Fig. 1C). The discrepancy between spectroscopic and crystallographic data prompted us to reexamine the Ni coordination and is explained by the effect of radiation on the oxidation state of Ni. It has been observed that radiation-induced changes of the metal center's oxidation state can take place during exposure to x-rays (26). The reduction of Ni(III) in the resting enzyme during x-ray data collection became evident from a difference Fourier omit map calculated with an initial fraction of data collected in a multiple-wavelength anomalous dispersion experiment (low-dose data set, see Materials and Methods). The map in Fig. 2 A shows electron density connecting the His-1 imidazolate through Nδ to the Ni ion, with an average distance of 2.63 Å and indicates a rotation around the Cβ–Cγ bond of the side chain (Fig. 2 A–D). This distance exceeds by ≈0.5 Å the commonly found bond length between Ni(III) and Nδ of imidazolates (23) and may indicate that reduction occurs already at low x-ray doses. Independent experimental confirmation of x-ray-induced reduction of Ni was obtained from density maps calculated with diffraction data assembled by merging reflections collected on 14 crystals, each contributing the initial 10 frames. The map confirms the liganding Nδ conformation and the above view of a fast onset of x-ray-induced reduction. Ni coordination by His-1 Nδ in the resting enzyme is, however, evident, and its disruption upon x-ray exposure becomes visible from maps calculated from an intermediate-dose data set collected on the same crystal (Fig. 2B): His-1 Nδ rotated ≈55° toward the carbonyl oxygen atom of Val-8. Fig. 2C shows the active site as initially observed for the high dose data set at 1.6-Å resolution, where His-1 Nδ rotated ≈60° with respect to the low-dose conformation. Based on the hydrogen-bonding pattern of the imidazolate nitrogen atoms, His-1 is believed to be double-protonated in the high-dose structure. The hydrogen-bonding pattern of His-1 Nε remains unaffected with respect to the low-dose crystal structure (Fig. 2D). In particular, Glu-17 may be regarded as an anchor for the imidazolate leaving the rotation about the Cβ–Cγ bond as the only degree of freedom in His-1 side-chain movements during catalysis.
Fig. 2.

σA-weighted 2 Fo – Fc electron density maps of the Ni ion environment. (A–C) Structures of subunit F captured at successively increasing x-ray doses. (A) The fifth ligand His-1 Nδ (2.5 Å distant to Ni here) is revealed at low x-ray exposure (map resolution 2.2 Å, 1.0 σ contour level). (B) After longer exposure of the same crystal as in A, the imidazolate ligation is disrupted. (C) Map at 1.6-Å resolution obtained from a different crystal as in A and B, applying a maximum total x-ray dose. The ligands show a distorted cis square-planar geometry, equaling the thiosulfate-reduced NiSOD. The average angle between planesdefined by N(His-1)-Ni-N(Cys-2) and S(Cys-2)-Ni-S(Cys-6) is 7.5°.(D) Superposition of models from A in green, B in magenta, and C in gray illustrates the His-1 imidazole rotation upon which Nδ reaches a distance of ≈2.9 Å to Val-8 O, thereby maintaining the hydrogen-bond triangle of His-1 Nε to Glu-17 Oε and Arg-47 Nη of the adjacent subunit. (E) Electron density map of thiosulfate-reduced NiSOD contoured at 1.1 σ showing the square-planar Ni(II) coordination. One thiosulfate-ion (S2O32–) per subunit is found 7–8 Å away from each metal center (subunit A). The precise bonding pattern of these ions varies among the 12 subunits in the asymmetric unit, indicating a high degree of disorder or low-binding specificity.
Sodium thiosulfate was used to mimic the reduction of the metal center on the first encounter with superoxide. The Ni(II) coordination as deduced from the 2.1-Å crystal structure of the thiosulfate-reduced NiSOD shows square-planar geometry (Fig. 2E), where His-1 is in the same conformation as observed in the 1.6-Å structure obtained after long exposure to X-rays (Fig. 2C). Other Ni to ligand distances are comparable with those observed in all other structures (Table 2).
Table 2. Ni ion bond lengths, Å.
| Bond | Low-dose big cell | Intermediate-dose big cell | High-dose big cell | High-dose small cell |
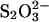 -reduced big cell -reduced big cell |
|---|---|---|---|---|---|
| Ni—His-1 Nδ | 2.63 | 3.69 | 3.87 | 3.96 | 3.81 |
| Ni—His-1 N | 2.11 | 2.12 | 2.06 | 2.07 | 2.17 |
| Ni—Cys-2 N | 1.93 | 1.94 | 1.93 | 1.94 | 1.99 |
| Ni—Cys-2 Sγ | 2.24 | 2.23 | 2.22 | 2.24 | 2.30 |
| Ni—Cys-6 Sγ | 2.26 | 2.25 | 2.20 | 2.18 | 2.29 |
Values are the average over the 12 and 6 independent subunits in the asymmetric unit of the big-cell and small-cell crystal forms, respectively.
Metal coordination by S ligands is a specific feature among known SODs; however, it is not uncommon in Ni-containing enzymes, especially for redox-active enzymes, including [NiFe]-hydrogenase (27), CO dehydrogenase (28), CO dehydrogenase/acetyl-CoA synthase (29, 30), and methyl coenzyme M reductase (31). Among Ni-containing proteins investigated so far, only redox-active enzymes feature thiolate ligation, which is found neither in the hydrolytically active urease nor in auxiliary proteins for Ni-related cellular tasks. Thiolate ligation was thus suggested to be required for the function of redox-active Ni centers at physiological redox potentials (12). The coordination of Ni by protein backbone nitrogen atoms as found in NiSOD has so far been observed very rarely in proteins. Recently, it was found in the active site (A-cluster) of a bifunctional CO dehydrogenase/acetyl-CoA synthase (29, 30). The acetyl-CoA synthase shows a Cys–Gly–Cys motif in which two Cys side chains and two backbone amide N atoms coordinate Ni(II) in square-planar geometry. This Ni ion is part of a [Fe4-S4]-Me-Ni cluster with still controversial nature of the bridging metal (Me) and is thought to be redox-inactive although its N2S2 ligand field is similar to that of redox-active Ni(II) in reduced NiSOD. The particular ligand field N3S2 (resting state) and N2S2 (reduced state) may be critical to NiSOD function because redox properties and the stability of the Ni(III) oxidation state depend on the type of ligands and the coordination geometry (32). The fact that O–2 can reduce and subsequently reoxidize the Ni center requires this center to possess a redox potential in between those of the couples (O–2/O2) and (O–2/H2O2), ≈0.3 V vs. normal hydrogen electrode. An important aspect of NiSOD's catalytic mechanism is the ability of the Ni(II/III) redox couple to reach this potential value by means of the Ni coordination described here.
Active-Site Environment. Access to the active site is first impeded by the side chains of Pro-5 and Tyr-9 and is then obstructed by a remarkable arrangement of the backbone nitrogen atoms from the active-site loop, forming in this way a small pocket on the enzyme's surface (Fig. 3). Calculation of solvent-accessible areas for active-site atoms reveals that the Ni ion and four of five ligands are essentially buried. A nonzero-accessible area was obtained solely for His-1 N, which amounts to only ≈1.1 Å2. This result suggests an outer sphere electron transfer between the superoxide and Ni ions. It should be noted, however, that the values for solvent-accessible areas of active-site atoms were derived from static structures and that structural flexibility of the enzyme upon substrate approach to the active site may render an inner sphere transfer possible (see ref. 33 for a discussion regarding Cu,ZnSOD). Both S ligands are protected from direct contact with substrate or product molecules, preserving the vulnerable thiolate ligands from oxidation, and thus reconciling the apparent implausibility of a Ni-thiolate complex as an agent of protection against oxidative stress. Channels leading to the hexamer's interior pass through the midpoint between two Ni sites (Fig. 1 A), but do not allow a direct connection between the solvent region and the metal centers (Fig. 3B).
Fig. 3.

Active-site accessibility. (A) Active-site environment from His-1 to Tyr-9 of the high-dose structure (Fig. 2C). Nitrogen atoms are labeled with the respective residue number. For Cys-2 to Gly-7, only backbone atoms are shown and oxygen atoms are omitted for clarity. Backbone nitrogen atoms from His-1, Asp-3, Cys-6, and Gly-7 separate the Ni ion and all other ligands (Cys-2 N, Cys-2 Sγ, and Cys-6 Sγ, right to the vertical line) from the solvent-accessible pocket (hosting two water molecules, left to the line). In all but the thiosulfate-reduced NiSOD structures, a water molecule (here W 828) is found close to the vacant axial position opposite the His-1 imidazole at ≈3.5 Å from the Ni ion, 3.3 Å from His-1 N, and hydrogen bonds to Cys-6 N. High temperature factors for this solvent molecule indicate elevated mobility. (B) Surface representation of thiosulfate-reduced NiSOD at the active-site loop and the innermost end of a channel that allows thiosulfate-ions to approach the Ni sites (only selected side chains are shown). The solvent-accessible pocket close to the Ni center is marked by an arrow and exhibits a bottleneck formed by Pro-5 and Tyr-9 ≈5 Å away from the Ni ion, conferring to NiSOD a selectivity for small molecules as substrate or inhibitors. A sulfate ion (not included in the surface calculation) from the crystallization liquor is found at the pocket's entrance in all described structures.
Electrostatic steering of substrate to the active site was addressed by calculating the electrostatic potential at the hexamer surface assuming all His side chains single-protonated (corresponding to physiological pH). The surface surrounding the active-site pocket does not show significant positively charged areas (Fig. 7, which is published as supporting information on the PNAS web site). The lack of a strong surface potential around the active site is in agreement with low ionic strength dependence of the catalytic rate constant for NiSOD (12), suggesting that the electrostatic steering of substrate to the active site is not essential for its fast catalytic rate, which is different than what has been observed for Cu,ZnSODs (3).
NiSOD is believed to act like other SODs according to the formal equations of the dismutation reaction,
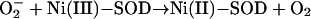 |
[1] |
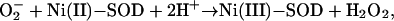 |
[2] |
where Ni(III)-SOD and Ni(II)-SOD represent the oxidized and reduced metal center. The formation of the product hydrogen peroxide requires two protons. Our crystal structures suggest some residues in the vicinity of the metal center as potential proton donors. Assuming that superoxide approaches the Ni(II) through the pocket, it may pick up one or both H+ from the main-chain nitrogens of the active-site loop (Fig. 3 A and B). At the entrance of the pocket, Tyr-9 Oη, Lys-64 Nζ, or water molecules of the protein's hydration shell are also available H+ donors. The existence of Tyr-9 in the vicinity of Ni is interesting because structurally unrelated Mn- and FeSODs also host a strictly conserved Tyr residue (Tyr-34 in E. coli) responsible for catalytic fine tuning (34). Mutations of Tyr-9 to aromatic residues reduces the enzyme activity, whereas substitutions with aliphatic residues abolish it (Table 1), suggesting that this residue is critical for structural stabilization by means of aromatic stacking as well as for fine tuning of catalysis.
Occurrence of NiSODs. NiSOD has to date been observed and characterized only in the Streptomyces genera (13). We find now that it is not confined to the Streptomyces genus but exists in several Actinomycetes, such as Micromonospora rosaria, Microtetraspora glauca, and Kitasatospora griseola. Their SODs show similar UV-visible and EPR spectra and almost perfect N-terminal sequence homology to S. seoulensis NiSOD (Fig. 8, which is published as supporting information on the PNAS web site), suggesting a common active-site structure. Moreover, by using blast (35), we detected four putative ORFs of unknown function from cyanobacteria, which show significant amino acid sequence homology to NiSOD from S. seoulensis (Fig. 4). The sequence alignment shows that these ORFs contain the His-1–Cys-2–Xaa–Xaa–Xaa–Cys-6 ligand motif (mature NiSOD numbering) and conserved residues (Asp-3, Tyr-9, and Arg-39), which are crucial for NiSOD activity. Secondary structure prediction of ORFs suggests the four-helix motif (Fig. 4), with perfect conservation of residues that stabilize the N terminus of the subunit by aromatic stacking interactions (Figs. 1B and 4). When mapping the per-residue sequence identity derived from the aligned ORFs onto the subunit structure of S. seoulensis NiSOD, it becomes evident that the N-terminal active-site loop and a number of residues forming the subunit interface are fully conserved. We therefore envisage that these ORFs are NiSODs in terms of structure and function, provided that the expression and posttranslational modification of N-terminal residues of these ORFs is feasible in cyanobacteria.
Fig. 4.

Sequence alignment of NiSOD with putative ORFs from cyanobacteria and secondary structure prediction. Sequences are given for S. seoulensis (S. seo), S. coelicolor (S. coe), Trichodesmium erythraeum IMS101 (T. ery), Synechococcus sp. WH 8102 (Synec), Prochlorococcus marinus subsp. Pastoris (P. ma1), and Prochlorococcus marinus str. MIT 9313 (P. ma2). Identical residues are marked with asterisks and similar residues are marked with colons or periods (strongly or weakly conserved groups). Predicted α-helices are highlighted in red, predicted β-sheets in cyan, and α-helices in S. seoulensis NiSOD (based on our structures) are shown as cylinders, colored as in Fig. 1B. Ni ligands in S. seoulensis NiSOD are highlighted in yellow, and residues involved in aromatic stacking at the N terminus are highlighted in magenta.
S. seoulensis NiSOD represents a third class of SODs, displays a distinct subunit fold, quaternary structure organization, and a distinct active-site structure with unprecedented Ni(III) as the catalytic metal. It adds another member to the small but growing number of Ni-metalloenzymes, offering the possibility to extend our knowledge of Ni-related biochemistry.
Supplementary Material
Acknowledgments
We thank G. Leonard and E. Mitchell of the European Synchrotron Radiation Facility (Grenoble, France) for excellent assistance with diffraction experiments and O. Carugo for helpful discussions and literature survey. This work was supported by a research grant from the Korea Science and Engineering Foundation and by a research fellowship of the BK21 project.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: SOD, superoxide dismutase.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 1Q0D, 1Q0F, 1Q0G, 1Q0K, and 1Q0M).
References
- 1.Halliwell, B. & Gutteridge, J. M. C. (1999) Free Radicals in Biology and Medicine (Oxford Univ. Press, New York).
- 2.Fridovich, I. (1995) Annu. Rev. Biochem. 64, 97–112. [DOI] [PubMed] [Google Scholar]
- 3.Bordo, D., Pesce, A., Bolognesi, M., Stroppolo, M. E., Falconi, M. & Desideri, A. (2001) in Handbook of Metalloproteins, eds. Messerschmidt, A., Huber, R., Poulos, T. & Wieghardt, K. (Wiley, Chichester, U.K.), pp. 1284–1300.
- 4.Stroupe, M. E., DiDonato, M. & Tainer, J. A. (2001) in Handbook of Metalloproteins, eds. Messerschmidt, A., Huber, R., Poulos, T. & Wieghardt, K. (Wiley, Chichester, U.K.), pp. 941–951.
- 5.Miller, A.-F. (2001) in Handbook of Metalloproteins, eds. Messerschmidt, A., Huber, R., Poulos, T. & Wieghardt, K. (Wiley, Chichester, U.K.), pp. 668–682.
- 6.Lynch, M. C. & Kuramitsu, H. K. (1999) Infect. Immun. 67, 3367–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Youn, H.-D., Youn, H., Lee, J.-W., Yim, Y.-I., Lee, J.-K., Hah, Y. C. & Kang, S.-O. (1996) Arch. Biochem. Biophys. 334, 341–348. [DOI] [PubMed] [Google Scholar]
- 8.Youn, H.-D., Kim, E.-J., Roe, J.-H., Hah, Y. C. & Kang, S.-O. (1996) Biochem. J. 318, 889–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim, E.-J., Kim, H.-P., Hah, Y. C. & Roe, J.-H. (1996) Eur. J. Biochem. 241, 178–185. [DOI] [PubMed] [Google Scholar]
- 10.Leclere, V., Boiron, P. & Blondeau, R. (1999) Curr. Microbiol. 39, 365–368. [DOI] [PubMed] [Google Scholar]
- 11.Folcher, M., Gaillard, H., Nguyen, L. T., Nguyen, K. T., Lacroix, P., Bamas-Jacques, N., Rinkel, M. & Thompson. C. J. (2001) J. Biol. Chem. 23, 44297–44306. [DOI] [PubMed] [Google Scholar]
- 12.Choudhury, S. B., Lee, J.-W., Davidson, G., Yim, Y.-I., Bose, K., Sharma, M. L., Kang, S.-O., Cabelli, D. E. & Maroney, M. J. (1999) Biochemistry 38, 3744–3752. [DOI] [PubMed] [Google Scholar]
- 13.Lee, J.-W., Roe, J.-H. & Kang, S.-O. (2002) Methods Enzymol. 349, 90–101. [DOI] [PubMed] [Google Scholar]
- 14.Kim, E.-J., Chung, H.-J., Suh, B., Hah, Y. C. & Roe, J.-H. (1998) Mol. Microbiol. 27, 187–195. [DOI] [PubMed] [Google Scholar]
- 15.Mulrooney, S. B. & Hausinger, R. P. (2003) FEMS Microbiol. Rev. 27, 239–261. [DOI] [PubMed] [Google Scholar]
- 16.Chun, J., Youn, H.-D., Yim, Y.-I., Lee, H., Kim, M.-Y., Hah, Y. C. & Kang, S.-O. (1997) Int. J. Syst. Bacteriol. 47, 492–498. [DOI] [PubMed] [Google Scholar]
- 17.Wuerges, J., Lee, J.-W., Kang, S.-O. & Djinovic Carugo, K. (2002) Acta Crystallogr. D 58, 1220–1223. [DOI] [PubMed] [Google Scholar]
- 18.Collaborative Computational Project 4 (1994) Acta Crystallogr. D 50, 760–763.15299374 [Google Scholar]
- 19.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905–921. [DOI] [PubMed] [Google Scholar]
- 20.Perrakis, A., Morris, R. & Lamzin, V. S. (1999) Nat. Struct. Biol. 6, 458–463. [DOI] [PubMed] [Google Scholar]
- 21.Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. (1991) Acta Crystallogr. A 47, 110–119. [DOI] [PubMed] [Google Scholar]
- 22.Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997) Acta Crystallogr. D 53, 240–255. [DOI] [PubMed] [Google Scholar]
- 23.Orpen, A. G., Brammer, L., Allen, F. H., Kennard, O., Watson, D. G. & Taylor, R. (1989) J. Chem. Soc. Dalton Trans. S1–S83.
- 24.Hubbard, S. J. & Thornton, J. M. (1993) naccess Computer Program (Dept. of Biochemistry and Molecular Biology, University College London, London).
- 25.Nicholls, A., Sharp, K. A. & Honig, B. (1991) Proteins 11, 281–296. [DOI] [PubMed] [Google Scholar]
- 26.Berglund, G. I., Carlsson, G. H., Smith, A. T., Szöke, H., Henriksen, A. & Hajdu, J. (2002) Nature 417, 463–468. [DOI] [PubMed] [Google Scholar]
- 27.Volbeda, A., Charon, M. H., Piras, C., Hatchikian, E. C., Frey, M. & Fontecilla-Camps, J. C. (1995) Nature 373, 580–587. [DOI] [PubMed] [Google Scholar]
- 28.Dobbek, H., Svetlitchnyi, V., Gremer, L., Huber, R. & Meyer, O. (2001) Science 293, 1281–1285. [DOI] [PubMed] [Google Scholar]
- 29.Doukov, T. I., Iverson, T. M., Seravalli, J., Ragsdale, S. W. & Drennan, C. L. (2002) Science 298, 567–572. [DOI] [PubMed] [Google Scholar]
- 30.Darnault, C., Volbeda, A., Kim, E. J., Legrand, P., Vernede, X., Lindahl, P. A. & Fontecilla-Camps, J. C. (2003) Nat. Struct. Biol. 10, 271–279. [DOI] [PubMed] [Google Scholar]
- 31.Ermler, U., Grabarse, W., Shima, S., Goubeaud, M. & Thauer, R. K. (1997) Science 278, 1457–1462. [DOI] [PubMed] [Google Scholar]
- 32.Hanss, J. & Krueger, H.-J. (1998) Angew. Chem. Int. Ed. Engl. 37, 360–363. [DOI] [PubMed] [Google Scholar]
- 33.Hart, P. J., Balbirnie, M. M., Ogihara, N. L., Nersissian, A. M., Weiss, M. S., Selverstone Valentine, J. & Eisenberg, D. (1999) Biochemistry 38, 2167–2178. [DOI] [PubMed] [Google Scholar]
- 34.Edwards, R. A., Whittaker, M. M., Whittaker, J. W., Baker, E. N. & Jameson, G. B. (2001) Biochemistry 40, 15–27. [DOI] [PubMed] [Google Scholar]
- 35.Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. (1990) J. Mol. Biol. 215, 403–410. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.