

Abstract
Although much is known about the physiological responses of many environmental stresses in tolerant animals, studies evaluating the regulation of stress-induced mechanisms that regulate the transitions to and from this state are beginning to explore new and fascinating areas of molecular research. Current findings have developed a general, but refined, view of the important molecular pathways contributing to stress-survival. However, studies utilizing newly developed technologies that broadly focus on genomic and proteomic screening are beginning to identify many new targets for future study. This minireview will provide a contextual overview on the use of DNA/RNA sequencing, microRNA annotation and prediction software, protein structure and function prediction tools, as well as methods of high-throughput protein expression analysis. We will also use select examples to highlight the existing use of these technologies in stress biology research. Such tools can be used in comparative stress biology in the characterization of animal responses to environmental challenges. Although there are many areas of study left to be explored, research in comparative stress biology will always be continuing as new technologies allow the further analysis of cell function, and new paradigms in gene regulation and regulatory molecules (such as microRNAs) are continuing to be discovered. Building upon the findings of past research, while utilizing new technologies in the appropriate manner, future studies can be carried out in new and exciting areas still unexplored. Proper use of rapidly developing technologies will help to create a complete understanding of the animal stress response and survival mechanisms utilized by many diverse organisms.
Keywords: Stress biology, Protein structure, RNA sequencing, Microarray, Multiplex
1. Introduction
Currently, the field of comparative animal biology is at the beginning of a large expansion of experimental knowledge as studies start to utilize new high-throughput technologies. To date, research has discovered much about the physiological responses of many tolerant animals to environmental stress [1–7], however new studies are broadly focused on genomic and proteomic screening and have been identifying many new targets for future study [8–10]. Many of these technologies should be intriguing to the comparative stress biologist, who now has available technology to assess the global expression of nearly all genes and proteins that contribute to survival in stress-tolerant animals [11]. Although there are many areas of study left to be explored, research in comparative and animal stress biology will always be continuing with the advancement of technologies that allow new insight into cell function, and new paradigms in gene regulation and regulatory molecules (such as microRNAs) are continuing to be discovered. Proper knowledge and use of genomic and proteomic-based technology will help to create a complete understanding of the stress response and survival mechanisms that are utilized by many diverse organisms.
Within the next few years the entire genome of many organisms, including those that display tolerances to extreme environmental conditions, will most likely be sequenced. For example, the genome of the anoxia/freeze-tolerant Western painted turtle (Chrysemys picta bellii) was only sequenced in 2013 and the hibernating thirteen-lined ground squirrel (Ictidomys tridecemlineatus) has been sequenced since 2008 [12]. With the current and future availability of genomic information, the prepared comparative biologist will be provided with a blue-print for protein structure, control domains and sites of post-translational modifications that are either conserved or perhaps unique in those organisms. The overall goal for global modeling of the cell is to better predict the behavior of biological systems. This type of research will have profound implications for the understanding of basic biology and improving future stress-tolerance of human systems.
As previously mentioned, being able to successfully utilize newly developed technologies and resources, researchers will be able to build upon previously explored areas of study. The end result will most likely be a deeper and extensive understanding of the biological processes that underlie natural mechanisms of animal stress tolerance. It should also be noted that the ability to analyze the stress response at a global level is not limited to the availability of a genome [13]. It is one of the goals of this minireview to provide a contextual overview of technologies and tools that can provide omic-level analysis, without the absolute need for an annotated genome. Several technologies have emerged in the recent years that allow researchers to quantitatively analyze the cellular response in a relatively short period of time and at low cost. These technologies include (1) the use of microarrays to examine the responses of mRNA [14], protein and microRNAs, (2) the use of RNA sequencing (RNASeq) to evaluate the state of transcription among all expressed genes [15], (3) multiplexed assays that have the ability to assess the expression of multiple analytes (mRNA, protein and enzyme activity) [16], and (4) the prediction of protein structure and function [17]. Below are brief overviews of each technology and its application to the field of comparative molecular biology.
2. Microarray analysis of gene and protein expression
Microarrays are widely available in the research marketplace, functioning as a solid-support for thousands of different sequences that are fixed at specific locations [18]. To date, there are a variety of microarray types and formats. Essentially, microarrays can be received as an advancement on end-point RT-PCR or immunoblotting as they have the ability to measure the expression of a very large number of genes (cDNA/oligo-based capture) or proteins (antibody-based capture) at the same time and within a single sample [19]. As a result, they are typically a chosen technology for experiments that require a large number of genes to be measured quickly or when sample amount is extremely limited for study. These arrays are also useful when discovery or initial characterization of a new model organism is necessary because they allow either the generation of project “leads” (heterologous screening) or a quantitative assessment of gene/protein expression when a homologous array is used (Box. 1) [20,21]. As microarrays can be used to examine the expression of hundreds (protein) or thousands (gene) of targets at once, it holds the promise to complete multiple years of RT-PCR or immunoblotting expression research (target-based) within days. However it is critical to note that the results obtained by microarray experiment need to be validated through other methods of expression analysis (ie. Immunoblotting (protein) or qRT-PCR (gene)). One must also realize that this technology is steadily changing and improving as new advances are being made to increase both array reproducibility and specificity. Nevertheless, at its current state this technology provides an excellent research tool to the comparative biologist to obtain complete expression data or a simple generation of project leads, using either homologous or heterologous arrays, that can be used to identify possible areas of future study. Ultimately, microarray-based studies promise to expand the knowledge of the cellular stress response, revealing patterns of coordinated gene expression and perhaps even uncovering entirely new stress-responsive cellular pathways. When combined with appropriate bioinformatic tools, microarray technology also aids in integrating target expression data with function at the cellular level, revealing hypotheses of how multiple targets may work together to produce a particular stress response to match a particular cellular need (such as metabolic adjustments, cytoskeletal reorganization, etc.) [22]. Outlined below is specific information regarding both DNA and protein microarrays.
Box 1.
Heterologous array: Using an array with sample from an animal that is different from the animal that the array was designed for (ex. testing for squirrel gene expression using a mouse cDNA microarray).
Homologous array: Using an array with samples from the same species that the array was designed for (ex. Using mouse samples on a mouse cDNA microarray for which it was designed).
2.1. Gene expression
Microarrays can be used to detect mRNA expression patterns comparatively within different stresses, organisms, tissues or time-points. The previous research from the Storey lab, using heterologous cDNA microarrays, has indicated that there may not be a large variety of genes involved in regulating the typical animal stress response [20]. This makes it critical to be able to detect “all-of-the-few” genes that play important roles, no matter how seemingly obscure. For a comparative biologist, an expression microarray experiment could be designed where gene expression data are generated over multiple stress points in multiple arrays and referenced to control conditions. Unfortunately, it must be noted that data obtained from cDNA microarray experiments do not yield sequence information and do not provide an indication of organism-specific novel genes or organism-specific “oddities” within the gene (such as mutation of splicing events that alter protein function). It is also important to note that two main types of DNA microarrays exist in today's marketplace, (1) oligomeric microarrays, and (2) cDNA microarrays. Oligomeric microarrays are spotted with synthesized oligos anywhere between 30 and 60 bp in length. Typically, these oligos are designed to have complementarity to the 3′ UTR (some companies differ, so you must check with your company of interest) and are often used because of their high stringency. By contrast, microarrays spotted with cDNA contain the complete transcript sequence. Classically, heterologous cDNA microarrays allow the highest degree of hybridization for use with new/unsequenced animals, because they are “forgiving” enough that small, poorly conserved regions of sequence do not dictate overall binding to the array. Overall, it is critical to check with each company to see what type of DNA microarray you are purchasing and to what location the probes are designed.
As an example of microarray use in comparative stress biology, one study employed the use of heterologous cDNA microarrays to determine the genes and mechanisms underlying the stress response associated with various confinement exposure lengths in gilthead sea bream (Sparus aurata) [23]. Another study used heterologous mouse cDNA microarrays to determine hibernation-responsive gene expression patterns in the brown adipose tissue of hibernating arctic ground squirrels [24]. This study identified 408 genes overexpressed during hibernation and 217 genes underexpressed during hibernation among the 11,670 annotated genes probed on the arrays. When mapping hibernation-responsive gene to GO categories, the TCA cycle, electron transport, ATP synthesis, fatty acid metabolism, and protein biosynthesis were identified as processes of importance to the hibernation cycle. Select results from the heterologous cDNA arrays were subsequently validated by qRT-PCR.
2.2. Protein expression
Antibody microarrays were conceived originally as miniaturized dot blots or immunoassays and are now rapidly becoming established as a powerful tool to assess widespread protein expression [25]. These microarrays make possible the parallel screening of thousands of unmodified or post-translationally modified proteins. In the microarray format, these experiments can be carried out with minimum use of materials, while generating large amounts of data from a single sample. When compared to the conventional use of gel electrophoresis and mass spectrometry for proteomic research, antibody microarrays are typically able to detect the proteins that are of lower abundance [26]. As low abundant proteins are often those of the greatest diagnostic interest (e.g. transcription factors), there is a need for highly selective and sensitive throughput technologies for protein detection, quantitation and differential expression analysis. For this reason, antibody-based microarrays are generating interest at the level of the comparative biologist [27]. It should be mentioned that although antibody microarrays offer the advantage of measuring the expression of multiple proteins within individual samples, there are disadvantages that should be noted. Of particular importance is that there is no separation of protein by molecular weight. Many antibodies used to detect a particular protein, often cross-react with other proteins within the same protein family or other proteins with similar detection epitopes. Studies utilizing antibody microarrays should keep this in mind and confirm all data with selective secondary immunoblotting.
3. RNA sequencing-based transcriptomics
RNA sequencing (commonly referred to as RNAseq) is a recently developed approach to transcriptome profiling that uses deep-sequencing technologies. RNAseq also provides more precise measurement of transcript expression levels (compared to DNA microarrays) and provides sequence information for the identified mRNA transcripts. Initially, Sanger sequencing of cDNA or EST libraries was used, but this approach has a relatively low throughput, is expensive and is generally not quantitative [28]. The development to tag-based methods of RNA sequencing allowed multiple samples to be sequenced in parallel, largely overcoming these issues [28]. However, tag-based sequencing methods are limited as they are only sequencing a portion of the transcript, ultimately limiting the use of traditional sequencing in the creation of a transcriptome.
Recently, the development of high-throughput RNAseq has provided a means to sequencing whole RNA transcripts, allowing the assembly and quantification of transcriptomes. The use of high-throughput RNAseq provides clear advantages over gene microarray studies as the analysis provides both the ability to sequence RNA and measure the dynamic expression of mRNA transcripts. Currently, RNAseq uses deep-sequencing technologies such as 454, illumina, SOLiD and HelicosBiotechnology (see Table 1 for comparison). In general, a population of total RNA is converted into a library of cDNA fragments with adaptors attached to one or both ends. Each adaptor-ligated transcript is then sequenced from one end (single-end sequencing) or both ends (pair-end sequencing), producing reads that are typically 30–400 bp in length [29]. The commonly used illumina sequencing process is similar in principle, but uses a solid phase bridge amplification method to create clusters of a specific gene before sequencing (Fig. 1). RNAseq reads are then aligned and mapped to a reference genome for further analysis, or assembled de novo without the genomic sequence (Fig. 2). Following the release of its genome sequence, RNAseq analysis has been used to determined mRNA expression during anoxia exposure in the Western painted turtle (C. picta bellii) [12]. To explore the transcriptomic basis of its anoxia tolerance, this study assembled an mRNA expression profile by sequencing poly A-enriched RNA isolated from the heart and brain (telencephalon) of normoxic and anoxic turtles. Differential gene expression significantly increased in the brain (19 genes) and heart (23 genes). Highly differentially expressed genes (> 10-fold; APOLD1, FOS, JUNB, ATF3, PTGS2, BTG1/2, and EGR1) were found to encode proteins that have been implicated in the control of cellular proliferation, cancers, and tumor suppression [12]. If a complete genome sequence is not available, a de novo transcriptome assembly may be constructed and used for mRNA expression analysis. However, researchers must consider all of the statistical concerns for this type of experimental design before undertaking this type of study as more reads are typically needed for a de novo assembly (see Box. 2).
Table 1.
Overview comparison of next-generation sequencing techniques.
| Platform | Method | Read length (bp) | Throughput |
|---|---|---|---|
| Roche 454 | Pyrosequencing | 400 | 400 Mb/run |
| Illumina/Solexa HiSeq | Reversible terminator chemistry | 2 × 100 | 600 Gb/run |
| ABI SOLiD | Ligation | 2 × 60 | 15 Gb/day |
| HelicosBiotechnology | Reversible terminator chemistry | 25–55 | 28 Gb/run |
| Roche 454 — GS Junior | Pyrosequencing | 400 | 50 Mb/run |
| Illumina/Solexa MiSeq | Reversible terminator chemistry | 2 × 150 | 1.0–1.4 Gb/run |
| ABI Iontorrent | H + ion selective transistor | – | 320 Mb/run |
Fig. 1.
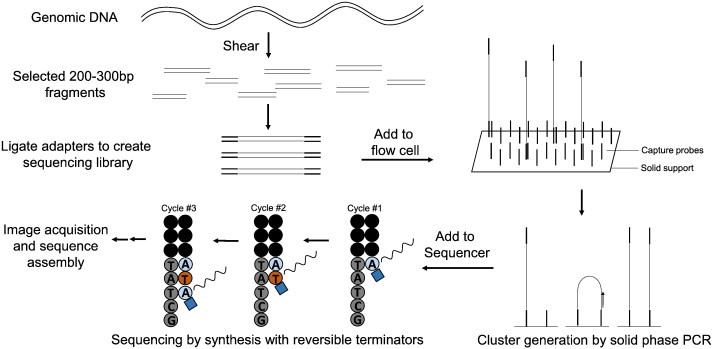
Overview of Illumina sequencing technology. Samples are initially fragmented and adapter sequences are ligated to both ends of the fragments. Adapted fragments are then randomly bound to the inside surface of the channels of a flow cell. Solid-phase bridge amplification is carried out creating large clusters of double stranded fragments. Sequencing is carried out by adding four labeled reversible terminators, primers, and DNA polymerase. Following laser excitation, the image (fluorophore corresponding to specific-bound nucleotide) is captured and the identity of the base is recorded.
Fig. 2.
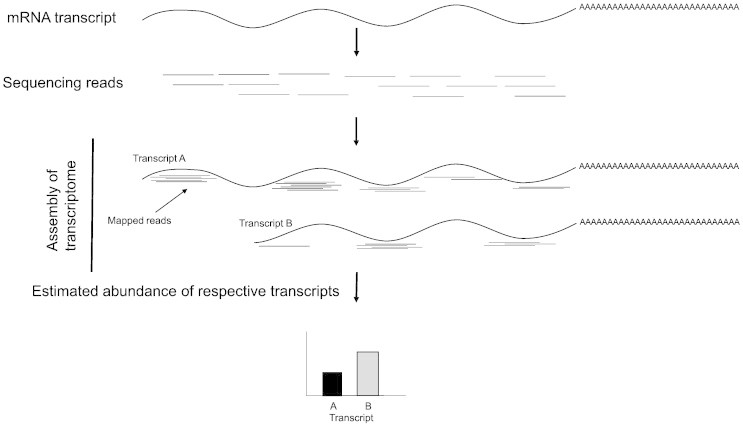
Overview of RNAseq transcriptome mapping for gene expression experiments.
Box 2. Statistical considerations for large scale transcriptome studies.
Proper assessment of sample sizes for microarray or RNAseq data is critical before beginning this type of massive parallel gene expression study. In order to identify the subset of results that is most likely to be biologically significant, it is necessary to address the problem of testing a large number of hypotheses (upward of 1.0 × 105). This is because, given typical levels of experimental and biological variation, we expect to find many genes showing differences in expression levels between samples “just by chance” or “between individuals” and not due to the environmental stress being studied. This is why nominal P-value cut-offs that we typically use in research (P < 0.01 or P < 0.05) are not practical in large scale genomic studies. The P-value depends on the effect size (i.e. expected fold change), the amount of biological variation from sample to sample and the sample sizes. To counteract the problem of testing many hypotheses, appropriate P-value cut-offs are determined to control for false discovery rates.
Because the P-value calculation depends on the amount of normal biological variation and sample sizes, it is important to address statistical analysis issues before beginning the study. As an extreme example, if there is only one control sample and one test sample (no replicates), it is not possible to determine the amount of ‘normal’ variation (biological and/or experimental), and therefore to calculate P-values. To choose an appropriate number of samples one needs to strike a suitable balance between the power to detect changes in expression, the cost of the experiment, and the amount of variation. For example, if the effects are likely to be small (low fold change like 1.2 or 2.0-fold compared to control values), then more samples will be needed to discover significant changes in expression. For example, let's assume that achieving a false discovery rate < 0.05 requires a nominal P-value < 0.00001. If a condition changes some trait value by a factor of 3-fold and if that trait has ~ 20% coefficient of variation (CV) due to biological variation, then one will have about 10% power to obtain a false detection rate < 0.05 with 5 replicates sampling each biological condition. With 8 replicates, the power increases to 50% (i.e. half of differentially regulated genes over 3-fold are expected to be detected). It is important to consider any statistical concerns before beginning this type of study. It is also important to mention that although multiple samples are required to reach statistical relevancy, output data from low sample numbers can be used for lead generation for downstream validation. In particular this latter type of experimental design will yield both an insight into potential gene changes, as well as provide species-specific sequence data that is of importance for organisms without a genomic sequence available (for review, see [31]).
Unlike microarray-based approaches, RNAseq experiments are not limited to detecting transcripts that correspond to an existing genomic sequence. For example, the detection of novel freeze-responsive genes such as FR10 and Li16 in the wood frog (Rana sylvatica), initially discovered by cDNA array, could not be possible using heterologous cDNA microarrays that have been prepared with cataloged genes from another organism. This makes RNAseq particularly attractive for non-model organisms with genomic sequences that are yet to be determined (de novo assembly). A second advantage of RNAseq is that it does not have an upper limit for quantification. Consequently, it has a large dynamic range of expression levels over which transcripts can be detected: a greater than 9000-fold range (not limited by fluorescence and the “hook” effect that plagues microarray analysis) [30]. By contrast, DNA microarrays lack sensitivity for genes expressed either at very low or very high levels and therefore have a much smaller dynamic range. RNAseq is also highly accurate for quantifying expression levels, as determined using quantitative RT-PCR [28]. Taking all of these advantages into account, RNAseq is the first sequencing-based method that allows the entire transcriptome to be surveyed in a very high-throughput and quantitative manner. However, it should be noted that bioinformatic analysis of RNAseq data is very intensive and typically must be done by the company servicing the project at an additional cost (typically doubling the cost of the experiment). However, it should be noted that open-source software has been recently developed to expedite and simplify the analysis of RNAseq data and de novo assembly (Trinity; http://trinityrnaseq.sourceforge.net/).
4. Discovery of microRNA sequence and identification of function
MicroRNAs are short (18–23 nt), non-coding RNAs that are known to have central roles in regulating the post-transcriptional expression of mRNA transcripts and have been shown to play an important role in the stress response [32]. A single microRNA (miRNA) is known to directly target hundreds of mRNAs [33,34]. Many human miRNAs (mature: 2578 & precursors: 1872) are released in the latest release of miRBase (Release v.20), yet similar numbers are sparse in non-human species and many still remain to be identified. In the past 10 years several groups have developed algorithms to identify targets for miRNA [35–37]. Most of the algorithms are mainly based on the conservation of the seed region and binding energy, but in the recent years many algorithms have incorporated expression profiles in their scoring function [38], which predicts the target more accurately. Before high-throughput identification of miRNA targets, many prediction tools were used, including TargetScan, miRanda, RNAhybrid, DIANA-microT, microInspector, and mirTarget2 [39–45]. The bioinformatics tools are still highly useful in validating microRNA targets in non-model organisms when gene sequences (including UTRs) are known. For example, both miR-15a and miR-16-1 are known to target cyclin D1 and regulate the cell cycle in humans [46]. Interest in the anoxic regulation of the cell cycle in tolerant turtles, prompted researchers to explore the possibility that miR-15a and miR-16-1 may regulate the turtle-specific cyclin D1 mRNA [46]. With no genomic information for the turtle at the time of study, researchers used 3′ rapid amplification of cDNA ends (RACE) to sequence the 3′ UTR of turtle cyclin D1. The ability of both miR-15a and miR-16-1 to target turtle cyclin D1 mRNA was then determined through a combined analysis using TargetScan and RNAhybrid (see Fig. 3). Unfortunately, if no gene sequence information is available for your specific miRNA:target interaction analysis must rely on heterologous analysis from the most closely related species with available genomic information [3,7,47–49].
Fig. 3.
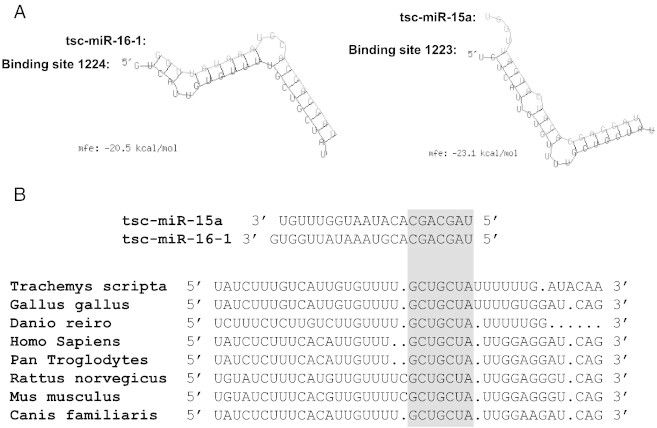
Binding of miR-16-1 and miR-15a to a conserved region of the turtle cyclin D1 mRNA. (A) Predicted binding structures from RNAhybrid. (B) Conservation analysis and seed-pairing identification by TargetScan. Figure modified from [46].
4.1. Identification from available genomic sequence
The prediction of novel miRNA from non-annotated genomic sequence has received considerable attention in the recent years. However, the vast majority of studies have focused on the human genome. The previous studies have shown that the specificity (the ability to correctly reject non-miRNA sequences) drops dramatically once human-trained methods are applied to other species [36]. Considering the expected ratio of true miRNA sequences to pseudo-miRNA hairpins is on the order of 1:1000, the use of cross-species prediction models with low specificity becomes useless for validation, as the number of false positives overwhelms the number of true positives. However, several newly developed methods and tools can be used to circumvent the issue of prediction specificity. Recognizing the problem of specificity, the HeteroMirPred was created for the identification of unannotated microRNA from genome sequences of non-human species [36,50]. This program attempts to address the non-human issue by using training data pooled from multiple species. As this software has been designed to operate across all eukaryotes, it suffers from its generalist prediction approach as it can commonly overlook known microRNAs. Currently, the most high throughout approach to microRNA sequence annotation involves the use of small RNA sequencing to target predictions back to the genome, greatly reducing the number of false positives that initially enter the prediction pipeline. MiRDeep2 was developed to discover active known or novel miRNA from deep sequencing data [51]. Using small RNAseq, MiRDeep2 map sequencing reads back to their location in the animal's genome. The program then extracts the surrounding nucleotides from the genome sequence to perform miRNA prediction. This method has been successfully used to identify the developmental response of 212 miRNA from soft-shell turtle embryos [52]. The combination of MiRDeep and RNAseq has also been successfully used for microRNA discovery in response to hibernation in the Arctic ground squirrel (Spermophilus parryii) [53]. Remarkably, this study found 200 ground squirrel miRNAs, including 18 novel miRNAs specific to the ground squirrel.
4.2. Complementary analysis of function
Hibernation research has now begun to highlight various adaptational roles for miRNAs. In particular, studies are beginning to move away from candidate-based miRNA analysis (i.e. whether a particular microRNA is able to regulate a specific target), and are beginning to address the ability of microRNA to collectively target and regulate cellular processes [3,4]. Typically, the gene and the miRNA expression data need to be co-related with the targets identified. This correlation could mostly be achieved by using pathway analysis tools, such as DIANA-micropath, Cytoscape and Pathway central [54–57]. For example, one of our own studies found that in response to torpor in little brown bats (Myotis lucifugus), differentially expressed microRNA in brain tissue converged on the common regulation on pathways of focal adhesion and axon guidance [3]. Interestingly, these same processes were also independently shown to be regulated during hibernation in the brain of the greater horseshoe bats (Rhinolophus ferrumequinum) [58].
5. Multiplex analysis
A multiplex assay is a type of laboratory procedure that simultaneously measures multiple analytes (up to 500) in a single assay. As this technology is under constant growth and change, this minireview will only outline on the principles of multiplex assays and highlight key technologies currently available at the time of publication.
Multiplex assays are widely used in functional genomics experiments that assay the state of a type of target (e.g. microRNA, mRNA, protein) within a single biological sample. Multiplexing assays work by performing multiple parallel reactions for different targets, greatly reducing the time needed to complete the analysis. Various companies are currently developing and refining multiplex technologies. Luminex xMAP technology is built on a flow cytometry platform that utilizes fluorescently tagged microspheres to detect the target analyte. Each color-coded tiny microsphere can be coated with a reagent specific to a particular bioassay, allowing the capture and detection of specific analytes from a sample. Within the analyzer, lasers excite the internal dyes that identify each microsphere particle, and also any reporter dye captured during the assay (Fig. 4). Currently, xMAP technology allows multiplexing of up to 500 unique assays within a single sample. The use of Luminex xMAP technology was recently used to determine the differential expression profiles of microRNA transcripts in response to dehydration stress in the tissues of Xenopus laevis [16]. Different from Luminex-based analysis, Mesoscale Discovery utilizes small antibody arrays, spotted onto the bottom of a microplate (available in 24-, 96-, and 384-well formats). In essence, the technology is at the cross section of Luminex and antibody microarrays, with the capability to analyze up to 100 spots per well. Like Luminex arrays, this technology also requires the use of specialized equipment. To date, several comparative studies have used Luminex technology to protein expression during hibernation [59,60]. For example, Luminex has recently been used to determine the activation of insulin signaling pathways pre-, post- and during hibernation in grizzly bears [59]. The technology has also been used to determine the activation response of immunological-response to white-nose syndrome, finding that bats showing visible signs of infection had significantly higher IL-4 expression when compared to bats without visible infection [60].
Fig. 4.
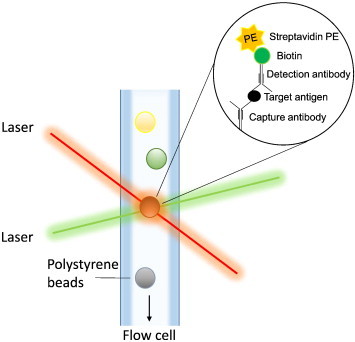
Luminex xMAP system. Luminex is based on microsphere bead technology that relies on flow cytometry and target-capture ligand binding.
6. Mass spectrometry
Mass spectrometry (MS) is an important and emerging technology for the characterization and sequencing of proteins. The technology works by ionizing compounds to generate charged peptide fragments and measuring their mass-to-charge ratios to identify amino acids sequences (for review see, [61]). For identification, proteins are enzymatically digested into smaller peptides using proteases (commonly trypsin, cutting sequences at lysine residues), after electrophoretic separation. The collection of peptide products is then introduced to the mass analyzer. When the characteristic pattern of peptides is used for the identification of the protein, the method is called peptide mass fingerprinting. If the identification is performed using the sequence data determined in MS analysis it is called de novo sequencing [62]. The use of MS/MS to identify unknown proteins may be of great interest to the comparative biologist. Apart from identifying the protein sequence of unknown proteins, mass spectrometry is also able to detect relative post-translation modifications such as phosphorylation, methylation and acetylation, among others [63]. Identification of modified amino acids that are both utilized and unique to a stress-tolerant animal may be of functional significance to the stress. One must keep in mind that this technique works best with highly abundant proteins (typically not proteins such as transcription factors) that are easily purified (such as many metabolic enzymes and structural proteins).
One growing field in mass spectrometry is the identification and dynamic changes of the phospho-proteome. Phosphorylated proteins are typically pre-fractionated and enriched prior to MS/MS, increasing the coverage of identification. Pre-fractionation is typically accomplished through the use of one of the two types of ion-exchange chromatography; (1) strong anionic ion-exchange (SAX), and (2) strong cationic ion-exchange (SCX). Following pre-fractionation, phosphorylated protein or peptides can be enriched by a variety of methods including (but not limited to); (1) immunoprecipitation by pan-specific antibodies, (2) pull-down by phospho-binding domains, (3) immobilized metal affinity chromatography (IMAC), (4) metal-oxide affinity chromatography (MOAC), and (5) Phos-Tag chromatography. A few examples of studies exploring phospho-proteomics in non-model organisms include phospho-proteome of chicken (Gallus gallus) embryo fibroblasts and of the mitochondria of hibernating thirteen-lined ground squirrels (I. tridecemlineatus) [64,65]. Recently, studies are beginning to explore the methyl-proteome by the enrichment of methylated protein through pan-methyl-arginine antibodies or methyl-lysine binding domains [66]. These types of enrichment methods, with wide-cross reactivity profiles, not only allow the ability to identify proteins in a whole cell complex, but also remain specificity in many non-human animals as amino acid sequence has little role in binding.
7. Analysis of protein structure and function
Several of the methods described in this minireview detail analysis that are used to identify new proteins or post-translational modifications. Importantly, when new or novel proteins are identified it is critical to determine, or predict, their functional role. While knockdown and overexpression analysis remains to be the ‘gold-standard’ in determining protein function, animals that do not have cell-lines or the genetic tools available, must resort to bioinformatics analysis to supplement and guide any molecular data (cellular localization, binding partners, etc.). Multiple tools exist to assist in these types of analysis. Once an amino acid sequence is obtained, the protein can initially be scanned for conserved domains with the NCBI Conserved Domain search resource (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). This analysis identifies any possible functional domains that exist in the protein (such as SH2 or ATP-binding domains), giving information of functional interactions and may guide further analysis. Too many resources currently exist for this Review to comprehensively provide an overview, however OpenPredictProtein (http://ppopen.informatik.tu-muenchen.de/) has curated a collection of valuable prediction tools that can be used on primary amino acid sequences [67]. Such tools include those for structural annotation (Solvent accessibility, transmembrane helices, protein disorder and flexibility, as well as disulfide bridges) and functional annotation (Gene ontology terms, subcellular localization and binding sites).
Occasionally, obtaining protein structure is necessary to determine more specific function information regarding the protein under study. As such, protein structure can be determined through several methods. If the protein is highly homologous to the existing body of protein crystal structures, SWISS-MODEL can be used to determine 3-dimensional (3D) protein structure [68]. Importantly, SWISS-MODEL is currently one of the most commonly used resources and provides information on the quality of prediction. For example, a study on the structural adaptations of aldolase enzyme that helps to drive glycolysis in anoxic turtles (Trachemys scripta elegans) used SWISS-MODEL to generate the structures of aldolase enzymes (ALDOA and ALDOB). These structures were then used to determine the mechanisms involved in substrate interactions compared to rabbit aldolase proteins, stating that differences in substrate binding and heterotetramer formation contribute to the higher activity of turtle aldolase (Fig. 5) [5]. When completely novel proteins are discovered, structures cannot be determined from homology-based methods and be predicted de novo. Several programs currently exist to facilitate de novo predictions, the most commonly used being I-TASSER (for proteins < 1500 amino acids) and QUARK (< 200 amino acids) [69]. To highlight the use of de novo structure prediction for completely novel proteins, a recent study used QUARK to determine the structure of two freeze-response proteins, FR10 and Li16, from the wood frog (Rana sylvatica) (Fig. 6) [17]. The ability to obtain structures for these novel proteins allowed researchers to model membrane interaction (PPM server; http://opm.phar.umich.edu/server.php) [70], leading to the hypothesis that FR10 was an excreted protein and Li16 may have functional roles in membrane-adaptation roles in response to freezing stress.
Fig. 5.
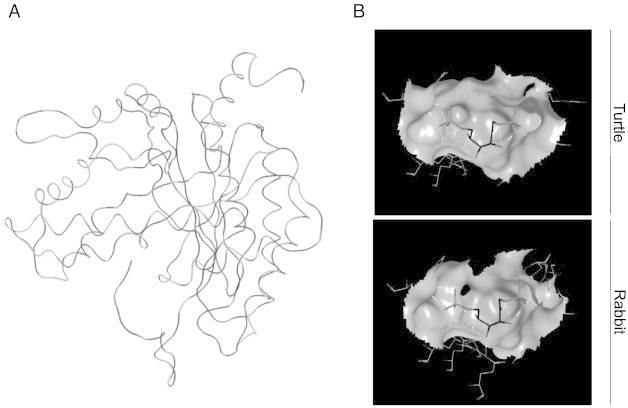
Identification and characterization of predicted turtle aldolase enzyme. (A) Overlaid tertiary structure of ALDOB from both rabbit (light) (1fdjA) and the turtle (dark) ALDOB protein predicted by SWISS-MODEL. (B) Predicted docking of fructose-1,6-bisphosphate on the active sites of both rabbit and turtle ALDOB enzyme. Ligand docking was performed with MOE Dock, employing Triangle Matcher as the placement and function London dG as the first scoring function. Figure modified from [5].
Fig. 6.
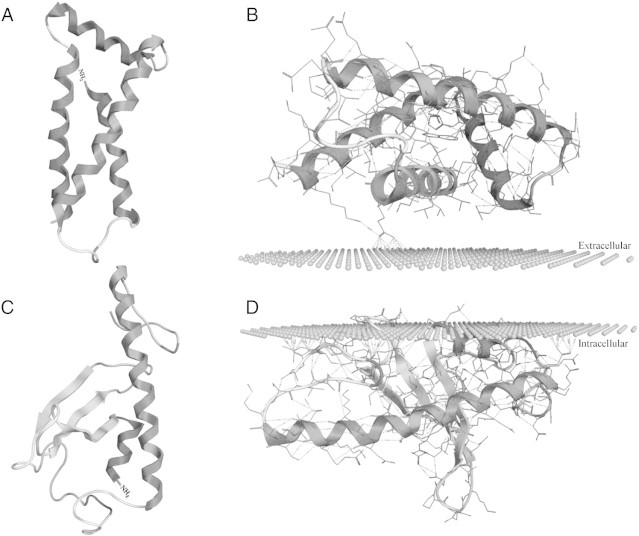
De novo protein modeling and prediction of function. (A) Predicted de novo protein structure of the novel freeze-responsive proteins, FR10 and Li16 from the freeze-tolerant wood frog (Rana sylvatica). Structures were predicted by the QUARK server and optimized by MOE software. (B) Membrane interactions of FR10 and Li16 based on PPM server prediction. Figure modified from [17].
8. Summary and outlook
The development of tools capable de novo assembly and predictions, introduces many new possibilities for comparative biologists to take part in “omic” studies and introduces the potential to discover novel proteins or genes with biologically-relevant function. Given the rich assortment of techniques and bioinformatic tools (many being open-source with GUIs) that are currently available and that have been refined for use non-human species, proper introduction and use of these tools in future research will help to discover and characterize the animal stress responses that are utilized by many diverse organisms.
References
- 1.Krivoruchko A., Storey K.B. Activation of the carbohydrate response element binding protein (ChREBP) in response to anoxia in the turtle Trachemys scripta elegans. Biochim Biophys Acta. 2014;14:00221–00229. doi: 10.1016/j.bbagen.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 2.Seibel B.A., Häfker N.S., Trübenbach K., Zhang J., Tessier S.N., Portner H.O. Metabolic suppression during protracted exposure to hypoxia in the jumbo squid, Dosidicus gigas, living in an oxygen minimum zone. J Exp Biol. 2014;217:2555–2568. doi: 10.1242/jeb.100487. [DOI] [PubMed] [Google Scholar]
- 3.Biggar K.K., Storey K.B. Identification and expression of microRNA in the brain of hibernating bats, Myotis lucifugus. Gene. 2014;544:67–74. doi: 10.1016/j.gene.2014.04.048. [DOI] [PubMed] [Google Scholar]
- 4.Biggar K.K., Wu C.W., Storey K.B. High-throughput amplification of mature microRNAs in uncharacterized animal models using polyadenylated RNA and stem-loop reverse transcription polymerase chain reaction. Anal Biochem. 2014;462:32–34. doi: 10.1016/j.ab.2014.05.032. [DOI] [PubMed] [Google Scholar]
- 5.Dawson N.J., Biggar K.K., Storey K.B. Characterization of fructose-1,6-bisphosphate aldolase during anoxia in the tolerant turtle, Trachemys scripta elegans: an assessment of enzyme activity, expression and structure. PLoS One. 2013;8:e68830. doi: 10.1371/journal.pone.0068830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang J., Biggar K.K., Storey K.B. Regulation of p53 by reversible post-transcriptional and post-translational mechanisms in liver and skeletal muscle of an anoxia tolerant turtle, Trachemys scripta elegans. Gene. 2013;513:147–155. doi: 10.1016/j.gene.2012.10.049. [DOI] [PubMed] [Google Scholar]
- 7.Maistrovski Y., Biggar K.K., Storey K.B. HIF-1α regulation in mammalian hibernators: role of non-coding RNA in HIF-1α control during torpor in ground squirrels and bats. J Comp Physiol B. 2012;182:849–859. doi: 10.1007/s00360-012-0662-y. [DOI] [PubMed] [Google Scholar]
- 8.Hampton M., Melvin R.G., Andrews M.T. Transcriptomic analysis of brown adipose tissue across the physiological extremes of natural hibernation. PLoS One. 2013;8:e85157. doi: 10.1371/journal.pone.0085157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun J., Mu H., Zhang H., Chandramouli K.H., Qian P.Y., Wong C.K. Understanding the regulation of estivation in a freshwater snail through iTRAQ-based comparative proteomics. J Proteome Res. 2013;12:5271–5280. doi: 10.1021/pr400570a. [DOI] [PubMed] [Google Scholar]
- 10.Hampton M., Melvin R.G., Kendall A.H., Kirkpatrick B.R., Peterson N., Andrews M.T. Deep sequencing the transcriptome reveals seasonal adaptive mechanisms in a hibernating mammal. PLoS One. 2011;6:e27021. doi: 10.1371/journal.pone.0027021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu Y., Shao C., Fedorov V.B., Goropashnaya A.V., Barnes B.M., Yan J. Molecular signatures of mammalian hibernation: comparisons with alternative phenotypes. BMC Genomics. 2013;14:567. doi: 10.1186/1471-2164-14-567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaffer H.B., Minx P., Warren D.E., Shedlock A.M., Thomson R.C., Valenzuela N. The western painted turtle genome, a model for the evolution of extreme physiological adaptations in a slowly evolving lineage. Genome Biol. 2013;14:R28. doi: 10.1186/gb-2013-14-3-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yates S.A., Swain M.T., Hegarty M.J., Chernukin I., Lowe M., Allison G.G. De novo assembly of red clover transcriptome based on RNA-Seq data provides insight into drought response, gene discovery and marker identification. BMC Genomics. 2014;15:453. doi: 10.1186/1471-2164-15-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fedorov V.B., Goropashnaya A.V., Tøien O., Stewart N.C., Chang C., Wang H. Modulation of gene expression in heart and liver of hibernating black bears (Ursus americanus) BMC Genomics. 2011;12:171. doi: 10.1186/1471-2164-12-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu S, Gao G, Palti Y, Cleveland BM, Weber GM and C.E. Rexroad. RNA-seq analysis of early hepatic response to handling and confinement stress in rainbow trout. PLoS ONE 9: e88492. [DOI] [PMC free article] [PubMed]
- 16.Wu C.W., Biggar K.K., Storey K.B. Dehydration mediated microRNA response in the African clawed frog Xenopus laevis. Gene. 2013;529:269–275. doi: 10.1016/j.gene.2013.07.064. [DOI] [PubMed] [Google Scholar]
- 17.Biggar K.K., Kotani E., Furusawa T., Storey K.B. Expression of freeze-responsive proteins, Fr10 and Li16, from freeze-tolerant frogs enhances freezing survival of BmN insect cells. FASEB J. 2013;27:3376–3383. doi: 10.1096/fj.13-230573. [DOI] [PubMed] [Google Scholar]
- 18.Lévêque N., Renois F., Andréoletti L. The microarray technology: facts and controversies. Clin Microbiol Infect. 2013;19:10–14. doi: 10.1111/1469-0691.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Git A., Dvinge H., Salmon-Divon M., Osborne M., Kutter C., Hadfield J. Systematic comparison of microarray profiling, real-time PCR, and next-generation sequencing technologies for measuring differential microRNA expression. RNA. 2010;16:991–1006. doi: 10.1261/rna.1947110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larade K., Storey K.B. Living without oxygen: anoxia-responsive gene expression and regulation. Curr Genomics. 2009;10:76–85. doi: 10.2174/138920209787847032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Degletagne C, Keime C, Rey B, Dinechin M, Forcheron F, Chuchana P, et al. Transcriptome analysis in non-model species: a new method for the analysis of heterologous hybridization on microarrays. BMC Genomics 11: 344. [DOI] [PMC free article] [PubMed]
- 22.Salleh A.H.M., Mohamad M.S., Deris S., Illias R. A review on pathway software based on microarray data interpretation. Int J Biosci Biotechnol. 2014;5:149–158. [Google Scholar]
- 23.Calduch-Giner J.A., Davey G., Saera-Vila A., Houeix B., Talbot A., Prunet P. Use of microarray technology to assess the time course of liver stress response after confinement exposure in gilthead sea bream (Sparus aurata L.) BMC Genomics. 2010;11:193. doi: 10.1186/1471-2164-11-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan J., Burman A., Nichols C., Alila L., Showe L.C., Showe M.K. Detection of differential gene expression in brown adipose tissue of hibernating arctic ground squirrels with mouse microarrays. Physiol Genomics. 2006;25:346–353. doi: 10.1152/physiolgenomics.00260.2005. [DOI] [PubMed] [Google Scholar]
- 25.Tu S., Jiang H.W., Liu C.X., Zhou S.M., Tao S.C. Protein microarrays for studies of drug mechanisms and biomarker discovery in the era of systems biology. Curr Pharm Des. 2014;20:49–55. doi: 10.2174/138161282001140113123707. [DOI] [PubMed] [Google Scholar]
- 26.Chandra H., Reddy P.J., Srivastava S. Protein microarrays and novel detection platforms. Expert Rev Proteomics. 2011;8:61–79. doi: 10.1586/epr.10.99. [DOI] [PubMed] [Google Scholar]
- 27.Rajala R.V.S. Phospho-site-specific antibody microarray to study the state of protein phosphorylation in the retina. J Proteomics Bioinform. 2008;1:242. doi: 10.4172/jpb.1000031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Z., Gerstein M., Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schatz M.C., Delcher A.L., Salzberg S.L. Assembly of large genomes using second-generation sequencing. Genome Res. 2010;20:1165–1173. doi: 10.1101/gr.101360.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagalakshmi U., Wang Z., Waern K., Shou C., Raha D., Gerstein M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li J., Witten D.M., Johnstone I.M., Tibshirani R. Normalization, testing, and false discovery rate estimation for RNA-sequencing data. Biostatistics. 2012;13:523–538. doi: 10.1093/biostatistics/kxr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Biggar K.K., Storey K.B. The emerging roles of microRNAs in the molecular responses of metabolic rate depression. J Mol Cell Biol. 2011;3:167–175. doi: 10.1093/jmcb/mjq045. [DOI] [PubMed] [Google Scholar]
- 33.Maziere P., Enright A.J. Prediction of microRNA targets. Drug Discov Today. 2007;12:452–458. doi: 10.1016/j.drudis.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 34.Friedman R.C., Farh K.K., Burge C.B., Bartel D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xue C., Li F., He T., Liu C., Li Y., Zhang X. Classification of real and pseudo microRNA precursors using local structure-sequence features and support vector machine. BMC Bioinformatics. 2005;6:310. doi: 10.1186/1471-2105-6-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y., Chen X., Jiang W., Li L., Li W., Yang L. Predicting human microRNA precursors based on an optimized feature subset generated by GA-SVM. Genomics. 2011;98:73–78. doi: 10.1016/j.ygeno.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 37.Lertampaiporn S., Thammarongtham C., Nukoolkit C., Kaewkamnerdpong B., Ruengjitchatchawalya M. Heterogeneous ensemble approach with discriminative features and modified-SMOTE bagging for pre-miRNA classification. Nucleic Acids Res. 2013;41:e21. doi: 10.1093/nar/gks878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Williamson V., Kim A., Xie B., McMichael G.O., Gao Y., Vladimirov V. Detecting miRNAs in deep-sequencing data: a software performance comparison and evaluation. Brief Bioinform. 2013;14:36–45. doi: 10.1093/bib/bbs010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lewis B.P., Burge C.B., Bartel D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 40.John B., Enright A.J., Aravin A., Tuschl T., Sander C., Marks D.S. MiRanda application: human microRNA targets. PLoS Biol. 2005;3:e264. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paraskevopoulou M.D., Georgakilas G., Kostoulas N., Vlachos I.S., Vergoulis T. DIANA-microT web server v5.0: service integration into miRNA functional analysis workflows. Nucleic Acids Res. 2013;41:W169–W173. doi: 10.1093/nar/gkt393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maragkakis M., Reczko M., Simossis V.A., Alexiou P., Papadopoulos G.L. DIANA-microT web server: elucidating microRNA functions through target prediction. Nucleic Acids Res. 2009;37:W273–W276. doi: 10.1093/nar/gkp292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rusinov V., Baev V., Minkov I.N., Tabler M. MicroInspector: a web tool for detection of miRNA binding sites in an RNA sequence. Nucleic Acids Res. 2005;33:W696–W700. doi: 10.1093/nar/gki364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X., El Naqa I.M. Prediction of both conserved and nonconserved microRNA targets in animals. Bioinformatics. 2008;24:325–332. doi: 10.1093/bioinformatics/btm595. [DOI] [PubMed] [Google Scholar]
- 45.Wang X. miRDB: a microRNA target prediction and functional annotation database with a wiki interface. RNA. 2008;14:1012–1017. doi: 10.1261/rna.965408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Biggar K.K., Storey K.B. Evidence for cell cycle suppression and microRNA regulation of cyclin D1 during anoxia exposure in turtles. Cell Cycle. 2012;11:1705–1713. doi: 10.4161/cc.19790. [DOI] [PubMed] [Google Scholar]
- 47.Biggar K.K., Dubuc A., Storey K.B. MicroRNA regulation below zero: differential expression of miRNA-21 and miRNA-16 during freezing in wood frogs. Cryobiology. 2009;59:317–321. doi: 10.1016/j.cryobiol.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 48.Kornfeld S.F., Biggar K.K., Storey K.B. Differential expression of mature microRNAs involved in muscle maintenance of hibernating little brown bats, Myotis lucifugus: a model of muscle atrophy resistance. Genomics Proteomics Bioinformatics. 2012;10:295–301. doi: 10.1016/j.gpb.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Biggar K.K., Kornfeld S.F., Maistrovski Y., Storey K.B. MicroRNA regulation in extreme environments: differential expression of microRNAs in the intertidal snail Littorina littorea during extended periods of freezing and anoxia. Genomics Proteomics Bioinformatics. 2012;10:302–309. doi: 10.1016/j.gpb.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yao Y., Ma L., Jia Q., Deng W., Liu Z., Zhang Y. Systematic characterization of small RNAome during zebrafish early developmental stages. BMC Genomics. 2014;15:117. doi: 10.1186/1471-2164-15-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Friedländer M.R., Mackowiak S.D., Li N., Chen W., Rajewsky N. MiRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40:37–52. doi: 10.1093/nar/gkr688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Z., Pascual-Anaya J., Zadissa A., Li W., Niimura Y., Huang Z. The draft genomes of soft-shell turtle and green sea turtle yield insights into the development and evolution of the turtle-specific body plan. Nat Genet. 2013;45:701–706. doi: 10.1038/ng.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu Y., Hu W., Wang H., Lu M., Shao C., Khaitovich P. Genomic analysis of miRNAs in an extreme mammalian hibernator, the Arctic ground squirrel. Physiol Genomics. 2010;42A:39–51. doi: 10.1152/physiolgenomics.00054.2010. [DOI] [PubMed] [Google Scholar]
- 54.Vlachos I.S., Kostoulas N., Vergoulis T., Georgakilas G., Reczko M., Maragkakis M. DIANA mirPath v. 2.0: investigating the combinatorial effect of microRNAs in pathways. Nucleic Acids Res. 2012;40:W498–W504. doi: 10.1093/nar/gks494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Papadopoulos G.L., Alexiou P., Maragkakis M., Reczko M., Hatzigeorgiou A.G. DIANA-mirPath: integrating human and mouse microRNAs in pathways. Bioinformatics. 2009;25:1991–1993. doi: 10.1093/bioinformatics/btp299. [DOI] [PubMed] [Google Scholar]
- 56.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shirdel E.A., Xie W., Mak T.W., Jurisica I. NAViGaTing the micronome — using multiple microRNA prediction databases to identify signaling pathway-associated microRNAs. PLoS One. 2011;6:e17429. doi: 10.1371/journal.pone.0017429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen J., Yuan L., Sun M., Zhang L., Zhang S. Screening of hibernation-related genes in the brain of Rhinolophus ferrumequinum during hibernation. Comp Biochem Physiol B Biochem Mol Biol. 2008;149:388–393. doi: 10.1016/j.cbpb.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 59.Nelson O.L., Jansen H.T., Galbreath E., Morgenstern K., Lauren J. Grizzly bears exhibit augmented insulin sensitivity while obese prior to a reversible insulin resistance during hibernation. Cell Metab. 2014;20:376–382. doi: 10.1016/j.cmet.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 60.Moore M.S., Reichard J.D., Murtha T.D., Nabhan M.L., Pian R.E., Ferreira J.S. Hibernating little brown myotis (Myotis lucifugus) show variable immunological responses to white-nose syndrome. PLoS One. 2013;8:e58976. doi: 10.1371/journal.pone.0058976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bensimon A., Heck A.J., Aebersold R. Mass spectrometry-based proteomics and network biology. Annu Rev Biochem. 2012;81:379–405. doi: 10.1146/annurev-biochem-072909-100424. [DOI] [PubMed] [Google Scholar]
- 62.Chen T., Kao M.Y., Tepel M., Rush J., Church G.M. A dynamic programming approach to de novo peptide sequencing via tandem mass spectrometry. J Comput Biol. 2001;8:325–337. doi: 10.1089/10665270152530872. [DOI] [PubMed] [Google Scholar]
- 63.Witze E.S., Old W.M., Resing K.A., Ahn N.G. Mapping protein post-translational modifications with mass spectrometry. Nat Methods. 2007;4:798–806. doi: 10.1038/nmeth1100. [DOI] [PubMed] [Google Scholar]
- 64.Chien K.Y., Blackburn K., Liu H.C., Goshe M.B. Proteomic and phosphoproteomic analysis of chicken embryo fibroblasts infected with cell culture-attenuated and vaccine strains of Marek's disease virus. J Proteome Res. 2012;11:5663–5677. doi: 10.1021/pr300471y. [DOI] [PubMed] [Google Scholar]
- 65.Chung D.J., Szyszka B., Brown J.C., Hüner N.P., Staples J.F. Changes in the mitochondrial phosphoproteome during mammalian hibernation. Physiol Genomics. 2013;45:389–399. doi: 10.1152/physiolgenomics.00171.2012. [DOI] [PubMed] [Google Scholar]
- 66.Carlson S.M., Gozani O. Emerging technologies to map the protein methylome. J Mol Biol. 2014 doi: 10.1016/j.jmb.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yachdav G., Kloppmann E., Kajan L., Hecht M., Goldberg T., Hamp T. PredictProtein — an open resource for online prediction of protein structural and functional features. Nucleic Acids Res. 2014;42:W337–W343. doi: 10.1093/nar/gku366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Y. Interplay of I-TASSER and QUARK for template-based and ab initio protein structure prediction in CASP10. Proteins. 2014;82:175–187. doi: 10.1002/prot.24341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lomize M.A., Pogozheva I.D., Joo H., Mosberg H.I., Lomize A.L. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 2012;40:D370–D376. doi: 10.1093/nar/gkr703. [DOI] [PMC free article] [PubMed] [Google Scholar]