

ABSTRACT
The essential metabolic enzyme CTP synthase (CTPsyn) can be compartmentalised to form an evolutionarily-conserved intracellular structure termed the cytoophidium. Recently, it has been demonstrated that the enzymatic activity of CTPsyn is attenuated by incorporation into cytoophidia in bacteria and yeast cells. Here we demonstrate that CTPsyn is regulated in a similar manner in Drosophila tissues in vivo. We show that cytoophidium formation occurs during nutrient deprivation in cultured cells, as well as in quiescent and starved neuroblasts of the Drosophila larval central nervous system. We also show that cytoophidia formation is reversible during neurogenesis, indicating that filament formation regulates pyrimidine synthesis in a normal developmental context. Furthermore, our global metabolic profiling demonstrates that CTPsyn overexpression does not significantly alter CTPsyn-related enzymatic activity, suggesting that cytoophidium formation facilitates metabolic stabilisation. In addition, we show that overexpression of CTPsyn only results in moderate increase of CTP pool in human stable cell lines. Together, our study provides experimental evidence, and a mathematical model, for the hypothesis that inactive CTPsyn is incorporated into cytoophidia.
Keywords: CTP synthase, cytoophidium, intracellular compartmentation, CTP, Drosophila, neurogenesis
INTRODUCTION
In recent years it has become apparent from protein localisation screens that an unprecedented number of metabolic enzymes are incorporated into novel cytoplasmic bodies (Narayanaswamy et al., 2009; Werner et al., 2009). The extent to which these aggregates represent biologically meaningful structures remains unclear; however there are well characterised examples in which the assembly of higher-order structures regulates flux through specific metabolic pathways. For example, it has been observed that six of the key enzymes involved in the de novo synthesis of purine nucleotides reversibly colocalise to discrete cytoplasmic bodies (purinosomes) in response to depletion of purine concentrations (An et al., 2008). It is thought that this aggregation occurs to facilitate substrate channelling between enzymes in the same pathway, although recent evidence has questioned whether these bodies are physiologically relevant (Zhao et al., 2014).
A distinct subset of cytoplasmic bodies has been identified in which enzymes are incorporated into filamentous structures. One of the earliest enzymes to be identified as subject to regulation by the assembly of monomers into higher-order structures is acetyl-CoA carboxylase (ACC). Purified ACC from animal tissues has been shown by electron microscopy to reversibly form filamentous structures upon incubation with its allosteric activator, citrate (Kleinschmidt et al., 1969). Conversely, incubation with malonyl CoA or Mg2+ ions, both inhibitors of ACC, caused rapid depolymerisation of citrate-induced filaments, indicating that polymerisation of ACC is required to upregulate enzymatic activity (Beaty and Lane, 1983a; Beaty and Lane, 1983b). One of the best studied enzyme filaments so far is the essential de novo pyrimidine biosynthesis enzyme, CTP synthase (CTPsyn). CTPsyn-containing structures have been termed cytoophidia due to their apparent snake-like morphology (from the Greek cyto, meaning cell; and ophidia, meaning serpents) (Liu, 2010). The filament forming properties of CTPsyn have been shown to be highly conserved having been demonstrated in bacteria (Ingerson-Mahar et al., 2010), yeast (Noree et al., 2010), Drosophila (Liu, 2010) and human cells (Carcamo et al., 2011; Chen et al., 2011). The conservation of this feature throughout evolution suggests that the ability of CTPsyn to assemble into filamentous structures confers an important biological function (for a review, see Liu, 2011).
The enzymatic activity of CTPsyn is regulated by all four nucleotides, through substrate concentration/energy generation (UTP, ATP) and allosteric interactions (CTP, GTP) (Levitzki and Koshland, 1971; Levitzki et al., 1971; Lieberman, 1956; Long and Pardee, 1967). ATP, UTP and CTP all promote assembly into the tetramer form of the enzyme, and have been shown to increase catalytic activity, whilst GTP acts as a positive allosteric activator of the glutaminase activity of the enzyme, although this has been shown to be non-essential in vitro (Aronow and Ullman, 1987; Endrizzi et al., 2004; Goto et al., 2004; Pappas et al., 1998; Robertson, 1995). Treatment of S. cerevisiae with CTP, an inhibitor of CTPsyn activity, was shown to increase filament formation (Noree et al., 2010). The compartmentalisation of CTPsyn has been reported to be influenced by a variety of different extrinsic factors. Treatment of S. cerevisiae with the non-specific kinase inhibitor staurosporine resulted in an increased abundance of cytoophidia, leading to the suggestion that cytoophidium formation may be dependent on phosphorylation of CTPsyn itself or of a critical regulator (Noree et al., 2010). These data must be interpreted with caution however, due to the broad effects of staurosporine which are known to cause toxicity (Tamaoki et al., 1986).
Here we report that CTPsyn is compartmentalised in response to nutrient stress in cultured Drosophila cells. Furthermore we show that this effect is also apparent in Drosophila tissues in vivo. We also show that dispersal of CTPsyn occurs in larval neuroblasts in vivo upon exiting quiescence, indicating that the formation of cytoophidia occurs just prior to neuronal stem cell activation. In addition, cytoophidia formation occurs in neuroblasts when young larvae are starved – a process that stalls larval growth and cell proliferation (Tennessen and Thummel, 2011). We then aimed to further understand the metabolic effects of CTPsyn compartmentalisation. To this end, we performed metabolomic profiling of 308 metabolites on adult flies and found that overexpressing CTPsyn does not change CTPsyn-related enzymatic activity, suggesting that cytoophidium formation aids metabolic stabilisation. Furthermore, we show that overexpression of CTPsyn only results in moderate increase of CTP pools in human stable cell lines. Finally, we report that point mutations at critical residues in the tetramer and dimer interfaces of CTPsyn have an effect on cytoophidia formation indicating that CTPsyn multimers may be incorporated into cytoophidia. Based on our experimental data, we propose a simple mathematical model to explain the dynamics of cytoophidium formation, which accurately predicts CTPsyn concentration dependent changes of intracellular CTP nucleotides.
MATERIALS AND METHODS
Cell culture
Drosophila S2R+ cell lines obtained from the Drosophila Genome Resource Centre (DGRC) were grown at 25°C in Schneider's medium with 10% heat-inactivated fetal bovine serum (FBS) following standard protocols. Human 293T cells were maintained in DMEM (Invitrogen, 11960) supplemented with 10% FBS (Bioindustry), glutamine 2 mM (Invitrogen, 25030) and 1% Penicillin–Streptomycin solution (Gibco, 15140). Cells were kept in a 37°C incubator with 5% CO2. For western blot assays, transfected 293T cells were selected with puromycin 10 µg/ml (InvivoGen, ant-pr-5b).
Transfection of Drosophila and human cells
Drosophila S2R+ cells were diluted to 1×106 cells/ml and left to settle on coverslips in a 24 well plate overnight. If cells were intended for immunostaining, autoclaved coverslips were added to wells for cells to adhere to. 1 µg of DNA with 3 µl of FuGENE HD was added to 50 µl of water and mixed by flicking. DNA-FuGENE mixture was incubated at room temperature for 20 minutes. The DNA-FuGENE solution was dropped evenly onto the S2R+ cells and left to transfect for 24 hours before preparing samples for immunofluorescence. Control wells containing no plasmid DNA or empty vector were included for each transfection experiment. To generate stable cell lines, 4.5 µg expression constructs were co-transfected with 0.5 µg pCopuro puromycin resistance plasmid (Addgene). Cell culture medium was supplemented with 2 µg/ml puromycin after 48 hours to select against non-transfected cells. Cultures were maintained in 2 µl/ml puromycin thereafter.
For transfection of human 293T cells, full length mouse CTP synthetase 1 cDNA sequence was cloned into pLVX-EF1alpha-AcGFP-N1 vector (Clontech, 631983). CTPsyn1-GFP and GFP vector were introduced into cells with jetPEI (polyplus) transfection reagents following manufacturer's protocol.
Immunofluorescence and microscopy
All cultured cells were fixed in 4% paraformaldehyde in PBS for 10 minutes and washed with PBT (1× PBS + 0.5% horse serum + 0.3% Triton X-100) before imaging. Drosophila larval tissues were obtained from age-matched animals raised on apple juice agar plates supplemented with wet yeast. All tissues were dissected into Grace's Insect Medium, fixed in 4% paraformaldehyde in PBS for 10 minutes and washed with PBT. Samples were incubated in primary and secondary antibodies consecutively overnight with intervening washes in PBT. Primary antibodies used were rabbit anti-CTPsyn (Santa Cruz 134457), mouse anti-miranda (gift from Fumio Matsuzaki). Rabbit anti-CTPS antibody (1:500, GeneTex, GTX105265) was used for staining in human cells. Secondary antibodies used were: donkey anti-rabbit Cy5 (Jackson 711-175-152), donkey anti-goat alexa 488 (molecular probes A11055), donkey anti-goat 549 (Jackson 711-585-152). Hoescht 33342 (1 µg/ml) was used to label DNA. All samples were imaged using a Zeiss LSM 510 META confocal microscope. Image processing and analysis was conducted using ImageJ.
Generation of CTPsyn expression constructs and mutants
A metallothionein (MT) promoter sequence was amplified from pMT/V5-HisA using primers containing adaptors with BglII and EcoRV digestion sites with Expand high fidelity DNA polymerase. The amplified promoter sequence and the pAVW destination vectors from the Drosophila gateway vector collection (obtained from the Drosophila Genomics Resource Centre, Bloomington, IN) were digested using BglII and EcoRV (NEB) and ligated together with the amplified pMT promoter to generate the pMT-VW expression vector. CTPsyn was cloned into pMT-VW using the Gateway recombination system (Invitrogen). Single base substitutions were made in the pMT-VW-CTPsyn expression vector (decribed above) using a quickchange PCR protocol. PCR primers were designed to include nucleotide substitutions of interest. PCR was performed on the pMT-VW-CTPsyn expression vector using Phusion high fidelity polymerase (NEB). Following amplification, template DNA was digested using Dpn1. Plasmid DNA was purified using a MinElute low volume PCR purification kit (Qiagen) and transformed into E. coli DH5α cells.
Drosophila genetics
To overexpress CTPsyn in various tissues, w ; pUASp-CTPsyn/Cyo flies (as described by Azzam and Liu, 2013) were crossed to the Act5c-Gal4 driver (Bloomington Drosophila stock centre) for ubiquitous expression. For neuroblast overexpression Pros-GAL4 was obtained from the putative-enhancer collection (Bloomington Drosophila Stock Centre [BDSC] at Indiana University, USA). Flies expressing UAS driven shRNA targeting AKT1 (P(KK100495)VIE-260B; Vienna Drosophila RNAi Center, Vienna) were crossed to flies with a GAL4 driver expressed in neuroblasts (Insu-GAL4, gift from Jürgen Knoblich) (AKT1 RNAi line previously characterised by Lee et al., 2013).
Analysis of Drosophila neuroblasts and imaginal tissues
w1118 larval CNS and imaginal discs were dissected for all wild-type experiments. Well-fed larvae were reared on apple juice plates with standard medium comprising of 80 g/l maize, 18 g/l dried yeast, 10 g/l soya flour 80 g/l malt extract, 40 g/l molasses, 8 g/l agar, 6.6 ml acid mix (comprising 50% propionic acid and 3.2% phosphoric acid)/l throughout larval life. For larvae on a restricted diet, larvae were removed at 5 hours and 24 hours for CNS and imaginal disc studies respectively, and placed on apple juice plates. CNS were imaged at 24 hours after larvae were removed from food (L2 stage) and imaginal discs were imaged 52 hours after removal from food (Stage L3). For larval starvation and re-feeding assays 1st instar larvae were starved for 1 day, then transferred to food and the number of neuroblasts containing cytoophidia were scored after 0, 1 and 5 hours. CNS and imaginal tissues were dissected, and fixed for 10 min in 4% paraformaldehyde. Immunofluorescence was performed using rabbit anti-CTP synthase y88 (sc-134457, 1:1000 Santa Cruz BioTech), rabbit anti-Miranda (1:200, gift from C. Doe,), guinea pig anti-Dpn (1:10,000, gift from J. B. Skeath), as previously described (Grice and Liu, 2011).
Western blotting
Cell lysates were run on a 12% polyacrylamide gel and transferred to a PVDF membrane (GE Healthcare). To prevent non-specific binding of antibody, membrane was incubated in blocking buffer (5% milk in PBS) for 1 hour in room temperature. After blocking, the membrane was incubated in primary antibodies overnight at 4°C. Primary antibodies: rabbit anti-CTPS IgG antibody (GeneTex, GTX105265) and mouse anti-αTubulin IgG (Sigma, T5168) were diluted in blocking buffer in 1:4000 and 1:10,000 dilution, respectively. For Drosophila western blots (supplementary material Fig. S3, Fig. S4B) rabbit anti-Histone H3 antibody (abcam ab1791) was used for the loading control (1:10,000). Following 3 washes with 1% Tween-20 in PBS, the membrane was incubated in secondary antibodies at room temperature for 1 hour. Secondary antibodies: HRP-conjugated anti-rabbit IgG (GeneTex, GTX77137) and anti-mouse IgG (PerkinElmer Life Sciences Inc., NEF822), were diluted in blocking buffer in 1:2000 and 1:20,000 dilution, respectively. The bound antibodies were detected with LuminataTM Forte Western HRP Substrate (Millipore, WBLUF0500) and X-ray film. Relative protein abundances were measured using imageJ.
Quantification of intracellular nucleotides
7×106 cells were washed with 1× phosphate buffer (pH 7.4) and lysed in 80% methanol. After centrifugation at 12,000 r.p.m. for 10 min at 4°C, supernatants were dried and kept at −80°C before injection. The pellet was resuspended in 200 µl water. 5 µl aliquots were injected onto a separation column (ACQUITY UPLC BEH C18, 1.7 µm, 2.1×100 mm reversed-phase column, Waters) with a flow rate of 0.5 ml min−1 and analysed with an Acquity Ultra Performance Liquid Chromatography (UPLC Waters) interfaced with PDA photodiode Array (Waters). Mobile phase A was 50 mM TEAA with 4 mM EDTA, pH 7.0 and mobile phase B, 100% acetonitrile. A programmed mobile phase-gradient was used during a 10-min run: 0 min, 0.1% B; 6.8 min, 2% B; 6.9 min, 0.1% B; 10 min, 0.1% B. The concentration of the four nucleotides ATP, UTP, CTP and GTP was quantified at 270 nm absorbance. Concentrations were determined by using calibration curves of the four nucleotides. The linearity gave a correlation coefficient of the linear regression curves greater than 0.99 for the four nucleotides. Peak areas were integrated, and the amounts calculated were as follows: ATP, 4.7 min; UTP, 2.7 min; CTP, 2.1 min, and GTP, 1.5 min.
Metabolomic profiling
Young adult female flies are fed with wet yeast for 2 days before the collection. The following three groups of flies were collected: 1) Control group – y,w flies fed with normal food; 2) Inhibition group – y,w flies fed with food containing 1 mg/ml DON to inhibit CTPsyn activity; 3) Overexpression group – globally overexpressing CTPsyn using an actin-GAL4 driver. These flies were subdivided into 30 subgroups with 100 flies in each subgroups. In total 3000 flies were collected for the metabolic profiling. Whole flies were frozen in liquid nitrogen and shipped to Metabolon, Inc (Durham, North Carolina, USA) for metabolomic profiling.
RESULTS
Cytoophidium assembly occurs in response to nutrient stress
It was previously shown that S. cerevisiae cells grown in media lacking glucose exhibited greater numbers of CTPsyn filaments (Noree et al., 2010). This effect could be reversed by addition of glucose. To see if the response to nutrient stress is evolutionarily conserved, we removed cell culture medium from Drosophila S2R+ cells in logarithmic growth and incubated with PBS. Cells were examined by immunofluorescence at various time points to determine the effect of nutrient stress on cytoophidium assembly. A significant increase in number of cytoophidia was seen after one hour, the shortest interval measured. Numbers of cells with visible cytoophidium formation reached a maximum of around 20%, after 4 hours in PBS (Fig. 1A). The observed response of cytoophidia to nutrient stress could be reversed by the re-addition of whole media (Fig. 1B). It remained unclear whether the increased cytoophidium formation displayed by nutrient restricted cells was a consequence of cell cycle arrest. We performed Edu staining to label S-phase cells in nutrient restricted cells to give an indication of the number of cells exiting the cell cycle. No significant change in Edu staining was seen following 5 hours of nutrient restriction, indicating that cell cycle arrest is not necessary for cytoophidium formation in cells undergoing nutrient restriction (Fig. 1C).
Fig. 1. Cytoophidium assembly occurs in response to nutrient stress.

(A) Mean number of cells containing visible cytoophidia increases in nutrient restricted cells. Significantly more cytoophidia are seen in S2R+ cells in PBS after just one hour. Control cells are maintained in complete Drosophila Schneider's medium. At least 100 cells were counted from ten sites per well from at least three independent replicates. (B) Cells with increased numbers of cytoophidia following nutrient restriction can be rescued by addition of whole media. Cells were examined two hours after re-introduction of whole media. (C) No significant change in EdU marked cells is observed after 5 hours, indicating that cell cycle arrest is not necessary for cytoophidia formation in S2 cells. (D) Response to nutrient restriction in imaginal disc tissue in vivo. Imaginal discs of L3 larvae display only diffuse cytoophidia when fed a normal diet. (E) In larvae raised on a nutrient restricted diet, the imaginal discs are smaller and cytoophidia are highly prevalent. Scale bars: 10 µm. ***P<0.001, **P<0.01, *P<0.05.
To investigate whether nutrient induced change in CTPsyn localisation was conserved in animal tissues in vivo, we raised larvae on food for 24 hours, transferred them onto minimal food for 52 hours, and then dissected. The time analysed corresponded to the third instar stage. Imaginal discs, including wing, leg, and haltere, display diffuse CTPsyn staining and exhibited no visible cytoophidia in well fed larvae (Fig. 1D). In nutrient restricted larvae, cytoophidia were abundant in the imaginal discs (eye, wing, leg and haltere) of third instar larvae, which were also reduced in size (Fig. 1D, showing imaginal discs from a nutrient restricted larva). In summary, CTPsyn localisation changes as part of an adaptive metabolic response to inactivate cytoplasmic CTPsyn during nutritional stress in vivo.
Redistribution of CTPsyn occurs during Drosophila neurogenesis
Having seen that cytoophidium formation was responsive to nutrient stress in vivo we asked whether cytoophidium formation may be required during normal developmental processes. The post embryonic neuroblasts of the Drosophila CNS have previously been shown to exhibit high levels of cytoplasmic (i.e. non-filamentous) CTPsyn (Chen et al., 2011). The majority of these neuroblasts remain in a quiescent state during the early 1st instar larval stage (Fig. 2A). When the larvae have the required access to food, neuroblasts exit quiescence and re-enter the cell cycle during late 1st (L1) and early 2nd instar (L2) stages (Chell and Brand, 2010; Sousa-Nunes et al., 2011; Truman and Bate, 1988). As the demand for nucleotides is upregulated in proliferating cells, we hypothesised that there may be a concomitant switch in cytoophidium assembly/disassembly. We dissected 1st and 2nd instar larvae and investigated CTPsyn distribution by immunofluorescence. In well fed larvae, CTPsyn can be observed in filaments in the early 3rd instar thoracic ganglion, however these filaments disappear as larvae develop through L2 and L3 stages (Fig. 2A,B). The cytoophidia appear on the ventral surface in cells corresponding to the regions where the quiescent neuroblasts reside (Fig. 2C). As neuroblasts exit quiescence they enlarge and begin to divide. Following this process CTPsyn filaments disappear, and CTPsyn is localised in the cytoplasm in a diffuse pattern (Fig. 2D).
Fig. 2. CTPsyn distribution in Drosophila larval post embryonic neuroblasts (pNbs).

(A) A schematic of the larval CNS showing the location of the pNbs. The box represents the section of the CNS represented in panels C and D. The majority of larval pNbs exit quiescence during the late L1 (1st instar) and early L2 (2nd instar) stages. This is characterised by pNb enlargement and the expression of markers such as Miranda (Mir). (B) Counts of pNbs containing cytoophidia in L2 early, L2 late and L3 larvae early reared under different feeding conditions (ND, normal diet; FR, food removed). n>10 animals per group. (C) CTPsyn localisation in early and late L2 when on a normal diet (see Materials and Methods). CTPsyn forms cytoophidia in quiescent pNbs (Mir negative). (D) As neuroblasts exit quiescence (late L2, normal diet) and begin to divide CTPsyn becomes diffuse. (E) CTPsyn localisation in late L2 when food is removed (late L2, food removed) after 24 hrs. CTPsyn aggregates into filaments when nutritional stresses are present. These images are representative of all 10 animals imaged. Scale bars: 10 µm.
Nutritional restriction during neurogenesis leads to cytoophidium formation
Neuroblasts continue to divide throughout larval life and CTPsyn remains diffuse in the neuroblast cytoplasm throughout this period (Chen et al., 2011). Nutritional stress during this period has been shown to affect neuroblast division and cell cycle entry. For example, when young larvae are starved before a critical weight period is reached (during the L3 stage), developmental progression and neurogenesis is halted (Tennessen and Thummel, 2011). To model nutritional stress, and to stall growth and neurogenesis, we transferred newly hatched larvae from basic food to apple juice plates, after 12 hours. We then reared the larvae on apple juice plates for 24 hours plates before dissecting. In these starved larvae, cytoophidia can be found in the majority of neuroblasts (Fig. 2E). We next looked to see how the re-addition of food, and thus the re-initiation of neurogenesis, affects the presence of cytoophidia (Fig. 3). 1st instar larvae were starved for 1 day, and then transferred to food. The number of neuroblasts containing cytoophidia was scored after 0, 1 and 5 hours (Fig. 3A). We found that re-feeding induced the dissociation of the cytoophidia in neuroblasts, which had accumulated during the starvation period (Fig. 3B,C). In addition, 5 hours after the re-addition of the normal diet, cytoophidia numbers were comparable with those seen in well fed L2 larvae. Furthermore, strong cytoplasmic staining was seen to reappear in the neuroblasts of these animals (Fig. 3C). These data again suggest that CTPsyn localisation changes as part of an adaptive metabolic response to inactivate cytoplasmic CTPsyn during nutritional stress in neuroblasts.
Fig. 3. Re-feeding starved larvae promotes cytoophidia disassembly.

(A) 1st instar larvae were starved for 1 day. Larvae were then transferred to food and the number of neuroblasts with cytoophidia were scored after 1 and 5 hours. (B) Re-feeding induces the dissociation of the cytoophidia in neuroblasts that accumulated during starvation. After 5 hours cytoophidia numbers are comparable with those seen in well fed L2 larvae. (C) Representative pictures CNS from larvae that have been re-fed for 0 hr, 1 hr and 5 hrs. These images are representative of >6 animals imaged. Scale bars: 10 µm.
To mimic nutritional stress, we also knocked-down the serine-threonine kinase AKT1 in neuroblasts using Insc-GAL4, UAS-AKT1-RNAi. During L1 and L2, normal food intake activates insulin signalling and the PI3K/AKT pathway. This in turn activates TOR, and represses the growth inhibitor Foxo, leading to the initiation of neuroblast division and cell proliferation (Fig. 4A) (Chell and Brand, 2010; Goberdhan and Wilson, 2003; Sousa-Nunes et al., 2011). Insc-GAL4, UAS-AKT1-RNAi neuroblasts have increased numbers of cytoophidia when compared to controls (Fig. 4B–D), suggesting that inactivation of the AKT1 pathway induces cytoophidia formation. The prolonged presence of cytoophidia in the neuroblast however, appears to have no detrimental effect. Overexpression of CTPsyn in the neuroblasts (Pros-GAL4, UAS-CTPsyn-A) generates extremely large cytoophidia, but this has no overt effect on overall development (supplementary material Fig. S1). In summary, cytoophidia are formed in neuroblasts that are quiescent, or in neuroblasts from the CNS of starved and developmentally stalled larvae. These results are consistent with the hypothesis that CTPsyn in cytoophidia is inactive.
Fig. 4. AKT1 knockdown induces Cytoophida formation.
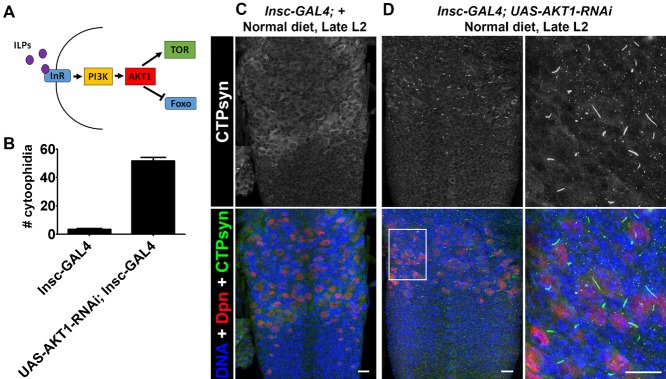
(A) When nutrients are available insulin-like peptides (ILPs) bind the insulin receptor (InR), activating the PI3K/AKT pathway. This in turn inhibits the growth inhibitor Foxo and activates TOR, leading to growth and division of cells, including neuroblasts. (B) AKT1 was knocked down in neuroblasts using Insc-GAL4, UAS-AKT1-RNAi to mimic nutritional stress. AKT1 knockdown induces cytophidia formation. (C) Insc-GAL4 controls displayed very few neuroblasts with cytophidia. (D) Cytophidia in Insc-GAL4, UAS-AKT1-RNAi neuroblasts. These images are representative of >6 animals imaged. Scale bars: 10 µm.
Either inhibition or overexpression of CTPsyn promotes cytoophidium assembly
We have previously shown that in human cells that the proportion of cells containing visible cytoophidia is low during logarithmic growth under standard conditions, with around 5% of cells displaying filaments (Chen et al., 2011). Filaments could be induced to form when cells were treated with the glutamine analogues 6-Diazo-5-oxo-L-norleucine (DON) and azaserine, which both act as competitive inhibitors of CTPsyn.
In order to investigate whether Drosophila cells displayed similar properties to human cell lines, S2R+ cells were treated with varying concentrations of DON. In untreated cells very low endogenous numbers of cytoophidia (≈3%) could be detected by immunofluorescence, whilst significant increases were observed in DON-treated cells as previously reported (supplementary material Fig. S2A–C). Furthermore, the response of S2R+ cells to DON was proportional to the concentration of drug applied as observed in human cells in vitro.
We have reported that only one isoform of Drosophila CTPsyn is able to form cytoophidia (Azzam and Liu, 2013). Ectopically expressing the cytoophidium-forming isoform of CTPsyn can induce cytoophidia in many tissues in Drosophila (Azzam and Liu, 2013). To see whether expression of CTPsyn would also induce cytoophidium formation in cell culture, CTPsyn was cloned into an expression vector under the control of a metallothionein promoter with a venus (EYFP) fluorescent tag (referred to as CTPsyn-YFP hereafter). This expression construct was transfected into Drosophila S2R+ cells and stable lines established in order to further investigate the formation of cytoophidia in vivo.
To determine whether expression of the CTPsyn-YFP transgene had an adverse effect on cell survival, cells were assayed for viability using a trypan blue dye exclusion assay. Loss of cell viability has been reported at concentrations exceeding 1 mM (Southon et al., 2004). Transgene expression was induced using 1 mM CuSO4 to achieve robust expression without affecting cell viability. No discernible difference in viability was observed in cells in which transgene expression was induced by CuSO4 for 8 hours, compared to uninduced or wild-type S2R+ cells indicating that CTPsyn overexpression has no adverse effect on the health of cell cultures (supplementary material Fig. S2E). Cytoophidium forming properties of the developed transgenic cell line were examined over time after CuSO4 induction, by fixing cells in paraformaldehyde and imaging at various timepoints. Cytoophidia were visible at very low levels after 4 hours post induction in less than 5% of cells. Cytoophidia were visible in 100% of cells after ten hours of incubation with CuSO4 (supplementary material Fig. S2D). The mean cytoophidia area increased with time. The average number of filaments per cell increased very rapidly between four and five hours, then decreased until eight hours, during which time the mean filament size increased significantly due to fusion of smaller cytoophidia into larger structures as previously reported (Gou et al., 2014) (supplementary material Fig. S2F). Together, these data indicate that cytoophidium formation is proportional to CTPsyn concentration.
Analysis of CTPsyn overexpression on metabolic profiles
In previous studies, we have shown that overexpression of CTPsyn or treatment with small molecule inhibitors is sufficient to induce abundant cytoophidia formation in Drosophila tissues in vivo (Azzam and Liu, 2013; Chen et al., 2011). In order to investigate the effect of altered CTPsyn concentrations and cytoophidia abundance on cellular physiology, we conducted metabolomic profiling on flies either overexpressing CTPsyn, treated with DON, or wild-type controls. We obtained global metabolome profiles covering 308 metabolites in three groups of Drosophila: flies with CTPsyn overexpression (Oe, as described by Azzam and Liu, 2013), wild-type flies treated with CTPsyn inhibitor DON (In), and wild-type flies with no treatment as a control (Wt).
Principle component analysis of metabolome data showed that the three groups are clearly distinct from each other in terms of their global metabolite levels (Fig. 5A). We then used a self-organizing map (SOM) to reveal the relative metabolite changes among the three groups for all metabolites. As shown in Fig. 5B, four major patterns emerge: low–low–high (I, upper left), high–low–low (II, upper right), low–high–high (III, lower left), and high–high–low (IV, lower right) in Oe, Wt, and In group respectively. We found that the metabolites involved in amino acid metabolism are enriched in pattern I (P = 0.005, Fisher's exact test), lysolipids are enriched in pattern II (P = 0.0008) and dipeptides are enriched in pattern IV (P = 0.0001).
Fig. 5. Global metabolomic profiling indicates negligible changes in CTPsyn activity in vivo.

(A) Principle component analysis shows that the samples of wild-type (Wt), CTPsyn inhibition (In), and CTPsyn overexpression (Oe) groups are clearly segregated. Two principal components (PC1 and PC2) are plotted and their proportions of variance are labeled. (B) The mean values of compounds in each state were normalized to (−1,1) and clustered into nine patterns by self-organizing mapping. The numbers of compounds in each pattern are showed on the top of the grids. The patterns that contain the most compounds are indicated by (I, II, III, V). (C) The heat map shows metabolites in major patterns that have significantly differential levels in three groups. Seventy-two metabolites in major patterns of (B) SOM (I, II, III, IV) were selected by one-way ANOVA with Bonferroni adjusted P<10−5. (D) The levels of glutamine in three groups are shown in the boxplot. Glutamine belongs to pattern I and is also colored in red in panel C.
To ensure that these metabolites show statistically significant differences among the three groups, we applied one-way ANOVA to further select the metabolites (Bonferroni adjusted P<10−5) belonging to the four major patterns (Fig. 5C). For the metabolites in patterns I and IV, the treatment of CTPsyn inhibition strongly influenced their levels compared to the control while the mutants with CTPsyn overexpression have no difference compared to the control. Glutamine belongs to pattern I and shows increased level in In compared to Wt and Oe (Fig. 5D). This can be explained by the fact that the inhibition of glutaminase activity of CTPsyn may lead to the accumulation of glutamine. The similar levels of glutamine between Oe and Wt groups suggest that the glutaminase activity of CTPsyn remains the same even after the overexpression of CTPsyn. These two major patterns support the idea that the CTPsyn sequestered in the cytoophidia filament has no CTPsyn-related enzymatic activity. For the metabolites in patterns II and III, the overexpression of CTPsyn strongly influenced their levels compared to the control and the group with CTPsyn inhibition. These groups of metabolites are unrelated to the canonical enzyme function of CTPsyn as they show no difference with the control after the inhibition of CTPsyn. These changes may be accounted for by the presence of Gal4 in the Oe group. However, some changes may be the result of metabolic functions of cytoophidia. In conclusion, our analysis of metabolome data supports the hypothesis that the formation of cytoophidia could sequester CTPsyn enzyme activity.
Overexpression of CTPsyn only moderately increases CTP pool in human cells
According to previous studies, manipulations of certain conditions, such as glutamine deprivation or inhibition of GTP synthesis with drugs, could stimulate cytoophidium formation in some cells (Calise et al., 2014; Gou et al., 2014). To investigate the function of the cytoophidium in mammalian cells, 293T cells were transfected with a construct of mouse CTP synthase 1-GFP fusion protein (mCTPsyn1-GFP) which is driven by the EF1α promoter. Expression of mCTPS1 promoted assembly of cytoophidia in 293T cells in normal culture condition (Fig. 6A,B). Cytoophidia in mCTPS1-GFP expressing cells were significantly longer than those in normal 293T cells with or without DON treatment (Fig. 6B,C; supplementary material Fig. S4), indicating that more CTPsyn proteins accumulated in cytoophidium structure in cells expressing mCTPsyn1. We generated three stable cell lines overexpressing mCTPsyn1-GFP at approximately 6–8-fold higher than endogenous levels. (Fig. 6D,E). However, the intracellular CTP concentration only increased to 2-fold higher than that of wild-type cells indicating that CTPsyn activity does not increase linearly with protein concentration (Fig. 6F).
Fig. 6. CTPsyn, cytoophidia and CTP production.

(A) Cytoophidia are undetectable in wild-type and GFP vector transfected human 293T cells. (B) The mCTPsyn1-GFP proteins were stably expressed by CTPsyn1 OE#1, CTPsyn1 OE#2 and CTPsyn1 OE#3 cell lines. Almost every cell contains one or more cytoophidia. (C) Quantification of length of cytoophidia expressed by three independent cell lines. (D) Western blot of CTPsyn1 protein in wild-type, and mCTPsyn1-GFP expressing 293T cell lines. (E) Quantification of the relative protein abundance of endogenous CTPsyn1 and exogenous mCTPsyn1-GFP in CTPsyn1 OE#1, CTPsyn1 OE#2 and CTPsyn1 OE#3 cell lines. (F) The relative CTP concentration in cell lysates of wild-type, and three CTPsyn1 overexpression 293T cell lines. Overexpression of CTPsyn moderately increased intracellular CTP concentration in human cells. (G) The fitting of total CTPsyn and CTP concentration using the non-linear relationship derived from our mathematical model (see text). The x axis is total CTPsyn and y axis is CTP concentration. Data points represent experimentally measured values in wild type and CTPsyn overexpressing human 293T cells. (H) Proposed model of cytoophidia assembly in response to nutrient or developmental conditions. Scale bars: 20 µm.
Mutations at the CTPsyn oligomer interfaces alter cytoophidia assembly
End product inhibition of CTPsyn by CTP is critical for the regulation of its enzymatic activity (Aronow and Ullman, 1987; Endrizzi et al., 2005; Long and Pardee, 1967). Previous experiments have shown that a mutation at the CTP binding site of CTPsyn in S. cerevisiae inhibited end product inhibition by CTP (Ostrander et al., 1998). Site-directed mutagenesis was used to construct a point mutation at the equivalent conserved CTPsyn residue (CTPsynE160K) in CTPsyn-YFP expression vector (supplementary material Fig. S2). When CTPsynE160K was transfected into Drosophila S2R+ cells, cytoophidium formation was seen to be completely disrupted, although diffuse cytoplasmic YFP could be seen, along with numerous punctate structures (supplementary material Fig. S5). Similar results have previously been reported in S. cerevisiae indicating that regulation of cytoophidium formation by end-product inhibition is an evolutionarily conserved phenomenon (Noree et al., 2010).
Allosteric binding of CTP has been shown previously to induce CTPsyn tetramer formation (Long and Pardee, 1967). It has also been demonstrated that treatment with the CTPsyn inhibitor, 6-Diazo-5-oxo-L-norleucine (DON), reduced the amount of CTPsyn tetramerisation in E. coli, which suggests that the tetramer form of the enzyme may be excluded from cytoophidia (Robertson, 1995). From these data it is unclear whether CTPsyn monomers, dimers or tetramers are incorporated into cytoophidia. We therefore decided to investigate the effect of mutations influencing residues at the tetramer or dimer interface of CTPsyn on cytoophidium assembly (supplementary material Fig. S4). We constructed point mutations in CTPsyn-YFP at the tetramer interface (CTPsynG151E and CTPsynR163H) and assayed for filament formation in S2R+ cells. Both CTPsynG151E and CTPsynR163H resulted in significantly longer cytoophidia when expression was induced in S2R+ cells compared to wild type protein controls (supplementary material Fig. S5). These data indicate that tetramer formation may not be necessary for CTPsyn to form cytoophidia. To determine whether CTPsyn dimer formation is necessary for cytoophidium formation we constructed point mutations at the monomer–monomer interface, predicted to disrupt dimer formation (CTPsynV114F and CTPsynM156I). Both CTPsynV114F and CTPsynM156I resulted in significantly shorter cytoophidia when expressed in S2R+ cells (supplementary material Fig. S5). Cytoophidium formation was not disrupted completely as in CTPsynE160K. It is unclear to us whether this is due to incomplete disruption of dimer formation in CTPsynV114F and CTPsynM156I. To determine whether changes in cytoophidia formation were due to changes in CTPsyn abundance, we performed western blots to detect the relative amounts of expressed proteins for each mutant. Comparison of relative CTPsyn abundances indicates that most of the mutant proteins did not result in an apparent change in protein abundance. However, CTPsynR163H and CTPsynE160K appeared to have significantly lower protein levels. CTPsynR163H displayed longer cytoophidia than wild type, despite lower protein abundance, whereas CTPsynE160K displayed significantly less cytoophidia and had clear morphological differences. Therefore, we conclude that CTPsyn increase is not a significant confounding factor in the interpretation of these phenotypes. Together these results imply that CTPsyn dimers may be the multimer subunits of cytoophidia, whilst tetramers may not necessarily be required for cytoophidium assembly.
A simple mathematical model of cytophidium formation
Two types of cytoophidia have been described in Drosophila germline cells, distinguished by their relative sizes; micro-cytoophidia and macro-cytoophidia (Liu, 2010). More recently, we have demonstrated that micro-cytoophidia can undergo multiple rounds of fusion to form macro-cytoophidia in mammalian cells (Gou et al., 2014). Here we developed a simple mathematical model to describe the incorporation of CTPsyn into cytoophidia. First, we only consider the micro-cytoophidia of size 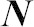 polymerised sequentially at both ends from CTPsyn n-mers (n = 2 for dimer and n = 4 for tetramer). The kinetic constants of nucleation and de-nucleation of n-mers in the filament are
polymerised sequentially at both ends from CTPsyn n-mers (n = 2 for dimer and n = 4 for tetramer). The kinetic constants of nucleation and de-nucleation of n-mers in the filament are 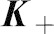 and
and 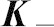 respectively. Under biochemical equilibrium, the numbers of filaments of length
respectively. Under biochemical equilibrium, the numbers of filaments of length 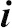 ,
, 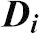 , satisfy:
, satisfy:
This leads to the exponential distribution of 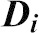 :
: 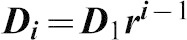 , where
, where 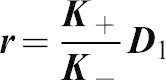 .
.
Here n-mers are formed from CTPsyn monomers with kinetic constants: 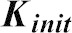 and
and 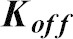 respectively. Therefore
respectively. Therefore 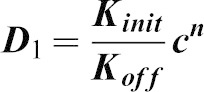 where
where 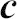 is the number of CTPsyn monomers. The total amount of CTPsyn is the sum of free CTPsyn monomers and those sequestered in filaments:
is the number of CTPsyn monomers. The total amount of CTPsyn is the sum of free CTPsyn monomers and those sequestered in filaments:
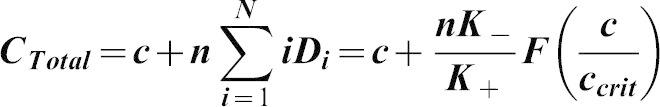 |
Here 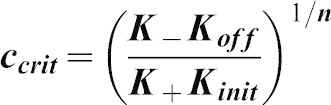 is the critical number of free CTPsyn monomers and
is the critical number of free CTPsyn monomers and 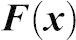 is
is
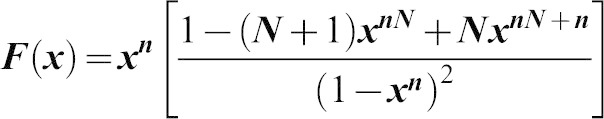 |
From the above equation, we can derive the relationship between free CTPsyn monomers and total amount of CTPsyn. For a cytoophidium of size  and CTPsyn protein of size
and CTPsyn protein of size  , we estimated
, we estimated 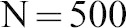 . When
. When 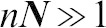 ,
, 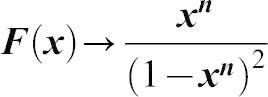 for
for 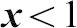 . When
. When 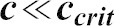 ,
, 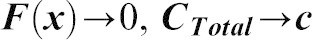 and all CTPsyn are in the form of monomers. When
and all CTPsyn are in the form of monomers. When 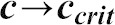 ,
, 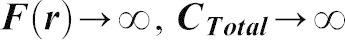 . In other words, c is kept close to
. In other words, c is kept close to 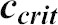 when
when 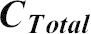 is significantly overexpressed.
is significantly overexpressed.
Our model can be readily generalised to include the CTPsyn in toroidal cytoophidia by considering the circularisation of linear cytoophidia as additional reversible biochemical reactions. Because circular cytoophidia are in equilibrium with linear cytoophidia of the same length and will not further increase in sizes, the inclusion of circular cytoophidia only leads to multiplying the amount of CTPsyn sequestered in filament by a constant. Furthermore, including the formation of macro-cytoophidia from the nucleation of micro-cytoophidia into our model does not change the nature of our result either. It is worth noting that cytoophidia only becomes visible after it is longer than a certain size. In our model the number of filaments exponentially decreases as the size increases. Therefore, the visible filament longer than the detection limit in a single cell can be very small. This explains 1–3 fibres per cells shown by our images while the majority of CTPsyn is sequestered into filaments below the limit of detection.
In order to determine whether our model was applicable to experimental data, we have fitted our model using the measured concentrations of CTP in wild type and CTPsyn overexpressing human 293T cells. We assumed that the CTP concentration is proportional to the concentration of catalytic active form of CTPsyn, tetramer 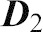 , and the cytophidium is formed by polymerization of dimers. According to our model, total CTPsyn and CTP concentration follow the relationship:
, and the cytophidium is formed by polymerization of dimers. According to our model, total CTPsyn and CTP concentration follow the relationship: 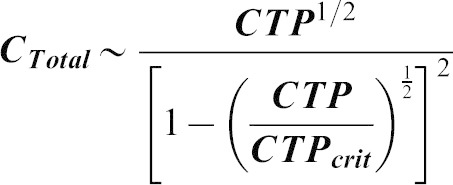 where
where 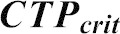 is the critical value of CTP. We fitted the data by non-linear regression (R2 = 0.96) (Fig. 6G).
is the critical value of CTP. We fitted the data by non-linear regression (R2 = 0.96) (Fig. 6G).
Our model predicts that CTP concentration does not increase linearly with total CTPsyn and reaches a plateau quickly when CTPsyn is overexpressed. This predicted upper limit of CTP (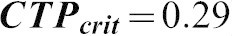 ) is only 3-fold higher than the measured CTP concentration (0.09) in wild type cells. This indicates that the level of CTP in the cells has been significantly buffered during CTPsyn overexpression due to cytophidium formation. This is because most CTPsyn enters into cytoophidia when CTPsyn is overexpressed. The amount of free CTPsyn monomers, dimers and tetramers are kept fairly constant but can be quickly released from the filament when CTPsyn is significantly depleted. This suggests that cytoophidia acts as a reservoir to tightly regulate the free active CTPsyn level to mediate cellular nucleotide homeostasis.
) is only 3-fold higher than the measured CTP concentration (0.09) in wild type cells. This indicates that the level of CTP in the cells has been significantly buffered during CTPsyn overexpression due to cytophidium formation. This is because most CTPsyn enters into cytoophidia when CTPsyn is overexpressed. The amount of free CTPsyn monomers, dimers and tetramers are kept fairly constant but can be quickly released from the filament when CTPsyn is significantly depleted. This suggests that cytoophidia acts as a reservoir to tightly regulate the free active CTPsyn level to mediate cellular nucleotide homeostasis.
DISCUSSION
We demonstrate that cytoophidia assemble during periods of cellular quiescence or low nucleotide demand as a mechanism to inhibit CTPsyn activity in cultured cells and in vivo. Our metabolomic data show that overexpressing CTPsyn has no overt effect on metabolic profiles, supporting the hypothesis that the formation of cytoophidia acts to sequester CTPsyn enzyme activity. Therefore cytoophidium formation is a physiologically relevant process during development and in mediating adaptive metabolic responses (Fig. 6H).
Polymerisation as a strategy for regulation of enzymatic activity
As the rate limiting enzyme in de novo CTP biosynthesis, CTPsyn occupies a central regulatory role in nucleotide metabolism. It is therefore unsurprising that it is subject to several complementary regulatory strategies. Not only is CTPsyn regulated by multimer assembly, end product inhibition and other allosteric regulators as previously discussed, it is also subject to phosphorylation by multiple kinases (Chang et al., 2007; Choi and Carman, 2007; Kassel et al., 2010). The discovery that CTPsyn activity is also regulated by assembly into higher-order cytoplasmic filaments adds a further level of sophistication to this already finely controlled metabolic pathway. It is unclear from the data presented here what the purpose of this extra level of enzymatic control is required for, however it is possible to speculate that cytoophidium assembly allows for CTPsyn activity to be rapidly up-regulated when required. The large surface area of a filamentous aggregate such as cytoophidia would allow for easy access by diffusible allosteric regulators in the cytoplasm, thereby facilitating rapid disassembly and upregulation. Punctate structures or vesicle bound enzymes may be subject to less rapid activation by similar mechanisms as the cytoplasmic surface area would be smaller.
Our mathematical model, and supporting data, indicate that cellular CTP pools may increase to a maximum of around 3-fold, regardless of the concentration of CTPsyn. We were unable to detect any overt cellular phenotypes arising from CTPsyn overexpression either in cell culture or in vivo models. Therefore, we hypothesise that up to a 3-fold increase in CTP levels has a negligible effect on normal cellular growth and development. This may be considered surprising, given the propensity for tumours to overexpress CTPsyn, and the efficacy of CTPsyn inhibitors as anti-neoplastic drugs. Further study will be required to elucidate whether the 3-fold CTP limit is overcome during tumourigenesis or whether these small changes are sufficient to impact cell growth in certain pathological states.
We have focused on the assembly of CTPsyn into cytoophidia in this study, however, it remains unclear the extent to which the novel mechanisms for enzymatic regulation suggested here apply to other recently identified filament forming enzymes. Screening of yeast GFP strain collections have identified at least four novel enzyme filaments which appear to be morphologically similar to cytoophidia, although their functions remain elusive (Noree et al., 2010). Moreover, it is unclear whether all filament forming enzymes are subject to enzymatic downregulation when assembled into cytoplasmic filaments. For example, it has long been known that polymerisation of acetyl Co-A carboxylase (ACC) upregulates enzymes activity, although it is unclear whether ACC forms equivalent macro-scale cytoplasmic structures (Beaty and Lane, 1983a; Beaty and Lane, 1983b; Kleinschmidt et al., 1969; Olivares et al., 2011). Recently, it was demonstrated that glutamine synthase is regulated in a similar manner, indicating that downregulation of activity through polymerisation may be a widespread mechanism by which enzymes are regulated (Petrovska et al., 2014).
Role of cytoophidium formation in neurodevelopment
It was previously shown that CTPsyn is highly expressed in third instar larval neuroblasts and exhibits a diffuse cytoplasmic localisation (Chen et al., 2011). Here we have shown that cytoophidia are highly prevalent in the quiescent neuroblasts of the early larval CNS. Having shown that CTPsyn activity is likely to be inhibited by incorporation into cytoophidia, we can now speculate that the disassembly of cytoophidia is necessary to synthesis critical nucleotides necessary for the transition of these cells from quiescence to proliferation after re-entering the cell cycle. It is not clear from these data however, whether the dispersal of CTPsyn is regulated by cell cycle regulators acting on CTPsyn itself (e.g. by phosphorylation) or through changes in concentrations of allosteric regulators previously shown to effect cytoophidium formation.
Highly regulated nucleotide metabolism is vital for the development of most metazoan tissues due to the essential requirement for de novo nucleotides for DNA synthesis. However, several inborn errors of nucleotide metabolism result in conditions with severe neurological symptoms whilst other tissues are seemingly less affected, such as hypoxanthine–guanine phosphoribosyl transferase (HGPRT) deficiency (Lesch–Nyhan disease) (Lesch and Nyhan, 1964). Therefore understanding the control of nucleotide metabolism in the developing CNS is of particular interest for understanding the mechanisms of hereditary pyrimidine enzymopathies.
The data presented here suggest that as well as being involved in the co-ordination of developmental processes, the compartmentalisation of CTPsyn into cytoophidia is involved in mediating adaptive metabolic responses to nutrient availability in tissue culture and in vivo. A protein localisation screen of yeast GFP strains previously identified an unprecedented number of proteins exhibiting cytoplasmic redistribution into punctae during nutrient restriction (Narayanaswamy et al., 2009). This indicates that CTPsyn is not the only protein to be regulated by spatial re-organisation in response to extrinsic signals. Further research will be required to determine whether cytoophidium dissociation is required for cells to re-enter the cell cycle following nutrient stress.
Conclusion
Although it has been demonstrated previously that CTPsyn activity seems to be intimately linked to its polymerisation state, it has been unclear whether cytoophidia contains catalytically active or inactive CTPsyn. Our study suggests that CTPsyn dimers may be the multimer subunits of cytoophidia, the incorporation of which acts to downregulate CTPsyn enzymatic activity. It is possible that the point mutations studied here, predicted to lie at the interfaces between the CTPsyn monomers have unintended effects on CTPsyn structure or monomer interactions, therefore further structural studies will be required in the future to confirm the presence of CTPsyn dimers within cytoophidia. However, our data are supported by previous observations that treatment with the CTPsyn inhibitor DON causes a reduction in tetramer formation in E. coli (Robertson, 1995). As DON results in drastically increased cytoophidia formation, it follows that tetramers are not incorporated into cytoophidia during drug treatment. During the preparation of this manuscript two groups presented similar data regarding the regulation of CTPsyn enzymatic activity by filament formation (Barry et al., 2014; Noree et al., 2014). Both studies broadly agree with the assertion that CTPsyn activity is downregulated by incorporation of monomers into cytoplasmic filaments through polymerisation. However, some controversy exists regarding the incorporation of multimers into the filament structure. Barry et al. have shown that the tetramer form of CTPsyn is incorporated into cytoplasmic filaments, whereas Noree et al. have suggested that dimers are the constituent components. This discrepancy could be explained by lack of conservation of filament forming properties in the model organisms studied. Our data provides some support for the fact that dimers are incorporated into cytoophidia. This discrepancy may be explained by a lack of conservation between prokaryotic and eukaryotic CTPsyn, as Barry et al. have focused on bacteria whereas Noree et al. and this manuscript have described yeast and metazoan models respectively. However, this explanation is unsatisfying due to the high sequence identity and conservation of function of CTPsyn. Further investigation into the assembly of eukaryotic cytoophidia will be required to resolve this issue.
Supplementary Material
Acknowledgments
We thank Zhaodong Wang and members of the Liu Lab for fruitful discussion.
Footnotes
Author Contributions: G.N.A., S.J.G., Q.-J.S. and J.-L.L. conceived this project; G.N.A., S.J.G., Q.-J.S., C.-C.C., L.-Y.S. and J.-L.L. designed the experiments; G.N.A., S.J.G., Q.-J.S., C.-C.C., G.A., P.-Y.W., L.F.-M., and J.-L.L. performed the experiments; G.N.A., S.J.G., Q.-J.S., C.-C.C., P.-Y.W., L.-M.P., L.-Y.S., J.Y. and J.-L.L. analysed the data; Y.X. and J.Y. provided the mathematical model; G.N.A., S.J.G., Q.-J.S., C.-C.C., L.-M.P., L.-Y.S., J.Y. and J.-L.L. wrote the paper.
Competing interests: The authors declare that they have no conflict of interest.
Funding
This work was supported by UK Medical Research Council (to J.-L.L.), Chinese Scholarship Council (to Q.-J.S.), and National Science Council of the Republic of China (NSC101-2811-B-182-017 to P.-Y.W. and NSC100-2311-B-182-001-MY3 to L.-M.P.). This works is also supported by both Chinese Academy of Sciences and German Max-Planck Society (to J.Y.).
References
- An S., Kumar R., Sheets E. D., Benkovic S. J. (2008). Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science 320, 103–106 10.1126/science.1152241 [DOI] [PubMed] [Google Scholar]
- Aronow B., Ullman B. (1987). In situ regulation of mammalian CTP synthetase by allosteric inhibition. J. Biol. Chem. 262, 5106–5112. [PubMed] [Google Scholar]
- Azzam G., Liu J. L. (2013). Only one isoform of Drosophila melanogaster CTP synthase forms the cytoophidium. PLoS Genet. 9, e1003256 10.1371/journal.pgen.1003256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry R. M., Bitbol A. F., Lorestani A., Charles E. J., Habrian C. H., Hansen J. M., Li H. J., Baldwin E. P., Wingreen N. S., Kollman J. M. et al. (2014). Large-scale filament formation inhibits the activity of CTP synthetase. eLife 3, e03638 10.7554/eLife.03638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaty N. B., Lane M. D. (1983a). Kinetics of activation of acetyl-CoA carboxylase by citrate. Relationship to the rate of polymerization of the enzyme. J. Biol. Chem. 258, 13043–13050. [PubMed] [Google Scholar]
- Beaty N. B., Lane M. D. (1983b). The polymerization of acetyl-CoA carboxylase. J. Biol. Chem. 258, 13051–13055. [PubMed] [Google Scholar]
- Calise S. J., Carcamo W. C., Krueger C., Yin J. D., Purich D. L., Chan E. K. (2014). Glutamine deprivation initiates reversible assembly of mammalian rods and rings. Cell. Mol. Life Sci. 71, 2963–2973 10.1007/s00018-014-1567-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carcamo W. C., Satoh M., Kasahara H., Terada N., Hamazaki T., Chan J. Y., Yao B., Tamayo S., Covini G., von Mühlen C. A. et al. (2011). Induction of cytoplasmic rods and rings structures by inhibition of the CTP and GTP synthetic pathway in mammalian cells. PLoS ONE 6, e29690 10.1371/journal.pone.0029690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y. F., Martin S. S., Baldwin E. P., Carman G. M. (2007). Phosphorylation of human CTP synthetase 1 by protein kinase C: identification of Ser(462) and Thr(455) as major sites of phosphorylation. J. Biol. Chem. 282, 17613–17622 10.1074/jbc.M702799200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chell J. M., Brand A. H. (2010). Nutrition-responsive glia control exit of neural stem cells from quiescence. Cell 143, 1161–1173 10.1016/j.cell.2010.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K., Zhang J., Tastan O. Y., Deussen Z. A., Siswick M. Y., Liu J. L. (2011). Glutamine analogs promote cytoophidium assembly in human and Drosophila cells. J. Genet. Genomics 38, 391–402 10.1016/j.jgg.2011.08.004 [DOI] [PubMed] [Google Scholar]
- Choi M. G., Carman G. M. (2007). Phosphorylation of human CTP synthetase 1 by protein kinase A: identification of Thr455 as a major site of phosphorylation. J. Biol. Chem. 282, 5367–5377 10.1074/jbc.M610993200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endrizzi J. A., Kim H., Anderson P. M., Baldwin E. P. (2004). Crystal structure of Escherichia coli cytidine triphosphate synthetase, a nucleotide-regulated glutamine amidotransferase/ATP-dependent amidoligase fusion protein and homologue of anticancer and antiparasitic drug targets. Biochemistry 43, 6447–6463 10.1021/bi0496945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endrizzi J. A., Kim H., Anderson P. M., Baldwin E. P. (2005). Mechanisms of product feedback regulation and drug resistance in cytidine triphosphate synthetases from the structure of a CTP-inhibited complex. Biochemistry 44, 13491–13499 10.1021/bi051282o [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goberdhan D. C., Wilson C. (2003). The functions of insulin signaling: size isn't everything, even in Drosophila. Differentiation 71, 375–397 10.1046/j.1432-0436.2003.7107001.x [DOI] [PubMed] [Google Scholar]
- Goto M., Omi R., Nakagawa N., Miyahara I., Hirotsu K. (2004). Crystal structures of CTP synthetase reveal ATP, UTP, and glutamine binding sites. Structure 12, 1413–1423 10.1016/j.str.2004.05.013 [DOI] [PubMed] [Google Scholar]
- Gou K. M., Chang C. C., Shen Q. J., Sung L. Y., Liu J. L. (2014). CTP synthase forms cytoophidia in the cytoplasm and nucleus. Exp. Cell Res. 323, 242–253 10.1016/j.yexcr.2014.01.029 [DOI] [PubMed] [Google Scholar]
- Grice S. J., Liu J. L. (2011). Survival motor neuron protein regulates stem cell division, proliferation, and differentiation in Drosophila. PLoS Genet. 7, e1002030 10.1371/journal.pgen.1002030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingerson-Mahar M., Briegel A., Werner J. N., Jensen G. J., Gitai Z. (2010). The metabolic enzyme CTP synthase forms cytoskeletal filaments. Nat. Cell Biol. 12, 739–746 10.1038/ncb2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassel K. M., Au D. R., Higgins M. J., Hines M., Graves L. M. (2010). Regulation of human cytidine triphosphate synthetase 2 by phosphorylation. J. Biol. Chem. 285, 33727–33736 10.1074/jbc.M110.178566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinschmidt A. K., Moss J., Lane D. M. (1969). Acetyl coenzyme A carboxylase: filamentous nature of the animal enzymes. Science 166, 1276–1278 10.1126/science.166.3910.1276 [DOI] [PubMed] [Google Scholar]
- Lauritsen I., Willemoës M., Jensen K. F., Johansson E., Harris P. (2011). Structure of the dimeric form of CTP synthase from Sulfolobus solfataricus. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 67, 201–208 10.1107/S1744309110052334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. G., Zhao N., Campion B. K., Nguyen M. M., Selleck S. B. (2013). Akt regulates glutamate receptor trafficking and postsynaptic membrane elaboration at the Drosophila neuromuscular junction. Dev. Neurobiol. 73, 723–743 10.1002/dneu.22086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesch M., Nyhan W. L. (1964). A Familial Disorder of Uric Acid Metabolism and Central Nervous System Function. Am. J. Med. 36, 561–570 10.1016/0002-9343(64)90104-4 [DOI] [PubMed] [Google Scholar]
- Levitzki A., Koshland D. E., Jr (1971). Cytidine triphosphate synthetase. Covalent intermediates and mechanisms of action. Biochemistry 10, 3365–3371 10.1021/bi00794a008 [DOI] [PubMed] [Google Scholar]
- Levitzki A., Stallcup W. B., Koshland D. E., Jr (1971). Half-of-the-sites reactivity and the conformational states of cytidine triphosphate synthetase. Biochemistry 10, 3371–3378 10.1021/bi00794a009 [DOI] [PubMed] [Google Scholar]
- Lieberman I. (1956). Enzymatic amination of uridine triphosphate to cytidine triphosphate. J. Biol. Chem. 222, 765–775. [PubMed] [Google Scholar]
- Liu J. L. (2010). Intracellular compartmentation of CTP synthase in Drosophila. J. Genet. Genomics 37, 281–296 10.1016/S1673-8527(09)60046-1 [DOI] [PubMed] [Google Scholar]
- Liu J. L. (2011). The enigmatic cytoophidium: compartmentation of CTP synthase via filament formation. BioEssays 33, 159–164 10.1002/bies.201000129 [DOI] [PubMed] [Google Scholar]
- Long C. W., Pardee A. B. (1967). Cytidine triphosphate synthetase of Escherichia coli B. I. Purification and kinetics. J. Biol. Chem. 242, 4715–4721. [PubMed] [Google Scholar]
- Narayanaswamy R., Levy M., Tsechansky M., Stovall G. M., O'Connell J. D., Mirrielees J., Ellington A. D., Marcotte E. M. (2009). Widespread reorganization of metabolic enzymes into reversible assemblies upon nutrient starvation. Proc. Natl. Acad. Sci. USA 106, 10147–10152 10.1073/pnas.0812771106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noree C., Sato B. K., Broyer R. M., Wilhelm J. E. (2010). Identification of novel filament-forming proteins in Saccharomyces cerevisiae and Drosophila melanogaster. J. Cell Biol. 190, 541–551 10.1083/jcb.201003001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noree C., Monfort E., Shiau A. K., Wilhelm J. E. (2014). Common regulatory control of CTP synthase enzyme activity and filament formation. Mol. Biol. Cell 25, 2282–2290 10.1091/mbc.E14-04-0912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares A., Trejo J. O., Arellano-Galindo J., Zuñiga G., Escalona G., Vigueras J. C., Marin P., Xicohtencatl J., Valencia P., Velázquez-Guadarram N. (2011). pep27 and lytA in Vancomycin-Tolerant Pneumococci. J. Microbiol. Biotechnol. 21, 1345–1351 10.4014/jmb.1105.05045 [DOI] [PubMed] [Google Scholar]
- Ostrander D. B., O'Brien D. J., Gorman J. A., Carman G. M. (1998). Effect of CTP synthetase regulation by CTP on phospholipid synthesis in Saccharomyces cerevisiae. J. Biol. Chem. 273, 18992–19001 10.1074/jbc.273.30.18992 [DOI] [PubMed] [Google Scholar]
- Pappas A., Yang W. L., Park T. S., Carman G. M. (1998). Nucleotide-dependent tetramerization of CTP synthetase from Saccharomyces cerevisiae. J. Biol. Chem. 273, 15954–15960 10.1074/jbc.273.26.15954 [DOI] [PubMed] [Google Scholar]
- Petrovska I., Nüske E., Munder M. C., Kulasegaran G., Malinovska L., Kroschwald S., Richter D., Fahmy K., Gibson K., Verbavatz J. M. et al. (2014). Filament formation by metabolic enzymes is a specific adaptation to an advanced state of cellular starvation. eLife 3, e02409 10.7554/eLife.02409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson J. G. (1995). Determination of subunit dissociation constants in native and inactivated CTP synthetase by sedimentation equilibrium. Biochemistry 34, 7533–7541 10.1021/bi00022a029 [DOI] [PubMed] [Google Scholar]
- Sousa-Nunes R., Yee L. L., Gould A. P. (2011). Fat cells reactivate quiescent neuroblasts via TOR and glial insulin relays in Drosophila. Nature 471, 508–512 10.1038/nature09867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southon A., Burke R., Norgate M., Batterham P., Camakaris J. (2004). Copper homoeostasis in Drosophila melanogaster S2 cells. Biochem. J. 383, 303–309 10.1042/BJ20040745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamaoki T., Nomoto H., Takahashi I., Kato Y., Morimoto M., Tomita F. (1986). Staurosporine, a potent inhibitor of phospholipid/Ca++dependent protein kinase. Biochem. Biophys. Res. Commun. 135, 397–402 10.1016/0006-291X(86)90008-2 [DOI] [PubMed] [Google Scholar]
- Tennessen J. M., Thummel C. S. (2011). Coordinating growth and maturation - insights from Drosophila. Curr. Biol. 21, R750–R757 10.1016/j.cub.2011.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truman J. W., Bate M. (1988). Spatial and temporal patterns of neurogenesis in the central nervous system of Drosophila melanogaster. Dev. Biol. 125, 145–157 10.1016/0012-1606(88)90067-X [DOI] [PubMed] [Google Scholar]
- Werner J. N., Chen E. Y., Guberman J. M., Zippilli A. R., Irgon J. J., Gitai Z. (2009). Quantitative genome-scale analysis of protein localization in an asymmetric bacterium. Proc. Natl. Acad. Sci. USA 106, 7858–7863 10.1073/pnas.0901781106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao A., Tsechansky M., Ellington A. D., Marcotte E. M. (2014). Revisiting and revising the purinosome. Mol. Biosyst. 10, 369–374 10.1039/c3mb70397e [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.