

Abstract
Probes that form covalent bonds with RNA molecules based on their chemical reactivity would advance our ability to study the transcriptome. We developed a set of electrophilic activity-based RNA probes designed to react with unusually nucleophilic RNAs. We used these probes to identify reactive genome-encoded RNAs, resulting in the discovery of a 42-nt catalytic RNA from an archaebacterium that reacts with a 2,3-disubstituted epoxide at N7 of a specific guanosine. Detailed characterization of the catalytic RNA revealed the structural requirements for reactivity. We developed this catalytic RNA into a general tool to selectively conjugate a small molecule to an RNA of interest. This strategy enabled up to 500-fold enrichment of target RNA from total mammalian RNA or from cell lysate. We demonstrated the utility of this approach by selectively capturing proteins in yeast cell lysate that bind to the ASH1 mRNA.
Introduction
Recent data suggesting that most of the genome is transcribed into functional, non-coding RNA,1 together with an increasing awareness of the complexity of RNA post-transcriptional regulation, have created the need for new tools to study and discover bioactive RNAs.2,3 Our knowledge of naturally occurring catalytic RNAs is limited to the ribosome and ten classes of phosphodiester-hydrolyzing RNAs.4,5 The two types of chemical reactions catalyzed by known biological ribozymes represents a small fraction of the impressive suite of transformations catalyzed by laboratory-evolved RNAs,6 possibly due to a lack of unbiased methods for detecting reactive cellular RNAs.
Chemical tools to discover novel functional RNAs lag behind the wealth of approaches available to investigate the proteome. Probes exploiting the ubiquity of unusually nucleophilic functional groups have found widespread use in activity-based protein profiling (ABPP)7,8 and self-labeling fusion proteins that are now widely used to study and manipulate proteins.9,10,11,12,13,14 ABPP uses electrophilic small-molecule “activity-based probes”, typically based on previously discovered irreversible enzyme inhibitors, linked to an affinity tag such as biotin. Enzymes that react with these probes are isolated and identified using the affinity tag. Two recent reports described the use of SELEX to isolate synthetic RNAs that react with α-halo acetamides,15,16 which have been used as mechanism-based inhibitors of serine proteases. The development and use of activity-based probes to identify naturally occurring, unusually reactive RNAs, however, remains unexplored. Current tools for studying RNA function instead rely predominantly on non-covalent binding between an RNA and its corresponding ligand or receptor.17,18,19,20
In this work we report the development of activity-based RNA probes and their initial application to identify unusually reactive genome-encoded RNAs (Fig. 1a). This method enables the unbiased identification of chemically reactive RNAs and complements current approaches that rely on secondary structure prediction or sequence homology with known RNAs. We identified several electrophilic small molecules tuned to be unreactive to most RNA molecules, but to react with RNAs containing unusually nucleophilic functional groups. We used these probes to perform the in vitro selection of unusually reactive genome-encoded RNA fragments, resulting in the identification of a novel 42-nt catalytic RNA from the thermophilic archaea Aeropyrum pernix. This RNA catalyzes the nucleophilic attack of a guanosine onto a disubstituted epoxide that is unreactive to other RNAs, resulting in irreversible C–N bond formation. After establishing a minimal reactive motif and performing a bioinformatic search for examples in other organisms, we identified numerous examples of this unusually reactive RNA encoded in the mouse genome.
Figure 1. Electrophilic probes for the discovery of unusually nucleophilic RNA.

(a) Electrophilic small molecules with carefully tailored reactivity selectively react with unusually nucleophilic RNAs. The reacted RNA species can be isolated using an affinity handle such as biotin linked to the probe. (b) Electrophiles were assayed by LC/MS for their ability to react irreversibly with an RNA pool of random sequence. Electrophiles capable of forming products with random RNA sequences, as evaluated by mass spectrometry, were deemed too reactive, and variants with attenuated reactivity were identified. An excessively electrophilic candidate probe (top) and an attenuated version (bottom) are shown. (c) Eight electrophiles of tuned electrophilicity that can serve as probes to identify unusually reactive RNAs. R = biotin.
Recognizing the potential utility of a self-labeling RNA that reacts with a bioorthogonal small molecule, we developed this catalytic RNA into a tool for selective and irreversible RNA modification. The resulting RNA enables the covalent conjugation of diverse small-molecules to an RNA of interest and provides a robust handle for selective RNA capture from total RNA or even from cell lysate. We applied this tool to capture the ASH1 mRNA and enrich three known ASH1 mRNA-binding proteins from yeast cell lysate.
Results
Development of Chemical Probes for Reactive RNAs
To develop activity-based chemical probes for RNA, we identified electrophilic small molecules capable of selectively and covalently labeling RNA functional groups with enhanced nucleophilicity, a feature shared among virtually all known RNA catalysts.4,15,21,22 We screened commercially available electrophilic small molecules for their inability to covalently modify an RNA pool of random sequence (N80), representing typical, non-reactive RNA. This RNA pool was incubated with each electrophile, then digested to mononucleotides by nuclease P1 and characterized by liquid chromatography/mass spectrometry (LC/MS).23 Electrophiles capable of efficiently modifying the random RNA pool (> 10% of mononucleotides modified) were deemed too reactive (Fig. 1b). Instead, we focused on probe candidates that were insufficiently electrophilic to react with the random RNA pool to a detectable extent (< 0.1%) (Fig. 1b).
Six electrophilic probes were identified from this screening approach: disubstituted epoxide 1, α-bromo acetamide 2,24 ester 3, thioester 4, acrylamide 5, and disubstituted α,β-unsaturated ketone 6 (Fig. 1c). In addition, we identified two small molecules that have no detectable reactivity with the random RNA pool and that have been previously used for protein activity-based profiling: α-chloro acetamide 7,15,25,26 and fluorophosphonate 8.27 Each of these eight probes was synthesized in a biotinylated form to enable streptavidin affinity capture of RNAs that form covalent bonds with the probes (Fig. 1c).
Identification of Epoxide-Reactive Catalytic RNAs
The eight biotinylated probes were combined into one cocktail and incubated with a pool of genome-derived RNA fragments from nine different organisms spanning all three kingdoms of life (Online Methods and Supplementary Information). Following five rounds of in vitro selection for binding to immobilized streptavidin, we observed enrichment of a substantial portion of the RNA pool and apparent convergence on discrete RNA species (Supplementary Results, Supplementary Fig. 1 and 2a). These results establish that unusually reactive RNA species exist within genome-encoded RNA pools and can be isolated using small-molecule probes of appropriate electrophilicity.
To identify the electrophile(s) that reacted with the enriched RNA, we performed one additional round of in vitro selection and then incubated the resulting post-round 6 RNA pool with each of the eight electrophilic probes. Streptavidin incubation and gel mobility shift assays revealed that one or more species of the post-round 6 RNA pool reacted with the disubstituted epoxide probe 1 (Supplementary Fig. 2b). Nuclease P1 digestion and LC/MS analysis resulted in a mass spectrum consistent with reaction of the epoxide probe with a guanosine nucleobase to yield the corresponding epoxide ring-opened product (Supplementary Fig. 2c).
To identify the reactive RNA, we subjected the round 6 DNA pool to high-throughput sequencing. We transcribed in vitro in the absence of flanking primer-binding sites the three most abundant round 6 library members and assayed their reactivity with epoxide probe 1 by gel mobility shift. The most abundant species (40% of reads) was a 125-nt fragment from two distinct regions of the B. fragilis genome, likely the result of ligation of two separate DNA fragments during construction of the genome-encoded RNA library. Although the full RNA reacted with epoxide probe 1, the individual genome-encoded fragments did not react with the probe. The second most abundant species (6.5% of reads) was a 52-nt fragment of the M. jannaschii genome. This RNA species showed only low levels of reactivity with the epoxide probe, suggesting that its reactivity with the probe is dependent on the specific sequence context of the selection. The third most abundant species (4.4% of reads) was a 63-nt fragment of the thermophilic archaea Aeropyrum pernix. This fragment catalyzed the epoxide ring-opening alkylation reaction in the presence or absence of the primer binding sites (Fig. 2a), indicating context independence.
Figure 2. A catalytic RNA from the A. pernix genome that reacts with a disubstituted epoxide.

(a) PAGE streptavidin gel mobility shift following incubation of random sequence RNA (“random”), the A. pernix species from Round 6 of the selection (“selected”), and the A. pernix fragment corresponding to the reference genome sequence (“genomic”) with epoxide probe 1 (1 mM) for 16 h at room temperature (1 µM RNA). The complete gel is shown in Supplementary Figure 12. (b) Secondary structure model of the minimized A. pernix catalytic RNA. The reactive guanosine (blue) was identified by RNAse T1 digestion and mass spectrometry. (c) Sequence logo based on high-throughput DNA sequencing of the RNA species surviving selection of a partially randomized RNA pool derived from the minimized 42-nt A. pernix catalytic RNA.
Characterization of the A. pernix Catalytic RNA
The A. pernix RNA sequence from the round 6 selection contained seven mutations or deletions compared with the A. pernix reference genome sequence (Supplementary Fig. 3).28 These mutations may have arisen from differences between the genomic DNA used to create the RNA pool and the reference genome, or from errors introduced during PCR amplification. To determine if these differences affect epoxide-opening activity, we generated an A. pernix fragment corresponding to the reference genome sequence, and observed no significant change in self-alkylation activity (Fig. 2a). Next we minimized the catalytic RNA by generating and assaying progressive 5’ and 3’ truncations (Supplementary Fig. 4), resulting in a minimized 42-nt catalytic RNA with activity similar to that of the round 6 sequence.
The minimized 42-nt A. pernix RNA exhibited a kcat of 1.6 × 10−3 min−1 and Km for epoxide probe 1 of ≤ 0.012 M (kcat/Km ≥ 0.13 min−1 M−1) (Supplementary Fig. 5). The kcat/Km for the epoxide reaction of the minimized 42-nt RNA is at least 1,900-fold higher than that of a 42-nt pool of random-sequence RNA, which showed no detectable reaction even after incubation with 8 mM 1 for 60 hours. This catalytic efficiency is comparable to that of many known in vitro selected catalytic RNAs,29,30,31,32 and represents the first reported example of a catalytic RNA capable of promoting epoxide ring opening.
LC/MS and NMR spectroscopic characterization of the product of the RNA-catalyzed epoxide ring opening revealed that N7 is the site of guanosine modification, similar to previous reports of DNA reacting with activated epoxides33,34 (Online Methods and Supplementary Note).
Sequence Requirements of the A. Pernix Catalytic RNA
Computational secondary structure prediction35 suggests that the A. pernix catalytic RNA folds into a stem-bulge-stem-loop structure (Fig. 2b). In order to probe this structural model and to identify the minimal sequence requirements for reactivity, we generated a partially randomized RNA pool derived from the minimized 42-nt A. pernix catalytic RNA and performed four rounds of reselection for epoxide reactivity (Supplementary Fig. 6a and Supplementary Note). The enriched RNA pool was reverse transcribed and analyzed by high-throughput sequencing, resulting in the sequence logo shown in Fig. 2c.36 The results are consistent with the predicted stem-bulge-stem model, reveal a highly conserved bulge region, and suggest that the 10-nt loop can be mutated without substantial loss of activity (Fig. 2b). This structural model was also supported by the results of site-directed mutagenesis experiments (Supplementary Table 2).
Structural Requirements of the Epoxide Probe
To identify RNA-probe interactions required for activity, we investigated the structural requirements of the disubstituted epoxide substrate by synthesizing and assaying a series of epoxide analogs. While removal of the biotin group did not affect probe reactivity, shortening the alkyl chain reduced activity (Fig. 3). Replacing the ester group with an amide similarly decreased reaction efficiency (Fig. 3). These results suggest that the RNA might form direct contacts with the ester group in our substrates. We also studied the effect of epoxide stereochemistry, and found that the cis epoxide reacted 22-fold faster with the RNA than the trans epoxide (Fig. 3b), demonstrating that the catalytic RNA is highly stereospecific with respect to the epoxide functional group. Together, these results suggest that catalysis is dependent on multiple, specific RNA-substrate interactions.
Figure 3. Epoxide substrate selectivity of the catalytic RNA.

(a) Epoxide analogues were tested to determine substrate specificity of the A. pernix catalytic RNA. Modification was analyzed by LC/MS following digestion into mononucleotides by nuclease P1. Relative reaction efficiencies, shown in parentheses, were determined by comparing the ion counts of unmodified GMP to modified GMP; all values are relative to the reaction efficiency of epoxide 9 (defined as 1.00). R = −(CH2)5NHBoc. (b) Reaction efficiency of the A. pernix catalytic RNA (1 µM) with trans-epoxide 1 (1.3 mM) over time. Time-course data show mean values ± s.d. for three replicates. (c) Epoxide probes that react efficiently with the optimized catalytic RNA as determined by LC/MS. (d) For azide-epoxide 14 and TAMRA-epoxide 16, RNA modification was also visualized by fluorescence imaging (λex = 532 nm, λem = 580 nm) following PAGE. Lane 1: background reactivity or binding of the DBCO-TAMRA probe with the catalytic RNA. Lane 2: incubation with azide-epoxide 14 followed by copper-free click reaction with DBCO-TAMRA. Lane 3: incubation with TAMRA-epoxide 16. The complete gel is shown in Supplementary Figure 13.
Epoxide-Opening RNAs Encoded by the Mouse Genome
To evaluate the potential existence of this catalytic RNA in other organisms, we developed a reactive minimal motif based on the sequence logo and mutational studies (Supplementary Fig. 6c) and performed bioinformatics searches for examples of the motif in the genomes of Saccharomyces cerevisiae, Escherichia coli, and mouse. Although no examples of this motif were discovered in the smaller S. cerevisiae and E. coli genomes, 233 occurrences were found in the mouse genome.
To establish the ability of the minimal motif to predict catalytic RNA activity and to test if the examples encoded by the mouse genome were bona fide epoxide-reactive RNAs, we transcribed in vitro 44 of the 233 mouse genome-encoded candidate catalytic RNAs, chosen at random, and assayed their reactivity with disubstituted epoxide 1 by gel mobility-shift assays. Of the 44 genomic variants that were tested, 14 (32%) showed substantial levels of reactivity with the epoxide compared to that of random RNA. These observations demonstrate that the minimal catalytic RNA motif can be used to identify other candidate catalytic RNAs and also establishes that multiple examples of this unusually nucleophilic RNA are encoded in the mouse genome.
Application for Selective RNA Modification
In contrast to the large number of available methods that effect the selective covalent modification of proteins,9,10 few techniques exist to selectively modify a genetically encoded RNA of interest.16 Such a method would provide a valuable tool for applications including live-cell RNA imaging,37,17,18 identification of RNA-binding proteins,19,20 and studies of RNA degradation.38,39,40 An ideal tool would enable efficient, selective, and bioorthogonal modification with a small-molecule probe capable of carrying diverse chemical functionality including affinity handles, azide- or alkyne-containing chemical handles, and fluorophores. We speculated that the self-labeling A. pernix RNA might serve as such a tool for selective RNA modification in complex biological samples. In order to improve the rate of the epoxide-RNA reaction to facilitate efficient covalent RNA modification, we performed seven rounds of highstringency reselections with decreasing epoxide incubation times on the partially randomized 42- nt A. pernix RNA library, resulting in an optimized catalytic RNA exhibiting a 5-fold faster rate of reaction (Supplementary Figs. 6, and 7).
Generality of the Epoxide Probe
Next we determined the ability of the epoxide probe to support catalytic RNA-mediated RNA modification with diverse chemical groups. Probes in which the biotin group was replaced with an azide, alkyne, carboxamide, alkyl chain, or tetramethylrhodamine (TAMRA) fluorophore were synthesized and tested for their ability to react with the optimized catalytic RNA (Fig. 3c and 3d). All of the probes assayed remained efficient RNA substrates, suggesting that this system can mediate a diverse range of RNA functionalization.
Catalytic RNA-Mediated RNA Enrichment
We integrated the above findings to test the ability of the optimized catalytic RNA when transcriptionally fused to an RNA of interest to enable enrichment of that RNA from a complex biological mixture. We cloned the optimized catalytic RNA at the 3’ end of the human 5S rRNA and generated the resulting RNA using T7 RNA polymerase. Following 8-hour incubation with epoxide probe 1, we evaluated modification of the RNA by gel mobility shift, revealing reaction efficiency (35%) similar to that of the unfused catalytic RNA (40%). This observation demonstrates that the optimized catalytic RNA can retain its activity when appended to an unrelated RNA of interest.
To establish the ability of the catalytic RNA to enrich an RNA of interest from total cellular RNA (Fig. 4a), we transfected the 5S rRNA–catalytic RNA construct into HEK 293T cells and isolated total RNA. As a control we used a construct in which the reactive G9 and base C35, predicted to pair with G9, were swapped to yield an inactive isomeric mutant catalytic RNA predicted to adopt the same secondary structure. Total cellular RNA was incubated for 5 hours with azide epoxide 14, followed by copper-free click chemistry using dibenzocyclooctyne–TAMRA (DBCO–TAMRA) to install a TAMRA fluorophore and PAGE analysis (Fig. 4b). While no TAMRA-bound 5S rRNA product was generated from the mutant catalytic RNA, we observed a strong fluorescent band of the expected size for the sample containing the active catalytic RNA (Fig. 4b). Repeating this experiment with a construct containing three consecutive copies of the catalytic RNA (5S rRNA–catalytic RNA3) resulted in 3.2-fold higher fluorescence signal (Fig. 4b).
Figure 4. Application of the epoxide-opening catalytic RNA to enrich RNAs of interest from total cellular RNA and to capture RNA-binding proteins.

(a) Transcriptional fusion of a self-labeling catalytic RNA to an RNA of interest may enable selective, covalent RNA modification in a complex biological sample. (b) Total RNA from HEK 293T cells was reacted with epoxide-azide 14, followed by DBCO-TAMRA. Total RNA was analyzed by PAGE and TAMRA-modified RNAs were visualized by fluorescence imaging. Lanes 1 and 6: in vitro transcribed catalytic RNA-fused 5S rRNA containing one or three copies of the catalytic RNA, respectively, rather than cellular RNA. Lanes 2 and 3: the inactive C9-G35 mutant RNA. Lanes 4–8: 5S rRNA fused to one copy (lanes 4–5) or three copies (lanes 6–8) of the active optimized catalytic RNA. Bands at the top of the gel result from incomplete removal of excess DBCO-TAMRA probe or background labeling of cellular rRNAs/mRNAs. The complete gel is shown in Supplementary Figure 14. (c) Western blot probing the presence of three known ASH1 mRNA-binding proteins (Puf6, Khd1, and She2) and one non-binding protein control (Guk1) in yeast cell lysate. Lanes 1 and 2: Lysate incubated overnight with streptavidin-coated magnetic beads only (lane 1) or pre-incubated with 5 µg of epoxide 1-modified ASH1-catalytic RNA (lane 2). Unbound proteins were washed away and captured proteins were eluted at 95 °C. Lane 3: Input lysate prior to incubation with beads. The complete gel is shown in Supplementary Figure 15.
To quantify the ability of the catalytic RNA to enrich an RNA of interest from total cellular RNA, we captured biotinylated RNA using immobilized streptavidin and quantified the amount of enriched catalytic RNA-fused 5S rRNA by reverse transcription-quantitative PCR (RT-qPCR). We isolated total RNA from HEK 293T cells transfected with a vector expressing the human 5S rRNA transcriptionally fused to either one or three copies of the catalytic RNA, or fused to the inactive mutant catalytic RNA. Total cellular RNA from each sample was incubated with epoxide probe 1 and captured with streptavidin-linked magnetic beads. Following on-bead reverse transcription, we performed qPCR to quantitate the extent to which the catalytic RNAfused transcript was enriched over an inactive transcript and observed 125- and 541-fold enrichment for transcripts fused to one and three copies of the catalytic RNA, respectively (Online Methods and Supplementary Table 1).
Next we attempted to selectively modify and isolate a catalytic RNA-fused transcript from total human cell lysate. HEK 293T cells expressing 5S rRNA–catalytic RNA transcripts were lysed and incubated with epoxide probe 1. Total RNA was isolated and the efficiency of reaction with the epoxide probe was quantified by RT-qPCR (following enrichment with immobilized streptavidin). The 5S rRNA transcript fused to one or three copies of the catalytic RNA was enriched 57-fold and 398-fold, respectively, over the inactive variant. These findings establish that catalytic RNA-fused transcripts are selectively modified even in mammalian cell lysate, enabling their facile isolation.
Unbiased Enrichment of RNA-Binding Proteins
The ability to selectively and covalently modify and immobilize an RNA of interest (Fig. 4a) in principle enables the rapid isolation of RNA-binding proteins.19,20 To demonstrate this capability, we transcribed in vitro the well-characterized yeast mRNA ASH141 with the optimized catalytic RNA inserted immediately upstream of the 3’-UTR. We immobilized the transcript by incubation with epoxide biotin probe 1 and capture with streptavidin-coated beads. We treated the immobilized ASH1 mRNA with yeast lysate from three strains individually expressing TAP-tagged proteins known to bind the ASH1 mRNA (Puf6, She2, and Khd1) and lysate from a fourth strain expressing TAP-tagged Guk1, which is not known to bind the ASH1 mRNA. Following extensive washing, binding proteins were eluted from the beads. Western blot revealed 29-fold average enrichment of the three known binding proteins relative to samples containing no RNA and no substantial enrichment of the non-binding protein Guk1 (Fig. 4c). These results demonstrate that the catalytic RNA–epoxide reaction enables site-specific biotinylation and immobilization of an mRNA of interest, followed by efficient capture of multiple proteins that bind the mRNA from cell lysate.
Discussion
The use of electrophilic chemical probes to discover unusually reactive RNAs provides an unbiased alternative to methods that rely on homology to known catalytic RNAs or on secondary structure prediction. To realize this potential, we identified electrophilic chemical groups with reactivity profiles biased towards reactive RNA modification, but insufficiently reactive to modify random RNA molecules. Although the reactivity of activated allylic or benzylic epoxides towards nucleic acids is well documented,33,34 to our knowledge the A. pernix catalytic RNA described in this work represents the first example of an RNA that catalyzes nucleophilic attack of an unactivated epoxide. The reaction occurs at a single guanosine base, resulting in C–N bond formation at N7.
Additional studies revealed that the A. pernix catalytic RNA requires only 42 nt for activity and likely adopts a stem-bulge-stem architecture with a stereospecific active site. Reselection using a partially randomized RNA pool established a sequence logo that was used to identify a minimal reactive motif and many naturally occurring transcripts capable of reacting with the epoxide. While many tested naturally occurring genome-encoded variants of the catalytic RNA proved to be active, it is possible that their activity is unrelated to any biological role. Elucidating their potential biological relevance, if any, will require future investigation.
The use of the transcriptionally fused catalytic RNA to modify an mRNA at a single position provides a unique tool for RNA labeling and the isolation of RNA-binding proteins. Standard approaches to isolating RNA-binding proteins involve in vitro transcription using biotinylated uracil,42 resulting in a heterogeneous mixture of biotinylated RNAs, or the use of a fused aptamer,19 which requires careful development of washing conditions to ensure maintenance of aptamer binding. Because the method described here establishes a robust covalent bond between the probe and the RNA, washing conditions can be vigorous; for example, RNA captured in this study survived successive washes with 8 M urea. The chemical orthogonality of the catalytic RNA-epoxide reaction is sufficient to enable selective covalent RNA modification in cell lysate, suggesting its value to studying RNAs of interest in native biological contexts. Moreover, the modularity of the epoxide probe suggests that cell-permeable analogues based on these developments may enable live-cell RNA imaging or RNA-protein crosslinking applications.
Online Methods
General
Unless otherwise noted, all starting materials were obtained from commercial suppliers and were used without further purification.
LC/MS Screen of RNA Modification by Small-Molecule Probes
A RNA pool of random sequence (N80) (1 µg), 150 mM NaCl, and 50 mM Na-HEPES, pH 7.4, was incubated at 65 °C for 5 minutes, then cooled at room temperature for 5 minutes. MgCl2 (10 mM) was then added to a total volume of 25 µL and the solution incubated for 10 minutes at room temperature, followed by addition of the small-molecule probe in DMSO (25 mM). After incubation at room temperature for 16 hours, the RNA was precipitated by addition of 5 µL of 3 M NaOAc and 180 µL of EtOH. Following EtOH precipitation, the RNA was taken up in 49 µL of 50 mM NH4OAc, pH 4.5. Nuclease P1 (0.5 U, Wako Chemicals) was added, and the solution was incubated at 37 °C for 1 hour. Following lyophilization, the powder was resuspended in 45 µL H2O.
RNA modifications were detected using negative-ion mode on a Waters Acquity ultra-performance liquid chromatography (UPLC) quadrupole TOF Premier mass spectrometer. LC was performed using a gradient from 0.1% (w/v) aqueous ammonium formate (A1) to methanol (B1) on an Acquity UPLC BEH C18 column (1.7 µm, 2.1 mm × 100 mm, Waters) at constant flow rate of 0.3 mL min−1. The mobile phase composition was: 100% A1 for 3 min; linear increase over 8 min to 100% B1; maintain at 100% B1 for 2 min; return to 100% A1 over 1 min. Electrospray ionization used a capillary voltage of 3 kV, a sampling cone voltage of 40 V, and a low-mass resolution of 4.7. The desolvation gas temperature was 300 °C, the flow rate was 800 L h−1, and the source temperature was 150 °C.
In Vitro Selection
Fragmented genomic DNA pools were constructed as described previously.43 DNA (0.2 µM) from the preceding round of the selection (or the starting pool for the first round) was incubated with 1X T7 RNA polymerase buffer (NEB), 1 mM of each rNTP, 5 mM DTT, and 28 µL of T7 RNA Polymerase (NEB) in 700 µL reaction volume for 10 hours at 37 °C. The transcription mixture was divided into two halves and 30 µL 4 M NaCl and 1 mL of EtOH was added to each half. Following EtOH precipitation, the RNA was purified on a 10% TBE-UREA PAGE Criterion gel (Bio-Rad; 240 V for 35 minutes). The excised gel containing the desired RNA was incubated in 300 mM NaCl (450 µL) at 4 °C overnight, at which point it was precipitated with ethanol and resuspended in 125 µL H2O.
The entire RNA pool was incubated at 37 °C for 15 minutes in 1X DNase buffer with 20 U DNase I (NEB). The solution was adjusted to 300 mM NaCl and 200 µL total volume and the RNA isolated by phenol/chloroform extraction, followed by EtOH precipitation. The resulting RNA pellet was resuspended in 100 µL H2O.
The reaction between small-molecule probe(s) and the RNA pool was performed as follows: the RNA pool (1 µM) in buffer containing 150 mM NaCl, 25 mM Na-HEPES, pH 7.4, was heated at 65 °C for 5 minutes, then allowed to cool for an additional 5 minutes. MgCl2 (10 mM) was then added for a total volume of 45 µL and the solution incubated for 10 minutes, followed by addition of 1.3 mM of the biotinylated small-molecule probe in DMSO. After incubation for the desired time, the RNA was precipitated by the addition of 5 µL of 3 M NaOAc and 180 µL of EtOH.
Dynabeads MyOne Streptavidin C1 coated beads (200 µg; Life Technologies) were prepared according to the instruction manual. The beads were resuspended in 20 µL of binding buffer (25 mM Na-HEPES, pH 7.4, 500 mM NaCl, 2.5 mM EDTA) and added to the RNA pellet. Following 25 minutes of room-temperature incubation on a rotation device, the beads were washed three times with 300 µL binding buffer, six times with 400 µL denaturing wash buffer (25 mM Na-HEPES, pH 7.4, and 5 mM EDTA in 8 M urea), three times with 300 µL H2O, and then resuspended in 41.2 µL of H2O.
To the RNA/bead solution was added 2 µM reverse transcription (RT) primer (GCCGCGAATTCACTAGTGATT). The solution was incubated at 65 °C for 5 minutes and room temperature for 3 minutes. 43.8 µL of RT solution (RT solution: 1.25 mM dNTPs (NEB), First Strand Buffer (Invitrogen), 230 mM DTT). Following removal of 19 µL of the mixture for a no-enzyme negative control, 4 µL of SuperScript III (Invitrogen) was added and the mixture incubated at 55 °C for 90 minutes.
The RT mixture was incubated with 35 µL of base-hydrolysis solution (80 mM Tris base, 15 mM EDTA, 1.25 M KOH) at 95 °C for 15 minutes and adjusted to pH 8.0 with 1 M HCl. 2 µL of this mixture was used in the subsequent PCR step using Taq DNA Polymerase (NEB).
Streptavidin Gel Mobility Shift Assays
The pellet resulting from EtOH precipitation of a reaction of RNA (750 ng) with a small-molecule probe was resuspended in 21 µL H2O. The RNA (7 µL) and streptavidin (NEB; 1 µg) were incubated at room temperature for 25 minutes and then combined with gel electrophoresis loading buffer. The sample was electrophoresed on a 10% PAGE denaturing gel (240 V, 35 minutes) and visualized following incubation with SYBR Green II (Life Technologies).
Kinetic Characterization of 42-nt A. Pernix Catalytic RNA
At substrate concentrations greater than 8 mM, low levels of RNA modification were observed, potentially due to substrate aggregation. To calculate an upper limit for the Km value, we fit a classic Michaelis-Menten equation to the experimentally derived data and additional values at very large substrate concentrations (more than 100-fold the calculated Km) that would be expected to result in complete RNA modification.
Analysis of High-Throughput Sequencing
To determine the fragment abundance in the round 5 cDNA library pool, the 80 bases after the constant primer sequence (TAGGCCGCGGGAATTCGATT) were identified. Sequences with Illumina Base quality scores of A through J at more than half of the positions across this 80-nt sequence were considered further. Sequences related by 12 or less mutations in this 80-nt sequence were binned and considered to have originated from the same fragment in the library. To determine the reselected variants from the partially randomized RNA pool derived from the minimized 42-nt A. pernix catalytic RNA, 42-nt sequences between the two constant primer sequences (TAGGCCGCGGGAATTCGATT and AATCACTAGTGAATTCGC) were identified. Only sequences with Illumina Base scores of B through J at more than half of the positions between the two primer sequences were considered further.
Determination of the Nucleotide Position of Epoxide-Catalytic RNA Modification
The canonical 42-nt A. pernix catalytic RNA and a variant featuring a single base substitution (G10 to A10) (see below) was incubated with epoxide-alkyne 14.
5’-GGCAAAG7A8G9N10G11CCCTGGGGTATGGAAGGGCTAGGCTCGTTGT-3’
Following EtOH precipitation, the RNA was resuspended in 50 mM NH4OAc, pH 6 (340 µL) and incubated with 1,500 U RNase T1 (Roche) at 37 °C for 1 hour. Following lyophilization, the powder was resuspended in 45 µL H2O and analyzed by LC/MS as described above.
RNase T1 cleaves the 3’-phosphodiester bond of unmodified guanosine nucleotides, but appears to have altered cleavage specificity for epoxide-modified G. Digestion of the canonical 42-nt A. pernix catalytic RNA (N10 = G), following modification by epoxide 9, yielded m/z fragments corresponding to GG–epoxide ([M−H]− m/z = 961.3) and AGG–epoxide ([M−H]− m/z = 1290.4). These ions are consistent with epoxide reaction at either G9 or G10 to yield the 3-nt, epoxide-modified fragment below (AGG–epoxide), which undergoes further partial digestion between A8 and G9 to yield the GG–epoxide fragment:
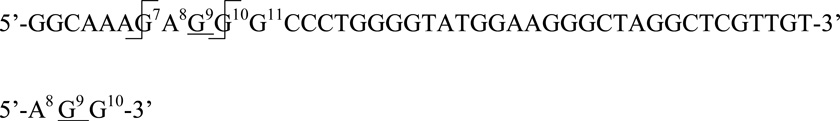
A similar analysis of the mutant in which N10 = A established epoxide reaction at G9. RNase T1 digestion yields fragments corresponding to GA–epoxide ([M−H]− m/z = 945.3), AGA–epoxide ([M−H]− m/z = 1274.4), GAG–epoxide ([M−H]− m/z = 1290.4), and AGAG–epoxide ([M−H]− m/z = 1619.5), were observed. The change in observed products establishes that modification occurred at a nucleotide adjacent to N10 and is consistent with reaction at G9. The observed ions can be explained by canonical RNase T1 cleavage at the 3’-phosphodiester bond of unmodified guanosine nucleotides to yield the 4-nt fragment below (AGAG–epoxide), followed by cleavage between A8/G9 to yield GAG–epoxide, between A10/G11 to yield AGA–epoxide, and at both A8/G9 and A10/G11 to yield GA–epoxide.
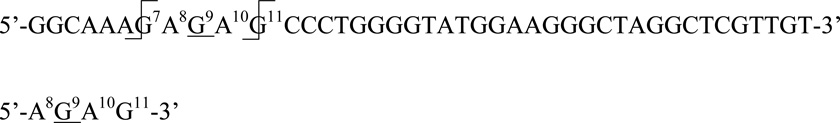
Synthesis and Characterization of the Authentic Chemical Standard
The epoxide substrate 9 and guanosine 5’-monophosphate disodium salt hydrate (GMP; Sigma Aldrich) were combined in equimolar quantities (0.2 mmole) in glacial acetic acid (1.5 mL) and heated at 37 °C for 7 hours.44 After allowing the mixture to cool, the acetic acid was removed at reduced pressure and the resulting residue was resuspended in H2O and purified by reverse-phase HPLC (Agilent 1200) using a C18 stationary phase column (Eclipse-XDB C18, 5 µm, 9.4 × 200 mm) and acetonitrile / triethylammonium acetate (0.1 M) gradient.
To cleave the ribose from the modified guanine base, the epoxide-GMP product was heated in 1 M HCl (2 mL) at 95 °C for 7 hours, which also resulted in hydrolysis of the ester functionality. The mixture was neutralized with ammonium hydroxide and then lyophilized. The resulting residue was resuspended in H2O and purified by HPLC.
Catalytic RNA Construct for RNA Labeling
The catalytic RNA was cloned into the anticodon loop of a tRNA scaffold.45 For the 5S rRNA, we employed a previously published construct containing a U5 transcription termination signal and the endogenous mammalian 5S promoter,46 analogous to a recent report detailing 5S rRNA imaging using a fused aptamer.47 The tRNA–optimized catalytic RNA sequence is as follows, with the optimized catalytic RNA in red:
The construct containing three tandem copies of the catalytic RNA was as follows:

Transfection of Mammalian Cells and Enrichment of Total RNA
Human embryonic kidney cells (HEK 293T) were obtained from ATCC and maintained in Dulbecco’s modified Eagle medium (DMEM, Life Technologies) supplemented with 10% (vol/vol) fetal bovine serum (FBS, Life Technologies) and penicillin/streptomycin (1X, Amresco). Cells at ~75% confluency were transfected 1 day after plating in 10 cm2 plates (Greiner Bio-One) with 50 µL Lipofectamine 2000 (Life Technologies) and 15 µg of plasmid DNA. 2–3 days following transfection, total RNA was isolated by addition of TRIzol (Life Technologies) and subsequent use of RNeasy Mini spin columns (Qiagen).
Labeling and Enrichment of Catalytic RNA-Fusion Transcripts in Total RNA
Total RNA from HEK 293T cells (10 µg) was incubated with the biotin-epoxide (1.3 mM) or the azide-epoxide (1.3 mM) as described above for 6 hours. Following EtOH precipitation, the azide-epoxide RNA was resuspended in 30 µL PBS and incubated for 1–3 hours with TAMRA-DBCO (15 µM; Click Chemistry Tools). The resulting solution was passed through two successive CENTRI•SEP Spin Columns (Princeton Separations) equilibrated with water and one RNEasy MinElute Column (Qiagen) to remove free TAMRA-DBCO. RNA concentration was quantified, normalized for all the samples, separated on a 5% PAGE-urea gel, and fluorescence was visualized using a Typhoon TRIO Variable Mode Imager (λex = 532 nm, λem = 580 nm).
For RT-qPCR quantification, the RNA pellet was resuspended in 50 µL H2O. RNA was incubated with 200 µg Dynabeads MyOne Streptavidin C1 coated beads (Life Technologies) in either binding buffer (the pulldown material; 1 µg RNA) or H2O (the no-pulldown material, 50 µg RNA). The samples were incubated at room temperature for 25 minutes, then washed three times with 300 µL binding buffer, six times with 500 µL denaturing wash buffer, and three times with 300 µL H2O. The beads were then resuspended in 10 µL H2O. On-bead reverse transcription was performed using 200 U Protoscript II (NEB) and 6 µM random primers (NEB) according to the manual. The RT mixture was incubated for 7 minutes at 25 °C and 75 minutes at 42 °C. RNase H (5 U; NEB) was then added and the sample incubated at 37 °C for 20 minutes. qPCR was performed using 1 µL of cDNA template and 24 µL of iTaq Universal SYBR Green Supermix qPCR mix (Bio-Rad). Quantitative PCR was performed on a CFX-96 Real-Time System with a C100 Thermocycler (Bio-Rad).
As described in the main text, enrichment values were determined by comparing the ΔCT of the catalytic RNA samples with the ΔCT of the inactive mutant RNA, where the ΔCT corresponds to the difference between the experimental sample and a control sample lacking streptavidin-linked bead capture. The ΔΔCT values were normalized by the ΔΔCT values of the housekeeping genes HPRT1 and tubulin. The average ΔCT values for each of the cDNAs of interest are shown in Supplementary Table 1.
Labeling and Enrichment of Catalytic RNA-Fusion Transcripts in Cell Lysate
HEK 293T cells were cultured in 15 cm2 plates as described above, washed once with 7.5 mL PBS, then removed from the plate in a total of 1 mL of PBS using a rubber-headed plate scraper. The cells were then lysed by passing through a QiaShredder spin column (Qiagen), followed by addition of 80 U murine RNase inhibitor (NEB). Cell lysate (15 µL) was combined with 1X PBS (215 µL), 20 mM NaCl, 25 mM Na-HEPES, pH 7.4, and MgCl2 (10 mM) and incubated at room temperature for 10 minutes, followed by addition of biotin-epoxide probe 1 (1.33 mM). The solution was incubated at room temperature for 6 hours. TRIzol LS (750 µL; Life Technologies) was added and RNA was isolated according to the TRIzol LS product instructions.
Pulldown of ASH1 mRNA-Binding Proteins
Catalytic RNA-fused ASH1 mRNA was transcribed in vitro following the protocol provided in the T7 High Yield RNA Synthesis Kit (NEB). The RNA was reacted with biotin-epoxide for 24 hours and then precipitated with ethanol as described above. The biotinylated mRNA was immobilized on 400 µg Dynabeads MyOne Streptavidin C1 coated beads (Life Technologies) as follows: yeast tRNA (100 µg/mL; Life Technologies), 10 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 5 µg of labeled RNA (total volume 49.5 µL) were incubated at 65 °C for 5 minutes and then cooled at room temperature for 5 minutes. MgCl2 (10 mM) was then added and the solution was incubated at room temperature for 10 minutes to enable RNA folding. The streptavidin-coated magnetic beads were prepared according to the product manual, but washed twice with 10 mM Tris-HCl, pH 7.4. The supernatant was removed and the beads resuspended in the RNA solution. The RNA-bead mixture was rotated at room temperature for 1 hour.
TAP-tagged yeast strains from the yeast genome-wide TAP-tagged library48 were cultured in YEPD medium penicillin/streptomycin (1X, Amresco), pelleted, and resuspended in 1.5 mL lysis buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 2 mM MgCl2, 0.5% v/v Triton-X 100, 1 mM DTT, and cOmplete, Mini, EDTA-free protease inhibitor (Roche)). The resuspended cells (900 µL) were combined with 0.5 mm silica-zirconia beads (Biospec Products) in a screw cap vial and bead beaten eight times at one-minute intervals on a Minibeadbeater (Biospec Products). After pelleting cellular debris, the supernatant (50 µL) was combined with the RNA-magnetic beads and the volume adjusted to 300 µL with lysis buffer. Solutions were incubated on a rotation device at 4 °C for 12 hours. The beads were then washed five times with 1 mL wash buffer (identical to lysis buffer but without protease inhibitor) and resuspended in 12 µL H2O and 4 µL load dye. After heating at 95 °C, the supernatant was loaded onto a 4–12% NuPAGE Bis-Tris Mini Gel (Life Technologies), which was run in MES buffer for 65 minutes at 150 V. Analysis was then performed by Western Blot.
Supplementary Material
Acknowledgements
This work was supported by NIH/NIGMS R01 GM065865 and the Howard Hughes Medical Institute. R.I.M. and S.M. were supported by a NIH National Research Service Award Postdoctoral Fellowship (F32GM099359 and F32GM101751). We thank Loyal Goff for assistance with bioinformatics analysis, Prof. David Engelke for providing the 5S rRNA plasmid, and Prof. Eranthie Weerapana for providing the fluorophosphonate probe. We are also grateful to Christoph Dumelin, Aaron Leconte, Lynn McGregor, David Thompson, and Dmitry Usanov for helpful discussions.
Footnotes
Author Contributions
R.I.M. designed the research, prepared materials, and performed experiments. J.P.G. and S.M. prepared materials and performed research. E.A.C. and W.I.L. prepared materials. D.R.L. designed and supervised the research. R.I.M. and D.R.L. wrote the manuscript.
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.Djebali S, et al. Landscape of Transcription in Human Cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kowtoniuk WE, et al. A Chemical Screen for Biological Small Molecule-RNA Conjugates Reveals CoA-Linked RNA. Proc. Natl. Acad. Sci. USA. 2009;106:7768–7773. doi: 10.1073/pnas.0900528106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dumelin CE, Chen Y, Leconte AM, Chen YG, Liu DR. Nat. Chem. Biol. 2012;8:913–919. doi: 10.1038/nchembio.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doudna JA, Cech TR. The Chemical Repertoire of Natural Ribozymes. Nature. 2002;418:222–228. doi: 10.1038/418222a. [DOI] [PubMed] [Google Scholar]
- 5.Fedor MJ, Williamson JR. The Catalytic Diversity of RNAs. Nat. Rev. Mol. Cell Bio. 2005;6:399–412. doi: 10.1038/nrm1647. [DOI] [PubMed] [Google Scholar]
- 6.Joyce GF. Forty Years of In Vitro Evolution. Angew. Chem. Int. Ed. 2007;46:6420–6436. doi: 10.1002/anie.200701369. [DOI] [PubMed] [Google Scholar]
- 7.Cravatt BF, Wright AT, Kozarich JW. Activity–Based Protein Profiling: From Enzyme Chemistry to Proteomic Chemistry. Annu. Rev. Biochem. 2008;77:383–414. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- 8.Sadaghiani AM, Verhelst SH, Bogyo M. Tagging and Detection Strategies for Activity–Based Proteomics. Curr. Opin. Chem Biol. 2007;11:20–28. doi: 10.1016/j.cbpa.2006.11.030. [DOI] [PubMed] [Google Scholar]
- 9.Hinner MJ, Johnsson K. How to Obtain Labeled Proteins and What to Do With Them. Curr. Opin. Biotechnol. 2010;21:766–776. doi: 10.1016/j.copbio.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 10.Jing C, Cornish VW. Chemical Tags for Labeling Proteins Inside Living Cells. Acc. Chem. Res. 2011;44:784–792. doi: 10.1021/ar200099f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.The electrophile N–methylisotoic anhydride has found widespread utility for RNA structure determination: Low, J.T. & Weeks, K.M. Shape–Directed RNA Secondary Structure Predicition. Methods. 2010;52:150–158. doi: 10.1016/j.ymeth.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baruah H, Puthenveetil S, Choi YA, Shah S, Ting AY. An Engineered Aryl Azide Ligase for Site-Specific Mapping of Protein–Protein Interactions through Photo–Cross–Linking. Angew. Chem. Int. Ed. 2008;47:701–7021. doi: 10.1002/anie.200802088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rutkowska A, Haering CH, Schultz C. A FlAsH–Based Cross–Linker to Study Protein Interactions in Living Cells. Angew. Chem. Int. Ed. 2011;50:12655–12658. doi: 10.1002/anie.201106404. [DOI] [PubMed] [Google Scholar]
- 14.Chidley C, Haruki H, Pedersen MG, Muller E, Johnsson KA. Yeast–Based Screen Reveals that Sulfasalazine Inhibits Tetrahydrobiopterin Biosynthesis. Nat. Chem. Biol. 2011;7:375–383. doi: 10.1038/nchembio.557. [DOI] [PubMed] [Google Scholar]
- 15.Ameta S, Jäschke A. An RNA Catalyst that Reacts with a Mechanistic Inhibitor of Serine Proteases. Chem. Sci. 2013;4:957–964. [Google Scholar]
- 16.Sharma AK, et al. Fluorescent RNA Labeling Using Self-Alkylating Ribozymes. ACS. Chem. Biol. 2014 doi: 10.1021/cb5002119. Advance online publication. [DOI] [PubMed] [Google Scholar]
- 17.Armitage BA. Imaging of RNA in Live Cells. Curr. Opin. Chem. Biol. 2011;15:806–812. doi: 10.1016/j.cbpa.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 18.Baker M. RNA Imaging In Situ. Nat. Methods. 2012;9:787–790. [Google Scholar]
- 19.Walker SC, Scott FH, Srisawat C, Engelke DR. RNA Affinity Tags for the Rapid Purification and Investigation of RNAs and RNA-Protein Complexes. Methods Mol. Biol. 2008;488:23–40. doi: 10.1007/978-1-60327-475-3_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McHugh CA, Russell P, Guttman M. Methods for Comprehensive Experimental Identification of RNA-Protein Interactions. Genome Biol. 2014;15:203–212. doi: 10.1186/gb4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doudna JA, Lorsch JR. Ribozyme Catalysis: Not Different, Just Worse. Nat. Struct. Mo. Biol. 2005;12:395–402. doi: 10.1038/nsmb932. [DOI] [PubMed] [Google Scholar]
- 22.Lilley DMJ, Eckstein F. Ribozymes and RNA Catalysis. Cambridge, UK: RSC Publishing; 2008. [Google Scholar]
- 23.Chen YG, Kowtoniuk WE, Agarwal I, Shen Y, Liu DR. LC/MS Analysis of Cellular RNA Reveals NAD-Linked RNA. Nat. Chem. Biol. 2009;5:879–881. doi: 10.1038/nchembio.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas JM, Perrin DM. Active Site Labeling of G8 in the Hairpin Ribozyme: Implications for Structure and Mechanism. J. Am. Chem. Soc. 2006;128:16540–16545. doi: 10.1021/ja063942y. [DOI] [PubMed] [Google Scholar]
- 25.Weerapana E, Simon GM, Cravatt BF. Disparate Proteome Reactivity Profiles of Carbon Electrophiles. Nat. Chem. Biol. 2008;4:405–407. doi: 10.1038/nchembio.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barglow KT, Cravatt BF. Discovering Disease-Associated Enzymes by Proteome Reactivity Profiling. Chem. Biol. 2004;11:1523–1531. doi: 10.1016/j.chembiol.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 27.Simon GM, Cravatt BF. Activity–Based Proteomics of Enzyme Superfamilies: Serine Hydrolases as a Case Study. J. Biol. Chem. 2010;285:11051–11055. doi: 10.1074/jbc.R109.097600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawarabayasi Y, et al. Complete Genome Sequence of an Aerobic Hyper-Thermophilic Crenarchaeon, Aeropyrum Pernix K1. DNA Res. 1999;6:83–101. doi: 10.1093/dnares/6.2.83. [DOI] [PubMed] [Google Scholar]
- 29.Lee N, Bessho Y, Wei K, Szostak JW, Suga H. Nat. Struct. Biol. 2000;7:28–33. doi: 10.1038/71225. [DOI] [PubMed] [Google Scholar]
- 30.Sengle G, et al. Novel RNA Catalysts for the Michael Reaction. Chem. Biol. 2001;8:459–473. doi: 10.1016/s1074-5521(01)00026-6. [DOI] [PubMed] [Google Scholar]
- 31.Fusz S, Eisenführ A, Srivatsan SG, Heckel A, Famulok MA. Ribozyme for the Aldol Reaction. Chem. Biol. 2005;12:941–950. doi: 10.1016/j.chembiol.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 32.Saran D, Nickens DG, Burke DHA. Trans Acting Ribozyme that Phosphorylates Exogenous RNA. Biochemistry. 2005;44:15007–15016. doi: 10.1021/bi051086h. [DOI] [PubMed] [Google Scholar]
- 33.Boysen G, Pachkowski BF, Nakamura J, Swenberg JA. The Formation and Biological Significance of N7-Guanine Adducts. Mutat. Res. 2009;678:76–94. doi: 10.1016/j.mrgentox.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hansen MR, Hurley LH. Pluramycins. Old Drugs Having Modern Friends in Structural Biology. Acc. Chem. Res. 1996;29:249–258. [Google Scholar]
- 35.Zuker M. Mfold Web Server for Nucleic Acid Folding and Hybridization Prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Machanick P, Bailey TL. MEME-ChIP: Motif Analysis of Large DNA Datasets. Bioinformatics. 2011;27:1696–1697. doi: 10.1093/bioinformatics/btr189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bao G, Rhee WJ, Tsourkas A. Fluorescent Probes for Live-Cell RNA Detection. Annu. Rev. Biomed. Eng. 2009;11:25–47. doi: 10.1146/annurev-bioeng-061008-124920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jao CY, Salic A. Exploring RNA Transcription and Turnover In Vivo by Using Click Chemistry. Proc. Natl. Acad. Sci. U.S.A. 2008;105:15779–15784. doi: 10.1073/pnas.0808480105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trcek T, Larson DR, Moldón A, Query CC, Singer RH. Single Molecule mRNA Decay Measurements Reveal Promoter-Regulated mRNA Stability in Yeast. Cell. 2011;147:1484–1497. doi: 10.1016/j.cell.2011.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rabani M, et al. Metabolic Labeling of RNA Uncovers Principles of RNA Production and Degradation Dynamics in Mammalian Cells. Nat. Biotechnol. 2011;29:436–442. doi: 10.1038/nbt.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cosma MP. Daughter-Specific Repression of Saccharomyces Cerevisiae HO: Ash1 is the Commander. EMBO Rep. 2004;5:953–957. doi: 10.1038/sj.embor.7400251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Langer PR, Waldrop AA, Ward DC. Enzymatic Synthesis of Biotin-Labeled Polynucleotides: Novel Nucleic Acid Affinity Probes. Proc. Natl. Acad. Sci. U.S.A. 1981;78:6633–6637. doi: 10.1073/pnas.78.11.6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Curtis EA, Liu DR. Discovery of Widespread GTP-Binding Motifs in Genomic DNA and RNA. Chem. Biol. 2013;20:521–532. doi: 10.1016/j.chembiol.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tretyakova NY, Sangaiah R, Yen TY, Swenberg JA. Synthesis, Characterization, and In Vitro Quantitation of N-7-Guanine Adducts of Diepoxybutane. Chem. Res. Toxicol. 1997;10:779–785. doi: 10.1021/tx970004q. [DOI] [PubMed] [Google Scholar]
- 45.Ponchon L, Dardel F. Recombinant RNA Technology: the tRNA Scaffold. Nature Meth. 2007;4:571–576. doi: 10.1038/nmeth1058. [DOI] [PubMed] [Google Scholar]
- 46.Paul CP, et al. Localized Expression of Small RNA Inhibitors in Human Cells. Mol. Ther. 2003;7:237–247. doi: 10.1016/s1525-0016(02)00038-2. [DOI] [PubMed] [Google Scholar]
- 47.Paige JS, Wu KY, Jaffrey SR. RNA Mimics of Green Fluorescent Protein. Science. 2011;333:642–646. doi: 10.1126/science.1207339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ghaemmaghami, et al. Global Analysis of Protein Expression in Yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.