

Abstract
The heme group in paramagnetic (S = 1/2) ferricytochromes c typically displays a markedly asymmetric distribution of unpaired electron spin density among the heme pyrrole β substituents. This asymmetry is determined by the orientations of the heme axial ligands, histidine and methionine. One exception to this is ferricytochrome c552 from Hydrogenobacter thermophilus, which has similar amounts of unpaired electron spin density at the β substituents on all four heme pyrroles. Here, determination of the orientation of the magnetic axes and analysis of NMR line shapes for H. thermophilus ferricytochrome c552 is performed. These data reveal that the unusual electronic structure for this protein is a result of fluxionality of the heme axial methionine. It is proposed that the ligand undergoes inversion at the pyramidal sulfur, and the rapid interconversion between two diastereomeric forms results in the unusual heme electronic structure. Thus a fluxional process for a metal-bound amino acid side chain has now been identified.
Class I cytochromes c (Cyts c) are electron-transfer proteins typically containing a single heme with His–Met axial ligation (1). The interaction between the axial Met and the heme iron is of interest because of the paucity of iron-thioether bonds in coordination chemistry (2) and the low intrinsic affinity of thioether for ferric iron (2–5). This low affinity is reflected in part by the finding that exogenous donor ligands can replace the Met axial ligand in oxidized mitochondrial Cyts c (6) and bacterial Cyts c2 (7). The ability of ligands to replace the axial Met in these proteins reflects not only a weak Fe(III)–S bond but also high conformational plasticity of the protein domain containing the axial Met in the oxidized protein (6–9). The mobility of the Met-containing domain in mitochondrial Cyts c is proposed to be a functionally important property in modulation of electron-transfer reorganization energy and binding to redox partners (10).
The role of the polypeptide chain in modulating the Fe–Met interaction also is expressed in species-dependent variations in the Met-ligand side-chain conformation. The most frequently observed axial Met conformations in class I Cyts c are shown in Fig. 1 and will be referred to herein as Met conformation A (Fig. 1 A) and B (Fig. 1B). These conformations are related to each other by inversion through the axial Met thioether sulfur. Most bacterial Cyts c8 (such as Cyts c551 from Pseudomonas aeruginosa, Pseudomonas stutzeri ZoBell strain, and P. stutzeri) display conformation A (11, 12, 14, 15). Conformation B is seen in mitochondrial class I Cyts c [i.e., horse Cyt c (h-Cyt c) and Saccharomyces cerevisiae iso-1-Cyt c], as well as bacterial Cyts c2 [i.e., Rhodospirillum rubrum and Rhodobacter capsulatus Cyts c2 (15–17)]. These different orientations of Met result in different heme electronic structures, and for the paramagnetic (S = 1/2) ferricytochromes this is reflected by distinct patterns of NMR hyperfine shifts (15, 18). For groups on the β pyrrole positions of the heme, such as the methyl groups at positions 1, 3, 5, and 8 (Fig. 1), the hyperfine shifting occurs primarily through a contact mechanism, which results from unpaired π electron spin density at heme substituents, causing polarization of the s orbitals. The π electron-density distribution in turn is determined by the orientation of the heme axial ligands (defined by the sulfur lone pair on the Met, and the π orbital normal to the ligand plane for His), where the filled ligand pπ orbital is oriented toward the pyrrole β substituents with the largest unpaired electron spin densities (18–20). Cyts c with Met in conformation A thus exhibit a pairwise ordering of heme methyl shifts with methyls 5 and 1 downfield of methyls 8 and 3 (shift ordering 5-CH3 > 1-CH3 > 8-CH3 > 3-CH3; Fig. 2A), whereas mitochondrial Cyts c (axial Met in conformation B) display a reversed pattern, with methyls 8 and 3 appearing downfield of methyls 5 and 1 (8-CH3 > 3-CH3 > 5-CH3 > 1-CH3; Fig. 2B) (18). In contrast with the methionine, the orientation of the axial His, aligned generally with the heme α–γ meso axis, does not vary substantially among Cyts c.
Fig. 1.

Heme axial Met side chain orientations and NOEs. Shown are orientations in Pa Cyt c551 (representative of the Cyts c8) (11) (A) and h-Cyt c (representative of mitochondrial Cyts c) (12) (B). (C) Two orientations proposed in this work for the axial Met in Ht Cyt c552. The plane of the axial His (not shown) lies approximately along the heme α–γ-meso axis in each case. NOESY cross peaks observed for the reduced proteins between the axial Met side chain and heme pocket amino acids (indicated in circles) or heme substituents are indicated with arrows. Connectivities in A and C are from this work; connectivities in B are from ref. 13. The Fisher heme-numbering system used in the text is indicated. P, propionate.
Fig. 2.

Downfield regions of 1H NMR (500-MHz) spectra of oxidized Pa Cyt c551 (50 mM sodium phosphate, pH 6.0, 300 K) (A), h-Cyt c (50 mM sodium phosphate, pH 7.0, 303 K) (B), Ht Cyt c552 (120 mM sodium acetate-d3, pH 5.0, 300 K) (C), Pa Cyt c551 (50 mM sodium phosphate, pH 6.0, 20% vol/vol CD3OD, 266 K) (D), Ht Cyt c552 (50 mM sodium phosphate, 20% vol/vol CD3OD, 266 K) (E). Heme methyl assignments are indicated (14, 21–24).
Two Cyts c with NMR spectra fitting neither of these archetypal patterns have been identified and have presented a puzzle in the Cyt c field (14, 15, 24). Reduced Nitrosomonas europaea (Ne) Cyt c552 has a three-dimensional structure and Met orientation similar to that in P. aeruginosa (Pa) Cyt c551 (25). However, oxidized Ne Cyt c552 displays an unusual heme methyl shift order (5-CH3 > 8-CH3, 3-CH3 > 1-CH3) and a highly compressed heme methyl shift range (4.2 ppm at 323 K; compare with 14.9 ppm for Pa Cyt c551) (14). Another unusual case is that of Hydrogenobacter thermophilus (Ht) Cyt c552. Despite high structural homology to Pa Cyt c551, Ht Cyt c552 has a reported Met orientation similar to that of the eukaryotic Cyts c (26). Regardless of this, the pattern of heme methyl shifts for Ht Cyt c552 differs from that seen in the eukaryotic Cyts c, with an 8-CH3 > 5-CH3 ∼ 3-CH3 > 1-CH3 ordering and, like Ne Cyt c552, a compressed shift range (6.2 ppm at 323 K; Fig. 2C) (24). Advances in understanding the relationship between heme protein molecular and electronic structure have allowed heme methyl shifts to be related to axial ligand geometries in low-spin ferric heme proteins (15, 27, 28). The heme methyl shift patterns of Ne and Ht Cyts c552, however, cannot be explained by any orientation of the axial ligands (14, 15, 24). This gap in our understanding of the relationship between heme protein molecular and electronic structure negatively impacts our ability to reliably use paramagnetic shift data to model and refine heme protein structures.
Here, analysis of NMR line shapes and of the orientation of the magnetic axes of Ht Cyt c552 is performed. The results reveal that the unusual hyperfine-shifted NMR spectrum of oxidized Ht Cyt c552 is a result of fluxionality of the axial methionine. This fluxional process is proposed to be an inversion through the methionine sulfur, resulting in rapid interconversion between the axial Met orientation typically seen in mitochondrial Cyts c and that seen in bacterial Cyts c8. Although it has precedent in inorganic and organometallic chemistry (29–32), such fluxional behavior has not been identified previously in a metalloprotein.
Materials and Methods
Protein Expression and Purification. Ht Cyt c552 was expressed and purified as described (24). To express Pa Cyt c551, a pET3c (ampicillin-resistant) vector (Novagen) containing the Pa Cyt c551 gene preceded by its periplasmic translocation sequence (pETPA) was used for cytochrome expression (33), and pEC86 was used to overexpress the ccm genes (34). Escherichia coli strain BL21(DE3) containing pETPA and pEC86 was cultured in LB supplemented with ampicillin (50 μg/ml) and chloramphenicol (50 μg/ml). The 25-ml culture in a 125-ml Erlenmeyer flask was shaken at 180 rpm, 37°C, for 8 h. This culture was used to inoculate 1 liter of LB in a 4-liter Erlenmeyer flask. The culture was shaken at 140 rpm at 37°C for 16 h, and cells were harvested by centrifugation. The protein purification procedure was as described (33).
NMR Spectroscopy. Proton NMR spectra were collected on a Varian INOVA 500-MHz spectrometer (operating at 499.839 MHz). Ht Cyt c552 samples [1–3 mM; Fe(III) form] were in 120 mM sodium acetate-d3/10% D2O, pH 5.0, and contained a 5-fold molar excess of K3[Fe(CN)6]. Pa Cyt c551 samples [1–3 mM; Fe(II) form] were in 50 mM sodium phosphate/10% D2O, pH 6.0. For preparation of reduced Pa Cyt c551, the protein sample was deoxygenated before addition of a 20- to 30-fold molar excess of Na2S2O4. For low-temperature (268–300 K) 1D 1H NMR spectra, oxidized protein samples were prepared in 50 mM sodium phosphate buffer, pH 6.0, with 10% D2O and 20% (vol/vol) CD3OD. Two-dimensional total correlation spectroscopy and NOESY spectra were collected at 300 K with 8,192 points in the F2 dimension, 512 increments in the F1 dimension, and a 30,000-Hz spectral width (for oxidized Ht Cyt c552), or 4,096 points in the F2 dimension, 512 increments in the F1 dimension, and a 12,000-Hz spectral width (for reduced Pa Cyt c551). The total correlation spectroscopy spin-lock time was 90 ms, and the NOESY mixing time was 100 ms. Presaturation was used to suppress the solvent signal. Assignments for 1H NMR chemical shifts were made according to standard procedures (35). Measurements of chemical shifts for reduced Pa Cyt c551 were guided by published assignments (36–38). Heme methyl shifts for Ht Cyt c552 at 323 K were determined by a 1D variable-temperature NMR experiment (300–344 K). T1 measurements at 284, 300, and 344 K on oxidized Ht Cyt c552 were made by the standard 180°–τ–90° pulse sequence with variable τ.
NMR Data Analysis. The 1-CH3 and 8-CH3 resonances in the NMR spectra of oxidized Ht Cyt c552 at 268, 271, 274, 279, 284, and 289 K were simulated by using the program windnmr v. 7.1.5 after processing the NMR spectra using nuts. The heme 3-CH3 and 5-CH3 resonances were excluded from analysis because of overlap. An uncoupled AB spin system was used to simulate spectra of two nuclei undergoing mutual chemical exchange. The following assumptions were made in the simulation. (i) The exchange was between Met conformations A and B (Fig. 1); (ii) a 1:1 population ratio of the two states was maintained; (iii) the inherent linewidth of Ht Cyt c552 in the absence of exchange is the same as that of the corresponding resonance in Pa Cyt c551 determined under identical conditions; and (iv) the chemical shift of the resonance in state A (Met conformation A) is the same as that of the corresponding resonance in Pa Cyt c551.¶ Values fit in the simulation were (i) values for the rate constant for the exchange (k), and (ii) the chemical shifts of state B (presumed to be Met conformation B). Motion of the axial His was not considered because covalent constraints strictly maintain its conformation in c-type cytochromes (39). The activation enthalpy for the process was determined from the slope of an Eyring plot of ln (k/T) vs. 1/T.
Pseudocontact shifts (δpc) for polypeptide protons in Ht Cyt c552 (assignments for reduced form from ref. 24; assignments for oxidized form measured here) and Pa Cyt c551 (assignments for reduced form measured here; assignments for oxidized form from ref. 33) were calculated from Eq. 1
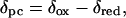 |
[1] |
where δox and δred are the respective chemical shifts of a particular nucleus in the Fe(III) (paramagnetic) and Fe(II) (diamagnetic) forms of the protein. This relationship assumes no redox-linked structure change and a contact shift equal to zero. The contact shift can be assumed to be zero for nuclei not on the heme or its ligands (28). Cyts c generally undergo a minimal amount of redox-linked structure change (5, 11, 16), and high-resolution crystal structures of Pa Cyt c551 confirm the validity of this assumption for this protein (11). Assigned protons with ambiguous positions in the 3D structure (i.e., geminal protons lacking stereospecific assignments) were excluded from analysis. Residue 59 was excluded from analysis in both proteins because a small redox-dependent conformational change is reported for this residue in Pa Cyt c551 (11).
Determination of Magnetic Axes. The structures of Pa Cyt c551 [x-ray crystal structure, Protein Data Bank (PDB) code 351C] (11) and of Ht Cyt c552 (NMR structure; PDB code 1AYG) (26) available in the PDB were used in searches for the parameters defining χ tensor orientation and anisotropy. Protons were added to the Pa Cyt c551 crystal structure by using the HBUILD module in charmm. To determine methyl proton coordinates, the position was averaged over one rotation. For Ht Cyt c552, the first conformation in the NMR ensemble was used as the reference structure. Each protein was placed in a molecular coordinate system with Fe at the origin, the +z axis perpendicular to the mean plane of the four heme pyrrole nitrogen atoms in the direction of the axial Met, the +x axis aligned with the pyrrole II N atom, and the +y axis aligned in the direction of pyrrole I N atom (Fig. 3).
Fig. 3.

In-plane molecular and magnetic axes. The molecular axes are indicated with solid arrows labeled x and y (the +z axis is normal to the heme plane, pointing toward the viewer). In-plane orientations of the mean plane of the two heme protein axial ligands measured from structures are indicated as Φ values and dashed lines. Orientations of the χxx axes, which are determined experimentally herein, are indicated by κ values and solid lines. Shown are the Φ and κ values for oxidized Pa Cyt c551 (A) and Ht Cyt c552 (B). The Φ values in A and B are determined from measurements on the crystal (11) (351C) and NMR (26) (1AYG) structures of these proteins, respectively. In C, the Φ value shown is based on averaging the ligand orientations measured from the crystal structures Pa Cyt c551 and for h-Cyt c (1HRC) (12). The crystal structure of h-Cyt c is used in C because of the poor definition of the axial Met-ligand angle in 1AYG.
Determinations of the magnetic axes from pseudocontact shift data proceeded generally as described (40), using an in-house program. After defining the molecular coordinate axes, the protein was rotated stepwise through the three Euler angles, α, β, and γ, by using the z-x-z convention. The Euler angles convert the molecular coordinate system to a coordinate system defined by the magnetic axes. Step sizes of 1.0, 0.5, and 1.0° were used for α, β, and γ, respectively, and an entire spherical search was performed. At each step, a linear least-squares fit of the set of experimental pseudocontact shift values (δpc,i) to Eq. 2 was performed,
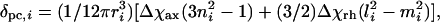 |
[2] |
where ri is the distance from the iron to atom i (determined from the three-dimensional structure), and li, mi, and ni are the direction cosines of the position vector of atom i (ri) with respect to the magnetic axes (28). The goodness of fit was assessed by calculating the sum-squared error between the calculated (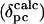 ) and experimental (
) and experimental (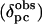 ) pseudocontact shifts.
) pseudocontact shifts.
Heme axial ligand orientation angles were determined from structures as follows. For Met, this angle is determined by projecting the bisector of the Met Cγ–Sδ–Cε angle onto the heme plane and taking a vector perpendicular to this projection. The orientation angle of Met is the angle between this vector and the heme x axis. For His, the orientation angle is the angle between the ligand imidazole plane and the xz plane of the molecular coordinate system. The average ligand-orientation angle is taken as the bisector of the acute angle formed by the His- and Met-ligand planes.
Results
NMR Line-Shape Analysis. Upon addition of CD3OD to samples, the heme methyl resonance linewidths of oxidized Ht Cyt c552 and Pa Cyt c551 increase (≈25% increase), and the lines shift slightly. The overall properties of the NMR spectra nevertheless indicate that folded protein conformations are maintained. This finding is similar to observations of oxidized h-Cyt c in 20% methanol (41). When temperature is decreased, the Ht Cyt c552 heme methyl resonances broaden substantially. Broadening to this degree is not observed in Pa Cyt c551 (Fig. 2). The T1 values for the four heme methyls of oxidized Ht Cyt c552 show little variation with temperature, despite the increase in linewidths (Table 1, which is published as supporting information on the PNAS web site). This observation indicates that the temperature-sensitive correlation time determining the increase in Ht Cyt c552 heme methyl linewidths as temperature is decreased is a chemical exchange time rather than an electronic relaxation time (41).
The shifts and linewidths of Ht Cyt c552 heme methyls 1 and 8 (24) were simulated by using dnmr to model exchange between axial Met conformations A and B (overlap of methyls 3 and 5 precluded their analysis). The simulated and experimental spectra at variable temperatures are shown in Fig. 4. At 274 K, the calculated chemical shifts for the heme methyls in state B are 34.7 ppm (8-CH3) and 8.6 ppm (1-CH3). These calculations are in the order of the measured values for oxidized h-Cyt c [38.4 ppm (8-CH3) and 6.3 ppm (1-CH3) at 274 K in 20% CD3OD], supporting the assumption that the Met is in conformation B in state B. Thus, these results support the hypothesis that the axial Met in Ht Cyt c552 is exchanging between conformations A and B on the NMR time scale. Consistent with this hypothesis, the heme methyl shifts of oxidized Ht Cyt c552 are nearly averages of those for a protein with a Met exclusively in conformation A (i.e., Pa Cyt c551) and for a protein with a Met exclusively in conformation B (i.e., h-Cyt c). For example, at 323 K (20 K above the fast-exchange limit; see Fig. 5, which is published as supporting information on the PNAS web site), averaging the respective heme methyl (8-CH3, 5-CH3, 3-CH3, and 1-CH3) shifts for oxidized h-Cyt c (32.4, 10.8, 30.1, and 7.6 ppm) (14) and Pa Cyt c551 (15.7, 29.0, 14.1, and 24.3 ppm) (14), the result is 24.0, 19.9, 22.1, and 16.0 ppm, which compares well with the measured shifts for Ht Cyt c552 (23.9, 22.5, 22.8, and 18.0 ppm).
Fig. 4.

Experimental (traces A, C, and E) and simulated (traces B, D, and F) 1H NMR spectra of oxidized Ht Cyt c552 (50 mM sodium phosphate, pH 6.0/20% vol/vol CD3OD). Temperatures are 284 K (A and B), 274 K (C and D), and 268 K (E and F). The resonances for only the two resolved methyls were simulated (8-CH3 and 1-CH3). Calculated exchange rates are 7.0 × 105 s–1 (284 K), 4.0 × 105 s–1 (274 K), and 2.0 × 105 s–1 (268 K). Not shown are spectra at 271 and 289 K, at which respective exchange rates of 2.2 × 105 s–1 and 8.0 × 105 s–1 are calculated.
The ΔH‡ for the axial Met fluxion was determined to be 59 ± 10 kJ/mol from Eyring analysis (see Fig. 6, which is published as supporting information on the PNAS web site). This activation enthalpy is similar to values determined for inversion at sulfur in transition metal complexes with thioether ligands (29–32).
Magnetic Axis Determinations. Assignments made for 1H NMR resonances in oxidized Ht Cyt c552 are reported in Table 2, which is published as supporting information on the PNAS web site. Proton resonance assignments for most heme substituents of oxidized Ht Cyt c552 were reported previously (24). Here, detection of connectivities between heme substituents and nearby amino acids (see Fig. 7, which is published as supporting information on the PNAS web site) confirms and extends those assignments.
A total of 155 and 163 pseudocontact shift values were determined for Ht Cyt c552 and Pa Cyt c551, respectively, according to Eq. 1, and are reported as Tables 3 and 4, which are published as supporting information on the PNAS web site. The results of the magnetic axes searches are summarized in Table 5, and plots of 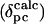 vs.
vs. 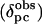 are shown in Fig. 8, both of which are published as supporting information on the PNAS web site. For both proteins, as expected for S = 1/2 hemes (28), the z axis is nearly perpendicular to the heme plane, as indicated by the small magnitude of the Euler angle β, which indicates the z axis tilt from the heme normal (5° for Pa Cyt c551 and –6.5° for Ht Cyt c552). When β is small, the in-plane rotation of the magnetic axes relative to the molecular axes is well defined by κ = α + γ, which is –12° for Pa Cyt c551 and –47° for Ht Cyt c552. This result for Pa Cyt c551 is in general agreement with literature values (323 K) of –26° (21) and –15° (42). To predict the orientation of the rhombic perturbation of S = 1/2 ferric heme electronic structure from heme–axial ligand interactions, the “counterrotation rule” can be used (28, 42, 43). In this formalism, if the mean axial ligand plane is oriented at an angle Φ from the N–Fe–N axis in the heme plane, the direction of the minimum χ value (χxx) would be at an angle κ = –Φ from that same axis (Fig. 3) (28, 42, 43).
are shown in Fig. 8, both of which are published as supporting information on the PNAS web site. For both proteins, as expected for S = 1/2 hemes (28), the z axis is nearly perpendicular to the heme plane, as indicated by the small magnitude of the Euler angle β, which indicates the z axis tilt from the heme normal (5° for Pa Cyt c551 and –6.5° for Ht Cyt c552). When β is small, the in-plane rotation of the magnetic axes relative to the molecular axes is well defined by κ = α + γ, which is –12° for Pa Cyt c551 and –47° for Ht Cyt c552. This result for Pa Cyt c551 is in general agreement with literature values (323 K) of –26° (21) and –15° (42). To predict the orientation of the rhombic perturbation of S = 1/2 ferric heme electronic structure from heme–axial ligand interactions, the “counterrotation rule” can be used (28, 42, 43). In this formalism, if the mean axial ligand plane is oriented at an angle Φ from the N–Fe–N axis in the heme plane, the direction of the minimum χ value (χxx) would be at an angle κ = –Φ from that same axis (Fig. 3) (28, 42, 43).
The average value of the ligand planes determined from the Pa Cyt c551 crystal structure is 16° (Fig. 3). If both axial ligands contribute equally to determining the in-plane orientation of the magnetic axes according to the counterrotation rule, the expected κ value thus is –16°, in good agreement with our experimental value of –12° as well as literature values (21, 42, 43). The in-plane magnetic axes for Ht Cyt c552 have a substantially different orientation (κ =–47°) from those in Pa Cyt c551. The predicted κ value based on the average ligand orientation angles for the family of 20 Ht Cyt c552 structures is –72°; the value based on only the first in the family of Ht Cyt c552 structures is also –72°. This value is in poor agreement with the experimental value of –47°. Notably, the experimental κ value for Ht Cyt c552 also is not in agreement with the value predicted based on the ligand orientations seen in crystallographically characterized h-Cyt c (–78°), to which the ligand orientations of Ht Cyt c552 were reported to be similar (26). Assuming the axial Met in Ht Cyt c552 samples the conformations A and B (spending 50% of its time in each of these conformations), a κ value of ≈–47° is predicted by applying the counterrotation rule to fluxional Ht Cyt c552, in excellent agreement with the experimental value of –47° (Fig. 3). This result is consistent with the proposal, based on line-broadening analysis, that the Ht Cyt c552 axial Met is fluxional, sampling conformations A and B.
Met Orientation in Reduced Proteins. The orientation of the axial Met in Fe(II) Cyts c can be evaluated by analysis of nuclear Overhauser effects (NOEs) between the axial Met and heme substituents, especially the meso protons (18). For Pa Cyt c551, which has its axial Met in orientation A, strong NOESY cross peaks are expected (and observed) between the axial Met ε-CH3 and the heme γ-meso and δ-meso protons, but not to the α-meso proton (Fig. 1 A). Cyts c with Met in conformation B have axial Met ε-CH3 in proximity of the heme α-meso and δ-meso, as well as the 2-thioether. NOEs are not expected from the axial Met ε-CH3 to the β-or γ-meso protons in that case (13, 18) (Fig. 1B). In the case of reduced Ht Cyt c552, NOEs characteristic of both orientations A and B are observed (Fig. 1C).∥ The pattern of NOEs observed for the axial Met side chain supports the proposal that the axial Met is fluxional in Ht Cyt c552 and suggests that this fluxionality is present in the reduced as well as the oxidized form. NOESY spectra showing connectivities to the axial Met ε-CH3 in the reduced form are shown in Fig. 9, which is published as supporting information on the PNAS web site.
Discussion
Fluxional behavior for an amino acid ligand in a metalloprotein has not previously been characterized to our knowledge. Nevertheless, fluxionality is common in coordination and organometallic chemistry. The particular fluxional process proposed to be occurring here, inversion at sulfur, is observed less frequently than inversion at second-row atoms such as nitrogen and oxygen because of the relatively high barrier for sulfur inversion (29). Inversion at sulfur may proceed through dissociative or nondissociative mechanisms. In the case of a molecule with a bond between the sulfur and a transition metal, the nondissociative mechanism is reported to be the more common (29). Additional studies are needed to determine definitively the nature of the fluxion in Ht Cyt c552 and its mechanism.
Comparison of the structure of Ht Cyt c552, which has a fluxional axial Met, and Pa Cyt c551, which is structurally homologous but not fluxional, provides some insight into factors promoting fluxionality in Ht Cyt c552. The most notable difference between the heme pocket structures of these proteins is the presence of a Gln residue in place of Asn at position 64 in Ht Cyt c552 (11, 26). Asn-64 is conserved in the Cyt c8 family and donates a hydrogen bond from its δ-NH2 group to the Met-61 δ-S atom (1, 11). The length of the Gln side chain in Ht Cyt c552 apparently does not position the Gln-64 ε-NH2 to hydrogen bond to the Met-61 δ-S, as reflected in the lack of a ring-current shift for either Gln-64 ε-NH2 proton; the shifts for reduced Ht Cyt c552 Gln-64 ε-NH2 protons are 8.81 and 6.37 ppm (26), which are in the expected range for a Gln or Asn side-chain NH2 (35). In contrast, the shifts for reduced Pa Cyt c551 Asn-64 δ-NH2 are 7.49 and 3.19 ppm (44). The unusually low shift of 3.19 ppm for one Asn-64 δ-NH2 proton suggests that it is influenced strongly by the heme ring current, placing it above the heme plane (note that no aromatic amino acids are in the vicinity of residue 64). We suggest that one or both of the following factors promotes axial Met fluxionality in Ht Cyt c552: (i) the absence of this hydrogen-bonding interaction raises the ground state energy in Ht Cyt c552 to allow fluxion to be observed, or (ii) the long Gln-64 side chain perturbs the heme pocket structure, causing crowding and inducing strain.
Ht Cyt c552 is not the only Cyt c to exhibit a compressed heme methyl shift range in the oxidized state. Oxidized Ne Cyt c552, also a member of the Cyt c8 structural family (25), has been reported to have compressed heme methyl shifts (14) that cannot be explained based on a single orientation of an axial His and Met (15). This observation suggests that heme axial Met fluxionality may be occurring in oxidized Ne Cyt c552, although additional studies are required to test this proposal. An unusual aspect of the Ne Cyt c552 heme pocket structure is the presence of a one-residue (Val) insertion in the axial Met-containing loop after position 64. This insertion leads to a rearrangement of this loop relative to that seen in other members of the Cyt c8 family, with the loop packing closer to the heme in Ne Cyt c552 (25). Like the presence of Gln at position 64 in Ht Cyt c552, the Val insertion in Ne Cyt c552 may lead to strain in the heme pocket, promoting fluxion. It is notable, however, that in reduced Ne Cyt c552, the reported NOEs are consistent with Met conformation A, and not B (25). It is possible that Met fluxion occurs in the oxidized, but not reduced form of Ne Cyt c552. Notably, as for Pa Cyt c551, NMR properties and the structure of Ne Cyt c552 demonstrate that the Asn residue at position 64 is indeed positioned to donate a hydrogen bond to Met-61 δ-S (25). Additional studies of both oxidation states of Ne Cyt c552 are needed to determine whether oxidation-state-dependent Met fluxion takes place in this protein.
Cyts c have been subjects of biophysical characterization for decades. Thus, the question of why such a fluxional process has not been noted previously in a Cyt c arises. It is important to note that, because of their wide availability, the bulk of the mutational and biophysical studies of Cyts c have been performed on eukaryotic species, rather than on bacterial species such as those studied here. We suggest that the Met fluxionality is modulated by heme pocket strain (32), and this is imposed by the unusually rigid character of the loop domain donating the axial Met in the bacterial Cyts c8, which is imparted by the presence of a polyproline region flanking the axial Met (33). This rigidity contrasts with the homologous loop in the eukaryotic Cyts c, which has high flexibility such that perturbations of the heme pocket are not likely to induce the strain proposed here to promote fluxionality (6, 9, 10, 33). The conformational plasticity of this loop in the eukaryotic proteins is reflected in the sensitivity of the Fe–S bond length to oxidation state. For S. cerevisiae iso-1-Cyt c, this bond length is 2.35 Å and 2.42 Å in the reduced and oxidized forms, respectively (16). In contrast, in Pa Cyt c551, the corresponding bond lengths are 2.35 and 2.36 Å (11) (note that these bond lengths are determined from x-ray crystal structures with resolutions ranging from 1.2 to 1.9 Å). Although the higher affinity of Met for Fe(II) relative to Fe(III) heme is an intrinsic property of the prosthetic group (4, 5), this translates to a substantial Fe–S bond-length change for the mitochondrial but not for the bacterial Cyt c. This observation supports the idea that the rigidity of the Met-donating loop is a particular property of the bacterial Cyts c8 that may induce strain at the heme site and in some cases lead to fluxional behavior of the axial Met.
The discovery of a fluxional heme axial Met in a thermophilic electron-transfer protein raises the question as to whether fluxionality has an effect on protein stability or function. The possibility of entropic stabilization of thermophilic proteins through folded-state effects has been discussed (45). The argument is that increasing folded-state entropy by promoting flexibility could enhance stability. Of course, entropy–enthalpy compensation is expected to yield a corresponding unfavorable increase in folded-state enthalpy (46). In the case of axial Met fluxion, however, if the Fe–S bond is not broken and other stabilizing interactions are not lost as a result of the Met motion, this may reduce the magnitude of any enthalpic compensation. Regarding the putative electron-transfer function of Ht Cyt c552, the effect of Met fluxion is not immediately apparent, in particular because any redox partners are unknown. One possibility is that it may influence directionality of electron transfer. Indeed, electron-spin delocalization patterns have been proposed to be optimized for transfer to physiological partners; however, the magnitude of any such effect is thought to be too small to have a substantial influence on rates (14). Systematic experimental studies addressing these questions, however, have yet to be reported. Regardless of any functional ramifications, these results should alert the structural biology and bioinorganic chemistry communities to the possibility of the existence of ligand fluxionality within metalloproteins.
Supplementary Material
Acknowledgments
We are grateful for generous gifts from Linda Thöney-Meyer (pEC86) and Francesca Cutruzzolá (pETPA) that allowed for successful protein expression. We also thank Maria Giulia Bigotti for helpful advice on Pa Cyt c551 expression and purification. This work was supported by National Institutes of Health Grant GM63170. K.L.B. thanks the Alfred P. Sloan Foundation for a research fellowship. L.Z. acknowledges a Robert and Marian Flaherty DeRight Graduate Fellowship, and B.S.R. acknowledges an Elon Huntington Hooker Graduate Fellowship and an Agnes M. and George Messersmith Fellowship.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: Cyt c, cytochrome c; Ht Cyt c552, Hydrogenobacter thermophilus Cyt c552; h-Cyt c, horse heart Cyt c; Ne Cyt c552, Nitrosomonas europaea Cyt c552; NOE, nuclear Overhauser effect; Pa Cyt c551, Pseudomonas aeruginosa Cyt c551.
Footnotes
This assumption is supported by the near reproduction of the Pa Cyt c551 heme methyl shifts in a mutant of Ht Cyt c552 (X.W. and K.L.B., unpublished work).
NOEs from the axial Met ε-CH3 to the heme α-meso, γ-meso, and δ-meso protons were reported in ref. 26; however, the text specifies the orientation to be orientation B, as does the structure.
References
- 1.Meyer, T. E. (1996) in Cytochrome c: A Multidisciplinary Approach, eds. Scott, R. A. & Mauk, A. G. (Univ. Sci. Books, Mill Valley, CA), pp. 33–99.
- 2.Murray, S. G. & Hartley, F. R. (1981) Chem. Rev. (Washington, D.C.) 81, 365–414. [Google Scholar]
- 3.Smith, M. & McLendon, G. (1981) J. Am. Chem. Soc. 103, 4912–4921. [Google Scholar]
- 4.Tezcan, F. A., Winkler, J. R. & Gray, H. B. (1998) J. Am. Chem. Soc. 120, 13383–13388. [Google Scholar]
- 5.Schejter, A. (1996) in Cytochrome c: A Multidisciplinary Approach, eds. Scott, R. A. & Mauk, A. G. (Univ. Sci. Books, Mill Valley, CA), pp. 335–345.
- 6.Sutin, N. & Yandell, J. K. (1972) J. Biol. Chem. 247, 6932–6936. [PubMed] [Google Scholar]
- 7.Dumortier, C., Holt, J. M., Meyer, T. E. & Cusanovich, M. A. (1998) J. Biol. Chem. 273, 25647–25653. [DOI] [PubMed] [Google Scholar]
- 8.Fetrow, J. S. & Baxter, S. M. (1999) Biochemistry 38, 4480–4492. [DOI] [PubMed] [Google Scholar]
- 9.Bai, Y., Sosnick, T. R., Mayne, L. & Englander, S. W. (1995) Science 269, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berghuis, A. M., Guillemette, J. G., McLendon, G., Sherman, F., Smith, M. & Brayer, G. D. (1994) J. Mol. Biol. 236, 786–799. [DOI] [PubMed] [Google Scholar]
- 11.Matsuura, Y., Takano, T. & Dickerson, R. E. (1982) J. Mol. Biol. 156, 389–409. [DOI] [PubMed] [Google Scholar]
- 12.Bushnell, G. W., Louie, G. V. & Brayer, G. D. (1990) J. Mol. Biol. 214, 585–595. [DOI] [PubMed] [Google Scholar]
- 13.Banci, L., Bertini, I., Huber, J. G., Spyroulias, G. A. & Turano, P. (1999) J. Biol. Inorg. Chem. 4, 21–31. [DOI] [PubMed] [Google Scholar]
- 14.Timkovich, R., Cai, M., Zhang, B., Arciero, D. M. & Hooper, A. B. (1994) Eur. J. Biochem. 226, 159–168. [DOI] [PubMed] [Google Scholar]
- 15.Shokhirev, N. V. & Walker, F. A. (1998) J. Biol. Inorg. Chem. 3, 581–594. [Google Scholar]
- 16.Berghuis, A. M. & Brayer, G. D. (1992) J. Mol. Biol. 223, 959–976. [DOI] [PubMed] [Google Scholar]
- 17.Yu, L. P. & Smith, G. M. (1990) Biochemistry 29, 2914–2919. [DOI] [PubMed] [Google Scholar]
- 18.Senn, H. & Wüthrich, K. (1985) Q. Rev. Biophys. 18, 111–134. [DOI] [PubMed] [Google Scholar]
- 19.Lee, K.-B., La Mar, G. N., Mansfield, K. E., Smith, K. M., Pochapsky, T. C. & Sligar, S. G. (1993) Biochim. Biophys. Acta 1202, 189–199. [DOI] [PubMed] [Google Scholar]
- 20.Santos, H. & Turner, D. L. (1993) Magn. Reson. Chem. 31, s90–s95. [Google Scholar]
- 21.Timkovich, R. & Cai, M. (1993) Biochemistry 32, 11516–11523. [DOI] [PubMed] [Google Scholar]
- 22.Moratal, J. M., Donaire, A., Salgado, J., Jiménez, H. R., Castells, J. & Piccioli, M. (1993) FEBS Lett. 324, 305–308. [DOI] [PubMed] [Google Scholar]
- 23.Santos, H. & Turner, D. L. (1986) FEBS Lett. 194, 73–77. [DOI] [PubMed] [Google Scholar]
- 24.Karan, E. F., Russell, B. S. & Bren, K. L. (2002) J. Biol. Inorg. Chem. 7, 260–272. [DOI] [PubMed] [Google Scholar]
- 25.Timkovich, R., Bergmann, D., Arciero, D. M. & Hooper, A. B. (1998) Biophys. J. 75, 1964–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hasegawa, J., Yoshida, T., Yamazaki, T., Sambongi, Y., Yu, Y., Igarashi, Y., Kodama, T., Yamazaki, K., Kyogoku, Y. & Kobayashi, Y. (1998) Biochemistry 37, 9641–9649. [DOI] [PubMed] [Google Scholar]
- 27.Banci, L., Bertini, I., Cavallaro, G. & Luchinat, C. (2002) J. Biol. Inorg. Chem. 7, 416–426. [DOI] [PubMed] [Google Scholar]
- 28.La Mar, G. N., Satterlee, J. D. & de Ropp, J. S. (2000) in The Porphyrin Handbook, eds. Kadish, K. M., Smith, K. M. & Ruilard, R. (Academic, New York), Vol. 5, pp. 185–298. [Google Scholar]
- 29.Toyota, S. (1999) Rev. Heteroatom Chem. 21, 139–162. [Google Scholar]
- 30.Shan, X. & Espenson, J. H. (2003) Organometallics 22, 1250–1254. [Google Scholar]
- 31.Tresoldi, G., Lo Schiavo, S., Lanza, S. & Cardiano, P. (2002) Eur. J. Inorg. Chem. 1, 181–191. [Google Scholar]
- 32.Canovese, L., Lucchini, V., Santo, C., Visentin, F. & Zambon, A. (2002) J. Organomet. Chem. 642, 58–63. [Google Scholar]
- 33.Russell, B. S., Zhong, L., Bigotti, M. G., Cutruzzolà, F. & Bren, K. L. (2003) J. Biol. Inorg. Chem. 8, 156–166. [DOI] [PubMed] [Google Scholar]
- 34.Arslan, E., Schulz, H., Zufferey, R., Künzler, P. & Thöny-Meyer, L. (1998) Biochem. Biophys. Res. Commun. 251, 744–747. [DOI] [PubMed] [Google Scholar]
- 35.Wüthrich, K. (1986) NMR of Proteins and Nucleic Acids (Wiley, New York).
- 36.Detlefsen, D. J., Thanabal, V., Pecoraro, V. L. & Wagner, G. (1990) Biochemistry 29, 9377–9386. [DOI] [PubMed] [Google Scholar]
- 37.Chau, M.-H., Cai, M. L. & Timkovich, R. (1990) Biochemistry 29, 5076–5087. [DOI] [PubMed] [Google Scholar]
- 38.Cai, M. & Timkovich, R. (1991) Biochem. Biophys. Res. Commun. 178, 309–314. [DOI] [PubMed] [Google Scholar]
- 39.Low, D. W., Gray, H. B. & Duus, J. Ø. (1997) J. Am. Chem. Soc. 119, 1–5. [Google Scholar]
- 40.Emerson, S. D. & La Mar, G. N. (1990) Biochemistry 29, 1556–1566. [DOI] [PubMed] [Google Scholar]
- 41.Burns, P. D. & La Mar, G. N. (1981) J. Biol. Chem. 256, 4934–4939. [PubMed] [Google Scholar]
- 42.Turner, D. L. (1995) Eur. J. Biochem. 227, 829–837. [DOI] [PubMed] [Google Scholar]
- 43.Shokhirev, N. V. & Walker, F. A. (1998) J. Am. Chem. Soc. 120, 981–990. [Google Scholar]
- 44.Timkovich, R. (1990) Biochemistry 29, 7773–7780. [DOI] [PubMed] [Google Scholar]
- 45.Lazaridis, T., Lee, I. & Karplus, M. (1997) Protein Sci. 6, 2589–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lumry, R. & Rajender, S. (1970) Biopolymers 9, 1125–1227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}