

Abstract
Purpose of review
To review the recent developments in understanding the pathophysiology of heparin-induced thrombocytopenia (HIT) and in applying this knowledge to the treatment of patients with suspected and proven HIT.
Recent findings
HIT pathophysiology is dynamic and complex. HIT pathophysiology is initiated by four essential components – heparin (Hep), platelet factor 4 (PF4), IgG antibodies against the Hep–PF4 complex, and platelet FcγRIIa. HIT is propagated by activated platelets, monocytes, endothelial cells, and coagulation proteins. Insights into the unique HIT antibody response continue to emerge, but without consensus as to the relative roles of B cells, T cells, and antigen-presenting cells. Platelet activation via FcγRIIa, the sine qua non of HIT, has become much better appreciated. Therapy remains challenging for several reasons. Suspected HIT is more frequent than proven HIT, because of the widespread use of Hep and the inadequacies of current diagnostic tests and scoring systems. In proven HIT, approved treatments reduce but do not eliminate thrombosis, and have substantial bleeding risk. Rational novel therapeutic strategies, directed at the initiating steps in HIT pathophysiology and with potential combinations staged over time, are in various phases of development.
Summary
Progress continues in understanding the breadth of molecular and cellular players in HIT. Translation to improved diagnosis and treatment is needed.
Keywords: FcγRIIa, heparin, heparin-induced thrombocytopenia, immune-mediated thrombocytopenia and thrombosis, platelet factor 4
INTRODUCTION
Heparin-induced thrombocytopenia (HIT) is an uncommon but devastating complication of heparin (Hep) therapy. Paradoxically, anticoagulant medication and thrombocytopenia manifest not as bleeding, but rather as limb and life-threatening thrombosis. The thrombotic events are multifocal, involving veins, arteries, and the microvasculature.
In Table 1, we provide a framework for understanding HIT in the context of the major human thrombotic disorders. We define immune-mediated thrombocytopenia and thrombosis (ITT) as intravascular activation of blood cells and endothelial cells by components of the innate and adaptive immune systems, resulting in platelet–fibrin thrombi in large and small vessels of arterial and venous beds, as well as in the microvasculature, often concurrently. Each class of thrombosis, including ITT, has an annual US incidence of disease in excess of 500 000 cases per year. HIT is a paradigm for the ITT disorders, in that we have an advanced appreciation of many of the molecular and cellular players. (Note: ITT is distinct from ITP, an auto-immune bleeding disorder in which antibodies to platelet surface glycoproteins cause accelerated platelet clearance.) We have found this framework to be useful in examining the mechanisms and models in pathophysiology, as well as in the rational design of novel therapeutics.
Table 1.
The four major classes of human thrombotic disorders
| Classes of thrombotic disorders | Selected examples |
|---|---|
| Atherothrombosis | Coronary artery thrombosis/myocardial infarction; ischemic stroke; mesenteric artery thrombosis; limb artery thrombosis |
| Deep-vein thrombosis and pulmonary embolism | Lower extremity; upper extremity; cerebral venous; abdomen |
| Immune-mediated thrombocytopenia and thrombosis | HIT; antiphospholipid syndrome; sepsis syndrome; thrombosis from therapeutic monoclonal antibodies; thrombotic thrombocytopenic purpura |
| MCCATS | Malformation, Cancer, Cardiac, Artificial surface, Trauma, Sickle cell disease |
HIT is one of the immune-mediated thrombocytopenia and thrombosis disorders.
HIT, heparin-induced thrombocytopenia.
PATHOPHYSIOLOGY
HIT is a dynamic and complex disorder. The initial steps of the most commonly recognized clinical form of HIT involve patient exposure to a form of Hep, followed by the development of IgG antibodies over 4–14 days directed to a complex of platelet factor 4 (PF4) and Hep. The IgG antibodies activate platelets via FcgRIIa. Thrombin is generated and platelet–fibrin thrombi are formed (Fig. 1).
FIGURE 1.

The multiple steps of HIT disorder. After immunization, there is initiation (upper left) culminating in IC-mediated platelet activation, then propagation (lower right) marked by the central roles of thrombin. Thrombin feeds back to enhance cellular activation and cleaves fibrinogen to fibrin. GAG, glycosaminoglycan; HIT, heparin-induced thrombocytopenia; IC, immune complex; PF4, platelet factor 4; ULC, ultralarge complex.
Since the essential initiating steps in HIT path-ophysiology were elucidated in the 1990s and early 2000s, attention has turned to the dissection of the complex steps encapsulated by the statements ‘IgG antibodies are formed to PF4/hep’ and ‘platelets are activated and thrombin is generated’.
Antibodies to the platelet factor 4 and heparin complex
Progress in the recent past has come from consideration of the origins of HIT antibody generation. Within days of treatment with Hep, IgG antibodies to Hep and PF4 are detected. The titers disappear for most patients after 90–120 days [1]. Some, but not all, HIT patients re-exposed to Hep manifest a detectable IgG again. Thus, the HIT IgG response is rightly called an atypical response. The outstanding questions are: what is the immunogen? What are the roles of antigen-presenting cells and T cells in the response, and which B cells produce the antibody?
It is generally accepted that the clinical manifestations of HIT are caused by antibodies that recognize an ultralarge complex (ULC) composed of Hep and PF4 tetramers. PF4 tetramers bind avidly to Hep and to cellular glycosaminoglycans (GAGs), an interaction that is central to the pathogenesis of HIT. Arepally and colleagues have used a murine immunization model to investigate the immunogen [2,3]. Their data are compatible with the immunogen PF4 and Hep having a different molar ratio than the activating ULC. It remains to be determined how these data reflect specific B-cells and antigen-presenting cells. Greinacher and colleagues have identified PF4 bound to certain bacteria as initiating the generation of HIT-like antibodies [4–7]. Others have concordant data [8■]. PF4 bound to nucleic acids may also be immunizing [9■,10], as may interaction with several polyanions [11■]; further studies are needed. Wang and colleagues recently reported that HIT antibody generation had features of lost tolerance [12■■]. They also identified marginal zone B-cells as a potential source of the pathologic IgG [13■].
HIT antibodies bind preferentially to PF4 when Hep is present over a narrow molar ratio of reactants and activate platelets through FcγRIIA. Several epitopes of PF4 are important for pathogenic antibody binding [14,15], including two distinct antigenic sites: site 1 – residues after the third cysteine residue (beginning with proline-37); and site 2 – residues in the amino terminus and proline-34 [14]. Formation of ULC involves charge neutralization of PF4 with GAG [2,16] and clustering of PF4 molecules [16,17]. These data support a model in which Hep clustered PF4 residues to form neoepitopes where pathologic antibodies bind. We used single molecule binding with optical tweezers to reveal the difference between binding of a nonpathogenic anti-PF4 antibody (RTO) vs. pathologic anti-PF4/Hep antibody (KKO) [18,19■]. Greinacher and colleagues recently used circular dichroism to identify a Hep-bound PF4 conformation that is recognized by HIT antisera, and to provide insights into the dependence on GAG length and charge [20]. Further work, including three-dimensional structural data, is required to definitively determine the nature of the neoepitopes created upon ULC formation.
The HIT antibodies in humans are polyclonal. In HIT mouse model systems, only the murine monoclonal antibody KKO has been proven to mirror the human HIT antibody. Asada et al. [21] identified a mouse IgG1 monoclonal HIT antibody used in ex-vivo HIT assays. Details of its preferential reactivity for PF4 and Hep over PF4 alone are needed. Progress in additional monoclonal HIT antibodies is anticipated.
As charge neutralization of PF4 by Hep is important for the formation of PF4–Hep complexes, Chudasama et al. [22] observed that other positively charged proteins, such as protamine, may similarly complex with Hep. Several groups have recently examined patients who have undergone cardiopulmonary bypass for antiprotamine–Hep antibodies and found that approximately 25–30% of such patients have demonstrable antibodies peaking 10–30 days after surgery, which can activate platelets [23–25]. These antibodies do not cross-react with PF4–Hep ULC, and demonstrate a preference for protamine–Hep over protamine alone [24,26]. The clinical significance of these antibodies is not clear, although Bakchoul et al. [24] found an increase in early arterial thromboembolic events compared with controls (odds ratio 21.6; 95% confidence interval, 2.9–161). Further studies are required to better define the clinical implications of these antibodies.
How exactly the antigenic ULC is decorated with IgG antibodies in the pathologic immune complex (IC), and the connection between the IgG density and orientation along the ULC responsible for Fcg receptor clustering on platelets and monocytes (see below) remain under investigation.
Platelet activation and thrombin generation
Propagation of HIT pathophysiologically follows from platelet activation (Fig. 2). Activation results in several consequences: platelets secrete more PF4, feeding back to create more antigen; platelets aggregate via activated integrin αIIbβ3; and platelets become procoagulant with a phosphatidylserine-positive surface for coagulation reactions and shedding of highly procoagulant microparticles [27–29].
FIGURE 2.
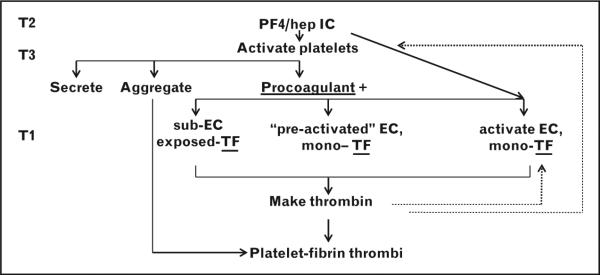
A detailed view of HIT disorder and therapy. There are three potential routes to TF-mediated thrombin generation, resulting in the final step of platelet–fibrin thrombi. On the left, the locus of current therapy directed at thrombin generation and action (T1) is compared with alternative and combination novel therapies geared to formation of the IC (T2) or inhibition of IC-mediated platelet activation (T3). Hep, heparin; HIT, heparin-induced thrombocytopenia; IC, immune complex; PF4, platelet factor 4; TF, tissue factor.
The HIT IC signals through FcγRIIa to activate platelets. We have used both rational biological candidate approaches and unbiased genomic methods to increase the understanding of FcgRIIa-mediated platelet activation. We have established that protein tyrosine kinase Syk is the major FcγRIIa signaling node [30]; CalDAG-GEF1, ADP signaling, and 12-LOX have emerged as major determinants of FcγRIIa/Syk-mediated activation [31,32]. Platelet FcgRIIa was recently shown to be a transmembrane signaling adapter for outside-in αIIbβ3 signaling, quite distinct from its role as an IC receptor [33■]. Translation of these findings to HIT could be of substantial value.
There is considerable interindividual variation in platelet activation in response to IC agonists. For example, the gold-standard assays for HIT, the serotonin release assay and the Hep-induced washed platelet activation test, rely on ‘good/highly reactive’ healthy donor platelets, the characteristics of which have been empirical [34]. Rollin et al. [35■] have used candidate-gene methods to explore variation in HIT platelet reactivity. Among their findings is identification of single-nucleotide polymorphisms (SNPs) in CD148, a membrane protein tyrosine phosphatase, that influences platelet reactivity. Scarparo et al. [36■] examined HIT candidate-gene polymorphisms in FcγRIIa, PECAM1, and FcγRIIIa. Of note, a combination of FcgRIIa and PECAM1 SNP genotypes was significantly associated with HIT thrombosis. We have examined mRNAs and miRNAs differentially expressed by 154 healthy donors in HIT-like platelet aggregation assays [37]. For the first time, we have unbiased data identifying putative determinants of IC-mediated platelet activation; functional validation studies are in progress.
Thrombin generation follows coagulation initiation, most likely by tissue factor. Any one or two of three pathways lead to tissue factor exposure (Fig. 2). With a preexisting wound, surgical or traumatic, subendothelial tissue factor may already be present. Inflammation or atherosclerosis may have ‘preactivated’ endothelium and monocytes presenting tissue factor. Also, IgG immune complexes activate monocytes and ECs [38–42]. As with platelets, antigenic complexes between PF4 and GAGs can form on the surface of monocytes [43]. In fact, the amount of antigenic complex formed on monocytes is greater than on platelets. Chemical depletion of monocytes from a mouse model of HIT demonstrated potentiation of thrombocytopenia, but significant blunting of thrombosis [43]. Further, HIT antigenic complexes, binding to FcgRI and FcγRIIa, transduce specific signals to activate monocytes. These data provide mechanisms by which HIT IC may be prothrombotic via interactions with monocytes.
Thrombin action is complex. Among its patho-physiological effects thrombin will feed back to activate platelets via PAR receptors, as well as cleave fibrinogen to fibrin. Platelet–fibrin thrombi formation is the final pathologic step (Fig. 2).
THERAPEUTICS
Current clinical practice will be summarized briefly first. In some HIT cases, thrombi form in macro-vascular beds with overt clinical symptoms, such as deep vein thrombosis/pulmonary embolism, myocardial infarction, stroke, or limb ischemia. In other cases, the thrombi are in the microvasculature, with effects that are overt clinically (e.g., adrenal thrombosis followed by hemorrhage and skin necrosis) or remain subclinical. When Hep is present and PF4–Hep complexes are formed, disease persists. Cell-surface GAGs also bind PF4, and as this complex is recognized by the anti-PF4/Hep IgG, platelets and leukocytes are still being activated. Thus, simple withdrawal of Hep does not end the ITT. A non-Hep parenteral anticoagulant has been the mainstay of treatment.
In April 2014, there was one drug approved by the Food and Drug Administration and European Medicines Agency (EMA) for the treatment of HIT, the direct thrombin inhibitor (DTI) argatroban, and one drug approved for percutaneous coronary interventions in HIT, bivalirudin (also a DTI). Lepirudin is no longer manufactured. In some nations, dana-paroid, a mix of highly and minimally sulfated GAGs, is approved. Argatroban has been noted to have multiple limitations in practice [23,44], so new treatments are of value. The Clinicaltrials.gov site listed 29 entries under ‘heparin-induced thrombocytopenia’ in April 2014. Only one new HIT therapy trial was recruiting, a study of oral FXa inhibitor rivaroxaban in HIT, NCT01598168.
Off-label use of fondaparinux has been the subject of expert opinion, clinical observation, and registries. Fondaparinux is a sulfated pentasaccharide that binds antithrombin, like unfractionated Hep and low molecular weight Heps. European Medicines Agency guidelines state, for ‘Patients with Heparin Induced Thrombocytopenia – Fondaparinux should be used with caution in patients with a history of HIT. The efficacy and safety of fondaparinux have not been formally studied in patients with HIT type II’ (www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000403/WC500027746.pdf). The American College of Chest Physicians guidelines [45], and a recent review [46], call for more studies on the risks and benefits of fondaparinux in HIT. Clinicaltrials. gov has two registries of fondaparinux use with observations of HIT (NCT01304238 and NCT01004939) [47]. The primary issues are the rare reports of HIT initiated by fondaparinux or failing to improve with fondaparinux [48,49]. In our opinion, use of fondaparinux or non-Hep anticoagulants outside of a clinical trial in HIT is a local decision, much like the selection of antimicrobials for fever and neutropenia has been local. Therapy for suspected HIT should be individualized. Treatment decisions require clinical judgment. Adverse consequences include persistent thrombocytopenia, new or extended thrombosis, or major hemorrhage. Considerations for the use of approved DTIs, off-label fondaparinux, or non-Hep anticoagulants in HIT are listed below. Considerations in the individualized treatment of suspected HIT are as follows:
-
(1)
specific patients receiving Hep, for example, cardiovascular, orthopedic, medically ill, etc.;
-
(2)
initial anticoagulation – use of unfractionated Hep, low molecular weight Hep, or fondaparinux;
-
(3)
indication – for treatment, primary prophylaxis, or secondary prophylaxis;
-
(4)
bleeding risk, including age, anatomical defects, or recent procedure;
-
(5)
likelihood of new or additional interventional procedure;
-
(6)
organ function (especially liver and kidney);
-
(7)
concurrent medications;
-
(8)
reversibility and half-life of the new anticoagulant;
-
(9)
likelihood of proven HIT among those suspected of HIT;
-
(10)
practice of proceeding to definitive HIT platelet activation assay;
-
(11)
costs and insurance reimbursement;
-
(12)
medicolegal environment for adverse consequences.
Recent clinical reports focus on estimating the proportion of suspected HIT patients that have true HIT, by use of an antibody detection method combined with one of the clinical scoring systems [50–60]. Currently, these approaches have inadequate positive predictive values to be generalizable [23]. More work is needed to make practical platelet activation assays more widely available in the general hospital clinical laboratory in a robust and timely way. Some progress in novel systems has been recently reported – multielectrode aggregometry [61], a lymphocyte cell line for HIT antibody activation [62■], determination of HIT-related platelet FcgRIIa proteolysis [63,64■], and a micro-patterned platelet activation assay [65■■].
Novel preclinical heparin-induced thrombocytopenia therapy
Every one of the approved, off-label, or clinical trial treatments is an anticoagulant geared to reduction of thrombin generation or action. Once HIT is established, thrombin is pathologic, or otherwise the approved DTIs would not work. However, continued thrombosis or major hemorrhage with DTIs remains problematic. It is unknown whether mono-therapy directed at thrombin, a late pathophysio-logical step, will be the basis for future approaches. Novel treatments targeted to earlier steps in the pathophysiology are in development (Fig. 2).
With our increased understanding of the structural nature of PF4–Hep complexes, there has been an interest in targeting these ULCs for the prevention and treatment of HIT. Therapeutics which prevent the formation of and promote ULC breakdown would represent a new approach [18,66,67].
A Hep-like molecule that has been investigated for disruption of PF4–GAG complexes is 2,3,-O-desulfated heparin (ODSH). ODSH lacks most of the anticoagulation activity of Hep, but retains many anti-inflammatory properties [68]. Joglekar et al. [66] compared PF4– Hep and PF4–ODSH complexes, and found that they both had similar light scattering and net charge properties, suggesting that both GAGs form somewhat similar complexes with PF4. However, PF4–Hep complexes formed in the presence of ODSH showed markedly reduced binding by antisera from HIT patients. It has also been demonstrated that, similar to danaparoid, ODSH decreases PF4 binding to platelets as well as activation of platelets by HIT antisera [67,68]. Taken together, these data suggest that some non-Hep GAGs may be of therapeutic value in HIT by disrupting the electrostatic interactions which drive ULC formation. Importantly, as the molar ratio of PF4 to GAG is important for the extent of complex formation and antibody recognition, empiric adjustment of dosing will be required to find the optimal dose at which the correct ratio will be achieved in vivo. A recent lucid summary of PF4 bioavailability in HIT was published, with implications for use of ODSH [34].
Another approach to disrupting ULCs is to antagonize PF4 tetramerization, which is a prerequisite for ULC formation. Our group screened over one million compounds in silico for their likelihood of binding to the dimer interface of PF4 [18]. Two of the candidate molecules inhibited tetramerization of PF4. Further, compounds PF4A01 and PF431-04 completely inhibited ULC formation and promoted the breakdown of preformed ULC. Importantly, PF4As inhibited ULC formation at all PF4 : Hep ratios tested, and both antagonists prevented cellular activation by ULC and HIT antibodies. Although potency (as measured by IC50) of these initial antagonists are in the micromolar range and we seek compounds with submicromolar potency, they represent proof of concept of this approach for the prevention and treatment of HIT.
Prevention of platelet activation by the HIT IC is another promising approach. Antiplatelet agents in the current use have not been shown to be beneficial when used alone, such as cox1 inhibitors, P2Y12 blockers, or αIIbβ3 blockers. However, we have used our mouse model of HIT to demonstrate that inhibition of Syk can safely and effectively prevent HIT [30]. We used the Portola compound PRT060318. Subsequent studies identified the Rigel compound R406 to block platelet activation by the HIT IC via FcgRIIa [69]. In more recent work, we are investigating other intracellular platelet signaling molecules for blocking FcγRIIa-mediated platelet activation, while preserving hemostasis. We are also exploring combination therapies directed at several points in the early pathophysiology, for example, with PF4 antagonists and Syk inhibitors, in vivo in the HIT mouse model.
CONCLUSION
HIT remains a challenging clinical problem. Current pathophysiology studies are focused on the origin of the antibody response, the nature of the antigenic complex and pathologic epitopes, the mechanisms of interindividual differences in platelet activation, and the roles of monocytes and endothelial cells. Progress in therapy is hampered by the challenges of inadequate positive predictive value of antibody detection and clinical scores in suspected HIT, very limited availability of practical platelet activation assays, and the paucity of new agents in human clinical trials.
KEY POINTS.
HIT is a complex and dynamic disorder, and a paradigm of the immune-mediated thrombocytopenia and thrombosis disorders.
HIT pathophysiology has an initiation phase, immunization to produce pathologic antibodies, then platelet activation by IgG–PF4–Hep immune complexes. The propagation phase feeds back to amplify the process and leads to thrombin generation culminating in platelet and fibrin thrombi.
HIT therapy needs improvement that could come from better diagnostics in the form of practical platelet activation assays, and from combinations of rational therapeutics targeting early and late steps in pathophysiology.
Acknowledgements
The authors wish to thank their laboratory and clinical teams at Thomas Jefferson University and Hospitals and at the University of Pennsylvania. Valuable insights have been provided by the co-investigators Mortimer Poncz, Lubica Rauova, Douglas Cines, Gowthami Arepally, and Adam Cuker (support from NIH P01HL110860 to S.McK., B.S.S.), Wolfgang Bergmeier (R01HL106009 to S.McK.), Michael Holinstat (R01HL114405 to S.McK.), and Paul Bray and Leonard Edelstein (Cardeza Foundation for Hematological Research). S.McK. received research support from Portola Pharmaceuticals.
Footnotes
Conflicts of interest
The authors have no other conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
■ of special interest
■■ of outstanding interest
- 1.Potschke C, Selleng S, Broker BM, Greinacher A. Heparin-induced thrombocytopenia: further evidence for a unique immune response. Blood. 2012;120:4238–4245. doi: 10.1182/blood-2012-04-419424. [DOI] [PubMed] [Google Scholar]
- 2.Suvarna S, Espinasse B, Qi R, et al. Determinants of PF4/heparin immunogenicity. Blood. 2007;110:4253–4260. doi: 10.1182/blood-2007-08-105098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suvarna S, Qi R, Arepally GM. Optimization of a murine immunization model for study of PF4/heparin antibodies. J Thromb Haemost. 2009;7:857–864. doi: 10.1111/j.1538-7836.2009.03330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greinacher A, Holtfreter B, Krauel K, et al. Association of natural antiplatelet factor 4/heparin antibodies with periodontal disease. Blood. 2011;118:1395–1401. doi: 10.1182/blood-2011-03-342857. [DOI] [PubMed] [Google Scholar]
- 5.Krauel K, Potschke C, Weber C, et al. Platelet factor 4 binds to bacteria, [corrected] inducing antibodies cross-reacting with the major antigen in heparin-induced thrombocytopenia. Blood. 2011;117:1370–1378. doi: 10.1182/blood-2010-08-301424. [DOI] [PubMed] [Google Scholar]
- 6.Krauel K, Weber C, Brandt S, et al. Platelet factor 4 binding to lipid A of Gram-negative bacteria exposes PF4/heparin-like epitopes. Blood. 2012;120:3345–3352. doi: 10.1182/blood-2012-06-434985. [DOI] [PubMed] [Google Scholar]
- 7.Arman M, Krauel K, Tilley DO, et al. Amplification of bacteria-induced platelet activation is triggered by FcgammaRIIA, integrin alphaIIbbeta3 and platelet factor 4. Blood. 2014;123:3155–3166. doi: 10.1182/blood-2013-11-540526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8■.Pongas G, Dasgupta SK, Thiagarajan P. Antiplatelet factor 4/heparin anti-bodies in patients with Gram negative bacteremia. Thromb Res. 2013;132:217–220. doi: 10.1016/j.thromres.2013.06.013. [This study confirms and extends the original observations of Greinacher and colleagues. PF4 bound to LPS is demonstrated to generate HIT-like antibodies.] [DOI] [PubMed] [Google Scholar]
- 9■.Jaax ME, Krauel K, Marschall T, et al. Complex formation with nucleic acids and aptamers alters the antigenic properties of platelet factor 4. Blood. 2013;122:272–281. doi: 10.1182/blood-2013-01-478966. [The discovery, pathologic role, and therapeutic implications of PF4 binding to nucleic acids are presented.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chong BH, Chong JJ. HIT: nucleic acid masquerading as heparin. Blood. 2013;122:156–158. doi: 10.1182/blood-2013-05-504126. [DOI] [PubMed] [Google Scholar]
- 11■.Brandt S, Krauel K, Gottschalk KE, et al. Characterisation of the conformational changes in platelet factor 4 induced by polyanions: towards in vitro prediction of antigenicity. Thromb Haemost. 2014;112 doi: 10.1160/TH13-08-0634. http://dx.doi.org/10.1160/TH13-08-0634. [This study examines the polyanion charge and length as contributing factors in the HIT antigen.] [DOI] [PubMed] [Google Scholar]
- 12■■.Zheng Y, Wang AW, Yu M, et al. B-cell tolerance regulates production of antibodies causing heparin-induced thrombocytopenia. Blood. 2014;123:931–934. doi: 10.1182/blood-2013-11-540781. [The investigators bring a new perspective to the immunizing events in HIT, with a view of B-cell tolerance breakdown.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13■.Zheng Y, Yu M, Podd A, et al. Critical role for mouse marginal zone B cells in PF4/heparin antibody production. Blood. 2013;121:3484–3492. doi: 10.1182/blood-2013-01-477091. [This study explores the unique HIT antibody response and presents evidence that marginal zone B-cells, rather than germinal center B-cells, may underlie the atypical response.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li ZQ, Liu W, Park KS, et al. Defining a second epitope for heparin-induced thrombocytopenia/thrombosis antibodies using KKO, a murine HIT-like monoclonal antibody. Blood. 2002;99:1230–1236. doi: 10.1182/blood.v99.4.1230. [DOI] [PubMed] [Google Scholar]
- 15.Ziporen L, Li ZQ, Park KS, et al. Defining an antigenic epitope on platelet factor 4 associated with heparin-induced thrombocytopenia. Blood. 1998;92:3250–3259. [PubMed] [Google Scholar]
- 16.Greinacher A, Gopinadhan M, Gunther JU, et al. Close approximation of two platelet factor 4 tetramers by charge neutralization forms the antigens recognized by HIT antibodies. Arterioscler Thromb Vasc Biol. 2006;26:2386–2393. doi: 10.1161/01.ATV.0000238350.89477.88. [DOI] [PubMed] [Google Scholar]
- 17.Rauova L, Poncz M, McKenzie SE, et al. Ultralarge complexes of PF4 and heparin are central to the pathogenesis of heparin-induced thrombocytopenia. Blood. 2005;105:131–138. doi: 10.1182/blood-2004-04-1544. [DOI] [PubMed] [Google Scholar]
- 18.Sachais BS, Litvinov RI, Yarovoi SV, et al. Dynamic antibody-binding properties in the pathogenesis of HIT. Blood. 2012;120:1137–1142. doi: 10.1182/blood-2012-01-407262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19■.Litvinov RI, Yarovoi SV, Rauova L, et al. Distinct specificity and single-molecule kinetics characterize the interaction of pathogenic and nonpathogenic antibodies against platelet factor 4-heparin complexes with platelet factor 4. J Biol Chem. 2013;288:33060–33070. doi: 10.1074/jbc.M113.481598. [Optical tweezer techniques are used to provide insights into the dynamics of PF4 clustering and the nature of epitopes of pathogenic HIT antibodies.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Block S, Greinacher A, Helm CA, Delcea M. Characterization of bonds formed between platelet factor 4 and negatively charged drugs using single molecule force spectroscopy. Soft Matter. 2014;10:2775–2784. doi: 10.1039/c3sm52609g. [DOI] [PubMed] [Google Scholar]
- 21.Asada R, Wanaka K, Walenga J, et al. Murine monoclonal antibody to platelet factor 4/heparin complexes as a potential reference standard for platelet activation assays in heparin-induced thrombocytopenia. Clin Appl Thromb Hemost. 2013;19:37–41. doi: 10.1177/1076029612453763. [DOI] [PubMed] [Google Scholar]
- 22.Chudasama SL, Espinasse B, Hwang F, et al. Heparin modifies the immunogenicity of positively charged proteins. Blood. 2010;116:6046–6053. doi: 10.1182/blood-2010-06-292938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee GM, Arepally GM. Heparin-induced thrombocytopenia. Hematology Am Soc Hematol Educ Program. 2013;2013:668–674. doi: 10.1182/asheducation-2013.1.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bakchoul T, Zollner H, Amiral J, et al. Antiprotamine-heparin antibodies: incidence, clinical relevance, and pathogenesis. Blood. 2013;121:2821–2827. doi: 10.1182/blood-2012-10-460691. [DOI] [PubMed] [Google Scholar]
- 25.Pouplard C, Leroux D, Rollin J, et al. Incidence of antibodies to protamine sulfate/heparin complexes in cardiac surgery patients and impact on platelet activation and clinical outcome. Thromb Haemost. 2013;109:1141–1147. doi: 10.1160/TH12-11-0844. [DOI] [PubMed] [Google Scholar]
- 26.Lee GM, Welsby IJ, Phillips-Bute B, et al. High incidence of antibodies to protamine and protamine/heparin complexes in patients undergoing cardio-pulmonary bypass. Blood. 2013;121:2828–2835. doi: 10.1182/blood-2012-11-469130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Warkentin TE, Hayward CP, Boshkov LK, et al. Sera from patients with heparin-induced thrombocytopenia generate platelet-derived microparticles with procoagulant activity: an explanation for the thrombotic complications of heparin-induced thrombocytopenia. Blood. 1994;84:3691–3699. [PubMed] [Google Scholar]
- 28.Hughes M, Hayward CP, Warkentin TE, et al. Morphological analysis of microparticle generation in heparin-induced thrombocytopenia. Blood. 2000;96:188–194. [PubMed] [Google Scholar]
- 29.Andre P, McKenzie SE, Bergmeier W. The parallel signaling pathways of phosphatidylserine (PS) exposure downstream of platelet FcgRIIa. Blood. 2013;122:3514. [Google Scholar]
- 30.Reilly MP, Sinha U, Andre P, et al. PRT-060318, a novel Syk inhibitor, prevents heparin-induced thrombocytopenia and thrombosis in a transgenic mouse model. Blood. 2011;117:2241–2246. doi: 10.1182/blood-2010-03-274969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stolla M, Stefanini L, Andre P, et al. CalDAG-GEFI deficiency protects mice in a novel model of Fcgamma RIIA-mediated thrombosis and thrombocytopenia. Blood. 2011;118:1113–1120. doi: 10.1182/blood-2011-03-342352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yeung J, Apopa PL, Vesci J, et al. 12-Lipoxygenase activity plays an important role in PAR4 and GPVI-mediated platelet reactivity. Thromb Haemost. 2013;110:569–581. doi: 10.1160/TH13-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33■.Zhi H, Rauova L, Hayes V, et al. Cooperative integrin/ITAM signaling in platelets enhances thrombus formation in vitro and in vivo. Blood. 2013;121:1858–1867. doi: 10.1182/blood-2012-07-443325. [The role of platelet FcgRIIa as a transmembrane adapter for outside-in integrin signaling is documented. The implications for HIT thrombus formation are under active investigation.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prechel MM, Walenga JM. Emphasis on the role of PF4 in the incidence, pathophysiology and treatment of heparin induced thrombocytopenia. Thromb J. 2013;11:7. doi: 10.1186/1477-9560-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35■.Rollin J, Pouplard C, Gratacap MP, et al. Polymorphisms of protein tyrosine phosphatase CD148 influence FcgammaRIIA-dependent platelet activation and the risk of heparin-induced thrombocytopenia. Blood. 2012;120:1309–1316. doi: 10.1182/blood-2012-04-424044. [This study is one of several by this group which has productively employed candidate-gene approaches with sizable well phenotyped patient cohorts to the biology of HIT. In this study, the role of polymorphisms in platelet protein tyrosine phosphatase CD148 is shown.] [DOI] [PubMed] [Google Scholar]
- 36■.Scarparo P, Lombardi AM, Duner E, et al. Heparin-induced thrombocytopenia: & the role of platelets genetic polymorphisms. Platelets. 2013;24:362–368. doi: 10.3109/09537104.2012.701026. [The value of a combination of candidate SNPs, namely in FcgRIIa and PECAM1, to HIT thrombosis is shown.] [DOI] [PubMed] [Google Scholar]
- 37.Abraham S, Andre P, Zhou Y, et al. Differential expression of microRNAs accompanies differential reactivity via platelet FcgammaRIIa in humans and transgenic mice. Blood. 2012;120:2165a. [Google Scholar]
- 38.Pouplard C, Iochmann S, Renard B, et al. Induction of monocyte tissue factor expression by antibodies to heparin–platelet factor 4 complexes developed in heparin-induced thrombocytopenia. Blood. 2001;97:3300–3302. doi: 10.1182/blood.v97.10.3300. [DOI] [PubMed] [Google Scholar]
- 39.Arepally GM, Mayer IM. Antibodies from patients with heparin-induced thrombocytopenia stimulate monocytic cells to express tissue factor and secrete interleukin-8. Blood. 2001;98:1252–1254. doi: 10.1182/blood.v98.4.1252. [DOI] [PubMed] [Google Scholar]
- 40.Kasthuri RS, Glover SL, Jonas W, et al. PF4/heparin-antibody complex induces monocyte tissue factor expression and release of tissue factor positive micro-particles by activation of FcgammaRI. Blood. 2012;119:5285–5293. doi: 10.1182/blood-2011-06-359430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tutwiler V, Ahn HS, Cines DB, et al. Microfluidic and flow cytometric studies support a central role of monocytes and coat platelets in the prothrombotic state in heparin-induced thrombocytopenia (HIT). Blood. 2011;118:531a. [Google Scholar]
- 42.Tutwiler V, Ahn HS, Fuentes R, et al. Fibrin generation in heparin-induced thrombocytopenia (HIT): pathomechanistic background for novel therapy and prophylaxis. Blood. 2012;120:635a. [Google Scholar]
- 43.Rauova L, Hirsch JD, Greene TK, et al. Monocyte-bound PF4 in the pathogenesis of heparin-induced thrombocytopenia. Blood. 2010;116:5021–5031. doi: 10.1182/blood-2010-03-276964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coventry DA, Webster NR. Heparin-induced thrombocytopenia and the health economic analysis of argatroban. Br J Anaesth. 2014;112:964–967. doi: 10.1093/bja/aeu011. [DOI] [PubMed] [Google Scholar]
- 45.Linkins LA, Dans AL, Moores LK, et al. American College of Chest Physicians. Treatment and prevention of heparin-induced thrombocytopenia: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141:e495S–e530S. doi: 10.1378/chest.11-2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kelton JG, Arnold DM, Bates SM. Nonheparin anticoagulants for heparin-induced thrombocytopenia. N Engl J Med. 2013;368:737–744. doi: 10.1056/NEJMct1206642. [DOI] [PubMed] [Google Scholar]
- 47.Schindewolf M, Steindl J, Beyer-Westendorf J, et al. Frequent off-label use of fondaparinux in patients with suspected acute heparin-induced thrombocytopenia (HIT) – findings from the GerHIT Multicentre Registry Study. Thromb Res. 2014 doi: 10.1016/j.thromres.2014.03.029. http://dx.doi.org/10.1016/j.thromres.2014.03.029. [DOI] [PubMed]
- 48.Ratuapli SK, Bobba B, Zafar H. Heparin-induced thrombocytopenia in a patient treated with fondaparinux. Clin Adv Hematol Oncol. 2010;8:61–65. [PubMed] [Google Scholar]
- 49.Bhatt VR, Aryal MR, Shrestha R, Armitage JO. Fondaparinux-associated heparin-induced thrombocytopenia. Eur J Haematol. 2013;91:437–441. doi: 10.1111/ejh.12179. [DOI] [PubMed] [Google Scholar]
- 50.Tanhehco YC, Rux AH, Sachais BS. Low-density lipoprotein apheresis reduces platelet factor 4 on the surface of platelets: a possible protective mechanism against heparin-induced thrombocytopenia and thrombosis. Transfusion. 2011;51:1022–1029. doi: 10.1111/j.1537-2995.2010.02911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tanhehco YC, Cuker A, Rudnick M, Sachais BS. Investigation of a potential protective mechanism against heparin-induced thrombocytopenia in patients on chronic intermittent hemodialysis. Thromb Res. 2013;131:244–248. doi: 10.1016/j.thromres.2012.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pierce W, Mazur J, Greenberg C, et al. Evaluation of heparin-induced thrombocytopenia (HIT) laboratory testing and the 4Ts scoring system in the intensive care unit. Ann Clin Lab Sci. 2013;43:429–435. [PubMed] [Google Scholar]
- 53.Cegarra-Sanmartin V, Gonzalez-Rodriguez R, Paniagua-Iglesias P, et al. Fondaparinux as a safe alternative for managing heparin-induced thrombocytopenia in postoperative cardiac surgery patients. J Cardiothorac Vasc Anesth. 2014 doi: 10.1053/j.jvca.2013.09.008. http://dx.doi.org/10.1053/j.jvca.2013.09.008. [DOI] [PubMed]
- 54.Beiras-Fernandez A, Kanzler I, Michel S, et al. Platelet factor 4-positive thrombi adhering to the ventricles of a ventricular assist device in patients with heparin-induced thrombocytopenia type II. Transplant Proc. 2013;45:2013–2016. doi: 10.1016/j.transproceed.2013.01.045. [DOI] [PubMed] [Google Scholar]
- 55.Matsuo T, Wanaka K, Walenga JM. Evaluation of circuit and AV fistula clotting and detection of anti-PF4/heparin complex antibodies in hemodialysis patients suspected of having heparin-induced thrombocytopenia. Clin Appl Thromb Hemost. 2013;19:73–78. doi: 10.1177/1076029612436676. [DOI] [PubMed] [Google Scholar]
- 56.Zhao D, Sun X, Yao L, et al. The clinical significance and risk factors of antiplatelet factor 4/heparin antibody on maintenance hemo-dialysis patients: a two-year prospective follow-up. PLoS One. 2013;8:e62239. doi: 10.1371/journal.pone.0062239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Althaus K, Hron G, Strobel U, et al. Evaluation of automated immunoassays in the diagnosis of heparin induced thrombocytopenia. Thromb Res. 2013;131:e85–e90. doi: 10.1016/j.thromres.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 58.Raschke RA, Curry SC, Warkentin TE, Gerkin RD. Improving clinical interpretation of the antiplatelet factor 4/heparin enzyme-linked immunosorbent assay for the diagnosis of heparin-induced thrombocytopenia through the use of receiver operating characteristic analysis, stratum-specific likelihood ratios, and Bayes theorem. Chest. 2013;144:1269–1275. doi: 10.1378/chest.12-2712. [DOI] [PubMed] [Google Scholar]
- 59.Garritsen HS, Probst-Kepper M, Legath N, et al. High sensitivity and specificity of a new functional flow cytometry assay for clinically significant heparin-induced thrombocytopenia antibodies. Int J Lab Hematol. 2014;36:135–143. doi: 10.1111/ijlh.12136. [DOI] [PubMed] [Google Scholar]
- 60.Solano C, Mutsando H, Self M, et al. Using HitAlert flow cytometry to detect heparin-induced thrombocytopenia antibodies in a tertiary care hospital. Blood Coagul Fibrinolysis. 2013;24:365–370. doi: 10.1097/MBC.0b013e32835cc17e. [DOI] [PubMed] [Google Scholar]
- 61.Galea V, Khaterchi A, Robert F, et al. Heparin-induced multiple electrode aggregometry is a promising and useful functional tool for heparin-induced thrombocytopenia diagnosis: confirmation in a prospective study. Platelets. 2013;24:441–447. doi: 10.3109/09537104.2012.724736. [DOI] [PubMed] [Google Scholar]
- 62■.Cuker A, Rux AH, Hinds JL, et al. Novel diagnostic assays for heparin-induced thrombocytopenia. Blood. 2013;121:3727–3732. doi: 10.1182/blood-2013-01-479576. [This study uses a lymphocyte cell line transfected with FcgRIIa to explore a generalizable activation assay for HIT.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gardiner EE, Karunakaran D, Arthur JF, et al. Dual ITAM-mediated proteolytic pathways for irreversible inactivation of platelet receptors: de-ITAM-izing FcgammaRIIa. Blood. 2008;111:165–174. doi: 10.1182/blood-2007-04-086983. [DOI] [PubMed] [Google Scholar]
- 64■.Nazi I, Arnold DM, Smith JW, et al. FcgammaRIIa proteolysis as a diagnostic & biomarker for heparin-induced thrombocytopenia. J Thromb Haemost. 2013;11:1146–1153. doi: 10.1111/jth.12208. [This study translates the findings of Gardiner and colleagues on activation-induced platelet FcgRIIa proteolysis into a candidate diagnostic for HIT.] [DOI] [PubMed] [Google Scholar]
- 65■■.Medvedev N, Palankar R, Krauel K, et al. Micropatterned array to assess the interaction of single platelets with platelet factor 4–heparin–IgG complexes. Thromb Haemost. 2014;111:862–872. doi: 10.1160/TH13-09-0752. [This study uses nanotechnology to explore HIT-related platelet activation. It is the underpinning of a candidate diagnostic assay.] [DOI] [PubMed] [Google Scholar]
- 66.Joglekar MV, Quintana Diez PM, Marcus S, et al. Disruption of PF4/H multimolecular complex formation with a minimally anticoagulant heparin (ODSH). Thromb Haemost. 2012;107:717–725. doi: 10.1160/TH11-11-0795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Krauel K, Hackbarth C, Furll B, Greinacher A. Heparin-induced thrombocytopenia: in vitro studies on the interaction of dabigatran, rivaroxaban, and low-sulfated heparin, with platelet factor 4 and anti-PF4/heparin antibodies. Blood. 2012;119:1248–1255. doi: 10.1182/blood-2011-05-353391. [DOI] [PubMed] [Google Scholar]
- 68.Rao NV, Argyle B, Xu X, et al. Low anticoagulant heparin targets multiple sites of inflammation, suppresses heparin-induced thrombocytopenia, and inhibits interaction of RAGE with its ligands. Am J Physiol Cell Physiol. 2010;299:C97–C110. doi: 10.1152/ajpcell.00009.2010. [DOI] [PubMed] [Google Scholar]
- 69.Lhermusier T, van Rottem J, Garcia C, et al. The Syk-kinase inhibitor R406 impairs platelet activation and monocyte tissue factor expression triggered by heparin-PF4 complex directed antibodies. J Thromb Haemost. 2011;9:2067–2076. doi: 10.1111/j.1538-7836.2011.04470.x. [DOI] [PubMed] [Google Scholar]