

Abstract
Background
Previously, we identified three loci affecting HDL-cholesterol levels in a screen for ENU-induced mutations in mice and discovered two mutated genes. We sought to identify the third mutated gene and further characterize the mouse phenotype.
Methods
We engaged, DNA sequencing, gene expression profiling, western blotting, lipoprotein characterization, metabolomics assessment, histology and electron microscopy in mouse tissues.
Results
We identify the third gene as Ampd2, a liver isoform of AMP Deaminase (Ampd), a central component of energy and purine metabolism pathways. The causative mutation was a guanine-to-thymine transversion resulting in an A341S conversion in Ampd2. Ampd2 homozygous mutant mice exhibit a labile hypercholesterolemia phenotype, peaking around 9 weeks of age (251 mg/dL vs. wildtype control at 138 mg/dL), and was evidenced by marked increases in HDL, VLDL and LDL. In an attempt to determine the molecular connection between Ampd2 dysfunction and hypercholesterolemia, we analyzed hepatic gene expression and found the downregulation of Ldlr, Hmgcs and Insig1 and upregulation of Cyp7A1 genes. Metabolomic analysis confirmed an increase in hepatic AMP levels and a decrease in allantoin levels consistent with Ampd2 deficiency, and increases in campesterol and β-sitosterol. Additionally, nephrotic syndrome was observed in the mutant mice, through proteinuria, kidney histology and effacement and blebbing of podocyte foot processes by electron microscopy.
Conclusion
In summary we describe the discovery of a novel genetic mouse model of combined transient nephrotic syndrome and hypercholesterolemia, resembling the human disorder.
Electronic supplementary material
The online version of this article (doi:10.1186/1476-511X-13-167) contains supplementary material, which is available to authorized users.
Keywords: LDL, HDL, AMP deaminase, B6, C3, Proteinuria, ENU
Introduction
Hypercholesterolemia is a major risk factor for developing coronary artery disease, the most common cause for heart disease and the leading cause of death worldwide. Coronary artery disease occurs when excess cholesterol in the bloodstream accumulates and forms plaques in the coronary arteries. This narrowing and hardening of the arteries can eventually lead to myocardial infarction. Inherited forms of hypercholesterolemia are present with very high circulating cholesterol levels. The most common type of inherited hypercholesterolemia is familial hypercholesterolemia, which occurs in roughly 1 in 500 people [1]. Known genes related to hypercholesterolemia include low density lipoprotein receptor (LDLR) [2], proprotein convertase subtilisin/kexin type 9 (PCSK9) [3], apolipoprotein B (APOB) [4] and low density lipoprotein receptor adaptor protein 1 (LDLRAP1) [5], among others.
A useful tool for discovering other causative genes involved in hypercholesterolemia is mouse genetics. Quantitative trait loci (QTL) analysis has been used to study the genetics of HDL-cholesterol (HDL-C) levels in humans and mice [6]. Phenotype-driven screens such as those using the highly potent mutagen N-ethyl-N-nitrosourea (ENU) have revealed novel genes and biology [7]. Specifically, genes associated with low HDL-C levels have been found by utilizing ENU [8], as well as genes associated with reduced cholesterol and triglyceride levels [9]. One advantage of ENU screens is that they induce point mutations, thereby allowing the identification of a single candidate gene responsible for the phenotype of interest. ENU screens can also generate an allelic series of mutations that display a range of effects from complete or partial loss of function to exaggerated function, and reveal gene functions in an unbiased manner [10]. We previously performed an ENU screen to identify lines of mice with altered lipid profiles [11]. One line of mice presented undetectable levels of HDL-C, caused by a missense mutation in the ATP binding cassette transporter A1 (Abca1) gene. The second line of mice which had very high levels of plasma total cholesterol and HDL-C was mapped to a missense mutation in CCAAT/enhancer binding protein α (C/ebpα). A third line of mice was characterized with a reduced penetrance labile hypercholesterolemia phenotype but the underlying mutation had not been identified.
In this study, we identify the mutation as a recessive loss of function missense mutation in the gene encoding adenosine monophosphate deaminase 2 (Ampd2). AMPD is the rate-limiting enzyme in the catabolism of adenosine monophosphate (AMP) to uric acid, for the elimination of nitrogenous waste [12]. It is also a component of the purine nucleotide cycle, responsible for the deamination of AMP to inosine monophosphate (IMP), which serves to replenish intermediates in the tricarboxylic (TCA) cycle when energy demands are high. In humans, there are at least four isoforms of AMPD. AMPD1 is the predominant form in skeletal muscle, AMPD2 is widely expressed in non-skeletal tissues, especially the liver, and AMPD3 is found in erythrocytes and is comprised of two isoforms, E1 and E2 [13].
We now complete the physical mapping and define the remaining causative mutation in our hypercholesterolemia mice [11]. We further characterize the Ampd2 mutant mice as having a transient hypercholesterolemia phenotype that also present with transient nephrotic syndrome (NS). Upon further investigation we find that the dyslipidemia noted in these mice parallels the proteinuria observed and thus represents a novel genetic mouse model of transient NS accompanied with the lipid abnormalities as characterized in the human disease.
Results
Identification of a mutation in Ampd2
We previously reported [11] the mapping of the mutation for the hypercholesterolemic mice to a 7 Mb region on chromosome 3, with 131 possible candidate genes identified. Forty five were selected for further mutation analysis based on functional relevance. After examining the exons and intronic junctions of these 45 candidate genes by direct sequencing, we identified a guanine-to-thymine transversion resulting in an alanine to serine conversion at amino acid 341 in Ampd2 (Figure 1A). Using exon capture and high-throughput sequencing we analyzed the region from D3Mit102 to rs13477320 in 2 affected animals, three ENU mutations were identified: the A341S missense mutation in Ampd2, and intronic mutations in Magi3 and Slc6a17 (data not shown). Western blotting illustrated the functional consequence of this mutation on Ampd2 protein expression. Complete loss of Ampd2 protein was observed in the livers of homozygous mutant mice (Ampd2m/m), with partial loss evident in the heterozygous mice (Ampd2+/m) relative to wild-type (+/+) (Figure 1B). Alanine 341 was conserved in all animal species including Caenorhabditis elegans, but not in plants and fungi (Figure 1C), yet was restricted to a highly conserved region of the protein.
Figure 1.
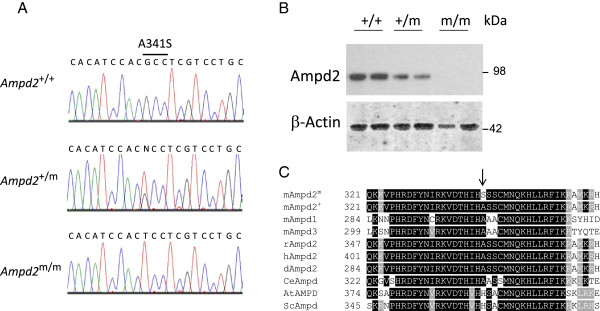
Identification of a loss-of-function mutation in Ampd2 in hypercholesterolemic mice. A) Sequencing chromatographs of Ampd2 in Ampd2 +/+, Ampd2 +/m and Ampd2 m/m mice. B) Liver immunoblot analysis of Ampd2 protein in Ampd2 +/+, Ampd2 +/m and Ampd2 m/m mice. C) Amino acid conservation of mouse Ampd2 residues 321–361 to Ampd2+, mouse Ampd1, mouse Ampd3, rat Ampd2, human Ampd2, dog Ampd2, Caenorhabditis elegans Ampd, Arabidopsis thaliana AMPD and Saccharomyces cerevisiae Ampd. Arrow indicates residue 341.
Lipid characterization of Ampd2mice
Lipid parameters were analyzed on Ampd2+/+, Ampd2+/m and Ampd2m/m mice at 9 weeks of age (Figure 2). This time point was chosen after the observation was made that these phenotypes were transient in subsequent mouse plasma collections at 9, 11 and 13 weeks [11] and confirmed the previously reported elevated total cholesterol levels in Ampd2m/m animals (Figure 2A). Additionally Ampd2m/m mice exhibited reduced penetrance, as evidenced by the appearance of some Ampd2m/m mice with normal total cholesterol levels (Figure 2A). Levels of HDL-C were also significantly elevated in Ampd2m/m mice (Figure 2B), mirroring the total cholesterol values for individual mice. No differences in triglyceride levels between Ampd2+/+, Ampd2+/m and Ampd2m/m mice were observed (Figure 2C). Analysis of ApoA1, the major protein component of HDL in plasma, showed a significant elevation in Ampd2m/m compared to Ampd2+/+ mice (Figure 2D) supporting the HDL-C data. Levels of ApoE were also significantly elevated in Ampd2m/m compared to Ampd2+/+ mice (Figure 2E). The elevation of ApoA1 and ApoE in Ampd2m/m mice was further evidenced by FPLC analysis of plasma lipoproteins (Figure 2F). As expected Ampd2m/m mice were found to have elevated HDL-C levels, in addition very low-density lipoprotein (VLDL) and low density lipoprotein (LDL) levels were increased relative to Ampd2+/+ mice.
Figure 2.
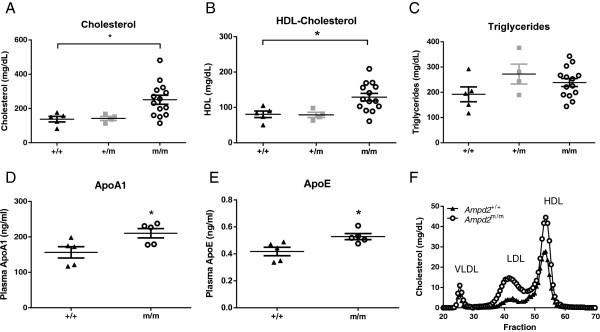
Lipid parameters of Ampd2 m/m , Ampd2 +/m and Ampd2 +/+ mice. Plasma total cholesterol (A), HDL-C (B), triglyceride (C), ApoA1 (D), ApoE (E) levels and FPLC lipoprotein analysis (F) of 9-week old mice. Individual mouse values are shown with means ± SEM. Statistical analyses were carried out by one-way ANOVA using a Tukey post test for panels A, B and C and by unpaired two-tailed t-tests for panels D and E; * p <0.05 vs. Ampd2 +/+.
Hepatic gene expression analysis of Ampd2mice
In an attempt to uncover a pathogenic mechanism for the observed hypercholesterolemia we focused our attention on liver lipid metabolism. An array of lipid metabolism genes were analyzed by quantitative RT-PCR in the livers of Ampd2+/+ and Ampd2m/m mice at 9 weeks of age (Figure 3A). Ldlr mRNA was decreased by 36%, which translated to reduced protein levels of Ldlr (Figure 3B), consistent with elevated LDL cholesterol levels (Figure 2F). Transcript levels of 3-hydroxy-3-methylglutaryl-CoA synthase (Hmgcs) were also 47% lower. The cholesterol and bile acid synthesis enzyme cholesterol 7 alpha-hydroxylase (Cyp7a1) mRNA was increased by 55% in Ampd2m/m mice. Insulin-induced gene 1 (Insig1) was decreased by 56% in Ampd2m/m mice. We confirmed normal expression of Ampd2, indicating the mutation had no effect on mRNA stability, whereas it likely resulted in protein instability (Figure 1B). We also confirmed normal expression of the broadly expressed paralog Ampd3, suggesting no overlapping compensation. In general it appeared that the changes in gene expression were a consequence of high cholesterol levels, and not causative, prompting us to employ additional technologies.
Figure 3.
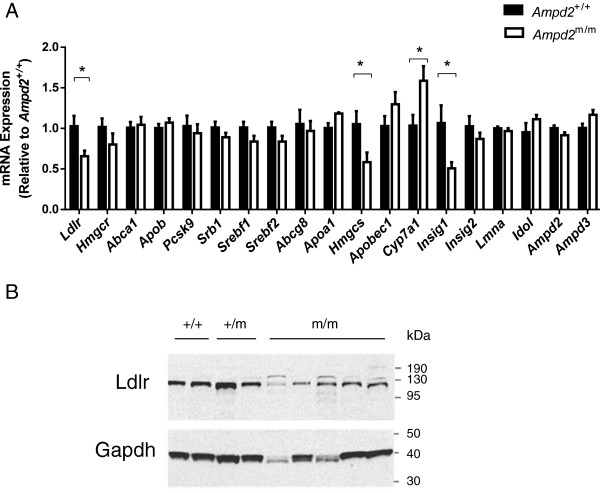
Gene expression analysis of Ampd2 m/m and Ampd2 +/+ mice. A) Liver mRNA levels were assessed by quantitative RT-PCR for an array of lipid metabolism genes on fed 9-week old Ampd2 +/+ and Ampd2 m/m mice (n = 5). Statistical analyses were carried out by unpaired two-tailed t-tests; * p <0.05 vs. Ampd2 +/+. B) Liver immunoblot analysis of Ldlr protein in Ampd2 +/+, Ampd2 +/m and Ampd2 m/m mice.
Metabolomic analysis of Ampd2deficient mice
We engaged metabolomics to assist in determining the mechanism of hypercholesterolemia in Ampd2m/m mice. Liver and plasma from 9-week old Ampd2+/+ and Ampd2m/m mice were collected for metabolite analysis. These analyses confirmed the cholesterol phenotype of the Ampd2m/m mice in both plasma and liver samples (Additional file 1: Table S1). Additionally cholesterol esters were elevated up to 5 fold. Allantoin, the terminal metabolite in the purine degradation pathway was significantly reduced in the livers from Ampd2m/m mice (Additional file 1: Table S1). The upstream metabolites AMP and ATP were significantly increased in Ampd2m/m mice (Additional file 1: Table S1 and Figure 4). Metabolomic analysis also demonstrated significant increases in plasma plant sterols including campesterol and β-sitosterol.
Figure 4.
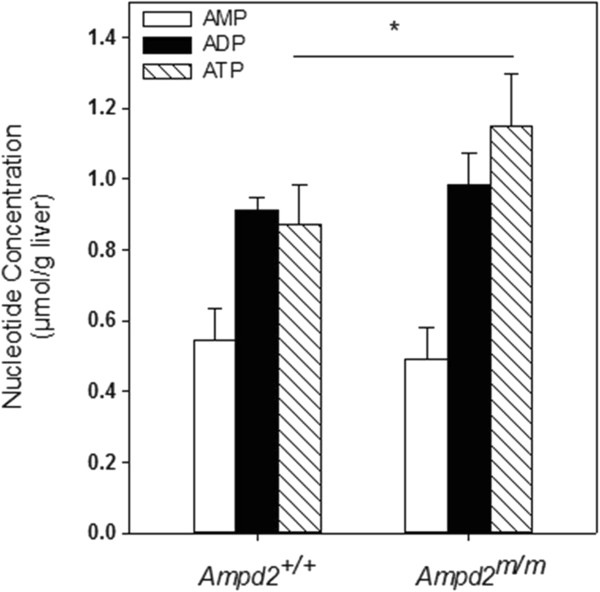
Hepatic AMP, ADP, and ATP levels in Ampd2 +/+ and Ampd2 m/m mice. Hepatic AMP, ADP, and ATP levels were determined by HPLC. Open bars represent AMP; solid bars - ADP and hatched bars - ATP. Statistical analysis was carried out by unpaired two-tailed t-tests, * p <0.05 vs. Ampd2 +/+.
Comparison of Ampd2deficiency on different genetic backgrounds
Initial gene mapping was carried out by outcrossing the ENU harboring C57BL/6 J (B6) genome to C3.SW-H2b/SnJ (C3) mice, and all studies presented to date have been using mice from heterozygous matings with this mixed genetic background. A homogeneous genetic background was achieved by backcrossing the Ampd2m/m mice to inbred B6 mice to obtain a pure B6 background. Ampd2+/+ and Ampd2m/m mice on a mixed B6/C3 background were compared to those on a pure B6 background with a focus on plasma cholesterol and urinalysis [14]. Ampd2m/m mice on both backgrounds exhibited dramatically elevated levels of urinary protein between 7 and 11 weeks of age, most evident in B6 mice (Figure 5B and 5E) than the mixed B6/C3 line, and showing significance throughout the course of the analysis. Hypercholesterolemia was evident in Ampd2m/m mice on both genetic backgrounds, with the mixed B6/C3 background showing higher absolute cholesterol levels, whereas B6 mice exhibited a milder but less variable phenotype. Additionally, the mixed background Ampd2m/m mice had consistently higher cholesterol levels vs. the Ampd2+/+ mice throughout the course of the study, peaking at 8–10 weeks of age, while the pure B6 Ampd2m/m mice had elevated cholesterol levels relative to the Ampd2+/+ mice only from 7–9 weeks of age (Figure 5A and 5D). Consistent with the presence of proteinuria and NS (see below), both backgrounds of Ampd2m/m mice displayed hypoalbuminemia (Figure 5C and 5F), and similarly to cholesterol levels, albumin levels attained statistical significance at a young age for both lines. Together these data in both genetic backgrounds revealed that the proteinuria observed temporally coincided with the cholesterol phenotypes, suggesting interplay between the traits.
Figure 5.
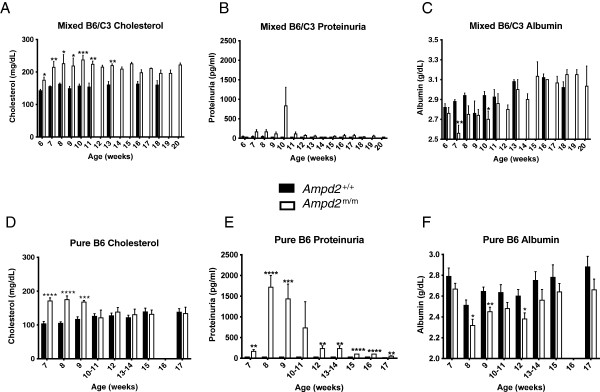
Temporal hypercholesterolemia and proteinuria in Ampd2 m/m mice in mixed B6/C3 and pure B6 genetic background. A and D) Plasma cholesterol level time course in Ampd2 +/+ and Ampd2 m/m mice. B and E) Proteinuria level time course in Ampd2 +/+ and Ampd2 m/m mice. C and F) Plasma albumin levels time course in Ampd2 +/+ and Ampd2 m/m mice on a mixed B6/C3 background and pure B6 background (n = 4-6). Statistical analyses were carried out by unpaired two-tailed t-tests; * p <0.05; ** p <0.01; *** p <0.001; **** p <0.0001 vs. Ampd2 +/+.
Confirmation of nephrotic syndrome in Ampd2m/mmice
To confirm the effect of the A341S mutation in the kidneys of Ampd2m/m mice we investigated Ampd2 protein abundance in both strains and verified that Ampd2 protein was absent in the liver and kidney of pure B6 Ampd2m/m mice (Figure 6A). NS was verified in Ampd2m/m mice by kidney histology (Figure 6B). Proteinaceous tubular casts and interstitial nephritis were observed in Ampd2m/m mice and not Ampd2+/+ mice (Figure 6B-D). By TEM, glomeruli of selected Ampd2m/m mice and Ampd2+/+ mice were examined to ascertain any ultrastructural differences (Figure 7). Glomeruli from Ampd2m/m mice showed effacement, fusion, and blebbing of podocyte foot processes, minimal to moderate thickening of the glomerular basement membrane, as well as denuding with subsequent pseudomembrane formation of the glomerular endothelium. While damage to foot processes, basal lamina and glomerular endothelium represents impairment of the glomerular filtration membrane, the subtle increase in cellularity of the mesangium, along with a predominant lymphocytic inflammatory infiltrate seen in Ampd2m/m mice, further supports changes secondary to glomerular dysfunction with ensuing proteinuria.
Figure 6.
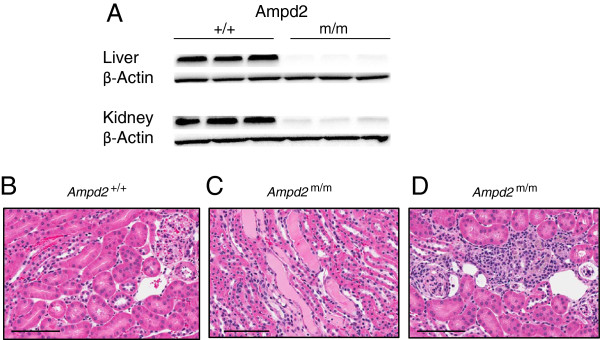
Ampd2 deficiency and histological features in Ampd2 m/m kidneys. A) Liver and kidney immunoblot analysis of Ampd2 protein in B6 Ampd2 +/+ and Ampd2 m/m mice. C) Proteinaceous tubular casts were observed in the kidneys of Ampd2 m/m mice. D) Interstitial nephritis was observed in the kidneys of Ampd2 m/m mice, but not in Ampd2 +/+ mice (B). Bar = 100 μm.
Figure 7.
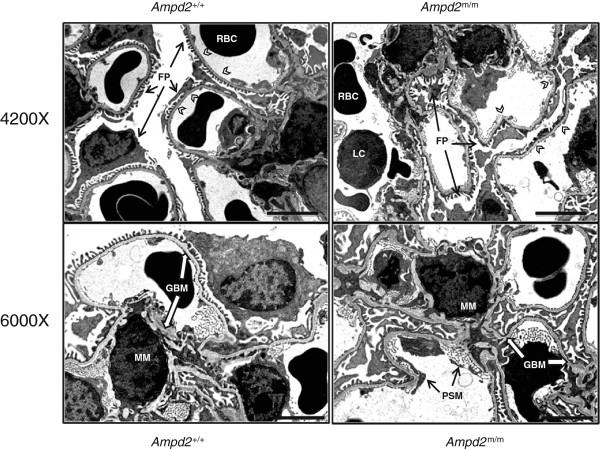
Electron micropscopic evaluation of the glomeruli from Ampd2 m/m mice. Glomeruli of Ampd2 m/m mice showed diffuse effacement, sloughing, and fusing of podocyte foot processes (FP). Extensive blebbing, fusing, and surface pseudomembrane formation (PSM) was evident along the glomerular tuft endothelium of Ampd2 m/m mice (white arrowheads). In addition to a minimal to moderate thickening of the basal lamina (GBM), there was a subtle increase in cellularity of the mesangial matrix (MM) and an increase in inflammatory cells, predominantly lymphocytes (LC) in Ampd2 m/m mice. RBC = Red Blood Cell. Upper photomicrographs 4200×; lower photomicrographs 6000×; Bar = 2 μm.
Effect of a potent AMPD/adenosine deaminase inhibitor on lipid and renal profiles of normal mice
Deoxycoformycin is a potent, but nonspecific inhibitor of AMPD/adenosine deaminase and is an approved chemotherapeutic drug. It dramatically decreases nucleotide metabolism and affects the cells ability to process DNA [15]. B6 mice were given a single injection of 10 mg/kg deoxycoformycin to determine the effect on plasma cholesterol and urinary protein levels. At 24 hours post injection, a rise in cholesterol as well as protein urea was seen in the plasma of deoxycoformycin treated mice (Additional file 2: Figure S1A and S1B). This data could suggest a link between dyslipidemia and NS when Ampd is inhibited, though the possibility that the response was an acute health reaction cannot be overlooked.
Discussion
In the present study, we identified a mutation in the Ampd2 gene that leads to NS and hypercholesterolemia. The combined phenotypes have not previously been reported in Ampd2 knockout mice [14]; minimal change kidney disease was observed in these mice, and it was speculated that Ampd2 function was critical for podocyte survival and related to defects in Inosine-5′-monophosphate (IMP). Furthermore, despite dyslipidemia and minimal change disease being commonly associated in human NS, our Ampd2m/m mice offer the first description of a mouse model to present both phenotypes. Further investigation will be needed to elucidate if these phenotypes are physiologically connected, or result independently from Ampd2 tissue-specific deficiency. Towards this end, restoring Ampd2 function in the liver or kidney of the Ampd2m/m mice could allow dissection of this relationship.
We identified a transient cholesterol phenotype in mixed B6/C3 Ampd2m/m mice, and have now confirmed that the renal phenotype is transient as well, temporally mirroring the cholesterol levels. In successive generations of the mixed B6/C3 Ampd2m/m mice we noticed less severe incidence of hypercholesterolemia and speculate that this could be due to the genetic drift of modifier alleles in the mixed B6/C3 background. To address this hypothesis, we generated pure backgrounds of Ampd2m/m mice on both B6 and C3 backgrounds. The pure B6 Ampd2m/m mice do have a penetrant but lesser phenotype for the cholesterol levels and a stronger phenotype for the renal parameter (Figure 5). Conversely, pure C3 Ampd2m/m mice showed no phenotype (data not shown). Together, this suggests that the mixed background can lead to a more severe phenotype, supporting the potential presence of modifier alleles and their epistatic effects on lipid traits [16].
The renal phenotype in the B6 Ampd2m/m mice was further characterized by TEM. The kidneys were collected when lipid and renal phenotypes were most severe. Consistent with the proteinuria observed in the urine and proteinaceous tubular casts seen on histopathology, Ampd2m/m mice were found to have damage to the glomerular filtration membrane at the ultrastructural level. Increases in inflammatory cells and in cellular debris also indicated progressive renal disease in Ampd2m/m mice.
It is of interest to understand how the loss of Ampd2 function led to alterations in lipid profiles and kidney function. Ampd plays roles both in the purine nucleotide cycle as well as the catabolism of AMP to uric acid. In the purine nucleotide cycle, three enzyme catalyzed reactions convert AMP to fumarate, in order to increase the concentration of TCA cycle intermediates [17]. The generation of fumarate provides skeletal tissue with its only source of intermediate substrate for the TCA cycle [18] and loss of Ampd function reduces fumarate production via the purine nucleotide cycle. This effect could result in changes in the concentrations of malate, oxaloacetate, citrate, isocitrate, α-ketoglutarate and succinate in the TCA cycle. Furthermore in the Ampd2m/m mice, malate and citrate levels were elevated while succinate levels were suppressed (Additional file 1: Table S1). These alterations in TCA cycle intermediates are not consistently decreased as could be expected by a defective purine nucleotide cycle by loss of Ampd2, suggesting that this aspect of hepatic Ampd2 does not underpin the lipid phenotype of the Ampd2m/m mice.
Ampd is also the rate limiting step in the degradation of AMP [19], a defect in Ampd would be expected to lead to an increase in AMP and a decrease in any intermediates downstream of AMP, including IMP, inosine, hypoxanthine, xanthine and uric acid. The metabolomic data showed significant reduction in the downstream uric acid catabolic intermediate, allantoin (Additional file 1: Table S1). If the hepatic deficiency of Ampd2 is the primary cause of the dyslipidemia, the defect in purine catabolism as here could provide a pathogenic mechanism. Along these lines Yang et al. [20] have reported that allantoin administration in DIO mice significantly improved dyslipidemia. Metabolomic analysis also demonstrated significant increases in plasma plant sterols including campesterol and β-sitosterol. These dietary derived sterols are eliminated from the body through hepatic biliary secretion using the ATP binding cassette G5/8 (ABCG5/8) transporter complex. No gene expression changes were observed for ABCG8 (Figure 3A) suggesting the accumulation of β-sitosterol and campesterol were due to defects in posttranslational ABCG5/8 transporter function. Thus, one mechanism that may lead to elevated plasma cholesterol observed in Ampd2m/m mice may be defects in ABCG5/8 transporter function.
The presence of dyslipidemia together with kidney abnormalities would normally be indicative of NS as the primary defect in the Ampd2m/m mice, as urinary protein loss is known to stimulate an increase in LDL synthesis by the liver [21]. However, the Ampd2m/m mice do not exhibit significantly elevated triglycerides (Figure 2C), a hallmark of dyslipidemia secondary to NS due to impaired clearance of chylomicrons and VLDL [21]. Furthermore induction of hypercholesterolemia during NS is primarily related to upregulation of HMG-CoA reductase activity [22]. An upregulation of Hmgcr was not observed in the liver of Ampd2m/m mice, and in fact, a trend of decreased gene expression was observed (Figure 3). Insulin-induced gene 1 (Insig1) was decreased by 56% in Ampd2m/m mice, possibly resulting in less binding of HMG CoA Reductase and increased cholesterol biosynthesis. Transcript levels of 3-hydroxy-3-methylglutaryl-CoA synthase (Hmgcs) were 47% lower in Ampd2m/m mice, possibly due to feedback inhibition caused by high levels of circulating plasma cholesterol, albeit only a minimal reduction in Srebp1 and 2 was observed. It has also been reported that rats with NS accompanied with hypercholesterolemia lack Cyp7a1 upregulation, while rats that have equally high hypercholesterolemia due to a high fat diet do not present with NS and show marked elevation of Cyp7a1 RNA levels [22, 23]. As shown in Figure 3, Ampd2m/m mice demonstrated a modest but significantly increased expression of Cyp7a1 mRNA, consistent with the dietary and not the NS induced lesion. This may be a compensatory mechanism to enhance cholesterol disposal through bile acid synthesis and secretion and switching to a different transporter in response to a defect in ABCG5/8 function. Other discrepancies between NS and dyslipidemia in Ampd2m/m mice and other rodent NS models exist. Zhou et al. [24] as well as others [25, 26] have reported that Ldlr protein abundance is significantly reduced in NS rats, with no concurrent reduction in Ldlr gene expression, whereas Ampd2m/m mice showed significant reductions in both. Shearer et al. [27] reported that in the nephrotic rat, proteinuria resulted in reduced HDL ApoE content. We found increases in both HDL-C and ApoE levels in our Ampd2m/m mice. Liu and Vaziri [28] report that NS induced LDLR deficiency is due to upregulation of PCSK9 and IDOL, both post-translational regulators of LDLR. We see no increased expression of either gene in the Ampd2m/m mice. Collectively, these data suggest that dyslipidemia is a direct consequence of Ampd2 deficiency, and not a consequence of NS. We propose that the pathogenic mechanism relates to defective catabolism of AMP to uric acid, including allantoin, versus impairment in the purine nucleotide cycle.
Recently, Akizu et al. [29] reported an early onset neurodegenerative condition resulting from mutations in AMPD2 and found that AMPD2 plays a critical role in the maintenance of guanine nucleotide pools in relation to adenosine derivatives on de novo purine synthesis. No neurological phenotypes were observed in Ampd2 knockout mice. Furthermore only Ampd2/3 double knockout mice exhibited slightly reduced brain size, and showed little evidence of the neuronal loss observed in the AMPD2 mutant human population. Nucleotide analysis of the double knockout brains showed increases in ATP nucleotide levels and a decrease in guanosine triphosphate, consistent with the increased ATP found in the liver of our Ampd2m/m mice. We also confirm a significant reduction in levels of guanine in Ampd2m/m mice; however our mice exhibited no obvious neurological phenotype.
In conclusion, we have identified a novel mouse model of transient hypercholesterolemia with concurrent NS associated with defective Ampd2 function. Ascertaining whether the two phenotypes are interrelated and the molecular mechanism of Ampd2 loss and hypercholesterolemia will require further investigation.
Materials and methods
Mouse breeding
Founder mice were identified at McLaughlin Research Institute [11] and subsequent generations were maintained at Charles River Laboratories to generate mixed C57BL/6 J (B6) C3.SW-H2b/SnJ (C3) animals. The Ampd2 line was backcrossed to B6 via MAX-BAX® technology, and 2 of the 100% B6 congenic animals were obtained for further heterozygous × heterozygous mating. Mice were cared for in accordance to the Guide for the Care and Use of Laboratory Animals, 8th Edition. Animals were single housed at an AAALAC accredited facility in non-sterile ventilated micro-isolator housing on corn cob bedding. All research protocols were approved by the (Amgen, Inc; Thousand Oaks) Institutional Animal Care and Use Committee. Animals had ad libitum access to pelleted feed (diet 2020X, Harlan Teklad; Indianapolis, IN) and water (reverse osmosis chlorinated water) via automatic watering system. Animals were maintained on a 12:12 hour light: dark cycle in rooms at 20–26°C, humidity maintained between 30-70% and had access to enrichment opportunities.
Plasma and urine collection and analysis
Blood samples were collected in EDTA plasma tubes via the retro-orbital sinus of conscious mice. Plasma total cholesterol, HDL-cholesterol, triglyceride and albumin levels were measured using the Olympus AU400e Chemistry Analyzer (Olympus America, Inc; Center Valley, PA) from plasma thawed on ice and diluted four-fold with saline. ApoA1 and ApoE values were determined by immunoassay (Linco Research; St. Charles, MO). Proteinuria was measured by dipping Albustix reagent strips directly into urine (Siemens Medical; Malvern, PA).
Deoxycoformycin studies
Commercial deoxycoformycin (Pentostatin; Tocris Bioscience; Bristol, UK) was dissolved in saline at a concentration of 25 mg/mL. Subsequent dilutions were made with saline and the mice were dosed via intraperitoneal injections at a concentration of 10 mg/kg. Baseline plasma samples were collected at 9 AM, immediately preceding a single dose of deoxycoformycin, and terminal plasma samples were collected 24 hours later.
Quantitative RT-PCR analyses
Total liver RNA was isolated using an RNeasy Mini kit (QIAGEN GmBH; Hilden, Germany) and A260 was measured using a Beckman Coulter DU 800 spectrophotometer. RT-PCR reactions were set up using QuantiTect Multiplex RT-PCR Kit (QIAGEN), and 100 ng RNA per reaction was used, with appropriate oligonucleotides and probes; Cyclophilin A used as the housekeeping gene (sequences available upon request). Reactions were run on an ABI Prism 7900HT temperature cycler (Life Technologies; Carlsbad, CA). Results were analyzed using SDS2.2.2 software (Life Technologies).
Lipoprotein analysis by fast performance liquid chromatography (FPLC)
Size-exclusion chromatography was performed by incorporating two Superose 6 10/300 GL columns (Amersham Biosciences; Piscataway, NJ) in tandem as described [30]. Fraction volumes of 275 μL were collected per well, from which 80 μL was removed and mixed with 120 μL of reagent from a colorimetric Cholesterol E Kit (Wako Chemicals USA; Inc., Richmond, VA) to measure cholesterol levels. To ensure linearity, a standard curve of pure cholesterol was included for each sample plate.
Metabolomics
Mass spectrometry-based metabolite profiling analysis was performed at Metanomics Health GmBH (Berlin, Germany) on plasma and liver samples processed and extracted using a proprietary methods yielding a lipid and polar fraction which were used for gas chromatography–mass spectrometry (GC-MS) and liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis, respectively. For GC-MS analysis the samples were sequentially derivatized before measurement. For LC-MS/MS analysis a proprietary technology was applied which allows target and high sensitivity Multiple Reaction Monitoring profiling in parallel to full screen analyses. The method has been validated in separate experiments to determine intra- and interday variability, blank portions and linearity of each metabolite comprising known and unknown signals. Thresholds for reliable determination of the metabolite have been evaluated and confirmed within the study. Liver metabolite data were normalized to the dry weight of samples. For plasma samples, this step was not necessary due to the application of the same volume for each plasma sample. Each metabolite was normalized against the median of the data obtained from the control animals (ratios vs. control). Two hundred and twenty four metabolites were measured in plasma, 152 metabolites with known structure. For liver samples, 574 metabolites were measured, 222 metabolites with known structure. Metabolites with statistically significant differences between genotypes are presented in Additional file 1: Table S1.
High performance liquid chromatography (HPLC) analysis of AMP, ADP, and ATP
Frozen (-80°C) mouse liver samples were homogenized at 10% (w/v) in acetonitrile, 10 mM KH2PO4 buffer pH 7.4 (3:1). The homogenate was centrifuged at 20,000 × g, 4°C, for 10 minutes and the supernatant centrifuged again at 20,000 × g, 4°C, for 10 minutes. The acetonitrile was removed by chloroform extraction three times. Final aqueous layer extract was diluted with 10 mM KH2PO4 Buffer, pH 7.4 to initial volume. Samples (20 μL) were analyzed using an Agilent 1100 Series HPLC equipped with a Hypersil Gold C-18, 250 × 4.6 mm 5 μm particle size HPLC column at a flow rate of 0.75 mL/minute using a 130 min step gradient protocol as follows: 10 minutes (100% A-10 mM tetrabutylammonium hydroxide, 10 mM KH2PO4, 0.125% methanol, pH 7.0), 3 minutes (80% A: 20% B- 2.8 mM tetrabutylammonium hydroxide, 100 mM KH2PO4, 30% methanol, pH 5.5), 10 minutes (70% A: 30% B), 12 minutes (55% Line A: 45% B), 11 minutes (40% A: 60% B), 9 minutes (35% A: 56% B), 10 minutes (25% A: 75% B), 30 minutes (100% B), 36 minutes (100% A). Spectra were collected at 206 nm and 260 nm for AMP (29.4 Retention Time), ADP (41.4 Retention Time), and ATP (49.1 Retention Time). Data was processed using Agilent Chemstation software using a standard curve for each analyte. The precision (%CV) of the assay was very good: 3.9% for AMP, 6.3% for ADP, and 3.9% for ATP with a limit of quantification of 0.1 μmol/g liver tissue.
Gene sequencing
PCR DNA fragments of candidate genes were subcloned into the pCR II vector using a TA cloning kit and sequenced with M13F and M13R primers or directly sequenced with the PCR primers on an ABI 3730xl DNA Analyzer. To genotype the guanine to thymine transversion of Ampd2, a BseR1 restriction digest was applied to a 173 bp PCR product amplified with the oligonucleotides 5′-GCACCTGCAGTCCTCATGTTTGTATAGC-3′, and 5′-GCTTGATGAAGCGCAGTAGATGTTTCTGG-3′.
Protein analysis
Approximately 20–40 mg of frozen liver or kidney were homogenized in NP40 lysis buffer with 1X of Complete Protease Inhibitors Cocktail (Roche; Indianapolis, IN). Protein lysates were resolved on 4-12% Bis-Tris polyacrylamide gels (Life Technologies). Separated proteins were transferred to PVDF membranes (Life Technologies), and blocked with Pierce Superblock. The blots were hybridized with antibodies directed against Ampd2 (QQ13; Santa Cruz Biotechnology, Inc. Dallas, TX), Ldlr (Cayman Chemical; Ann Arbor, MI), Gapdh (IMGENEX; San Diego, CA) or β-actin (Sigma-Aldrich) overnight at 4°C with gentle shaking. The membranes were washed 3 times with TBS-T, incubated with secondary antibody conjugated to HRP at room temperature for 1.5 hours, and washed another 3 times in large volume changes of TBS-T. The blots were processed with SuperSignal West Dura substrate (Thermo Fisher Scientific, Inc. Rockford, IL), and visualized on a Fluorochem HD2 imager (ProteinSimple, Santa Clara, California).
Histological analysis
Samples of kidney were fixed by immersion in 10% neutral buffered formalin for 24 hours, trimmed into cassettes, processed routinely to paraffin embedment, sectioned at 5 μm, stained with hematoxylin and eosin and periodic acid methenamine silver [31] and examined histologically.
Electron microscopy
A portion of the renal cortex was collected for transmission electron microscopy (TEM), minced into 1 mm cubes and placed into 2.5% glutaraldehyde fixative. Following fixation, the tissues were processed for TEM. Samples were first rinsed in 0.1 M Sorenson’s Phosphate buffer, post-fixed in 2% osmium tetroxide, embloc stained with uranyl acetate, after which the tissues were dehydrated through a graded series of alcohol and acetone then 100% Spurr’s resin and cured at 65°C. Semi-thin sections (500 nm sections stained with Toluene blue) of each resin block were examined and four resin blocks/animal were chosen for thin sectioning (80–90 nm, gold sections placed on a 100 μm mesh copper grid). The four grids per animal were examined on both a JEM 100 kV (JEOL USA; Inc. Peabody, MA) and a Tecnai 110 kV (TEI; Hillsboro, OR) microscope with ten digital photomicrographs shot for each resin block thereby providing an adequate number of photomicrographs for comparison between the genotypes.
Statistical analysis
In some graphs individual mouse values are shown with means ± SEM. In other graphs only the means ± SEM are displayed. Statistical analyses were carried out by one-way ANOVA using a Tukey post test comparing with three or more groups. For statistical analyses comparing two groups unpaired two-tailed t-tests for used; * p <0.05; ** p <0.01; *** p <0.001; **** p <0.0001 vs. Ampd2+/+.
Electronic supplementary material
Additional file 1: Table S1: Metabolomic analysis of significantly altered metabolites in liver and plasma in Ampd2 m/m and Ampd2 +/+ mice. Values represent the fold change. Statistical analyses were carried out by unpaired two-tailed t-tests. (DOCX 28 KB)
Additional file 2: Figure S1: Effects of deoxycoformycin, a potent inhibitor of AMPD/adenosine deaminase in B6 mice. Cholesterol (A) as well as urinary protein levels (B) were elevated 24 hours post injection. Statistical analyses were carried out by unpaired two-tailed t-tests; * p <0.05 ***; p <0.001 vs. saline. (PDF 73 KB)
Footnotes
Competing interests
All authors declare that they do not have any competing or financial interests. JH, CML, WGR, JRT, JL, PAC, KWK, MMV, and DJL are employees of Amgen, Inc.
Authors’ contributions
TJ, WGR, SAK, GC, DJL and MMV conceived and designed the experiments. JH, CL, MC, WB, TG, PAC, JB, KK and CC performed the experiments. JRT, JL and DJL analyzed the data. JH. and DJL wrote the paper. All authors read and approved the final manuscript.
Contributor Information
Joan Helmering, Email: joan.helmering@amgen.com.
Todd Juan, Email: todd.juan@gmail.com.
Chi Ming Li, Email: chi-ming.li@amgen.com.
Mark Chhoa, Email: nucleuss001@yahoo.com.
Will Baron, Email: cloneman@att.net.
Tibor Gyuris, Email: gyuris_tibor@yahoo.com.
William G Richards, Email: brichard@amgen.com.
James R Turk, Email: jim.turk@amgen.com.
Jeff Lawrence, Email: jeff.lawrence@amgen.com.
Patrick A Cosgrove, Email: pat.cosgrove@amgen.com.
Jim Busby, Email: jbusby@amgen.com.
Ki Won Kim, Email: kkim@amgen.com.
Stephen A Kaufman, Email: sierratreeline@gmail.com.
Connie Cummings, Email: connie@ultrapathimaging.com.
George Carlson, Email: gac@mri.montana.edu.
Murielle M Véniant, Email: mveniant@amgen.com.
David J Lloyd, Email: dlloyd@amgen.com.
References
- 1.Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J Clin Invest. 2003;111:1795–1803. doi: 10.1172/JCI200318925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ishibashi S, Brown MS, Goldstein JL, Gerard RD, Hammer RE, Herz J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest. 1993;92:883–893. doi: 10.1172/JCI116663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich D, Derré A, Villéger L, Farnier M, Beucler I, Bruckert E, Chambaz J, Chanu B, Lecerf JM, Luc G, Moulin P, Weissenbach J, Prat A, Krempf M, Junien C, Seidah NG, Boileau C. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–156. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- 4.Tybjaerg-Hansen A, Steffensen R, Meinertz H, Schnohr P, Nordestgaard BG. Association of mutations in the apolipoprotein B gene with hypercholesterolemia and the risk of ischemic heart disease. N Engl J Med. 1998;338:1577–1584. doi: 10.1056/NEJM199805283382203. [DOI] [PubMed] [Google Scholar]
- 5.Soutar AK, Naoumova RP. Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat Clin Pract Cardiovasc Med. 2007;4:214–225. doi: 10.1038/ncpcardio0836. [DOI] [PubMed] [Google Scholar]
- 6.Leduc MS, Lyons M, Darvishi K, Walsh K, Sheehan S, Amend S, Cox A, Orho-Melander M, Kathiresan S, Paigen B, Korstanje R. The mouse QTL map helps interpret human genome-wide association studies for HDL cholesterol. J Lipid Res. 2011;52:1139–1149. doi: 10.1194/jlr.M009175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mohr M, Klempt M, Rathkolb B, de Angelis MH, Wolf E, Aigner B. Hypercholesterolemia in ENU-induced mouse mutants. J Lipid Res. 2004;45:2132–2137. doi: 10.1194/jlr.M400236-JLR200. [DOI] [PubMed] [Google Scholar]
- 8.Aljakna A, Choi S, Savage H, Hageman Blair R, Gu T, Svenson KL, Churchill GA, Hibbs M, Korstanje R. Pla2g12b and Hpn are genes identified by mouse ENU mutagenesis that affect HDL cholesterol. PLoS One. 2012;7:e43139. doi: 10.1371/journal.pone.0043139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pitman JL, Bonnet DJ, Curtiss LK, Gekakis N. Reduced cholesterol and triglycerides in mice with a mutation in Mia2, a liver protein that localizes to ER exit sites. J Lipid Res. 2011;52:1775–1786. doi: 10.1194/jlr.M017277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salinger AP, Justice MJ. Mouse mutagenesis using N-Ethyl-N-Nitrosourea (ENU) CSH Protoc. 2008;2008:pdb prot4985. doi: 10.1101/pdb.prot4985. [DOI] [PubMed] [Google Scholar]
- 11.Juan T, Véniant MM, Helmering J, Babij P, Baker DM, Damore MA, Bass MB, Gyuris T, Chhoa M, Li CM, Ebeling C, Amato J, Carlson GA, Lloyd DJ. Identification of three loci affecting HDL-cholesterol levels in a screen for chemically induced recessive mutations in mice. J Lipid Res. 2009;50:534–545. doi: 10.1194/jlr.M800471-JLR200. [DOI] [PubMed] [Google Scholar]
- 12.Szydlowska M, Roszkowska A. Expression patterns of AMP-deaminase isozymes in human hepatocellular carcinoma (HCC) Mol Cell Biochem. 2008;318:1–5. doi: 10.1007/s11010-008-9773-x. [DOI] [PubMed] [Google Scholar]
- 13.Gross M. Molecular biology of AMP deaminase deficiency. Pharm World Sci. 1994;16:55–61. doi: 10.1007/BF01880656. [DOI] [PubMed] [Google Scholar]
- 14.Toyama K, Morisaki H, Cheng J, Kawachi H, Shimizu F, Ikawa M, Okabe M, Morisaki T. Proteinuria in AMPD2-deficient mice. Genes Cells. 2012;17:28–38. doi: 10.1111/j.1365-2443.2011.01568.x. [DOI] [PubMed] [Google Scholar]
- 15.Spiers AS. Deoxycoformycin (pentostatin): clinical pharmacology, role in the chemotherapy of cancer, and use in other diseases. Haematologia (Budap) 1996;27:55–84. [PubMed] [Google Scholar]
- 16.Su Z, Li Y, James JC, McDuffie M, Matsumoto AH, Helm GA, Weber JL, Lusis AJ, Shi W. Quantitative trait locus analysis of atherosclerosis in an intercross between C57BL/6 and C3H mice carrying the mutant apolipoprotein E gene. Genetics. 2006;172:1799–1807. doi: 10.1534/genetics.105.051912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gibala MJ, MacLean DA, Graham TE, Saltin B. Anaplerotic processes in human skeletal muscle during brief dynamic exercise. J Physiol. 1997;502(Pt 3):703–713. doi: 10.1111/j.1469-7793.1997.703bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gibala MJ, Gonzalez-Alonso J, Saltin B. Dissociation between muscle tricarboxylic acid cycle pool size and aerobic energy provision during prolonged exercise in humans. J Physiol. 2002;545:705–713. doi: 10.1113/jphysiol.2002.028084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saint-Marc C, Pinson B, Coulpier F, Jourdren L, Lisova O, Daignan-Fornier B. Phenotypic consequences of purine nucleotide imbalance in Saccharomyces cerevisiae. Genetics. 2009;183:529–538. doi: 10.1534/genetics.109.105858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang TT, Chiu NH, Chung HH, Hsu CT, Lee WJ, Cheng JT. Stimulatory effect of allantoin on imidazoline I (1) receptors in animal and cell line. Horm Metab Res. 2012;44:879–884. doi: 10.1055/s-0032-1312624. [DOI] [PubMed] [Google Scholar]
- 21.Trevisan R, Dodesini AR, Lepore G. Lipids and renal disease. J Am Soc Nephrol. 2006;17:S145–S147. doi: 10.1681/ASN.2005121320. [DOI] [PubMed] [Google Scholar]
- 22.Vaziri ND. Molecular mechanisms of lipid disorders in nephrotic syndrome. Kidney Int. 2003;63:1964–1976. doi: 10.1046/j.1523-1755.2003.00941.x. [DOI] [PubMed] [Google Scholar]
- 23.Liang KH, Oveisi F, Vaziri ND. Gene expression of hepatic cholesterol 7 alpha-hydroxylase in the course of puromycin-induced nephrosis. Kidney Int. 1996;49:855–860. doi: 10.1038/ki.1996.118. [DOI] [PubMed] [Google Scholar]
- 24.Zhou Y, Zhang X, Chen L, Wu J, Dang H, Wei M, Fan Y, Zhang Y, Zhu Y, Wang N, Breyer MD, Guan Y. Expression profiling of hepatic genes associated with lipid metabolism in nephrotic rats. Am J Physiol Renal Physiol. 2008;295:F662–F671. doi: 10.1152/ajprenal.00046.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han S, Vaziri ND, Gollapudi P, Kwok V, Moradi H. Hepatic fatty acid and cholesterol metabolism in nephrotic syndrome. Am J Transl Res. 2013;5:246–253. [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L, Shearer GC, Budamagunta MS, Voss JC, Molfino A, Kaysen GA. Proteinuria decreases tissue lipoprotein receptor levels resulting in altered lipoprotein structure and increasing lipid levels. Kidney Int. 2012;82:990–999. doi: 10.1038/ki.2012.244. [DOI] [PubMed] [Google Scholar]
- 27.Shearer GC, Newman JW, Hammock BD, Kaysen GA. Graded effects of proteinuria on HDL structure in nephrotic rats. J Am Soc Nephrol. 2005;16:1309–1319. doi: 10.1681/ASN.2004080644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu S, Vaziri ND. Role of PCSK9 and IDOL in the pathogenesis of acquired LDL receptor deficiency and hypercholesterolemia in nephrotic syndrome. Nephrol Dial Transplant. 2014;29:538–543. doi: 10.1093/ndt/gft439. [DOI] [PubMed] [Google Scholar]
- 29.Akizu N, Cantagrel V, Schroth J, Cai N, Vaux K, McCloskey D, Naviaux RK, Van Vleet J, Fenstermaker AG, Silhavy JL, Scheliga JS, Toyama K, Morisaki H, Sonmez FM, Celep F, Oraby A, Zaki MS, Al-Baradie R, Faqeih EA, Saleh MA, Spencer E, Rosti RO, Scott E, Nickerson E, Gabriel S, Morisaki T, Holmes EW, Gleeson JG. AMPD2 regulates GTP synthesis and is mutated in a potentially treatable neurodegenerative brainstem disorder. Cell. 2013;154:505–517. doi: 10.1016/j.cell.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garber DW, Kulkarni KR, Anantharamaiah GM. A sensitive and convenient method for lipoprotein profile analysis of individual mouse plasma samples. J Lipid Res. 2000;41:1020–1026. [PubMed] [Google Scholar]
- 31.Phillips CL, Pfeiffer BJ, Luger AM, Franklin CL. Novel collagen glomerulopathy in a homotrimeric type I collagen mouse (oim) Kidney Int. 2002;62:383–391. doi: 10.1046/j.1523-1755.2002.00451.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1: Metabolomic analysis of significantly altered metabolites in liver and plasma in Ampd2 m/m and Ampd2 +/+ mice. Values represent the fold change. Statistical analyses were carried out by unpaired two-tailed t-tests. (DOCX 28 KB)
Additional file 2: Figure S1: Effects of deoxycoformycin, a potent inhibitor of AMPD/adenosine deaminase in B6 mice. Cholesterol (A) as well as urinary protein levels (B) were elevated 24 hours post injection. Statistical analyses were carried out by unpaired two-tailed t-tests; * p <0.05 ***; p <0.001 vs. saline. (PDF 73 KB)