

Abstract
Background
The Syrian hamster (golden hamster, Mesocricetus auratus) is gaining importance as a new experimental animal model for multiple pathogens, including emerging zoonotic diseases such as Ebola. Nevertheless there are currently no publicly available transcriptome reference sequences or genome for this species.
Results
A cDNA library derived from mRNA and snRNA isolated and pooled from the brains, lungs, spleens, kidneys, livers, and hearts of three adult female Syrian hamsters was sequenced. Sequence reads were assembled into 62,482 contigs and 111,796 reads remained unassembled (singletons). This combined contig/singleton dataset, designated as the Syrian hamster transcriptome, represents a total of 60,117,204 nucleotides. Our Mesocricetus auratus Syrian hamster transcriptome mapped to 11,648 mouse transcripts representing 9,562 distinct genes, and mapped to a similar number of transcripts and genes in the rat. We identified 214 quasi-complete transcripts based on mouse annotations. Canonical pathways involved in a broad spectrum of fundamental biological processes were significantly represented in the library. The Syrian hamster transcriptome was aligned to the current release of the Chinese hamster ovary (CHO) cell transcriptome and genome to improve the genomic annotation of this species. Finally, our Syrian hamster transcriptome was aligned against 14 other rodents, primate and laurasiatheria species to gain insights about the genetic relatedness and placement of this species.
Conclusions
This Syrian hamster transcriptome dataset significantly improves our knowledge of the Syrian hamster's transcriptome, especially towards its future use in infectious disease research. Moreover, this library is an important resource for the wider scientific community to help improve genome annotation of the Syrian hamster and other closely related species. Furthermore, these data provide the basis for development of expression microarrays that can be used in functional genomics studies.
Introduction
The Syrian hamster (golden hamster, Mesocricetus auratus) has recently been used as an experimental rodent model for important infectious diseases including Ebola and other viral hemorrhagic fevers [1]–[8]. For instance, Syrian hamsters infected with mouse-adapted Ebola virus (EBOV) manifest many of the clinical and pathological findings observed in EBOV-infected non-human primates (NHPs) and humans, including systemic viral replication, suppression of the innate immune response, an uncontrolled inflammatory response, and disseminated intravascular coagulation syndrome [9]. The Syrian hamster is emerging as a promising model for leishmaniasis [10] and dyslipidaemia research [11], [12]. The Syrian hamster is also an important animal model in neurosciences research [13], [14]. For instance, this species has been widely used in the studies of circadian rhythms [15], cardiomyopathy [16], aggression [17], reproduction [18], and sensory systems [19].
Genotyping of Mesocricetus auratus is currently under way at the Broad Institute (NCBI-BioProject accession: PRJNA77669) but not yet published. So far, only 860 cDNA sequences from the Syrian hamster are available in the NCBI-dbEST database [20], where 728 sequences have been collected in the context of testis organs [21] and 125 sequences have been collected in the context of embryonic cells [22]. More recently, while Schmucki et al. analyzed the liver transcriptome of the Syrian hamster with a focus on lipid metabolism [23] the data is not publicly available as of this writing.
Drafts of the genome and transcriptome of Chinese hamster ovary (CHO) cells have recently been published [24], [25], although it should be noted that CHO cells represent cells in an immortalized condition and therefore will likely contain genetic mutations not present in natural conditions. The current release of the CHO cell draft genome is composed of 109,152 scaffolds and 265,786 contigs representing a total length of 2,318,115,958 nucleotides. Preliminary gene annotation of the CHO cell genome was performed using vertebrate experimental data and cross-species comparisons. The current release of the CHO cell transcriptome comprises 121,636 transcript fragments representing a total length of 179,731,611 nucleotides. More recently, Lewis et al. compared the genome of CHO cells and the genome of the Chinese hamster obtained from tissues, and they showed a significant proximity between these different conditions [26]. Further efforts will be continued regarding the update of the CHO and Chinese hamster genomes and transcriptomes.
The aims of our study were: (i) to provide to the scientific community a large panel of annotated mRNA sequences from the Mesocricetus auratus transcriptome; (ii) to provide new biological insights and knowledge about the Mesocricetus auratus species; and (iii) to use this data to allow the design of a future gene expression microarray. Here we sequenced a normalized 3′ mRNA fragment primed cDNA library produced from pooled RNA isolated from the major organs of adult female Syrian hamsters following strategies in common-use described elsewhere [27], [28]. We reasoned that pooling a large variety of different organs of animals will provide a large pool of mRNA fragments to sequence and annotate. Sequencing reads were de novo assembled into contigs. The combined contig and unassembled read (singleton) dataset, designated as the Syrian hamster transcriptome, was annotated based on the mouse and rat transcriptomes. We identified the most highly covered and the most highly expressed transcripts in our Syrian hamster transcriptome and performed a functional enrichment analysis to identify which canonical pathways and biological functions were most significantly represented. In order to contribute to the annotation efforts of the Chinese hamster species, we aligned our Syrian hamster transcriptome to the current version of the CHO cell genome and transcriptome. Finally, we aligned our Syrian hamster transcriptome to 14 other primate species and analyzed the genomic divergence of our transcripts in order to gain insights into the genomic evolution of the Syrian hamster.
Results
Sample collection and sequencing of a cDNA library produced from female Syrian hamster organs
The brains, lungs, spleens, kidneys, livers, and hearts were collected from three adult female Syrian hamsters. Total RNAs were isolated, pooled, and contaminating genomic DNA removed. Following adaptor ligation, cDNAs were 3′ fragment-sequenced on a Roche 454 GS FLX Titanium instrument. The sequencing generated 1,283,840 reads with an average length of 344 bases. Reads were trimmed for quality and reads shorter than 40 bases were discarded, resulting in 1,212,395 sequence reads available for further assembly and analysis. Figure 1A shows the length distribution of reads before assembly. Consistent with most of the publicly available transcriptome libraries [29], we observed that our reads ranged between 200 and 600 nucleotides in length.
Figure 1. Histograms showing the length distribution of the reads and the length distribution of the singletons and contigs.

(A) The length distribution of the reads is shown in a gray histogram. Bins of the histogram have been set to 50 nucleotides. The lengths of the reads range from 40 to 631, with a median length of 387 and a mean length of 352. The reads represents a total of 426,683,712 nucleotides bases. (B) The length distribution of the 111,796 singletons is shown in a red histogram while the length distribution of the 62,482 contigs is shown in a blue histogram. Bins of the histograms have been set to 25 nucleotides. The lengths of the singleton sequences range from 50 to 614, with a median length of 187 and a mean length of 265. The lengths of the contig sequences range from 50 to 4,054, with a median length of 473 and a mean length of 487. Our Syrian hamster transcriptome represents a total of 60,117,204 nucleotides bases.
Library assembly
Quality-filtered reads were assembled into contigs. Resulting contigs and unassembled reads (singletons) were quality filtered and contigs or singletons shorter than 50 bases were discarded. Among the 1,212,395 reads, 62,482 contigs and 111,796 singletons were generated. Figure 1B shows the length distributions of the 174,278 combined contig/singleton dataset. The lengths of the singletons ranged from 50 to 614, with a median length of 187.50 bases. The lengths of the contigs ranged from 50 to 4,054, with a median length of 473.50 bases. We observed that most of the reads ranging between 75 and 400 nucleotides were assembled. Short reads are subject to noise and have low quality scores, making them more difficult to assemble. On the other hand, larger reads are difficult to assemble in this context because our library was targeted against 3′ mRNA priming. The final dataset (contigs plus singletons) represents a total of 60,117,204 nucleotides and is designated as the Syrian hamster transcriptome.
Library annotation
The Syrian hamster transcriptome was aligned to the mouse and rat transcriptome references ( Table 1 ). Amongst the 174,278 contigs and singletons, 41,651 (23.90%) were significantly aligned (expected value cutoff of 10) to the mouse transcriptome and 26,258 (15.07%) were significantly aligned to the rat transcriptome. Of these, 11,648 transcripts (representing 9,562 genes) contained functional annotation in the mouse transcriptome, and 7,223 transcripts (representing 7,137 genes) were functionally annotated in the rat transcriptome ( Table 1 ). Therefore, 11,648 Syrian sequence fragments or transcripts are now annotated by way of homology with the mouse genome.
Table 1. Transcriptome references and alignment statistics.
| Species name | # of genes | # of transcripts | # and % of alignments | # and % of mapped genes | # and % of mapped transcripts |
| Mouse (Mus musculus) | 38,293 | 92,484 | 41,651 (23.90%) | 9,562 (22.96%) | 11,648 (12.59%) |
| Rat (Rattus norvegicus) | 26,405 | 29,189 | 26,258 (15.07%) | 7,137 (27.18%) | 7,223 (24.75%) |
| Chinese Hamster Ovary cells (Cricetulus griseus) | NA | 121,636* | 7,845 (4.50%) | NA | 4,390 (3.61%) |
| Chimpanzee (Pan troglodytes) | 28,012 | 29,160 | 2,884 (1.65%) | 1,631 (56.55%) | 1,643 (5.63%) |
| Ferret (Mustela putorius furo) | 23,811 | 23,963 | 16,169 (9.28%) | 4,169 (25.78%) | 4,187 (17.47%) |
| Gorilla (Gorilla gorilla gorilla) | 29,216 | 35,727 | 8,319 (4.77%) | 2,733 (32.85%) | 2,735 (7.66%) |
| Guinea pig (Cavia porcellus) | 25,028 | 26,129 | 15,014 (8.61%) | 4,050 (26.97%) | 4,155 (15.90%) |
| Human (Homo sapiens) | 62,316 | 213,551 | 23,020 (13.21%) | 5,409 (23.50%) | 7,254 (3.40%) |
| Kangaroo rat (Dipodomys ordii) | 26,405 | 29,189 | 2,103 (1.21%) | 1,252 (59.53%) | 1,252 (4.29%) |
| Macaque (Macaca mulatta) | 30,246 | 44,725 | 13,792 (7.91%) | 3,804 (27.58%) | 4,163 (9.31%) |
| Orangutan (Pongo abelii) | 28,443 | 29,447 | 15,331 (8.80%) | 3,929 (25.63%) | 3,952 (13.42%) |
| Pig (Sus scrofa) | 25,322 | 30,586 | 10,910 (6.26%) | 2,978 (27.30%) | 3,051 (9.98%) |
| Pika (Ochotona princeps) | 23,028 | 23,028 | 1,575 (0.90%) | 989 (62.79%) | 989 (4.29%) |
| Rabbit (Oryctolagus cuniculus) | 23,394 | 28,188 | 4,344 (2.49%) | 1,946 (44.80%) | 2,007 (7.12%) |
| Shrew (Sorex araneus) | 19,134 | 19,139 | 1,330 (0.76%) | 759 (57.07%) | 759 (3.97%) |
| Squirrel (Ictidomys tridecemlineatus) | 22,398 | 23,572 | 7,730 (4.44%) | 2,723 (35.23%) | 2,733 (11.59%) |
| Tree Shrew (Tupaia belangeri) | 20,820 | 20,824 | 1,786 (1.02%) | 1,091 (61.09%) | 1,091 (5.24%) |
For each transcriptome reference used in this study, the name of the species, the number of genes available, and the number of transcripts available are indicated.
*The number of available transcripts indicated for the Chinese hamster ovary cells represents the number of available transcript fragments available and not the number of distinct transcripts. Moreover, for each transcriptome reference used in this study, the number of aligned contigs and singletons, the number of mapped transcripts and the number of mapped genes are indicated. The percentages of mapped transcripts and mapped genes relative to the total number of transcripts and genes available on the transcriptome references are provided. Moreover the percentage of alignments relative to the total number of contigs and singletons in our library (174,278) is also provided.
We also investigated the positioning of the mRNA encoded contigs and singletons of our Syrian hamster transcriptome against other species' different transcript regions such as, 5′ untranslated regions (5′ UTR), coding regions, or 3′ untranslated regions (3′ UTR). With respect to the mouse transcriptome reference, 4,314 fragments of our Syrian hamster transcriptome (10.36%) aligned to 5′ UTRs, while 6,493 fragments of our dataset (15.59%) aligned to coding regions. In addition, 26,764 fragments of the Syrian hamster transcriptome (64.26%) aligned to 3′ UTRs ( Figure 2A ). A further 4,080 fragments of the Syrian hamster transcriptome (9.80%) aligned between 5′ UTRs, coding regions, and 3′ UTRs of the mouse transcriptome. Based on the rat transcriptome reference, 521 fragments of the Syrian hamster transcriptome (1.98%) aligned to 5′ UTRs while, 5,568 fragments of the Syrian hamster transcriptome (21.20%) aligned to coding regions. In addition, 13,371 fragments of the Syrian hamster transcriptome (50.92%) aligned to 3′ UTRs ( Figure 2B ). Finally, a total of 6,798 fragments of the Syrian hamster transcriptome (25.89%) aligned between 5′ UTRs, coding regions, and 3′ UTRs of the rat transcriptome. As expected from the experimental design of our library, the majority of our Syrian hamster transcriptome sequences aligned to 3′ UTRs of mouse and rat annotated transcripts.
Figure 2. Pie diagrams showing the alignment positions of the contigs and singletons on the mouse and rat transcript regions.

(A) Pie diagram showing the distribution of alignment positions of the 41,651 contigs and singletons on the mouse transcripts regions (5′ UTR, coding region, 3′ UTR, or inter-region). (B) Pie diagram showing the distribution of alignment positions of the 26,258 contigs and singletons on the rat transcripts regions. For each species and transcript region the number and percentage of aligned sequences are indicated.
Regarding the publicly available mouse and rat genome datasets, 45,804 of our Syrian hamster transcriptome fragments aligned to either the mouse or rat transcriptomes, and 22,105 of these same sequences aligned to both transcriptomes simultaneously, suggesting commonly occurring transcripts. Our Syrian hamster transcriptome dataset was 65.10% similar to the mouse transcriptome and 64.46% similar to the rat transcriptome. These similarities increased to 74.48% and 74.26% for the mouse and rat respectively, when comparisons were restricted to coding regions-only within those two reference genomes.
In the mouse genome, we found that 214 of those transcripts mapped at 90% of their lengths to either contigs or singletons in the Syrian hamster transcriptome (Table S1). Among these highly covered mouse transcripts were genes associated with a range of cellular activities involving, but not limited to inflammation, cell death, metabolism, and initiation of translation. These results suggest that highly covered transcripts, representative of a wide variety of cellular processes, were obtained through our methodology.
Over-expressed sequence reads and over-represented canonical pathways
In order to obtain further biological insight into our Syrian hamster transcriptome, we next identified over-expressed genes based on the number of individual reads that mapped to mouse-annotated genes ( Table 2 ). We found that 20 mouse genes contained at least 600 x read depth, and 49 mouse genes contained at least 500 fold read depth.
Table 2. List of the top 50 expressed genes in the library.
| Ensembl Gene ID | Associated Gene Name | Description | Count |
| ENSMUSG00000028647 | Mycbp | c-myc binding protein | 1120 |
| ENSMUSG00000020594 | Pum2 | pumilio 2 (Drosophila) | 1017 |
| ENSMUSG00000008575 | Nfib | nuclear factor I/B | 945 |
| ENSMUSG00000022010 | Tsc22d1 | TSC22 domain family, member 1 | 895 |
| ENSMUSG00000062078 | Qk | quaking | 861 |
| ENSMUSG00000078578 | Ube2d3 | ubiquitin-conjugating enzyme E2D 3 | 795 |
| ENSMUSG00000026621 | Mosc1 | MOCO sulphurase C-terminal domain containing 1 | 710 |
| ENSMUSG00000028161 | Ppp3ca | protein phosphatase 3, catalytic subunit, alpha isoform | 707 |
| ENSMUSG00000028790 | Khdrbs1 | KH domain containing, RNA binding, signal transduction associated 1 | 695 |
| ENSMUSG00000006740 | Kif5b | kinesin family member 5B | 684 |
| ENSMUSG00000031627 | Irf2 | interferon regulatory factor 2 | 682 |
| ENSMUSG00000036781 | Rps27l | ribosomal protein S27-like | 660 |
| ENSMUSG00000026655 | Fam107b | family with sequence similarity 107, member B | 658 |
| ENSMUSG00000006373 | Pgrmc1 | progesterone receptor membrane component 1 | 652 |
| ENSMUSG00000060961 | Slc4a4 | solute carrier family 4 (anion exchanger), member 4 | 641 |
| ENSMUSG00000024750 | Zfand5 | zinc finger, AN1-type domain 5 | 639 |
| ENSMUSG00000028788 | Ptp4a2 | protein tyrosine phosphatase 4a2 | 634 |
| ENSMUSG00000019943 | Atp2b1 | ATPase, Ca++ transporting, plasma membrane 1 | 605 |
| ENSMUSG00000097347 | AC121292.1 | 603 | |
| ENSMUSG00000004980 | Hnrnpa2b1 | heterogeneous nuclear ribonucleoprotein A2/B1 | 600 |
| ENSMUSG00000093904 | Tomm20 | translocase of outer mitochondrial membrane 20 homolog (yeast) | 593 |
| ENSMUSG00000068823 | Csde1 | cold shock domain containing E1, RNA binding | 586 |
| ENSMUSG00000020315 | Spnb2 | spectrin beta 2 | 579 |
| ENSMUSG00000068798 | Rap1a | RAS-related protein-1a | 579 |
| ENSMUSG00000020390 | Ube2b | ubiquitin-conjugating enzyme E2B | 570 |
| ENSMUSG00000026064 | Ptp4a1 | protein tyrosine phosphatase 4a1 | 570 |
| ENSMUSG00000020053 | Igf1 | insulin-like growth factor 1 | 569 |
| ENSMUSG00000027706 | Sec62 | SEC62 homolog (S. cerevisiae) | 553 |
| ENSMUSG00000064373 | Sepp1 | selenoprotein P, plasma, 1 | 549 |
| ENSMUSG00000014956 | Ppp1cb | protein phosphatase 1, catalytic subunit, beta isoform | 538 |
| ENSMUSG00000007850 | Hnrnph1 | heterogeneous nuclear ribonucleoprotein H1 | 536 |
| ENSMUSG00000031207 | Msn | moesin | 518 |
| ENSMUSG00000020152 | Actr2 | ARP2 actin-related protein 2 | 515 |
| ENSMUSG00000022261 | Sdc2 | syndecan 2 | 514 |
| ENSMUSG00000047187 | Rab2a | RAB2A, member RAS oncogene family | 512 |
| ENSMUSG00000004936 | Map2k1 | mitogen-activated protein kinase kinase 1 | 510 |
| ENSMUSG00000026576 | Atp1b1 | ATPase, Na+/K+ transporting, beta 1 polypeptide | 506 |
| ENSMUSG00000022234 | Cct5 | chaperonin containing Tcp1, subunit 5 (epsilon) | 504 |
| ENSMUSG00000001175 | Calm1 | calmodulin 1 | 502 |
| ENSMUSG00000069662 | Marcks | myristoylated alanine rich protein kinase C substrate | 490 |
| ENSMUSG00000017776 | Crk | v-crk sarcoma virus CT10 oncogene homolog (avian) | 484 |
| ENSMUSG00000038014 | Fam120a | family with sequence similarity 120A | 484 |
| ENSMUSG00000036478 | Btg1 | B cell translocation gene 1, anti-proliferative | 483 |
| ENSMUSG00000027177 | Hipk3 | homeodomain interacting protein kinase 3 | 478 |
| ENSMUSG00000043991 | Pura | purine rich element binding protein A | 474 |
| ENSMUSG00000022283 | Pabpc1 | poly(A) binding protein, cytoplasmic 1 | 471 |
| ENSMUSG00000031342 | Gpm6b | glycoprotein m6b | 471 |
| ENSMUSG00000050608 | Minos1 | mitochondrial inner membrane organizing system 1 | 471 |
| ENSMUSG00000018446 | C1qbp | complement component 1, q subcomponent binding protein | 469 |
| ENSMUSG00000026568 | Mpc2 | mitochondrial pyruvate carrier 2 | 461 |
For each of the top 50 expressed genes in the library, based on the mouse annotations, the Ensembl mouse gene identified, the associated gene name, description, and the number of count (number of time that the genes have been mapped by the reads) are indicated.
Most of the mouse genes showing high read depth were annotated as being involved in fundamental cellular processes such as cell morphology and organization, cell cycle progression, cell function and maintenance, transcription, protein synthesis and turnover, cell death, and molecular transport. Genes associated with cell type or tissue-specific functions were not significantly over-represented, consistent with our method of generating cDNA reads from pooled, multiple organ tissues. Our aim in this study was to sequence and annotate a large number of hamster mRNA 3′ fragments as a preliminary effort towards generation of an expression array, our observation that the distribution of reads were across common cellular functions, suggests our assembly is not overly biased against a specific cell or tissue type.
We also performed a functional enrichment analysis of our Syrian hamster transcriptome. Based on the list of 9,562 mouse genes that were mapped to our contigs and singletons, we identified the over-represented canonical pathways in our library ( Table 3 ). “Protein ubiquitination” ( Figure 3A , p-value = 1.99E-18) and “molecular mechanisms of cancer” ( Figure 3B , p-value = 5.01E-14) were the two most over-represented canonical pathways. However, there was also significant enrichment of many other canonical pathways related to biochemical, cellular, and disease-associated cellular processes. These included a multitude of signaling pathways, including RhoGTPase, protein kinase A, integrin, Rac, ERK/MAPK, mTOR, PI3K/Akt, PTEN, insulin, WNT/b-catenin, growth factor (VEGF, NGF, HGF, FGF, GM-CSF), and cellular junction signaling pathways. All of these pathways are biologically essential for intra- and intercellular communication and have known pleiotropic effects on transcription and translation, cellular proliferation, development, differentiation, cytoskeletal dynamics, cellular morphology, cell death, metabolism, and host responses to stress or infection. Consistent with this data, we also observed enrichment of functional categories associated with these biological activities ( Table 3 ). The biological functions associated with “cardiovascular system development and function” (p-values range from 1.05E-03 to 4.15E-17) and “nervous system development and function” (p-values range from 1.29E-03 to 1.46E-19) were statistically over-represented.
Table 3. Functional enrichment of the mouse genes mapped by our transcriptome assembly.
| Rank | Biological Function [p-value range] | Canonical pathway (p-value) |
| 1 | Organismal Surviva [1.11E-03 – 4.03E-26] | Protein Ubiquitination Pathway (1.99E-18) |
| 2 | Nervous System Development and Function [1.29E-03 – 1.46E-19] | Molecular Mechanisms of Cancer (5.01E-14) |
| 3 | Organ Morpholog [1.32E-03 – 4.20E-19] | Integrin Signaling (3.16E-13) |
| 4 | Tissue Morphology [1.08E-03 – 1.07E-18] | EIF2 Signaling (3.98E-12) |
| 5 | Cardiovascular System Development and Function [1.05E-03 – 4.15E-17] | Epithelial Adherens Junction Signaling (2.51E-11) |
List of the top 5 biological functions and the top 5 canonical pathways found as statistically over-represented based on the list of 9,546 mouse genes mapped by our transcriptome assembly. The range of p-values is indicated for the biological functions and the p-value is indicated for each canonical pathways.
Figure 3. Schematic representation of the top two over-represented canonical pathways in our transcriptome assembly.

(A) Representation of the “Protein Ubiquitination” canonical pathway. (B) Representation of the “Molecular Mechanisms of Cancer” canonical pathway. Both pathways have been generated based on mouse annotations. Transcripts involved in these pathways are indicated by different node shapes and associations are indicated by different edge shapes. Legends for the different nodes and edges are given in Figure S1. For both pathways, transcripts present in our library are indicated in gray. Associated p-values showing the statistical over-representation significance of the canonical pathways are also indicated.
Comparison with the Chinese hamster species
In order to contribute to the annotation efforts for the Chinese hamster (Cricetulus griseus) species, we aligned our Syrian hamster transcriptome to the current draft versions of the CHO cell genome and its transcriptome ( Table 1 ).
We found that 7,845 fragments in our Syrian hamster transcriptome aligned to the CHO cell transcriptome ( Table 1 ) and 85,652 aligned to the CHO cell genome (Table S2). On the other hand, 4,390 transcript fragments from the CHO cell dataset mapped to the Syrian hamster transcriptome ( Table 1 ). Our aligned Syrian hamster transcriptome showed 85.14% similarity with the CHO cell transcriptome, an expectedly higher value than what we saw for the same comparison with the mouse and rat transcriptomes.
Cross-species comparison
In order to obtain further insights about the genomic evolution of the Syrian hamster we aligned our Syrian hamster transcriptome to 14 other transcriptomes, all of which are publicly available on the Ensembl database [30] ( Table 1 ). This compendium of transcriptome references included the human (Homo sapiens), chimpanzee (Pan Troglodytes), gorilla (Gorilla gorilla gorilla), macaque (Macaca mulatta), and orangutan (Pongo abelii) sequences, as well as the ferret (Mustela putorius furo), guinea pig sequences (Cavia porcellus), and pig (Sus scrofa). As expected, the greatest number of aligned sequences occurred with the mouse and rat species transcriptomes ( Table 1 ). The human and the non-human primate species also showed high numbers of aligned sequences, possibly due to the current high quality assembly and annotation of those genomes. The CHO, ferret, pig, rabbit (Oryctolagues cuniculus), and squirrel (Ictidomys tridecemlineatus) species showed intermediary numbers of aligned sequences, while the guinea pig, kangaroo rat (Dipodomys ordii), pika (Ochotona princeps), shrew (Sorex araneus) and tree shrew (Tupaia belangeri) had the lowest numbers of aligned sequences. Of 174,278 Syrian hamster transcriptome fragments 50,433 aligned to at least one transcript reference while 61 fragments from our dataset aligned in common across all of these transcriptome references. Importantly, 76,175 of our Syrian hamster transcriptome fragments did not align to any of the 17 transcriptomes tested, nor to the CHO cell genome. It is important to note that some of the variability seen in our transcriptome comparisons may be due to differences in genome quality, assembly and annotation for the reference genomes tested.
Figure 4A is a distogram showing the results of our analysis of transcript sequences shared in common. The kangaroo rat, pika, shrew, and tree shrew had the lowest amount of commonly aligned sequences, amongst themselves and with the other species. The mouse and rat species showed the highest number of aligned sequences, presumably because of both their relatedness and genome quality/completeness.
Figure 4. Distogram showing the commonly mapped transcripts and phylogenetic tree showing the divergences amongst the different species.
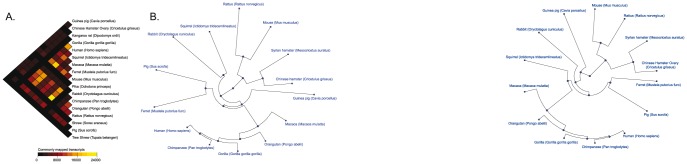
(A) Distogram showing the number of transcripts commonly mapped by the Syrian hamster transcriptome between the different species used in this study. Each cell of the distogram represents the number of transcripts commonly mapped by two different species using a gradient color. (B) Phylogenetic tree showing the genomic divergence between a subset of the different species used in this study. Each leaf of the tree represents a different species and the distances of the edges are proportional to the genomic distances between the species. Genomic distances have been calculated based on the list of 611 Syrian hamster contigs and singletons that have been commonly aligned on the transcriptome references of the 13 species having the highest number of commonly aligned sequences.
We then investigated the evolutionary divergence between the Syrian hamster and the 13 species with the largest numbers of mapped sequences and the largest degrees of shared sequences (i.e. excluding the pika, kangaroo rat, shrew, tree shrew). We found that 611 transcriptomic fragments (Table S3) have been significantly aligned on the transcriptome references of these 13 most related species and we constructed a phylogenetic tree ( Figure 4B ).The Syrian hamster transcriptome branched most closely with the CHO genome as expected. The mouse and rat transcriptome clustered together and close to the Syrian hamster and CHO cluster, as expected. All the primate species formed a super group, while the ferret and pig transcriptomes clustered together as the rabbit and squirrel transcriptomes. Consistent with a recently published study [31], we observed that the genomic divergence between the Syrian and Chinese hamsters is comparable to the divergence seen between the rat and mouse. Also, as expected, we observed that the Guinea pig does not cluster with the rodent species [32], [33].
Discussion
Here we present the assembly and analysis of a Syrian hamster transcriptome derived from the pooled RNAs from brains, lungs, spleens, kidneys, livers, and hearts of three adult females. The 3′ poly-T primed cDNAs that were sequenced on a long read-format Roche 454 were assembled into contigs, such that 22,105 of these contigs or singletons were annotated based on homology with both the mouse and rat transcriptomes, while 45,804 contigs or singletons were annotated based upon homology to one or the other mouse or rat transcriptomes. We identified 214 quasi-complete transcript sequences based on homology with mouse mRNAs and their annotations. In addition, we aligned our Syrian hamster transcriptome to the CHO cell transcriptome in order to further annotate our hamster species, and we observed a transcriptome similarity of 85.14% between the two.
When compared to a large compendium of transcriptome references, comprised of rodent, primate, and laurasiatheria species, using 661 Syrian hamster transcriptome fragments that aligned in common, the Syrian hamster transcriptome was found to be evolutionarily closest to the CHO genome and in close proximity to the mouse and rat species. The branch pattern and branch length between the Syrian and Chinese hamster transcriptomes was found to be similar to that observed between the mouse and the rat species. This observation was also described by Ryu et al. [31], but those previous efforts focused on mitochondrial gene sequences for their phylogeny analysis.
In the Syrian hamster transcriptome, we were able to identify a number of genes involved in a broad spectrum of fundamental biological processes. In addition to the 214 quasi-complete transcripts, identified based on mouse annotations and the most highly expressed transcripts, functional analysis of the entire set of sequence fragments in the Syrian hamster transcriptome that mapped to mouse genes revealed that a number of critical biological pathways are well-represented, including many related to key processes that are potentially perturbed or induced during infection. Among the most significantly enriched canonical pathways were several involved with protein synthesis, turnover, and antigen processing (protein ubiquitination, EIF2 signaling), metabolism and stress responses (mitochondrial dysfunction, NRF2-mediated oxidative stress response, PI3K/Akt, and mTOR signaling), and inflammatory and immune responses (production of NO and reactive oxygen species by macrophages, CXCR4 signaling, IL-1 signaling, and IL-3 signaling). The aim of this study was to collect and annotate a large panel of transcripts regardless of tissue origin. These observations suggest that we have generated a representative transcriptome of the Syrian hamster. Therefore this transcriptome data could be used to generate a biologically meaningful first-generation expression DNA microarray for analysis of Syrian hamster response to disease, including those infectious agents known to alter immune and pro-inflammatory responses. Mechanisms of transcriptome regulation in the Syrian hamster, by way of these important pathways can now be monitored and analyzed further.
Only ∼20% of the fragments in the Syrian hamster transcriptome aligned to the mouse and rat transcriptomes and even less aligned to the CHO cell transcriptome. This low percentage is due in part to species specificity, alignment stringency, but also to the fact that transcriptome references are far from being completely known and annotated. For instance, some classes of non-coding transcripts are now increasingly recognized as major components of regulation, and are widely expressed, but are poorly characterized and annotated. The transcriptome references that we used mainly contain known and annotated transcripts and our assembly may contain many expression contigs and singletons currently unknown and un-annotated in these other genomes.
The CHO cell genome is a useful tool for further improving the quality of our Syrian hamster transcriptome annotation for functional genomics work [24], [25]. CHO cells have been used in a variety of genetic, cell biology, and pharmacology studies. They also are the mammalian cell line of choice for producing large quantities of recombinant proteins in large amounts or in or industrial laboratory settings. Although Chinese and Syrian hamsters are phylogenetically distinct within the rodent subfamily Cricetinae [34], [35], our data confirm that they are more closely related to one another as compared to other muroid rodents.
Through our work, we have increased the number of contig sequences available in the public domain for the Syrian hamster from 860 to 174,278, where 50,433 (28.93% of the Syrian hamster transcriptome) aligns to at least one transcriptome reference. Moreover, 85,652 (49.14% of the Syrian hamster transcriptome) fragments have aligned to the draft CHO genome, leading to an overall total of 98,103 (56.29%) annotated Syrian hamster transcripts. As a note, the work performed by Schmucki et al in [23] focused on transcriptome analysis of lipid metabolism in the golden hamster liver, and no contigs or other sequences have been released to the public domain to date.
With additional funding, future plans are in place for Illumina-based RNA sequencing using paired-end technology to add and improve on our current contig assembly. These efforts will improve our coverage of the Syrian hamster transcriptome, as well as permit more comprehensive and robust phylogenetic comparisons with other species. These combined efforts will lead to a better understand of the Syrian hamster transcriptome under a variety of infectious agent models related to human disease and pathogenesis.
Conclusions
The Syrian hamster is becoming an increasingly popular model for a variety of diseases, in particular, diseases known to infect non-human primates and humans. This Syrian hamster transcriptome discussed here represents a critical step forward in providing the tools necessary for advancing functional genomics in this important animal model.
Material and Methods
Animal housing
All hamsters were housed in individually ventilated cages (IVCs). All hamsters are co-housed, unless scientifically justified and approved by the Institutional Animal Care and Use Committee (IACUC) or deemed necessary for veterinary reasons. Housing density is determined by the guidelines outlined in the Guide for the Care and Use of Laboratory Animals and the Association for the Assessment and Accreditation of the Laboratory Animal Care, International (AAALAC). Food and sterile or acidified water were provided ad libitum. Hamster diets were consist of pellets containing a variety of foods such as grains and dried vegetables along with some seeds. Water was provided by either water bottles or water pouches. The light/dark cycle was 14 hours light, 10 hours dark.
RNA extraction
Three adult female Syrian hamsters were euthanized (exsanguinated while under isoflurane sedation) and six tissues – liver, lung, heart, brain, kidney, and spleen – were harvested from each hamster. All animal studies conformed to the guidelines set forth by the National Institutes of Health (NIH) and were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at Rocky Mountain Laboratories, Division of Intramural Research, National Institute of Allergy and Infectious Diseases, NIH. One hundred mg of hamster tissue was homogenized with a Qiagen TissueLyzer II (Qiagen, Valencia, CA) in 1 mL Trizol (Invitrogen, Carlsbad, CA) following manufacturer's recommendations. To each aliquot 200 µL of 1-bromo-3-chloropropane (Sigma-Alrich) was added, the mixture was vortexed for 15 seconds and centrifuged at 4°C at 16,000x for 15 minutes. The aqueous phase was removed and passed through a Qiagen QiaShredder column to fragment remaining gDNA in the sample. The Qiagen AllPrep DNA/RNA 96 method was then performed including on-column Dnase 1 treatment to obtain high quality RNA with no genomic DNA contamination (Qiagen, Valencia, CA). RNA yield was determined by spectrophotometry (A260/A280) and RNA quality was determined using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). The average RNA integrity number (RIN) for all 18 RNAs (3 animals times 6 tissues) was 6.4. An RNA aliquot from each organ of each animal was pooled and a total of 170 µg of RNA was prepared for sequencing.
Library construction and 454 sequencing
The eighteen total RNA samples (6 tissues times 3 animals) were pooled equally into one pool. The total RNA pool underwent additional cleaning using the mirVana isoltation kit following manufacturer's recommendations (Ambion). Poly A RNA cDNA was synthesized according to a standard protocol using an oligo(dT)-linker primer for first strand synthesis. The N0 cDNA was PCR amplified during 18 cycles using a high fidelity DNA polymerase. Normalization was carried out by one cycle of denaturation and renaturation of the cDNA, resulting in N1-cDNA. Reassociated ds-cDNA was separated from the remaining ss-can (normalized cDNA) by passing the mixture over a hydroxylapatite column. After hydroxylapatite chromatography, the ss-cDNA was amplified with 15 PCR cycles. For 454 sequencing, cDNA in the size range of 500–700 bps was eluted from a preparative agarose gel. An aliquot of the size fractionated cDNA was analyzed on a 1.5% agarose gel. 454 adaptors were ligated to the size fractionated N1 cDNA and 3′ fragment sequenced on a Roche 454 using GS FLX technology with Titanium series chemistry following manufacturer's recommendations.
GS FLX sequencing generated 1,283,840 reads with an average length of 344 bases. Raw reads were trimmed for quality and reads shorter than 40 bases were discarded. The sequencing resulted in 1,212,395 reads of a total length 426,683,712 bases.
Library assembly
The trimmed and filtered reads was assembled using MIRA [36] (version 3) with the following parameters: mira –job = denovo,est,accurate,454 454_SETTINGS -CL:qc = no:cpat = yes -AL:mo = 40:mrs = 90. MIRA assembly produced 62,567 contigs and 125,228 singletons. There were 85 contigs and 13,432 singletons discarded due to poor quality (repetitive or poly-T sequence) or short read length (<50 bases), resulting in 62,482 contigs and 111,796 singletons for a total of 174,278 Syrian hamster transcriptome sequences totaling 60,117,204 bases.
Transcriptome and genome references
The transcriptome references used in this study were retrieved from the Ensembl Database [30] via the Biomart interface. Transcriptome references used in this study were obtained from the release 71 of the Ensembl database. The draft version of the CHO genome and transcriptome were retrieved from the Pre Ensembl website.
Alignments
Syrian hamster transcriptome sequences were aligned to transcriptome references using BLAST [37]. An Expect value cutoff parameter of 10 was used and alignments results were filtered in order to only keep sequences aligned at least at 80%.
Similarities of the assembled library with the transcriptome references
The similarities to the mouse, rat and other transcriptome references were calculated based on BLAST results. For all Syrian hamster transcriptome sequences that aligned to the transcriptomes, we calculated the ratio between the total number of correct nucleotide matches and the total combined length of our Syrian hamster transcriptome, which is 60,117,204 bases.
Identification of over-represented canonical pathways and biological functions
Functional enrichment of canonical pathways and biological functions was performed using Ingenuity Pathways Analysis (Ingenuity Systems, Inc.). Canonical pathways refer to pathways curated by Ingenuity as part of its knowledgebase, based on extensive characterization in the peer-reviewed literature published using human, mouse, and rat experimental models. These typically represent common properties of a particular signaling module, mechanism, or pathway. IPA examines differentially expressed transcripts in the context of known biological functions, mapping each gene identifier to its corresponding molecule in the Ingenuity Pathways Knowledge Base (IPKB). For all analyses, the p-values – representing the statistical over-representation significance – were generated using the right-tailed Fisher's Exact Test [38] and were adjusted using the Benjamini-Hochberg Multiple Testing correction [39].
Distrogram construction
The distogram represented in figure 4A was constructed using the “squash” package [40] of the R suite [41]. This representation is a color-coded, rotated triangular matrix indicating the distance between every pair of species in term of number of aligned sequences shared.
Phylogenetic tree construction
To construct the phylogenetic tree, we used the 611 transcriptomic sequences that have been significantly aligned on the transcriptome reference of the 13 species having the largest numbers of mapped sequences and the largest degrees of shared sequences. Sequences and matched transcripts were aligned using the Needleman-Wunsch multiple alignment algorithm [42], using the multialign function in MATLAB (open and extend gap penalties have been taken into consideration). Genomic divergences between sequences were calculated using the Jukes-Cantor method [43], based on the ‘NUC44’ scoring matrix. Indel mismatches have not been taken into consideration for the computation of genomic divergences. The phylogenetic tree was constructed by using the neighbor-joining method (NJ) [44].
Supporting Information
Legend for the IPA canonical pathways representations. Figure showing the annotations of the different node and edge shapes in the representations of the canonical pathways obtained from Ingenuity Pathway Analysis (IPA)
(PDF)
List of highly-covered transcripts by the Syrian hamster transcriptome. Table showing the 214 highly-covered transcripts (transcripts mapped at least at 90% by the contigs and singletons) based on the mouse annotations. For each highly-covered transcript, the Ensembl Mouse gene identified, the associated gene name as well as the gene description is indicated.
(TXT)
Alignment positions of the Syrian hamster transcriptome over the Chinese Hamster Ovary cell genome. Table showing the alignment positions of the Syrian hamster transcriptome sequences to the draft of the Chinese hamster genome. We found that 85,652 contigs and singletons of our library have been aligned on the Chinese Hamster Ovary cell genome draft. For each aligned Syrian hamster transcriptome sequence, the CHO genome segment, the start alignment position, the end alignment position, and the strand are indicated.
(TXT)
List of sequences used to infer the phylogenic tree. Table providing the list of contigs and singletons used to construct the phylogenic tree shown in figure 4. All the 611 contigs and singletons in this table have been significantly aligned on the transcriptomes of species having the highest number of commonly aligned sequences.
(TXT)
Acknowledgments
The authors thank Marcus J. Korth for valuable feedback on the manuscript.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. The unassembled reads have been uploaded on the NCBI-SRA database [45] and are available via the BioProject SRP032520. Moreover, we released the annotated transcriptome assembly in LabArchives (http://www.labarchives.com/) and are available via the DOI http://dx.doi.org/10.6070/H4FQ9TKP.
Funding Statement
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health and the NIAID Regional Centers of Excellence (U54 AI081680) to MGK. The funders were not involved in the study design, data collection and analysis, or preparation of the manuscript. NIAID was solely involved in the decision to publish.
References
- 1. Wahl-Jensen V, Bollinger L, Safronetz D, de Kok-Mercado F, Scott DP, et al. (2012) Use of the Syrian hamster as a new model of ebola virus disease and other viral hemorrhagic fevers. Viruses 4: 3754–3784 10.3390/v4123754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Xiao SY, Guzman H, Zhang H, Travassos da Rosa AP, Tesh RB, (n.d.) West Nile virus infection in the golden hamster (Mesocricetus auratus): a model for West Nile encephalitis. Emerg Infect Dis 7: 714–721 10.3201/eid0704.010420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tesh RB, Guzman H, da Rosa AP, Vasconcelos PF, Dias LB, et al. (2001) Experimental yellow fever virus infection in the Golden Hamster (Mesocricetus auratus). I. Virologic, biochemical, and immunologic studies. J Infect Dis 183: 1431–1436 10.1086/320199 [DOI] [PubMed] [Google Scholar]
- 4. Aguilar P V, Barrett AD, Saeed MF, Watts DM, Russell K, et al. (2011) Iquitos virus: a novel reassortant Orthobunyavirus associated with human illness in Peru. PLoS Negl Trop Dis 5: e1315 10.1371/journal.pntd.0001315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shinya K, Makino A, Tanaka H, Hatta M, Watanabe T, et al. (2011) Systemic dissemination of H5N1 influenza A viruses in ferrets and hamsters after direct intragastric inoculation. J Virol 85: 4673–4678 10.1128/JVI.00148-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roberts A, Lamirande EW, Vogel L, Jackson JP, Paddock CD, et al. (2008) Animal models and vaccines for SARS-CoV infection. Virus Res 133: 20–32 10.1016/j.virusres.2007.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Safronetz D, Zivcec M, Lacasse R, Feldmann F, Rosenke R, et al. (2011) Pathogenesis and host response in Syrian hamsters following intranasal infection with Andes virus. PLoS Pathog 7: e1002426 10.1371/journal.ppat.1002426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zivcec M, Safronetz D, Haddock E, Feldmann H, Ebihara H (2011) Validation of assays to monitor immune responses in the Syrian golden hamster (Mesocricetus auratus). J Immunol Methods 368: 24–35 10.1016/j.jim.2011.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ebihara H, Zivcec M, Gardner D, Falzarano D, LaCasse R, et al. (2013) A Syrian golden hamster model recapitulating ebola hemorrhagic fever. J Infect Dis 207: 306–318 10.1093/infdis/jis626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gomes-Silva A, Valverde JG, Ribeiro-Romão RP, Plácido-Pereira RM, DA-Cruz AM (2013) Golden hamster (Mesocricetus auratus) as an experimental model for Leishmania (Viannia) braziliensis infection. Parasitology: 1–9. doi:10.1017/S0031182012002156. [DOI] [PubMed]
- 11. Briand F (2010) The use of dyslipidemic hamsters to evaluate drug-induced alterations in reverse cholesterol transport. Curr Opin Investig Drugs 11: 289–297. [PubMed] [Google Scholar]
- 12. Castro-Perez J, Briand F, Gagen K, Wang S-P, Chen Y, et al. (2011) Anacetrapib promotes reverse cholesterol transport and bulk cholesterol excretion in Syrian golden hamsters. J Lipid Res 52: 1965–1973 10.1194/jlr.M016410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morin LP, Wood RI (2001) A stereotaxic atlas of the golden hamster brain. Academic Press San Diego:
- 14.Van Hoosier GL, McPherson CW (1987) The Laboratory Hamsters. Access Online via Elsevier.
- 15. Monecke S, Brewer JM, Krug S, Bittman EL (2011) Duper: a mutation that shortens hamster circadian period. J Biol Rhythms 26: 283–292 10.1177/0748730411411569 [DOI] [PubMed] [Google Scholar]
- 16. Nigro V, Okazaki Y, Belsito A, Piluso G, Matsuda Y, et al. (1997) Identification of the Syrian hamster cardiomyopathy gene. Hum Mol Genet 6: 601–607. [DOI] [PubMed] [Google Scholar]
- 17. Ricci LA, Schwartzer JJ, Melloni RH (2009) Alterations in the anterior hypothalamic dopamine system in aggressive adolescent AAS-treated hamsters. Horm Behav 55: 348–355 10.1016/j.yhbeh.2008.10.011 [DOI] [PubMed] [Google Scholar]
- 18. Chelini MOM, Palme R, Otta E (2011) Social stress and reproductive success in the female Syrian hamster: endocrine and behavioral correlates. Physiol Behav 104: 948–954 10.1016/j.physbeh.2011.06.006 [DOI] [PubMed] [Google Scholar]
- 19. Rawji KS, Zhang SX, Tsai Y-Y, Smithson LJ, Kawaja MD (2013) Olfactory ensheathing cells of hamsters, rabbits, monkeys, and mice express α-smooth muscle actin. Brain Res 1521: 31–50 10.1016/j.brainres.2013.05.003 [DOI] [PubMed] [Google Scholar]
- 20. Boguski MS, Lowe TM, Tolstoshev CM (1993) dbEST–database for “expressed sequence tags”. Nat Genet 4: 332–333 10.1038/ng0893-332 [DOI] [PubMed] [Google Scholar]
- 21. Oduru S, Campbell JL, Karri S, Hendry WJ, Khan SA, et al. (2003) Gene discovery in the hamster: a comparative genomics approach for gene annotation by sequencing of hamster testis cDNAs. BMC Genomics 4: 22 10.1186/1471-2164-4-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Landkocz Y, Poupin P, Atienzar F, Vasseur P (2011) Transcriptomic effects of di-(2-ethylhexyl)-phthalate in Syrian hamster embryo cells: an important role of early cytoskeleton disturbances in carcinogenesis? BMC Genomics 12: 524 10.1186/1471-2164-12-524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schmucki R, Berrera M, Küng E, Lee S, Thasler WE, et al. (2013) High throughput transcriptome analysis of lipid metabolism in Syrian hamster liver in absence of an annotated genome. BMC Genomics 14: 237 10.1186/1471-2164-14-237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu X, Nagarajan H, Lewis NE, Pan S, Cai Z, et al. (2011) The genomic sequence of the Chinese hamster ovary (CHO)-K1 cell line. Nat Biotechnol 29: 735–741 10.1038/nbt.1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hammond S, Swanberg JC, Kaplarevic M, Lee KH (2011) Genomic sequencing and analysis of a Chinese hamster ovary cell line using Illumina sequencing technology. BMC Genomics 12: 67 10.1186/1471-2164-12-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lewis NE, Liu X, Li Y, Nagarajan H, Yerganian G, et al. (2013) Genomic landscapes of Chinese hamster ovary cell lines as revealed by the Cricetulus griseus draft genome. Nat Biotechnol 31: 759–765 10.1038/nbt.2624 [DOI] [PubMed] [Google Scholar]
- 27. Liu H, Wang T, Wang J, Quan F, Zhang Y (2013) Characterization of liaoning cashmere goat transcriptome: sequencing, de novo assembly, functional annotation and comparative analysis. PLoS One 8: e77062 10.1371/journal.pone.0077062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ji P, Liu G, Xu J, Wang X, Li J, et al. (2012) Characterization of common carp transcriptome: sequencing, de novo assembly, annotation and comparative genomics. PLoS One 7: e35152 10.1371/journal.pone.0035152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nagaraj SH, Gasser RB, Ranganathan S (2007) A hitchhiker's guide to expressed sequence tag (EST) analysis. Brief Bioinform 8: 6–21 10.1093/bib/bbl015 [DOI] [PubMed] [Google Scholar]
- 30. Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, et al. (2013) Ensembl 2013. Nucleic Acids Res 41: D48–55 10.1093/nar/gks1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ryu SH, Kwak MJ, Hwang UW (2013) Complete mitochondrial genome of the Eurasian flying squirrel Pteromys volans (Sciuromorpha, Sciuridae) and revision of rodent phylogeny. Mol Biol Rep 40: 1917–1926 10.1007/s11033-012-2248-x [DOI] [PubMed] [Google Scholar]
- 32. D'Erchia AM, Gissi C, Pesole G, Saccone C, Arnason U (1996) The guinea-pig is not a rodent. Nature 381: 597–600 10.1038/381597a0 [DOI] [PubMed] [Google Scholar]
- 33. Graur D, Hide WA, Li WH (1991) Is the guinea-pig a rodent? Nature 351: 649–652 10.1038/351649a0 [DOI] [PubMed] [Google Scholar]
- 34. Romanenko SA, Volobouev VT, Perelman PL, Lebedev VS, Serdukova NA, et al. (2007) Karyotype evolution and phylogenetic relationships of hamsters (Cricetidae, Muroidea, Rodentia) inferred from chromosomal painting and banding comparison. Chromosome Res 15: 283–297 10.1007/s10577-007-1124-3 [DOI] [PubMed] [Google Scholar]
- 35. Trifonov VA, Kosyakova N, Romanenko SA, Stanyon R, Graphodatsky AS, et al. (2010) New insights into the karyotypic evolution in muroid rodents revealed by multicolor banding applying murine probes. Chromosome Res 18: 265–275 10.1007/s10577-010-9110-6 [DOI] [PubMed] [Google Scholar]
- 36. Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Müller WEG, et al. (2004) Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res 14: 1147–1159 10.1101/gr.1917404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215: 403–410 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 38. Fisher RA (1922) On the Interpretation of χ2 from Contingency Tables, and the Calculation of P. J R Stat Soc 85: 87–94 10.2307/2340521 [DOI] [Google Scholar]
- 39. Benjamini Y, Hochberg Y (1995) Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J R Stat Soc Ser B 57: 289–300 10.2307/2346101 [DOI] [Google Scholar]
- 40.Eklund A (2012) squash: Color-based plots for multivariate visualization. Available: http://cran.r-project.org/package=squash.
- 41. R Development Core Team R (2011) R: A Language and Environment for Statistical Computing. R Found Stat Comput 1: 409 10.1007/978-3-540-74686-7 [DOI] [Google Scholar]
- 42. Needleman SB, Wunsch CD (1970) A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol 48: 443–453. [DOI] [PubMed] [Google Scholar]
- 43.Jukes TH, Cantor CR (1969) Evolution of Protein Molecules. Munro HN, editor Academy Press.
- 44. Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4: 406–425. [DOI] [PubMed] [Google Scholar]
- 45. Kodama Y, Shumway M, Leinonen R (2012) The Sequence Read Archive: explosive growth of sequencing data. Nucleic Acids Res 40: D54–6 10.1093/nar/gkr854 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Legend for the IPA canonical pathways representations. Figure showing the annotations of the different node and edge shapes in the representations of the canonical pathways obtained from Ingenuity Pathway Analysis (IPA)
(PDF)
List of highly-covered transcripts by the Syrian hamster transcriptome. Table showing the 214 highly-covered transcripts (transcripts mapped at least at 90% by the contigs and singletons) based on the mouse annotations. For each highly-covered transcript, the Ensembl Mouse gene identified, the associated gene name as well as the gene description is indicated.
(TXT)
Alignment positions of the Syrian hamster transcriptome over the Chinese Hamster Ovary cell genome. Table showing the alignment positions of the Syrian hamster transcriptome sequences to the draft of the Chinese hamster genome. We found that 85,652 contigs and singletons of our library have been aligned on the Chinese Hamster Ovary cell genome draft. For each aligned Syrian hamster transcriptome sequence, the CHO genome segment, the start alignment position, the end alignment position, and the strand are indicated.
(TXT)
List of sequences used to infer the phylogenic tree. Table providing the list of contigs and singletons used to construct the phylogenic tree shown in figure 4. All the 611 contigs and singletons in this table have been significantly aligned on the transcriptomes of species having the highest number of commonly aligned sequences.
(TXT)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. The unassembled reads have been uploaded on the NCBI-SRA database [45] and are available via the BioProject SRP032520. Moreover, we released the annotated transcriptome assembly in LabArchives (http://www.labarchives.com/) and are available via the DOI http://dx.doi.org/10.6070/H4FQ9TKP.