

Abstract
We have shown earlier that Kir2 channels are suppressed by the elevation of membrane cholesterol. Moreover, it is also well known that activation of Kir channels is critically dependent on a regulatory phospholipid, phosphatidylinositol-4,5-bisphosphate (PIP2). In this study we examined the cross-talk between cholesterol and PIP2 in the regulation of Kir2 channels. The strength of Kir2–PIP2 interactions was assessed by acute sequestering of PIP2 with neomycin dialyzed into cells through a patch pipette while simultaneously recording whole cell currents. Consistent with a reduction in PIP2 levels, dialysis of neomycin resulted in a decrease in Kir2.1 and Kir2.3 current amplitudes (current rundown), however, this effect was significantly delayed by cholesterol depletion for both types of channels suggesting that cholesterol depletion strengthens the interaction between Kir2 channels and PIP2. Furthermore, mutation of Kir2.1 that renders the channels' cholesterol insensitive abrogated cholesterol depletion-induced delay in the current rundown whereas reverse mutation in Kir2.3 has the opposite effect. These observations provide further support for the functional cross-talk between cholesterol and PIP2 in regulating Kir2 channels. Consistent with these observations, there is a significant structural overlap between cytosolic residues that are critical for the sensitivity of Kir2 channels to the two lipid modulators but based on recent studies, there is little or no overlap between cholesterol and PIP2 binding sites. Taken together, these observations suggest that cholesterol and PIP2 regulate the channels through distinct binding sites but that the signals generated by the binding of the two modulators converge.
Keywords: Ion channel regulation, Cholesterol binding sites, PIP2 binding sites
1. Introduction
Inwardly rectifying K+ channels (Kir) are a major class of potassium channels that are ubiquitously expressed in numerous cell types and play central roles in regulating membrane potential and K+ homeostasis [1,2]. A series of studies from our laboratory (eg. [3–5]) and from other investigators (eg. [6–10]) showed that Kir2 channels are regulated by two lipid modulators that are integral part of the plasma membrane. These include cholesterol, a major lipid component of the plasma membrane constituting 10 to 45% of total lipids [11], and phosphatidylinositol 4,5-bisphosphate (PIP2), a minor lipid component of the plasma membrane constituting about 1% of the lipids in the inner leaflet [12], as described in detail in several of our recent reviews [13–15]. Our studies have shown that most Kir channels are cholesterol sensitive with the predominant effect being cholesterol-induced suppression of channel function [3–5]. Focusing on Kir2 channels, a major sub-family of the Kir family, we showed that cholesterol sensitivity of the channels can be attributed to stabilizing the channels in the closed state [3,16] and not to the interaction of the channels with caveolin (Cav-1) [17]. Moreover, we identified several structural domains that confer cholesterol sensitivity to the channels: a “cholesterol sensitivity belt” that confers cholesterol sensitivity to the channels by regulating the impact of cholesterol on channel gating [18], a “two-way molecular switch” that allows switching of cholesterol sensitivity of the channels by altering interactions between critical residues in the cytosolic domain [19] and functional links between the N- and the C-termini [20]. Furthermore, based on studies on cholesterol sensitivity of bacterial homologues of Kir channels reconstituted into liposomes, we demonstrated that cholesterol regulates Kir channels through direct sterol–protein interactions [21,22]. Similarly, a later study showed that purified Kir2 channels are also sensitive to cholesterol [10]. Finally, we identified two novel putative cholesterol binding sites in Kir2.1 channels that are found in non-annular sites in between the transmembrane helices of the channels [23].
In contrast to cholesterol, the dominant effect of PIP2 on Kir channels is activation that is attributed to stabilizing the channels in the open state [7–10]. PIP2 was also found to directly interact with Kir channels via electrostatic interactions between the negatively charged phosphate groups of the phosphate head group and positively charged amino acids in the cytoplasmic domains of the channel (reviewed by [14,24–26]). Indeed, PIP2 binding sites have been recently identified by solving the structure of Kir2.2 channel crystallized with the PIP2 [27].
The effects of the two modulators on Kir function are clearly distinct: changes in membrane cholesterol regulate purified bacterial Kir in the absence of PIP2 [21,22] and sequestering of cellular PIP2 does not affect cholesterol sensitivity of Kir2.1 in mammalian cells [16]. Conversely, PIP2 was shown to activate purified Kir2 channels in the absence of cholesterol [28]. However, there is also accumulating evidence that there is significant functional interaction between cholesterol and PIP2 in regulating Kir2 channels. Specifically, several residues are shown to play important roles in the sensitivities of the channels to both cholesterol and PIP2 [16,19]. Furthermore, we showed earlier that cholesterol depletion results in a delay in the rundown of the channels' activity in response to neomycin [16] that is known to sequester PIP2, an observation that suggests that cholesterol affects Kir–PIP2 interactions. In this study, we provide further evidence that cholesterol regulates Kir–PIP2 interactions and analyze the structural overlap between the residues responsible for cholesterol and PIP2 sensitivities of the channels.
2. Materials and methods
2.1. Cells, cholesterol modulation and transfection
Chinese hamster ovary (CHO) K1 cell line was maintained as previously described [16]. Cholesterol depletion was performed by exposing the cells to 5 mM methyl-β-cyclodextrin (MβCD) which results in ~ 50–60% decrease in the level of membrane cholesterol [4]. MβCD and cholesterol were purchased from Sigma Chemical (St. Louis, MO). Cells were transiently co-transfected with Kir2.x constructs and eGFP (cmv-pcDNA3.1-GFP-TOPO, Invitrogen, Carlsbad, CA) using Lipofectamine (Gibco-BRL, Gaithersburg, MD) according to the manufacturer's instructions. The experiments were performed 24–48 h after transfection.
2.2. Kir2.x constructs and electrophysiology
Kir2.x constructs were co-transfected with eGFP (cmv-pcDNA3.1-GFP-TOPO, Invitrogen, Carlsbad, CA) using Lipofectamine (Gibco-BRL) according to the manufacturer's instructions. Kir2.1-HA and Kir2.3-HA with the HA tags (influenza hemagglutinin epitope: YPYDVPDYA) inserted into the extracellular domains of the channels were inserted in a pGW1 vector. Both HA-Kir constructs were a gift of Dr. Carol Vandenberg. Recordings were performed in whole-cell of the standard patch clamp technique [29]. Pipettes were pulled (SG10 glass, 1.20 mm ID, 1.60 mm; Part# FPENNU1.20ID1.60OD, Richland Glass, Richland, NJ) to give a final resistance of 2–6 MΩ. These pipettes generated high resistance seals without fire polishing. A saturated salt agar bridge was used as reference electrode. Currents were recorded using an EPC9 amplifier (HEKA Electronik, Lambrecht, Germany) and accompanying acquisition and analysis software (Pulse & PulseFit, HEKA Electronik, Lambrecht, Germany). The external recording solution contained (in mM): 150 NaCl, 6 KCl, 10 HEPES, 1.5 CaCl2, 1 MgCl2, and 1 EGTA, pH 7.3. The pipette solutions contained (in mM): 145 KCl, 10 HEPES, 1 MgCl2, 4 ATP, 1 EGTA, and pH 7.3. Current was monitored by 500 ms linear voltage ramps from − 160 mV to + 60 mV at an interpulse interval of 5 s. The holding potential for both protocols was − 60 mV. Pipette and whole cell capacitances were automatically compensated. Whole cell capacitance and series resistance were compensated and monitored throughout the recording. Rs was compensated by 60–90% with the compensation being limited by the stability of the patch. Current density was calculated by normalizing the currents recorded from individual cells to the capacitance of the same cell.
2.3. Statistical analysis
Statistical significance was evaluated using a Standard t-test assuming two-tailed distributions with unequal variance. All experiments were performed with 5–10 cells for each experimental condition.
3. Results
3.1. Impact of cholesterol depletion on Kir2.1–PIP2 and Kir2.3–PIP2 interactions
3.1.1. Kir2.1
Earlier studies assessed the functional interactions between Kir channels and PIP2 by over-expressing the channels in Xenopus oocytes and measuring the rundown of the current in excised inside-out macropatches with and without perfusing PIP2 to the cytosolic side of the membrane (eg. [7,8]). These studies showed that Kir currents rapidly decreased after the excision of the patch but could be reactivated by perfusion of PIP2. Furthermore, the rate of recovery depended on the strength of the Kir–PIP2 interactions and could be affected by specific mutations or by using different PIP2 analogs. Thus, measuring PIP2-dependent current rundown or recovery was established as a method to assess the strength of Kir–PIP2 interactions (eg. [7,8]). In our recent study, we used an alternative approach of dialyzing neomycin, an agent known to sequester PIP2 by binding to its headgroups and restricting its ability to interact with proteins [30,31], into the patch pipette in whole-cell configuration [16]. As expected, dialyzing neomycin into the pipette resulted in Kir2.1 rundown expressed in Chinese hamster ovary cells (CHO) [16]. We also showed that cholesterol depletion resulted in a significant delay in the current rundown of Kir2.1. Since a faster rundown is interpreted as a weaker Kir–PIP2 interaction [8], these data led us to hypothesize that cholesterol depletion strengthens Kir2.1–PIP2 interactions [16]. It is important to note that since the currents are normalized to the currents recorded at time 0, a delay in neomycin-induced current rundown cannot be attributed to an increase in local PIP2 concentration. To further characterize this observation, we first show here that neomycin-induced rundown of Kir2.1 current is concentration-dependent. Fig. 1A (upper family of traces) shows typical Kir2.1 whole cell currents in CHO cells at time 0 before the start of the dialysis and 60 s after establishing whole-cell configuration that allows neomycin to diffuse into the cytosol. Fig. 1B shows that the effect is concentration-dependent with 1 μM of neomycin having no effect while concentrations of 10 and 100 μM neomycin induce significant rundown. Also, as was shown in our previous study described above, depletion of the cells of cholesterol using 5 mM MβCD results in a significant delay in the rundown suggesting that cholesterol depletion results in strengthening of Kir2.1–PIP2 interactions (Fig. 1C). To further verify that this effect is specific for PIP2, a complementary series of experiments was performed by dialyzing PIP2 antibodies into the cell. The rundown in this case is significantly slower than with neomycin, which most likely reflects a slower diffusion of the antibodies into the cell but, more importantly, cholesterol depletion has the same effect of slowing the rundown process, which points to strengthening Kir2.1–PIP2 interactions (Fig. 1D). This experiment also excludes the possibilities that the observed delay in the current rundown can be due to the effect of cholesterol on the neomycin access to the membrane or PIP2-independent effects of neomycin on channel function. The possibility that the effect of MβCD on neomycin-induced current rundown can be independent of cholesterol depletion was excluded in our previous study by comparing MβCD and MβCD saturated with cholesterol [16].
Fig. 1.
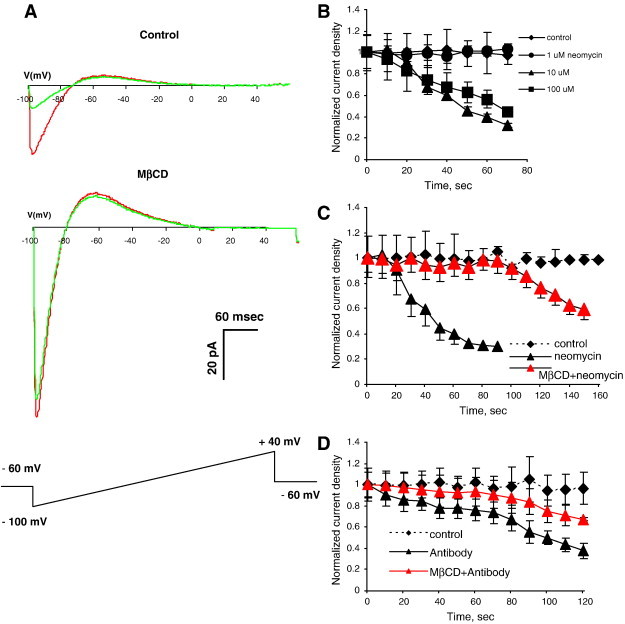
Impact of cholesterol depletion on Kir2.1–PIP2 interactions.
A. Representative Kir2.1 current traces before (red) and after (green) dialyzing neomycin into the cell through a patch pipette. The upper family of traces was recorded in a cell in normal cholesterol conditions and the lower family of traces recorded from a cell depleted of cholesterol by pre-exposure to 5 mM MβCD for 1 h. B: The time courses of Kir2.1 rundown in response (1–100 μM) neomycin. C: The time courses of Kir2.1 rundown in response 10 μM neomycin in control and cholesterol depleted cells. D: The time courses of Kir2.1 rundown in response anti-PIP2 antibodies in control and cholesterol depleted cells (all data show means + SEM, n = 5–10 cells per condition).
3.1.2. Kir2.3
Qualitatively similar effects were observed with Kir2.3 channels (Fig. 2). As expected, since Kir2.3 is known to have a weaker affinity to PIP2 than Kir2.1, the dose response of the current rundown in response to neomycin was shifted to lower concentrations of neomycin with 1 μM neomycin leading to significant rundown (Fig. 2A). More importantly, we show that, similar to Kir2.1, cholesterol depletion resulted in a significant delay in Kir2.3 current rundown when cells were dialyzed with 1 μM neomycin (Fig. 2B). Also, the delay in Kir2.3 rundown was less pronounced than that in Kir2.1 channels suggesting that the impact of cholesterol depletion on Kir2.3 channels is weaker than that on Kir2.1 channels. This observation is indeed consistent with our previous studies showing that Kir2.3 is less sensitive to cholesterol than Kir2.1 [3].
Fig. 2.
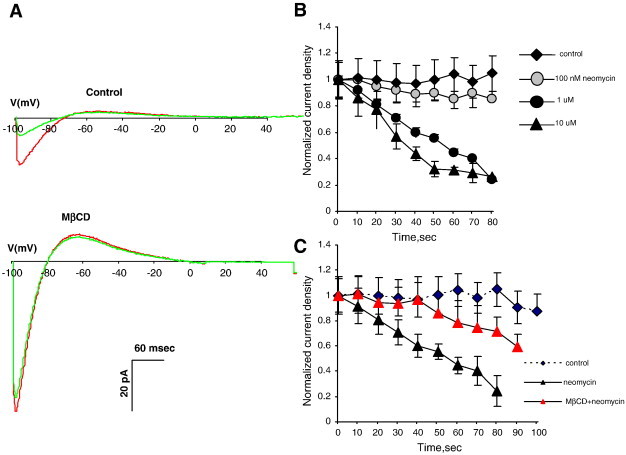
Impact of cholesterol depletion on Kir2.3–PIP2 interactions.
A. Representative Kir2.1 current traces before (red) and after (green) dialyzing neomycin into the cell through a patch pipette in a control cell (upper traces) and in a cholesterol depleted cell (lower family). B: The time courses of Kir2.1 rundown in response (100 nM–10 μM) neomycin. C: The time courses of Kir2.1 rundown in response 1 μM neomycin in control and cholesterol depleted cells (all data show means + SEM, n = 5–10 cells per condition).
3.2. Reverse mutations Kir2.1 L222I and Kir2.3 I214L Have opposite effects on cholesterol depletion-induced delays of Kir2.1/Kir2.3 rundown
Our earlier studies showed that a single residue substitution of leucine at position 222 to isoleucine abrogates the sensitivity of Kir2.1 channels to cholesterol whereas the reverse substitution of an isoleucine at a corresponding position of Kir2.3 to leucine increases cholesterol sensitivity of Kir2.3 channels [16]. Here, we tested the impact of these reverse mutations on neomycin-induced current rundown. Our new data show that L222I substitution abrogates the effect of cholesterol depletion on the kinetics of neomycin-induced Kir2.1 current rundown (Fig. 3A). As was shown in Fig. 1, the rundown of the WT Kir2.1 is strongly delayed by cholesterol depletion (see triangle symbols) but the time courses of L222I Kir2.1 rundown in control and cholesterol depleted cells are identical (circles), indicating that cholesterol depletion has no effect on the strength of the interaction of Kir2.1 L222I mutants with PIP2. A loss of the delay effect is consistent with the loss of cholesterol sensitivity of Kir2.1 L222I described above.
Fig. 3.
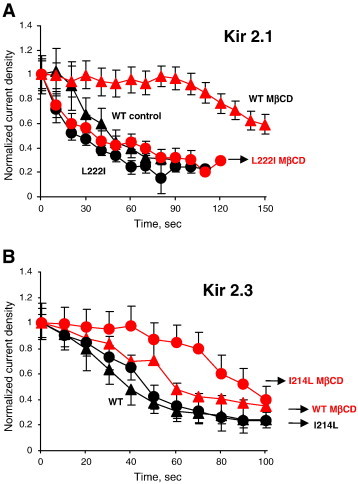
Impact of reverse mutations Kir2.1 L222I and Kir2.3 I214L on cholesterol dependence of Kir–PIP2 interactions.
A: The time courses of Kir2.1 WT and Kir2.1-L222I rundown in response 10 μM neomycin in control and cholesterol depleted cells. B: The time courses of Kir2.3 WT and Kir2.3-I214L rundown in response 1 μM neomycin in control and cholesterol depleted cells (all data show means + SEM, n = 5–10 cells per condition).
Conversely, I214L substitution enhances the effect of cholesterol depletion on the kinetics of neomycin-induced Kir2.3 rundown (Fig. 3B). More specifically, we show here that whereas in the Kir2.3 WT channel cholesterol depletion resulted in a relatively small delay in the neomycin-induced current rundown (triangles), the same treatment resulted in a much stronger delay in the neomycin-induced current rundown of the Kir2.3 I214L mutant (circles). Again, these observations are consistent with the increased sensitivity of Kir2.3 I214L to cholesterol compared to the WT Kir2.3 channel that was demonstrated in our earlier studies [16]. Taken together, data shown in Figs. 3A and B suggest that cholesterol plays an important role in determining the strength of Kir–PIP2 interactions.
4. Discussion
Several studies demonstrated that in mammalian cells PIP2 concentrates in low-density membrane fractions suggesting that it resides in cholesterol-rich membrane domains [32,33]. Furthermore, it was also shown that cholesterol depletion abolishes the localization of PIP2 to cholesterol-rich membrane fractions [34]. Consistent with these observations, we showed that cholesterol depletion results in substantial changes in the localization of fluorescently tagged PIP2 in aortic endothelial cells leading from punctuate to uniform distribution, which in turn has a significant effect on membrane–cytoskeleton adhesion [35]. Cholesterol depletion was also suggested to inhibit PIP2 turnover and hydrolysis (Kwik et al., 2003). Thus, since both cholesterol and PIP2 are known to regulate Kir channels, it is plausible to expect that the two effects would be mutually dependent.
One possibility that we explored in our earlier studies is that cholesterol may regulate Kir channels by regulating their access to PIP2. Indeed, if cholesterol depletion disperses PIP2 from cholesterol-rich to cholesterol-poor domains, it might increase the local concentration of PIP2 near the channels in cholesterol-poor domains and increase Kir activity. Our observations, however, show that this is not likely to be the case because sequestering PIP2 has no effect on cholesterol sensitivity of Kir2.1 channels [16]. Clearly, an increase in PIP2 hydrolysis also cannot contribute to the sensitivity of the channels to cholesterol because, as described above, cholesterol depletion increases Kir2 activity whereas loss of membrane PIP2 results in the loss in channel function [14]. In this study, we demonstrate an alternative possibility that cholesterol depletion results in strengthening of Kir2–PIP2 interactions, as assayed by the rundown of the current in response to PIP2 sequestration. This effect is consistent with the fact that both cholesterol depletion and the interaction of the channels with PIP2 result in an increase in Kir2 currents. The effect of cholesterol depletion-induced strengthening of Kir2–PIP2 interactions is observed for two major members of the Kir2 subfamily, Kir2.1 and Kir2.3. Furthermore, a single-residue mutation in Kir2.1 that was shown earlier to abrogate cholesterol sensitivity of the channel and decrease Kir2.1–PIP2 interactions also abrogates the effect of cholesterol on Kir2.1–PIP2 interactions whereas a reverse mutation in Kir2.3 increases cholesterol sensitivity of the channels and strengthens the effect of cholesterol on Kir2.3–PIP2 interactions.
Is it then possible that cholesterol sensitivity of Kir2 channels might be fully attributed to regulation of Kir2–PIP2 interactions? Our data does not support this possibility. First, our recent studies showed that cholesterol regulates bacterial Kir channels reconstituted in liposomes in the absence of PIP2 and that this effect critically depends on Kir-cholesterol binding [21,22]. Even though specific effects of PIP2 on Kir2.1 and KirBac1.1 are different, the observations with purified KirBac1.1 channels demonstrate that cholesterol can interact with Kir channels directly. It is not possible, however, to test whether this is also true for mammalian Kir2 channels because these channels require PIP2 for activation. We have also shown earlier that in contrast to cholesterol depletion, cholesterol enrichment had no effect on Kir2.1–PIP2 interactions indicating that this mechanism does not contribute to the suppression of Kir current in cholesterol-enriched cells [16]. We conclude, therefore, that strengthening of Kir2–PIP2 interactions may contribute to the sensitivity of mammalian Kir2 channels to cholesterol depletion but cannot fully account for the sensitivity of the channels to cholesterol.
Comparing the structural requirements for the sensitivity of the channels to the two lipid modulators provides further insights into the crosstalk between the sensitivities of Kir2.1 channels to cholesterol and to PIP2 (Fig. 4A). Earlier studies identified a series of residues in the cytosolic domains of the channels that affect the strength of the interactions between Kir2.1 and PIP2: specifically, three “PIP2-sensitive residues” were identified in the N-terminus (K50, H53, R67), five residues were identified at the interface between the transmembrane domain 2 (TM2) and the C-terminus (K182, K185, K187, K188 and R189) and eleven were identified in the C-terminus including the CD loop (N216, R218, K219 and L222), βD loop (R228), EF loop (N251), and G-loop (G300, V302, E303, C311 and R312) [19,36,37]. Furthermore, several of these residues, or more precisely, the corresponding residues in Kir2.2 channels, that reside on the interface between the TM and cytosolic domains were found to interact directly with PIP2 in crystallography studies [27]. We tested a number of the “PIP2 sensitive residues” for their role in cholesterol sensitivity of the channels and found that there is a 60% overlap between the two sets: out of 10 “PIP2-sensitive residues” tested for their sensitivity to cholesterol, 6 residues (H53, N216, K219, L222, N251, C311) were also found to be important for cholesterol sensitivity of the channels and 4 residues (K182, K185, K187, R228) were not [16,18,19]. It is also interesting to note that among the 4 residues that are important for the PIP2 sensitivity but not for cholesterol sensitivity of the channels, the majority are part of the PIP2 binding site. A significant (60%) overlap between the residues that are important for the channel sensitivity to both modulators strongly suggests a common mechanism.
Fig. 4.
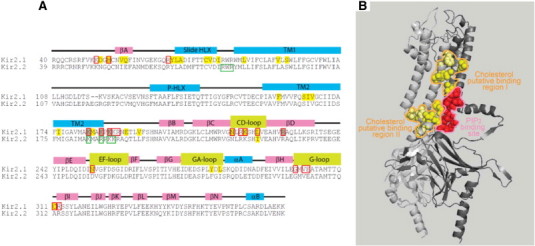
A: Overlap between residues involved in cholesterol or PIP2 sensitivity of Kir2.1. The figure shows sequence alignment of the Kir2.1 and Kir2.2 channels with key secondary structural elements labeled above the sequence. Residues that were found to affect the cholesterol sensitivity of the channel in functional studies are highlighted yellow. Red boxes highlight residues that were found to play an important role in channel-PIP2 interactions in functional studies. Yellow-filled red boxes highlight residues whose mutation affects both channel–PIP2 interactions and the sensitivity of the channel to cholesterol. Light brown-filled red boxes filled in light brown highlight residues whose mutation to glutamine affects channel–PIP2 interactions but does not affect the sensitivity of the channel to cholesterol. Green boxes highlight residues with direct bonding interactions to PIP2 as determined using crystallography. B: A lack of overlap between the PIP2-binding site and putative cholesterol-binding sites in Kir2.2. A ribbon presentation of two adjacent subunits (Gray and light gray) of the crystal structure of Kir2.2 (PDB ID 3spi) showing the binding site of PIP2 in the channel (red balls). Also shown are the corresponding Kir2.2 residues to Kir2.1 residues that form two putative cholesterol binding sites in Kir2.1 based on functional data and molecular modeling (yellow balls — direct interaction; light yellow balls — secondary effect within 4 Å from directly interacting residues).
What is the possible mechanism of the structural overlap between the residues that confer the sensitivities of the channels to cholesterol and to PIP2? One obvious possibility would be an overlap of the two binding sites. This possibility would be consistent with the opposite effects of cholesterol and PIP2 on channel function because if cholesterol was to prevent PIP2 from binding to the channel, it would result in the inhibition of the current and vice versa, cholesterol depletion would result in increased Kir–PIP2 binding and increase in channel activity. However, structural information does not support this possibility: First, from the analysis described above, we see that the overlap between the “PIP2 sensitive” and “cholesterol-sensitive” residues actually excludes residues that interact with PIP2 directly. Indeed, as shown in Fig. 4B, in agreement with earlier functional data for Kir2.1 [24,36–38], the binding site of PIP2 in Kir2.2 channels is formed of positively charged residues at the interface between the cytosolic and the TM domains [27]. The equivalent site in Kir2.1 is adjacent to the two putative cholesterol binding sites found in Kir2.1 in our recent studies but does not overlap with them [23]. The first putative cholesterol-binding site is located between the TM helices of two adjacent channel subunits whereas the second one is located at the interface between the cytosolic and transmembrane domains of the channel. The corresponding residues in Kir2.2 that form these two putative cholesterol binding sites in Kir2.1 are depicted in Fig. 4B. These data indicate that the two modulators bind to different adjacent non-overlapping sites of the channels and suggest that functional cross-talk between them may be due to local interactions between the adjacent binding sites as well as due to the overlap in the intramolecular transduction pathway that leads from modulator binding to channel gating. Interestingly, a similar possibility that two modulators may bind to distinct binding sites but then converge in their regulatory function was demonstrated in our recent study of the cross talk between cholesterol and caveolin in regulating Kir2.1 channels [17].
In conclusion, we propose the following model: Earlier studies suggested that PIP2 binding to the interface between the TM and the cytosolic domains may act as a “glue” between the cytosolic tails of the channels exerting a mechanical force for the channels to open [24]. We propose here that binding of cholesterol in between the helices of the TM domain exerts the opposite effect making it more difficult for the TM helices to move into the open conformation of the channels, thus stabilizing the channels in the closed state. This working hypothesis is demonstrated in Fig. 5 when cholesterol binds between the TM helices of the channels, it interferes with the tilt of the TM2 domain necessary for the channel opening and weakens the strength of the interaction between the channel and PIP2 (Fig. 5: left). It is also important to note, however, that cholesterol does not necessarily prevent PIP2 from binding to the channel. In contrast, removal of cholesterol from its binding sites in the Kir2 channel allows the TM2 domain to tilt into an open conformation and enhances PIP2 binding at the interface between the cytosolic and TM domains of the channels that stabilizes the channels in the open state (Fig. 5: right). Clearly, however, more studies are needed to test this hypothesis.
Fig. 5.
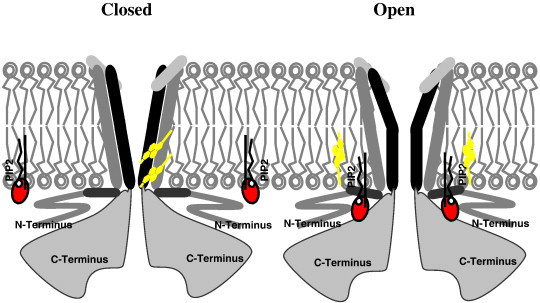
Schematic model of cholesterol and PIP2 interactions with Kir2 channels.
It is also important to note that the opposite effects of cholesterol and PIP2 have been reported for multiple types of ion channels. Indeed, similar to Kir2.1 channels, the dominant effect of cholesterol on multiple types of ion channels, including other types of Kir and several types of voltage-gated K+, Na+ and Ca+ 2 channels is to suppress channel activity [14], whereas the most common effect of PIP2 is to increase channel activity of the same channels [39]. Little is known however about the functional cross-talk between the two lipid modulators in regulating other types of ion channels. Our current study proposes a new model for the interplay between these two modulators in regulating ion channel function.
Acknowledgments
We thank Mr. Gregory Kowalsky for his help in the preparation of the figures and Ms. Catherine Osborn for helping in preparing the revision. The work was supported by the National Institutes of Health grants HL073965 and HL083298 (to I. Levitan) and a Scientist Development Grant (11SDG5190025) from the American Heart Association (to A. Rosenhouse-Dantsker).
References
- 1.Kubo Y. International Union of Pharmacology. LIV. Nomenclature and molecular relationships of inwardly rectifying potassium channels. Pharmacol Rev. 2005;57(4):509–526. doi: 10.1124/pr.57.4.11. [DOI] [PubMed] [Google Scholar]
- 2.Hibino H. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010;90(1):291–366. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- 3.Romanenko V.G. Cholesterol sensitivity and lipid raft targeting of Kir2.1 channels. Biophys J. 2004;87:3850–3861. doi: 10.1529/biophysj.104.043273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tikku S. Relationship between Kir2.1/Kir2.3 activity and their distribution between cholesterol-rich and cholesterol-poor membrane domains. Am J Physiol Cell Physiol. 2007;293:C440–C450. doi: 10.1152/ajpcell.00492.2006. [DOI] [PubMed] [Google Scholar]
- 5.Rosenhouse-Dantsker A. Comparative analysis of cholesterol sensitivity of Kir channels: role of the CD loop. Channels. 2010;4:63–66. doi: 10.4161/chan.4.1.10366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang C.L., Feng S., Hilgemann D.W. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by GBY. Nature. 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- 7.Rohacs T. Distinct specificities of inwardly rectifying K + channels for phosphoinositides. J Biol Chem. 1999;274(51):36065–36072. doi: 10.1074/jbc.274.51.36065. [DOI] [PubMed] [Google Scholar]
- 8.Du X. Characteristic interactions with PIP2 determine regulation of Kir channels by diverse modulators. J Biol Chem. 2004:M403413200. doi: 10.1074/jbc.M403413200. [DOI] [PubMed] [Google Scholar]
- 9.Enkvetchakul D., Jeliazkova I., Nichols C.G. Direct modulation of Kir channel gating by membrane phosphatidylinositol 4,5-bisphosphate. J Biol Chem. 2005;280(43):35785–35788. doi: 10.1074/jbc.C500355200. [DOI] [PubMed] [Google Scholar]
- 10.D'Avanzo N. Enantioselective protein–sterol interactions mediate regulation of both prokaryotic and eukaryotic inward rectifier K < sup > + </sup > channels by cholesterol. PLoS One. 2011;6(4):e19393. doi: 10.1371/journal.pone.0019393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yeagle P.L. Cholesterol and the cell membrane. Biochim Biophys Acta. 1985;822:267–287. doi: 10.1016/0304-4157(85)90011-5. [DOI] [PubMed] [Google Scholar]
- 12.McLaughlin S. PIP(2) and proteins: interactions, organization, and information flow. Annu Rev Biophys Biomol Struct. 2002;31:151–175. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- 13.Levitan I. Cholesterol and ion channels, in cholesterol binding and cholesterol transport proteins. In: Harris J.R., editor. Springer Science; 2010. pp. 509–549. [Google Scholar]
- 14.Rosenhouse-Dantsker A., Mehta D., Levitan I. Regulation of ion channels by membrane lipids. Compr Physiol. 2012;2:31–68. doi: 10.1002/cphy.c110001. [DOI] [PubMed] [Google Scholar]
- 15.Levitan I., Singh D.K., Rosenhouse-Dantsker A. Cholesterol binding to ion channels. Front Physiol. 2014;5 doi: 10.3389/fphys.2014.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Epshtein Y. Identification of a C-terminus domain critical for the sensitivity of Kir2.1 channels to cholesterol. Proc Natl Acad Sci U S A. 2009;106:8055–8060. doi: 10.1073/pnas.0809847106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han H. Silencing of Kir2 channels by caveolin-1: cross-talk with cholesterol. J Geophys Res. 2014 Sep 15;592(Pt 18):1025–4038. doi: 10.1113/jphysiol.2014.273177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenhouse-Dantsker A., Logothetis D.E., Levitan I. Cholesterol sensitivity of KIR2.1 is controlled by a belt of residues around the cytosolic pore. Biophys J. 2011;100(2):381. doi: 10.1016/j.bpj.2010.11.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosenhouse-Dantsker A. Distant cytosolic residues mediate a two-way molecular switch that controls the modulation of inwardly rectifying potassium (Kir) channels by cholesterol and phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) J Biol Chem. 2012;287(48):40266–40278. doi: 10.1074/jbc.M111.336339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenhouse-Dantsker A. Cholesterol sensitivity of KIR2.1 depends on functional inter-links between the N and C termini. Channels. 2013;7:303–312. doi: 10.4161/chan.25437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh D.K. Direct regulation of prokaryotic Kir channel by cholesterol. J Biol Chem. 2009;284(44):30727–30736. doi: 10.1074/jbc.M109.011221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh D.K. Cholesterol regulates prokaryotic Kir channel by direct binding to channel protein. Biochim Biophys Acta (BBA) — Biomembr. 2011;1808(10):2527. doi: 10.1016/j.bbamem.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosenhouse-Dantsker A. Identification of novel cholesterol-binding regions in Kir2 channels. J Biol Chem. 2013;288(43):31154–31164. doi: 10.1074/jbc.M113.496117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Logothetis D.E. Phosphoinositide-mediated gating of inwardly rectifying K(+) channels. Pflugers Arch. 2007;455(1):83–95. doi: 10.1007/s00424-007-0276-5. [DOI] [PubMed] [Google Scholar]
- 25.Hilgemann D.W. On the physiological roles of PIP(2) at cardiac Na + Ca2 + exchangers and K(ATP) channels: a long journey from membrane biophysics into cell biology. J Physiol. 2007;582:903–909. doi: 10.1113/jphysiol.2007.132746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suh B.C., Hille B. PIP2 is a necessary cofactor for ion channel function: how and why? Annu Rev Biophys. 2008;37:175–195. doi: 10.1146/annurev.biophys.37.032807.125859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hansen S.B., Tao X., MacKinnon R. Structural basis of PIP2 activation of the classical inward rectifier K + channel Kir2.2. Nature. 2011;477(7365):495. doi: 10.1038/nature10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D'Avanzo N. Direct and specific activation of human inward rectifier K + channels by membrane phosphatidylinositol 4,5-bisphosphate. J Biol Chem. 2010;285(48):37129–37132. doi: 10.1074/jbc.C110.186692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hamill O.P. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391(2):85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 30.Downes C.P., Michell R.H. The polyphosphoinositide phosphodiesterase of erythrocyte membranes. Biochem J. 1981;198:133–140. doi: 10.1042/bj1980133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kwik J. Membrane cholesterol, lateral mobility, and the phosphatidylinositol 4,5-bisphosphate-dependent organization of cell actin. Proc Natl Acad Sci U S A. 2003;100(24):13964–13969. doi: 10.1073/pnas.2336102100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pike L., Casey L. Localization and turnover of phosphatidylinositol 4,5-bisphospate in caveolin-enriched membrane domains. J Biol Chem. 1996;271(43):26453–26456. doi: 10.1074/jbc.271.43.26453. [DOI] [PubMed] [Google Scholar]
- 33.Golub T., Caroni P. PI(4,5)P2-dependent microdomain assemblies capture microtubules to promote and control leading edge motility. J Cell Biol. 2005;169(1):151–165. doi: 10.1083/jcb.200407058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pike L.J., Miller J.M. Cholesterol depletion delocalizes phosphatidylinositol bisphosphate and inhibits hormone-stimulated phosphatidylinositol turnover. J Biol Chem. 1998;273(35):22298–22304. doi: 10.1074/jbc.273.35.22298. [DOI] [PubMed] [Google Scholar]
- 35.Hong Z. How cholesterol regulates endothelial biomechanics? Front Vasc Biol. 2012;3:426. doi: 10.3389/fphys.2012.00426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soom M. Multiple PIP2 binding sites in Kir2.1 inwardly rectifying potassium channels. FEBS Lett. 2001;490:49–53. doi: 10.1016/s0014-5793(01)02136-6. [DOI] [PubMed] [Google Scholar]
- 37.Lopes C.M. Alterations in conserved Kir channel–PIP2 interactions underlie channelopathies. Neuron. 2002;34:933–944. doi: 10.1016/s0896-6273(02)00725-0. [DOI] [PubMed] [Google Scholar]
- 38.Logothetis D.E., Lupyan D., Rosenhouse-Dantsker A. Diverse Kir modulators act in close proximity to residues implicated in phosphoinositide binding. J Physiol. 2007:133157. doi: 10.1113/jphysiol.2007.133157. [jphysiol.2007] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Logothetis D.E. Channelopathies linked to plasma membrane phosphoinositides. Pflugers Arch. 2010;460:321–341. doi: 10.1007/s00424-010-0828-y. [DOI] [PMC free article] [PubMed] [Google Scholar]