

Abstract
Poly(arginine) mimics bearing long hydrophobic side chains adopt stable helical conformation and exhibit helix-related cell-penetrating properties. Elongating polypeptide backbone length and increasing side chain hydrophobicity further increase the helicities of poly(arginine) mimics. They show superior cell membrane permeability up to two orders of magnitude higher than that of HIV-TAT peptide and excellent DNA and siRNA delivery efficiencies in various mammalian cells.
Introduction
Cell-penetrating peptides (CPPs) are oligopeptides consisting of 10-30 amino acid residues that have excellent membrane permeability.1 When integrated with delivery systems, they facilitate the intracellular delivery of various cargos, including small molecules, macromolecules (e.g., proteins and nucleic acids) and nanoparticles.2-13 CPPs typically have a large number of arginine (Arg) residues in their primary structures, and the guanidinium groups of the Arg residues are crucial to the penetration efficiencies of CPPs because of their interactions with the sulfate groups of glycosaminoglycans localized on cell membranes.14,15 An example of such guanidine-rich CPPs is HIV-TAT, an 11-mer peptide containing 6 Arg residues.5,16 In addition to the critical roles of guanidine groups, peptide conformation and hydrophobic content also have significant effect on CPP’s penetration efficiencies.5,17-20 Several well-known CPPs, such as Pep-1, MPG, TP10, and melittin, either adopt inherent helical structures or form helices in the cell membranes, presenting a rigid amphiphilic structure to interact with the lipid bilayers to promote membrane permeation.8,18,20-23 A large body of data on CPP translocation show that the formation of trans-membrane helix in CPPs is essential for stabilizing their membrane interactions and promoting their cellular uptake.24-26 Increasing the hydrophobicity of the side chains and/or the backbone of CPPs and CPP mimics have also been reported to promote their interaction with phospholipids and facilitate their translocation in a “self-activated” manner.6,27-29
Oligo- and polyarginines are structurally the simplest CPP mimics with Arg as the only building block and can be readily prepared. However, they adopt random coil conformation in aqueous solution or when associated with phospholipid membranes due to the strong side chain charge repulsion and lack of hydrophobic or amphiphilic structure.30 Thus, their membrane permeability mainly relies on the electrostatic interaction with lipid membranes mediated by their guanidinium charge groups. Guanidine-rich CPP mimics with various backbones, such as peptoid,31 β-peptide,32 oligocarbamate,29,33 and even non-peptidyl synthetic polymers,27,28,34-36 have been reported. They have enhanced hydrophobicity but still lack the capability to adopt helical structures. It would thus be interesting to integrate both helicity and hydrophobicity into the design of guanidine-rich CPPs to potentially develop CPPs with unprecedented, superior membrane permeability. In this study, we tested this hypothesis by developing a class of helical poly(arginine) mimics (HPRMs) bearing guanidinium groups and long, hydrophobic side chains, and demonstrated that these HPRMs had superior membrane activities, up to two orders of magnitude higher than that of TAT, and remarkable DNA and siRNA delivery capabilities.
Poly(arginine) adopts random coil conformation at physiological pH due to the pendant guanidine charge repulsion. Only at pH higher than 12.5 when the pendant guanidinium groups are completely deprotonated, poly(arginine) with sufficient long backbone may adopt helical conformation.37 We therefore first aimed to develop poly(arginine) mimics that could adopt stable helix. A 57-mer poly(γ-(5-aminohexyl)-L-glutamate) (PAHG57) (Scheme 1), a poly-L-lysine (PLL) analogue with its positively charged side-chain amine groups placed 11 σ-bonds away from the peptide backbone, has reduced helical surface charge density and thus side chain charge repulsion.38 Consequently, PAHG57 adopts stable α-helical conformation (45% helicity) at physiological pH, as opposed to the random coil conformation of PLL under the same condition.38 Arg+, with delocalized charge to α-carbon distance of ≈ 4-6 σ-bonds, has helical propensity similar to Lys+ with charge to α-carbon distance of 5 σ-bonds (Scheme 1),39 and poly(arginine) has slightly higher helical content than poly(lysine) of similar molecular weights.40 We hypothesized that a poly(arginine) analogue with side chain guanidinium groups placed with significant distance from the peptide backbone would also adopt stable α-helical conformation.
Scheme 1.
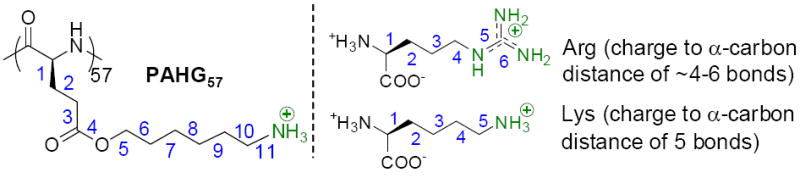
Structure of PAHG57, Arg, and Lys.
Results and discussion
To prove this hypothesis, we synthesized P1 (Table 1) via ring-opening polymerization (ROP) of γ-chloroalkyl L-glutamic acid N-carboxylanhydrides (L-Glu-NCA) followed by introduction of guanidinium groups via the azide-alkyne Huisgen cycloaddition, the so-called “click” chemistry (Scheme 2).41 P1 had guanidinium groups at the termini of the side chains that were 13 σ-bond away from the polypeptide backbone (Table 1). As expected, it adopted right-handed α-helical conformation with 30% helicity,42 evidenced by the characteristic double minima at 208 nm and 222 nm in the CD spectrum (Figure 1A). The helicity of P1 remained unchanged from pH 1 to 9 (Figure 1B), suggesting that the structural property of P1 would be well maintained against pH changes under physiological intracellular conditions (e.g., in both acidic endolysosomal compartments and slightly basic cytosol). The molar ellipticity of P1 at 222 nm was independent of the P1 concentration, suggesting that P1 remained in monomeric form (Supplementary Figure S23A).
Table 1.
HPRMs with Various Molecular Weights, Structures, and Side-Chain Lengths.
| HPRM | Monomer | Mn a | Mw/Mn b | DP c | X d | Helicity (%) |
|---|---|---|---|---|---|---|
| P1 | L-Cl-Glu NCA | 17,600 | 1.11 | 51 | 1 | 30.0 |
| P2 | D-Cl-Glu NCA | 18,700 | 1.17 | 54 | 1 | 31.6 |
| P3 | D,L-Cl-Glu NCA | 18,000 | 1.16 | 52 | 1 | -- |
| P4 | L-Cl-Glu NCA | 23,900 | 1.10 | 69 | 1 | 32.0 |
| P5 | L-Cl-Glu NCA | 19,400 | 1.25 | 50 | 4 | 55.7 |
| P6 | L-Cl-Glu NCA | 22,900 | 1.26 | 55 | 6 | 64.9 |
| P7 | L-Cl-Glu NCA | 29,900 | 1.11 | 72 | 6 | 69.8 |
| P8 | L-Cl-Glu NCA | 4,200 | 1.16 | 10 | 6 | 50.7 |
Number average molecular weight;
Molecular weight distribution;
Degree of polymerization (DP);
Number of methylene groups.
Scheme 2.
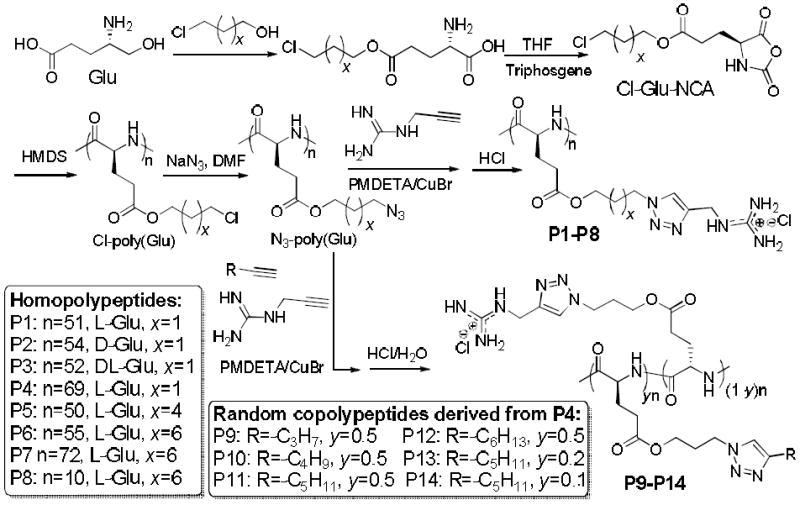
Synthesis of HPRMs (P1-P14).
Fig. 1.
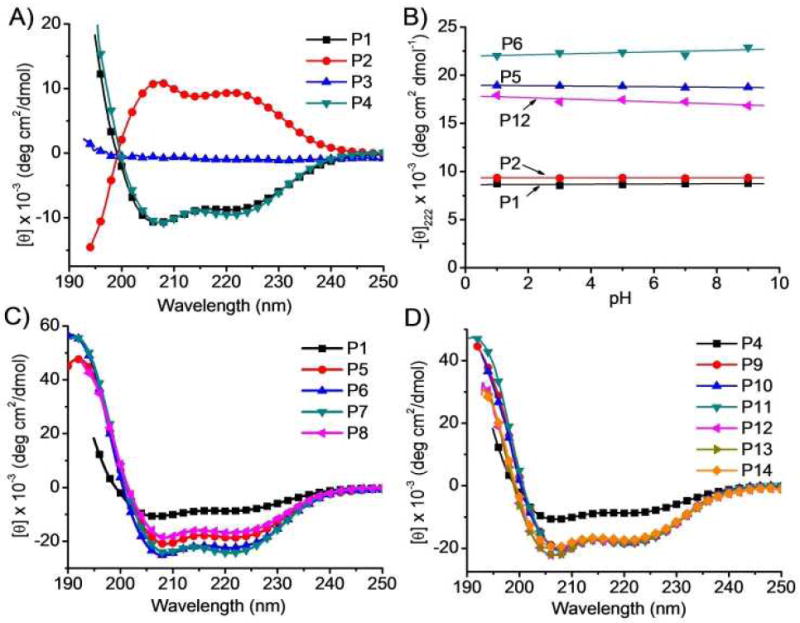
CD spectra of HPRMs in water (A, C, D). Molar ellipticities of selected HPRMs at 222 nm as a function of pH (B).
By labelling the HPRMs with rhodamine (RhB), we first compared the membrane permeability of P1 against TAT and Arg9 in HeLa, 3T3-L1, and Raw 264.7 cells that represented carcinoma cells, fibroblasts, and macrophages, respectively. After incubation with cells for 2 h at 37 °C, the intracellular concentration of CPPs in the cell lysate was quantified by spectrofluorimetry following removal of surface-bound polypeptides by washing with PBS-heparin.43 P1 was efficiently internalized into HeLa cells with an uptake level of 7.5 ± 0.6 μg/mg cellular protein (Figure 2A), which is 13.6 and 7.0 times higher than TAT (0.5 ± 0.1 μg/mg) and Arg9 (1.1 ± 0.1 μg/mg), respectively. Interestingly, the helical sense has no effect on cell penetration. P2, a polymer having exactly the same chemical structure and molecular weight (MW) as P1 but prepared with D-Glu NCA (Table 1), showed left-handed α-helical conformation with helicity (32%, Figure 1A) and membrane permeability (7.1 ± 0.6 μg/mg, Figure 2A) almost identical to P1. Nevertheless, P3, a random coil analogue of P1 and P2 prepared from the racemic D,L-Glu NCA, showed a 7-fold lower permeability (1.1 ± 0.3 μg/mg) than P1 and P2 (Figure 2A), which was comparable to that of TAT and Arg9. Such case also held true for Raw 264.7 and 3T3-L1 cells (Supplementary Figure S24-25). P4, a P1 analogue with higher MW, showed helicity and membrane permeability similar to P1 (Figure 2A).
Fig. 2.
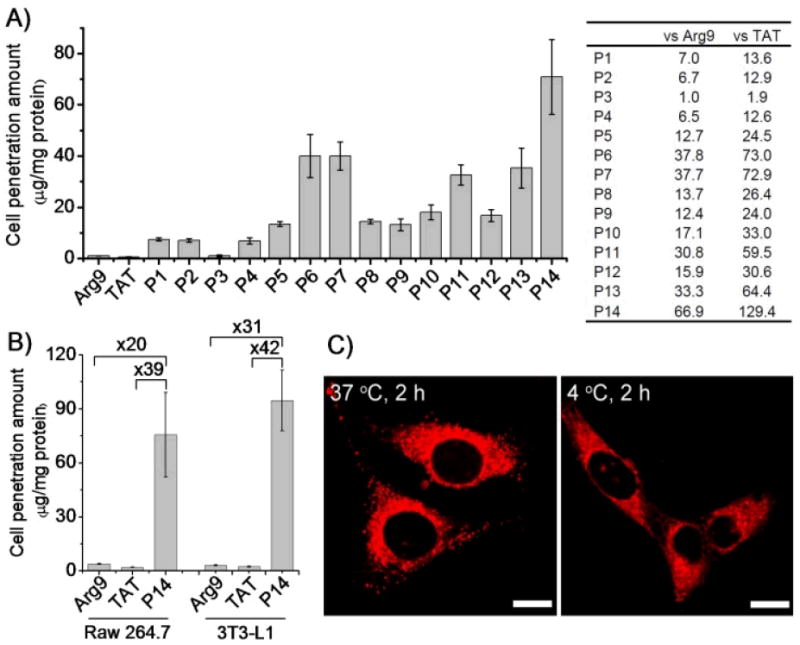
HPRMs mediate effective cell penetration. (A) Uptake of RhB-HPRMs in HeLa cells (20 μg/mL, left) and fold of increment over Arg9 and TAT (right) following incubation at 37 °C for 2 h. (B) Uptake levels of the top-performing RhB-P14 (20 μg/mL) in 3T3-L1 and Raw 264.7 cells following incubation at 37 °C for 2 h. (C) CLSM images of HeLa cells incubated with RhB-P14 at 37 °C or 4 °C for 2 h. Bar = 20 μm.
Increasing the hydrophobicity of peptide side chains can enhance their intramolecular hydrophobic interactions in aqueous solution,38 resulting in improved helical contents and stability. To verify if this holds true in the design of HPRMs, we prepared P5 (x = 4) and P6 (x = 6), analogues of P1 (x = 1) with increased hydrophobic side chain length and a guanidinium-to-backbone distance of 16 and 18 σ-bonds, respectively (Scheme 2). As expected, the helicities of P5 and P6 increased to 56% and 65%, respectively, from the 30% of P1 (Figure 1C). Their cell penetration levels also increased to 13.4 ± 1.0 and 40.0 ± 8.4 μg/mg (Figure 2A), respectively, following the same trend of their helicity increment. P7, a P6 analogue with a 40% elongated backbone (Scheme 2), showed slightly increased helicity (70%) and comparable membrane permeability (39.9 ± 5.5 μg/mg), 72.9 and 37.7 times more permeable than TAT and Arg9, respectively (Figure 2A). P8, a P6 analogue with a very short backbone (DP = 10), had a remarkably high helicity (51%) given its low MW. This very short but helical HPRM had approximately the same length as TAT (11 mer) and Arg9 (9 mer) but was 26.4 and 13.7 times more permeable than TAT and Arg9 in HeLa cells, respectively (Figure 2A). The excellent membrane permeability of P8 was also observed in Raw 264.7 and 3T3-L1 cells (Supplementary Figure S24-25), substantiating the importance of the integrated effect of helicity and side chain hydrophobicity on CPP’s membrane permeability.
Random copolymers containing both guandinium and hydrophobic pendant groups have been reported to show improved membrane activities compared to guanidinium-bearing homopolymers, presumably due to further increased hydrophobicity.27,28 We thus went on to study if further increased cell-penetrating property can be achieved in HPRMs through similar strategy. P9-P14, random copolymer analogues of P4, were prepared (Scheme 2, Table 2) and their membrane activities were evaluated (Figure 2). When the alkyl content was fixed at 50 mol% in the random co-polypeptides with the alkyl chain length (R group) being gradually increased from −C3H7 to −C6H13 (P9-P12), a significant increase of their helicities was observed compared to P4 (from 32% for P4 to 52-55% for P9-P12, Table 1 and 2, Figure 1D). P9-P12 all showed enhanced membrane permeability compared to P4, with P11 being the best among these four polymers (32.6 ± 3.9 μg/mg, Figure 2A) and approximately 5 times more membrane permeable than P4. By decreasing the content of the alkyl block of P11 to 20 mol% (P13) and 10 mol% (P14), the co-polypeptide helicity remained unchanged while the membrane permeability was further improved presumably due to increased cationic charge density. P14 showed a superior membrane permeability of 70.9 ± 14.5 μg/mg (Figure 2A), approximately 10, 67, and 129 times more permeable than P4, Arg9, and TAT, respectively. These results indicated the necessity to maintain a proper balance between the cationic charge and the hydrophobic domain in the design of CPP mimics. Additionally, the higher MW of P14 in comparison to TAT and Arg9 also enables better association with cell membranes to aid stronger membrane permeability. P14 was also found to be the most membrane active HPRM in Raw 264.7 and 3T3-L1 cells, outperforming TAT by 39 and 42 times and Arg9 by 20 and 31 times, respectively (Figure 2B; Supplementary Figure S24-25). Confocal laser scanning microscopy (CLSM) observation accordantly revealed extensive internalization of RhB-P14 in HeLa cells (Figure 2C).
Table 2.
HPRMs with Various Hydrophobic Side Groups.a
| Polymer | R | y b | Helicity (%) |
|---|---|---|---|
| P9 | n-C3H7– | 0.5 | 53.9 |
| P10 | n-C4H9– | 0.5 | 54.7 |
| P11 | n-C5H11– | 0.5 | 54.0 |
| P12 | n-C6H13– | 0.5 | 51.9 |
| P13 | n-C5H11– | 0.2 | 53.5 |
| P14 | n-C5H11– | 0.1 | 54.6 |
The DP and polydispersity of the polypeptides are 69 and 1.10;
The molar content of hydrophobic alkyl side-groups.
Upon identifying the cell penetration properties of these HPRMs, we selected the top-performing P14 to further elucidate the membrane permeation mechanisms. We first evaluated the cell uptake level at 4°C when energy-dependent endocytosis was inhibited. Cell fixation will cause artificial uptake and re-localization of the internalized CPPs, and thus earlier studies on fixed cells featured the direct translocation mechanism while later studies on live, non-fixed cells indicated the presence of a non-endocytotic mechanism. To avoid the artifact of cell fixation, we visualized the uptake of RhB-P14 in live cells and noted that RhB-P14 could still be extensively internalized in HeLa cells at 4°C (Figure 2C).44 A further quantitative analysis also revealed that the cell uptake level was reduced by only 30~50% at 4°C compared to that at 37 °C (Figure 3A and Supplementary Fig. S26), indicating that majority of the polypeptides traversed the cell membrane via the energy-independent, non-endocytotic pathway. By monitoring the cell internalization in the presence of various endocytic inhibitors, we probed the involvement of different endocytic pathways. Uptake of RhB-P14 was significantly reduced by caveolae inhibitors, genistein and methyl-β-cyclodextrin (mβCD) (Figure 3A and Supplementary Fig. S26).45-47 Comparatively, chlorpromazine and wortmannin that respectively inhibited the clathrin- and macropinocytosis-mediated endocytosis showed unappreciable inhibitory effect.45-47 It was thus indicated that P14 was endocytosed via caveolae- rather than clathrin- or macropinocytosis-mediated pathways.
Fig. 3.
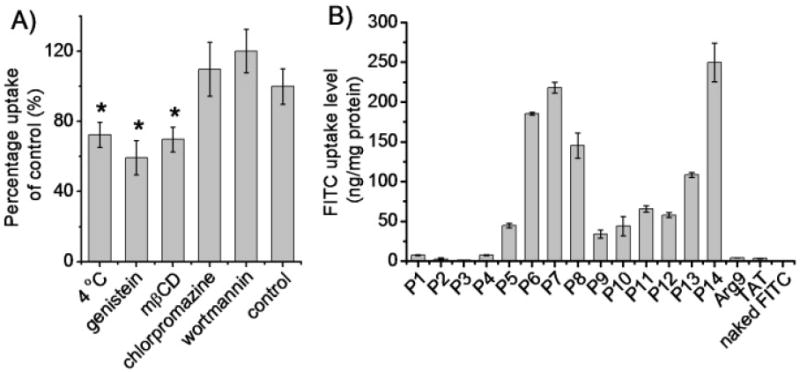
HPRMs enter cells via caveolae-mediated endocytosis and non-endocytosis. (A) Uptake level of RhB-P14 in HeLa cells in the presence of various endocytic inhibitors (n=3). (B) Uptake level of FITC in HeLa cells following co-incubation with polypeptides for 2 h at 37 °C (n=3).
Since majority of the P14 enters the cells via non-endocytotic translocation, we further investigated the HPRM-induced pore formation on cell membranes by studying the cellular internalization of fluorescein (FITC), a hydrophilic and membrane-impermeable fluorescent molecule.48,49 FITC was negligibly taken up by HeLa cells while treatment with HPRMs led to an notable increase in the FITC uptake level by 1-2 orders of magnitude (Figure 3B), substantiating that HPRMs were able to induce pore formation on cell membranes to allow extensive diffusion of FITC into the cells. The pore-forming capacities of HPRMs accorded well with their cell penetration levels (Figure 2A and 3B), which further demonstrated that high helicities and hydrophobic alkyl domains were favorable for polypeptides to mediate effective membrane penetration via the pore formation mechanism. In a direct comparison with TAT and Arg9, the most membrane-active P14 mediated two-order-of-magnitude higher efficiency for FITC uptake. MTT assay revealed that HPRMs with higher penetration capacities (P6, P7, P11, P13, and P14) exhibited cytotoxicity at high concentration (100 μg/mL, Supplementary Fig. S27), which might be attributed to their excessively strong pore formation capabilities that compromised the membrane integrity. However, none of the test polypeptides induced appreciable cytotoxicity at the concentration used for the uptake study (20 μg/mL) (Supplementary Fig. S27), indicating that the distinguished cell penetration and pore formation properties of the HPRMs were not based on compromised cell viability.
Upon identifying P14 as the top-performing cell-penetrating HPRM, we were motivated to further explore its potential as molecular transporters to mediate intracellular delivery of various cargos. To this end, plasmid DNA encoding luciferase (pCMV-Luc) and siRNA duplexes towards tumor necrosis factor α (TNF-α) were selected as representative DNA and siRNA molecules, and the gene transfection as well as gene silencing efficiencies mediated by P14 were evaluated. Because of its polycationic nature, P14 was able to condense the anionic DNA and siRNA at N/P ratios higher than 5 and 15, respectively (Supplementary Fig. S28). Flow cytometry analyses revealed that after incubation with P14/YOYO-1-DNA complexes (N/P ratio of 15) or P14/FAM-siRNA complexes (N/P ratio of 30) for 4 h, over 95% of the cells (HeLa, 3T3-L1, Raw 264.7) had dramatically taken up YOYO-1-DNA while over 85% of the Raw 264.7 cells internalized FAM-siRNA (Supplementary Fig. S29-30). In comparison, Arg9, TAT, poly-L-arginine (PLR), and Lipofectamine™ 2000 (LPF2000) showed much weaker capability in delivering DNA/siRNA molecules intracellularly. CLSM observation further revealed separation of YOYO-1-DNA or FAM-siRNA from Lysotracker Red-stained endosome/lysosomes following 4-h treatment with the P14/YOYO-1-DNA or P14/FAM-siRNA complexes (Supplementary Fig. S31-32), indicating that P14 could effectively avoid the endosomal entrapment, one of the most critical barriers against non-viral gene and siRNA delivery. In comparison, internalized Arg9/YOYO-1-DNA and Arg9/FAM-siRNA complexes were co-localized well with Lysotracker Red, which suggested its inability to escape endosomal entrapment. Hence, P14/DNA complexes mediated effective luciferase expression in the three tested cell types (Figure 4A), among which 3T3-L1 and Raw 264.7 cells have been reported to be difficult-to-transfection.9,50 Similarly, P14/siRNA complexes also triggered notable TNF-α knockdown in Raw 264.7 cells (Figure 4B). In consistency with the cell uptake trend, P14 notably outperformed the commercial CPPs (Arg9, TAT, PLR) and demonstrated an improvement over LPF2000 by 1-2 orders of magnitude with respective to gene transfection and silencing efficiencies.
Fig. 4.
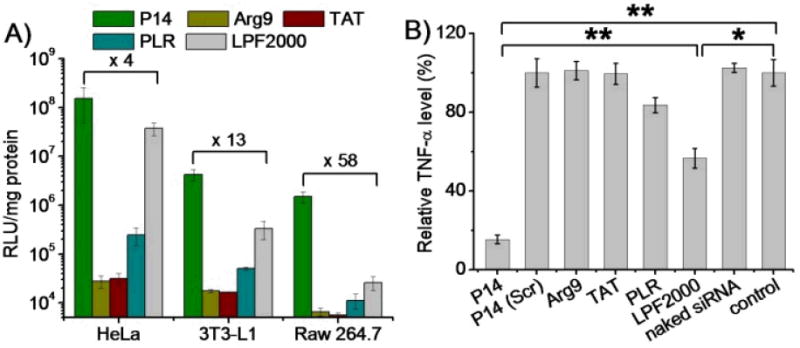
P14 mediates effective DNA/siRNA delivery in mammalian cells. (A) Transfection efficiencies of P14/DNA complexes (N/P ratio = 15) in HeLa, 3T3-L1, and Raw 264.7 cells. Arg9, TAT, poly(L-arginine) (PLR), and lipofectamine™ 2000 (LPF2000) at the N/P ratio of 15, 15, 10, and 5 served as controls, respectively. (B) Silencing efficiency of P14/siRNA complexes (N/P ratio = 30) towards LPS-induced TNF-α production in Raw 264.7 cells. Results were expressed as percentage TNF-α level of untreated cells (control), and were compared to Arg9, TAT, PLR, and LPF2000 at the N/P ratio of 20, 20, 15, and 7.5, respectively.
Conclusions
In summary, we developed a new family of cationic, α-helical, poly(arginine) mimics with excellent cell penetration efficiencies. We unraveled their structure-activity relationship which indicated that the helical secondary structure, the polypeptide backbone length, and the presence of hydrophobic domains collectively contributed to the membrane activities of these new CPPs that were closely related to the pore formation mechanisms on cell membranes. By either elongating the linker between polypeptide backbone and guanidinium side groups or extending the alkyl side chains, we observed remarkably augmented cell penetration efficiencies of these poly(arginine) mimics, outperforming classical CPPs such as TAT and Arg9 by 1-2 orders of magnitude. These helical CPPs demonstrated high capacity and efficiency in delivering DNA and siRNA to mammalian cells to mediate effective gene transfection and silencing, outperforming commercial transfection reagent LPF2000 by 1-2 orders of magnitude. This study provides a simple strategy to transform conventional polypeptides to functional materials with superior cell-penetrating and molecular transporting capabilities.
Supplementary Material
Acknowledgments
J.C. acknowledges support from the NSF (CHE-1153122) and the NIH (NIH Director’s New Innovator Award 1DP2OD007246 and 1R21EB013379).
Footnotes
Supporting Information
Electronic Supplementary Information (ESI) available: Synthesis and characterization (1H NMR, GPC) of monomers and polypeptides, molar ellipticity of polypeptides, cell uptake in Raw 264.7 and 3T3-L1 cells, MTT assay, gel retardation assay, flow cytometry analyses, CLSM images, and transfection efficiencies at different N/P ratios. See DOI: 10.1039/b000000x/
Notes and references
- 1.Fonseca SB, Pereira MP, Kelley SO. Adv Drug Deliver Rev. 2009;61:953–964. doi: 10.1016/j.addr.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 2.Mandal D, Shirazi AN, Parang K. Angewandte Chemie International Edition. 2011;50:9633–9637. doi: 10.1002/anie.201102572. [DOI] [PubMed] [Google Scholar]
- 3.Hsu T, Mitragotri S. Proceedings of the National Academy of Sciences. 2011;108:15816–15821. doi: 10.1073/pnas.1016152108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kwon EJ, Liong S, Pun SH. Molecular Pharmaceutics. 2010;7:1260–1265. doi: 10.1021/mp1000668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meade BR, Dowdy SF. Advanced Drug Delivery Reviews. 2008;60:530–536. doi: 10.1016/j.addr.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geihe EI, Cooley CB, Simon JR, Kiesewetter MK, Edward JA, Hickerson RP, Kaspar RL, Hedrick JL, Waymouth RM, Wender PA. P Natl Acad Sci USA. 2012;109:13171–13176. doi: 10.1073/pnas.1211361109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eguchi A, Meade BR, Chang YC, Fredrickson CT, Willert K, Puri N, Dowdy SF. Nat Biotechnol. 2009;27:567–U110. doi: 10.1038/nbt.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morris MC, Depollier J, Mery J, Heitz F, Divita G. Nat Biotechnol. 2001;19:1173–1176. doi: 10.1038/nbt1201-1173. [DOI] [PubMed] [Google Scholar]
- 9.McNaughton BR, Cronican JJ, Thompson DB, Liu DR. P Natl Acad Sci USA. 2009;106:6111–6116. doi: 10.1073/pnas.0807883106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kondo E, Saito K, Tashiro Y, Kamide K, Uno S, Furuya T, Mashita M, Nakajima K, Tsumuraya T, Kobayashi N, Nishibori M, Tanimoto M, Matsushita M. Nat Commun. 2012;3 doi: 10.1038/ncomms1952. [DOI] [PubMed] [Google Scholar]
- 11.Jewell CM, Jung JM, Atukorale PU, Carney RP, Stellacci F, Irvine DJ. Angew Chem Int Edit. 2011;50:12312–12315. doi: 10.1002/anie.201104514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drappier C, Wirotius AL, Bathany K, Ibarboure E, Condassamy O, Garanger E, Lecommandoux S. Polym Chem-Uk. 2013;4:2011–2018. [Google Scholar]
- 13.Zhang K, Fang HF, Chen ZY, Taylor JSA, Wooley KL. Bioconjugate Chem. 2008;19:1880–1887. doi: 10.1021/bc800160b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wender PA, Galliher WC, Goun EA, Jones LR, Pillow TH. Adv Drug Deliver Rev. 2008;60:452–472. doi: 10.1016/j.addr.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Naik RJ, Chandra P, Mann A, Ganguli M. Journal of Biological Chemistry. 2011;286:18982–18993. doi: 10.1074/jbc.M111.227793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rudolph C, Plank C, Lausier J, Schillinger U, Müller RH, Rosenecker J. Journal of Biological Chemistry. 2003;278:11411–11418. doi: 10.1074/jbc.M211891200. [DOI] [PubMed] [Google Scholar]
- 17.Deshayes S, Morris MC, Divita G, Heitz F. Cell Mol Life Sci. 2005;62:1839–1849. doi: 10.1007/s00018-005-5109-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daniels DS, Schepartz A. Journal of the American Chemical Society. 2007;129:14578–14579. doi: 10.1021/ja0772445. [DOI] [PubMed] [Google Scholar]
- 19.Lattig-Tunnemann G, Prinz M, Hoffmann D, Behlke J, Palm-Apergi C, Morano I, Herce HD, Cardoso MC. Nat Commun. 2011;2 doi: 10.1038/ncomms1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith BA, Daniels DS, Coplin AE, Jordan GE, McGregor LM, Schepartz A. J Am Chem Soc. 2008;130:2948–2949. doi: 10.1021/ja800074v. [DOI] [PubMed] [Google Scholar]
- 21.Kolusheva S, Shahal T, Jelinek R. Biochemistry-Us. 2000;39:15851–15859. doi: 10.1021/bi000570b. [DOI] [PubMed] [Google Scholar]
- 22.Mano M, Henriques A, Paiva A, Prieto M, Gavilanes F, Simoes S, De Lima MCP. J Pept Sci. 2007;13:301–313. doi: 10.1002/psc.842. [DOI] [PubMed] [Google Scholar]
- 23.Ruzza P, Calderan A, Guiotto A, Osler A, Borin G. J Pept Sci. 2004;10:423–426. doi: 10.1002/psc.558. [DOI] [PubMed] [Google Scholar]
- 24.Morris MC, Deshayes S, Heitz F, Divita G. Biol Cell. 2008;100:201–217. doi: 10.1042/BC20070116. [DOI] [PubMed] [Google Scholar]
- 25.Deshayes S, Heitz A, Morris MC, Charnet P, Divita G, Heitz F. Biochemistry-Us. 2004;43:1449–1457. doi: 10.1021/bi035682s. [DOI] [PubMed] [Google Scholar]
- 26.Veldhoen S, Laufer SD, Trampe A, Restle T. Nucleic Acids Res. 2006;34:6561–6573. doi: 10.1093/nar/gkl941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Som A, Tezgel AO, Gabriel GJ, Tew GN. Angew Chem Int Edit. 2011;50:6147–6150. doi: 10.1002/anie.201101535. [DOI] [PubMed] [Google Scholar]
- 28.Som A, Reuter A, Tew GN. Angew Chem Int Edit. 2012;51:980–983. doi: 10.1002/anie.201104624. [DOI] [PubMed] [Google Scholar]
- 29.Patil KM, Naik RJ, Rajpal, Fernandes M, Ganguli M, Kumar VA. J Am Chem Soc. 2012;134:7196–7199. doi: 10.1021/ja210026m. [DOI] [PubMed] [Google Scholar]
- 30.Law M, Jafari M, Chen P. Biotechnology Progress. 2008;24:957–963. doi: 10.1002/btpr.13. [DOI] [PubMed] [Google Scholar]
- 31.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. Proc Natl Acad Sci U S A. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith BA, Daniels DS, Coplin AE, Jordan GE, McGregor LM, Schepartz A. Journal of the American Chemical Society. 2008;130:2948–2949. doi: 10.1021/ja800074v. [DOI] [PubMed] [Google Scholar]
- 33.Wender PA, Rothbard JB, Jessop TC, Kreider EL, Wylie BL. J Am Chem Soc. 2002;124:13382–13383. doi: 10.1021/ja0275109. [DOI] [PubMed] [Google Scholar]
- 34.Cooley CB, Trantow BM, Nederberg F, Kiesewetter MK, Hedrick JL, Waymouth RM, Wender PA. J Am Chem Soc. 2009;131:16401. doi: 10.1021/ja907363k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kolonko EM, Pontrello JK, Mangold SL, Kiessling LL. J Am Chem Soc. 2009;131:7327–7333. doi: 10.1021/ja809284s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kolonko EM, Kiessling LL. J Am Chem Soc. 2008;130:5626. doi: 10.1021/ja8001716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hayakawa T, Kondo Y, Yamamoto H. Bulletin of the Chemical Society of Japan. 1969;42:1937–1941. doi: 10.1246/bcsj.42.479. [DOI] [PubMed] [Google Scholar]
- 38.Lu H, Wang J, Bai YG, Lang JW, Liu SY, Lin Y, Cheng JJ. Nat Commun. 2011;2 doi: 10.1038/ncomms1209. [DOI] [PubMed] [Google Scholar]
- 39.Pace CN, Scholtz JM. Biophys J. 1998;75:422–427. doi: 10.1016/s0006-3495(98)77529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holzwarth G, Doty P. Journal of the American Chemical Society. 1965;87:218–228. doi: 10.1021/ja01080a015. [DOI] [PubMed] [Google Scholar]
- 41.Engler AC, Lee HI, Hammond PT. Angew Chem Int Ed. 2009;48:9334–9338. doi: 10.1002/anie.200904070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morrow JA, Segall ML, Lund-Katz S, Phillips MC, Knapp M, Rupp B, Weisgraber KH. Biochemistry. 2000;39:11657–11666. doi: 10.1021/bi000099m. [DOI] [PubMed] [Google Scholar]
- 43.McNaughton BR, Cronican JJ, Thompson DB, Liu DR. Proc Natl Acad Sci U S A. 2009;106:6111–6116. doi: 10.1073/pnas.0807883106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmidt N, Mishra A, Lai GH, Wong GCL. Febs Lett. 2010;584:1806–1813. doi: 10.1016/j.febslet.2009.11.046. [DOI] [PubMed] [Google Scholar]
- 45.Zhao X, Yin L, Ding J, Tang C, Gu S, Yin C, Mao Y. Journal of Controlled Release. 2010;144:46–54. doi: 10.1016/j.jconrel.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 46.McLendon PM, Fichter KM, Reineke TM. Molecular Pharmaceutics. 2010;7:738–750. doi: 10.1021/mp900282e. [DOI] [PubMed] [Google Scholar]
- 47.Khalil IA, Kogure K, Akita H, Harashima H. Pharmacological Reviews. 2006;58:32–45. doi: 10.1124/pr.58.1.8. [DOI] [PubMed] [Google Scholar]
- 48.Gabrielson NP, Lu H, Yin LC, Li D, Wang F, Cheng JJ. Angew Chem Int Edit. 2012;51:1143–1147. doi: 10.1002/anie.201104262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ter-Avetisyan G, Tünnemann G, Nowak D, Nitschke M, Herrmann A, Drab M, Cardoso MC. Journal of Biological Chemistry. 2009;284:3370–3378. doi: 10.1074/jbc.M805550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun P, Zhong M, Shi X, Li Z. Journal of Drug Targeting. 2008;16:668–678. doi: 10.1080/10611860802201381. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.